 Zoe Ingold
Zoe Ingold Gideon Grogan
Gideon Grogan Benjamin R. Lichman
Benjamin R. Lichman- 1York Structural Biology Laboratory, Department of Chemistry, University of York, York, United Kingdom
- 2Centre for Novel Agricultural Products, Department of Biology, University of York, York, United Kingdom
Deoxypodophyllotoxin synthase (DPS) is a 2-oxoglutarate (2-OG) dependent non-heme iron (II) dioxygenase that catalyzes the stereoselective ring-closing carbon-carbon bond formation of deoxypodophyllotoxin from the aryllignan (−)-yatein. Deoxypodophyllotoxin is a precursor of topoisomerase II inhibitors, which are on the World Health Organization’s list of essential medicines. Previous work has shown that DPS can accept a range of substrates, indicating it has potential in biocatalytic processes for the formation of diverse polycyclic aryllignans. Recent X-ray structures of the enzyme reveal possible roles for amino acid side chains in substrate recognition and mechanism, although a mutational analysis of DPS was not performed. Here, we present a structure of DPS at an improved resolution of 1.41 Å, in complex with the buffer molecule, Tris, coordinated to the active site iron atom. The structure has informed a mutational analysis of DPS, which suggests a role for a D224-K187 salt bridge in maintaining substrate interactions and a catalytic role for H165, perhaps as the base for the proton abstraction at the final rearomatization step. This work improves our understanding of specific residues’ contributions to the DPS mechanism and can inform future engineering of the enzyme mechanism and substrate scope for the development of a versatile biocatalyst.
1 Introduction
2-Oxoglutarate (2-OG) dependent dioxygenases (2-ODDs) are a large family of non-heme iron (II) dependent enzymes that catalyze a wide range of reactions across both primary and secondary metabolism (Islam et al., 2018). 2-ODDs are major contributors to the chemical diversity of plant specialized metabolism, typically catalyzing hydroxylation reactions but also a variety of oxidative reactions including demethylations, halogenations and bond migrations (Hagel and Fachini, 2019; Mitchell and Weng, 2019). Notable examples include those involved in the biosynthesis of huperzine A, where three 2-ODDs catalyze desaturation, ring cleavage and isomerisation; and in the biosynthesis of acutumine, which involves dechloroacutumine halogenase, the first plant-derived halogenase to be characterized (Kim et al., 2020; Nett et al., 2021).
2-ODDs are widely used in industrial biocatalysis, with perhaps the best known of these being the free proline hydroxylases, which are used in the industrial scale hydroxylation of this amino acid (Peters and Buller, 2019). There is an increasing interest in expanding the biocatalytic range of this enzyme superfamily and deoxypodophyllotoxin synthase (DPS) is an ideal candidate to explore this. First reported by Sattely and co-workers, DPS is responsible for the key C-C bond forming step in the biosynthetic route to podophyllotoxin; a lignin derived compound that acts as the synthetic precursor to drugs such as etoposide and teniposide, both of which are inhibitors of topoisomerase II and are included on the World Health Organisation (WHO)’s list of essential medicines (Lau and Sattely, 2015; Decembrino et al., 2021). The enzyme catalyzes the stereoselective ring closure of the precursor (−)-yatein 1, to give the tetracyclic deoxypodophyllotoxin 2 (Figure 1) (Lau and Sattely, 2015).
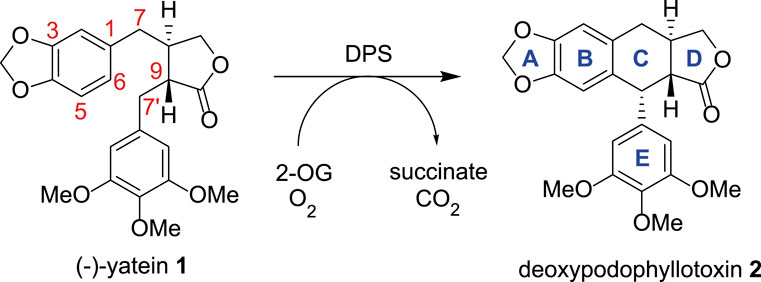
FIGURE 1. The conversion of (−)-yatein 1 to deoxypodophyllotoxin 2 catalyzed by DPS, with oxygen and 2-oxoglutarate acting as co-substrates.
Currently, podophyllotoxin is isolated directly from Mayapple (Podophyllum hexandrum), which is slow growing and has a limited environmental range, making it less than ideal as a source for the large-scale synthesis of etoposide (Decembrino et al., 2021). Elucidation of the podophyllotoxin biosynthetic pathway has led to the development of synthetic biology platforms for its production through heterologous expression of biosynthetic genes including DPS. These platforms may eventually provide an alternative source of podophyllotoxin (Eljounaidi and Lichman, 2020; Decembrino et al., 2021). Sattely and co-workers were able to combine the expression of deoxypodophyllotoxin pathway genes from Mayapple with metabolic flux reprogramming in the plant chassis Nicotiana benthamiana, leading to the accumulation of 35 mg g-1 (−)-deoxypodophyllotoxin and 1 mg g-1 etoposide aglycone, levels eight times greater than those found in the native plant (Schultz et al., 2019; Kim et al., 2022). In addition, Urlacher and co-workers have demonstrated production of deoxypodophyllotoxin from matairesinol in E. coli through a five-step multi-enzyme cascade, starting with a substrate concentration of 78 mg mL-1 and achieving a 98% yield of the final product (Decembrino et al., 2020; Decembrino et al., 2021).
The one-step oxidative, stereoselective ring closing reaction catalyzed by DPS has considerable synthetic utility (Li et al., 2019). Notably, equivalent chemical routes typically form the non-natural isodeoxypodophyllotoxin diastereomers, which have different bioactivities (Alizadeh et al., 2017). There is therefore considerable interest in the potential of DPS as an in vitro biocatalyst for the stereoselective formation of diverse aryllignans. Furthermore, an understanding of the DPS mechanism will enable protein engineering to expand the scope of the reaction and explore the surrounding chemical space.
Using DPS expressed in E. coli, Chang and co-workers first showed that DPS would tolerate substrate analogs without the methoxy groups on the southern aromatic ring (Chang et al., 2019). They also demonstrated that hydroxylated products at the benzylic 7′ position could result if the substrate were deuterated at this position, implicating this carbon as the site for hydrogen atom abstraction in the mechanism (Figure 2A) (Chang et al., 2019). Fuchs, Kroutil and co-workers further investigated the product distribution of DPS catalysis with a series of substrates. In addition to performing a gram-scale synthesis of 2 from racemic 1 using DPS, they showed that different products were formed when the substrate had a hydroxyl group on the C-7 atom with the opposite configuration. Hence, in two kinetic resolution processes, rac-3 was transformed to the C7′-hydroxylated product 4 (Figure 2B), but rac-5, with the etoposide configuration for the hydroxyl, was cyclized to product 6 (Figure 2C; Lazzarotto et al., 2019). They also showed that the three methoxy groups of ring E were required for good activity. C7′-hydroxylation activity was observed with various changes to the benzodioxole moiety (rings A and B), including substitution by phenyl, p-chlorophenyl or even naphthyl groups. However, a cyclic product was only observed with an anisyl group, with the methoxy moiety para to the new cyclic C-C bond. Renata and co-workers subsequently showed that DPS could be used for preparative scale transformation of yatein analogs in yields of 40%–80%, with extended scope for variety in ring E, with substitution to a benzodioxole giving the product (−)-polygamain in 36% yield (Li et al., 2019).
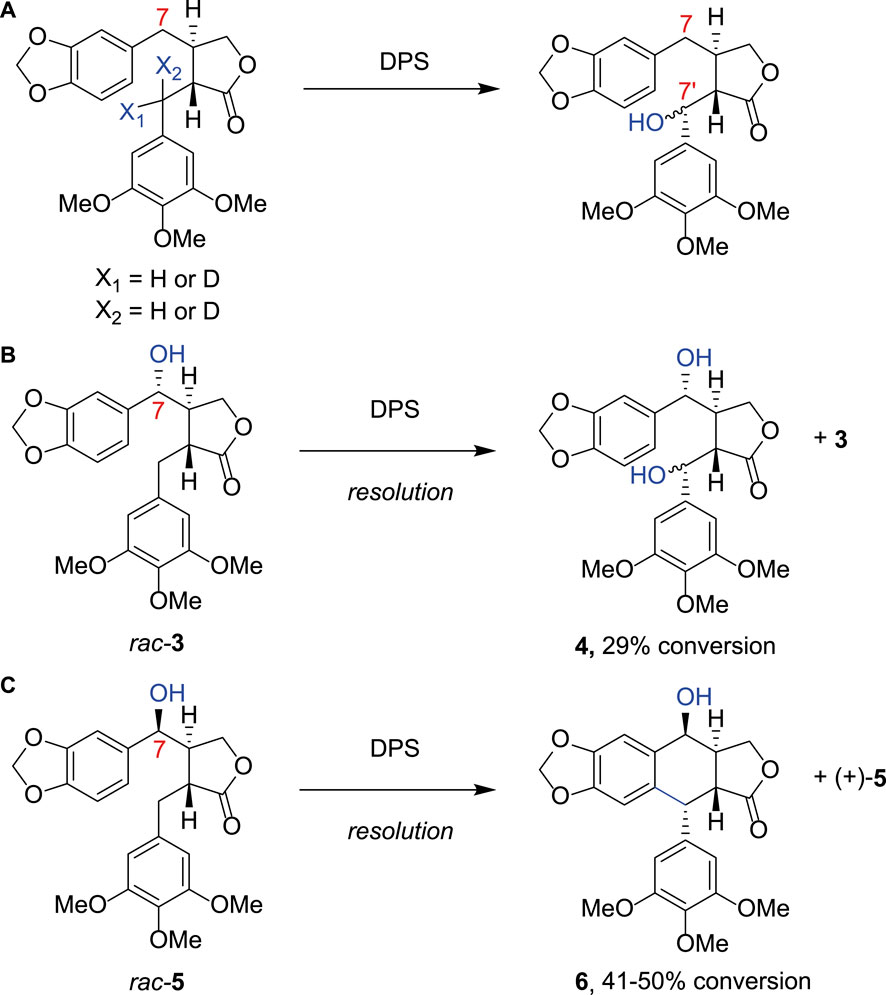
FIGURE 2. Alternative DPS substrates. (A) Using substrate analogues deuterated at either position X1 or X2 Chang et al. produced a product hydroxylated at the C7′ position, indicating that this is the most likely site of hydrogen atom abstraction from (−)-yatein by DPS; (B) Fuchs et al. further investigated the substrate scope of DPS demonstrating that when a hydroxyl group was present at the C7 atom rac- 3 was transformed to the C7‘-hydroxylated product 4; (C) rac-5, with the etoposide configuration for the hydroxyl, was cyclized to product 6.
Both the mechanism and substrate scope of DPS would be better explained with structural information, which would also facilitate subsequent engineering, and so we set out to obtain an X-ray crystal structure of DPS. During this period, Chang and co-workers presented crystal structures of DPS in complex with 2-oxoglutarate (PDB 7E37) and racemic yatein and succinate (7E38) at resolutions of 2.09 and 2.05 Å respectively (Tang et al., 2022). DPS displayed the canonical double stranded α-helix fold (DSBH) adopted by members of the superfamily, and in the active site, the expected 2-His-1-carboxylate facial triad for iron coordination was observed, with contributions from the side chains of H184, D186 and H239.
The structure in complex with yatein, in addition to experiments performed with substrate analogs, supported the hypothesis that ring cyclisation occurs via hydrogen abstraction from the C7′-benzylic position of the southern substituent, followed by electron transfer to form a carbocation at this position. The resulting ring closure for forming ring C is facilitated by the substrate adopting a U-shaped conformation within the active site that positions the C6 atom of the benzodioxole (ring B) close to the carbocation. The aromaticity of the benzodioxole ring is thought to be restored via proton abstraction from C6 by an as yet unidentified basic residue within the active site (Tang et al., 2022).
Despite the availability of the structure of DPS, a mutational analysis that investigates the contribution of identified amino acid side chains to binding and catalysis has not been presented. In this work, we present an additional structure of DPS, refined to an improved resolution of 1.41 Å, and in complex with the buffer molecule Tris (hydroxymethyl)aminomethane (Tris). The structures together have permitted the identification of a number of active site residues for mutation. In addition to effects on enzyme stability and activity, our results suggest that H165 is a plausible candidate for the base that catalyzes the final step of ring cyclisation in DPS.
2 Materials and methods
The enzyme substrate (−)-yatein was purchased from Sigma-Aldrich (≥98.0% pure based on HPLC). Deoxypodophyllotoxin was purchased from Toronto Research Chemicals (>95% pure based on HPLC). Unless otherwise stated all other chemicals were bought from either Fisher Scientific, Apollo Scientific or Merck (Sigma Aldrich) at standard purity.
2.1 Expression of DPS
The DPS gene (Supplementary Section S1.1) was purchased from GenScript in a pET-21a (+) vector, inserted between NdeI and XhoI restriction sites, and provided as a freeze-dried plasmid. This was rehydrated and used to transform BL21 (DE3) (New England BioLabs) cells via heat shock at 42°C for 45 s, then 500 μL of Luria Bertani (LB) media were added and the suspension was incubated at 37°C for 1 h with shaking at 180 r.p.m. 250 μL of this culture was then added to LB agar plates containing 100 μg mL-1 ampicillin and these were incubated at 37°C overnight. Single colonies were selected and inoculated into 10 mL LB broth containing 100 μg mL-1 ampicillin. These cultures were incubated for 18 h at 180 rpm at 37°C with shaking and then transferred to 2.5 L flasks containing 1 L LB broth with 100 μg mL-1 ampicillin sealed with foam bungs. These cultures were incubated at 37°C until they reached an absorbance value of 0.8–1.0 at 600 nm, upon which they were induced with the addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). Following induction, the cultures were left to grow at 16°C for 18 h with shaking at 180 r.p.m. Cells were then harvested using a Lynx6000 centrifuge (Thermo Scientific) at 5,422 × g for 30 min. The pellets were resuspended in tris buffer C (200 mM Tris-HCl, 200 mM NaCl, pH 7.4) or MOPS buffer C (200 mM MOPS, 100 mM NaCl, pH 7.4), depending upon the experiment, with one cOmplete Mini EDTA-Free Protease Inhibitor tablet (Roche) per 1 L of culture. Resuspended cells were lysed using a cell disruptor homogenizer (Constant Systems Ltd.) at a pressure of 26 kPsi. Lysed cells were centrifuged at 28,928 × g, 4°C for 1 h and the supernatant retained for enzyme purification. Tris buffers were used for crystallization experiments. Upon solving the structure of DPS the buffers were switched to MOPS buffers so that tris in the active site did not potentially interfere with the activity or stability of the enzyme.
2.2 Purification of DPS
The cell supernatant was filtered through a 0.45 μm PES filter before being loaded onto a 5 mL FF HisTrap column (GE Healthcare) pre-loaded with nickel sulfate. DPS was eluted using an imidazole gradient from Buffer A (100 mM Tris-HCl, 20 mM imidazole, 200 mM NaCl, pH 7.4) to Buffer B (200 mM Tris-HCl, 300 mM imidazole, 100 mM NaCl, pH 7.4). Fractions containing DPS, as determined by SDS-PAGE, were combined and concentrated to a volume of 2 mL using 10,000 Da MWCO PES centrifugal concentrators. This was loaded onto a Superdex gel filtration S75 16/60 column and eluted using Buffer C (200 mM Tris-HCl, 200 mM NaCl, pH 7.4, Supplementary Section S1.2). Following elution, fractions were analysed using SDS-PAGE (Supplementary Section S1.2) and those containing DPS were combined. The concentration was determined using the A280 value from an Implen Nanophotometer NP80 and an extinction co-efficient of 35,120 M-1 cm-1 (ATT Bioquest Protein Concentration Calculator) and adjusted as required.
2.3 Crystallization of DPS
Screening of crystallization conditions was performed using commercially available INDEX (Hampton Research), PACT premier and CSSI/II (Molecular Dimensions) screens in 96-well sitting drop trays. Crystals of the DPS-Tris complex were grown using WT-DPS concentrated to 20 mg mL-1 in mother liquor composed of 0.1 M Bis-Tris, 28% w/v PEG monomethyl ether 2000 at a pH of 6.5. The mother liquor to protein solution ratio was 1:1. Crystals were harvested directly into liquid nitrogen with nylon CryoLoops™ (Hampton Research), using mother liquor without any further cryoprotectant.
2.4 X-ray data collection and refinement
The dataset described in this manuscript was collected at the Diamond Light Source, Didcot, Oxfordshire, United Kingdom on beamline I04. Data were processed and integrated using XDS (Kabsch, 2010) and scaled using SCALA (Evans, 2006) included in the Xia2 (Winter, 2010) processing system. Data collection statistics are provided in Table 1. The crystal of DPS in complex with tris(hydroxymethylaminomethane) (henceforth Tris) was obtained in space group P21, with one molecule in the asymmetric unit. The solvent content in the crystals was 42.6%. The structure of DPS was solved by molecular replacement using MOLREP (Vagin and Teplyakov, 1997) using the monomer of auxin dioxygenase DAO (6KUN) as the model. The structure was built and refined using iterative cycles in Coot (Emsley and Cowtan, 2004) and REFMAC (Murshudov et al., 1997). Following building and refinement of the protein and water molecules in each structure complex, residual density was observed in the omit maps at the active site, which was modelled and refined as Tris plus one additional water molecule. Refinement statistics for the structure are presented in Table 1. The coordinates and structure factors for DPS-Tris have been deposited in the PDB with accession code 8CI9.
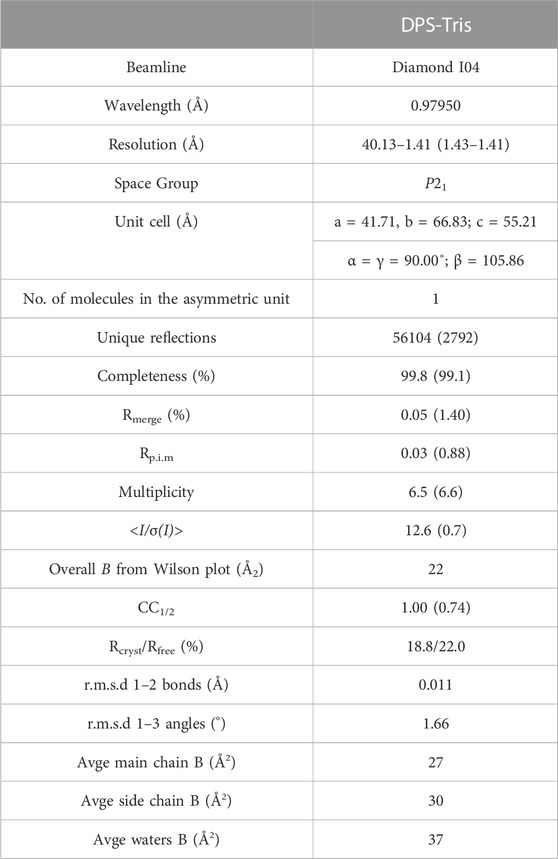
TABLE 1. Data collection and refinement statistics for DPS in complex with Tris (PDB id: 8CI9). Numbers in brackets refer to data for highest resolution shells.
2.5 Expression and purification of DPS mutants
For D186G, H239A, S182A and L181A the DPS-pET21a (+) construct was used as a template and overlapping primer pairs containing the point mutation were designed as listed in Supplementary Table S1 (Supplementary Section S1.3). They were purchased from IDT and Q5 ot Start High Fidelity 2X Master Mix was used as per the protocol in Supplementary Table S2 (New England Biosciences). The sequences were verified using Eurofins Genomics Supreme Run Tubes. The mutants were expressed and purified as for the wild-type enzyme for HPLC, described in the protocol above.
For M167A, H165A and D224A, mutagenesis was achieved using a Quikchange Site Directed Mutagenesis Kit (Agilent Technologies) with the primers shown in Supplementary Table S3 and protocol in Supplementary Table S4. The sequences were verified using Eurofins Genomics Supreme Run Tubes. The mutants were expressed and purified as described for the WT enzyme, except 100 mM MOPS was used in buffers A and B in place of Tris, no centrifugal concentration was required and no size-exclusion chromatography was performed.
2.6 Activity assays
The activities of wild-type DPS and mutants were evaluated using HPLC by measuring deoxypodophyllotoxin peak area compared to a standard curve. The reactions (250 µL) contained DPS (1.2 μM), iron sulphate (10 μM), 2-OG (1.0 mM), (−)-yatein (0.2 mM), sodium ascorbate (1.0 mM) in MOPS Buffer B. They were formulated in two parts (125 µL), with part 1 containing DPS, iron sulfate and 2-OG and part 2 (−)-yatein and sodium ascorbate. After 5 min incubation (25°C, 500 rpm) the two parts were combined, mixed by pipette for 7 s and returned to the incubator. Samples of 50 μL were removed at 1 min intervals and quenched with equal volumes of acetonitrile. These were then centrifuged at 16,000 × g for 5 min and the supernatant transferred to HPLC vials. The HPLC analysis was performed on an Agilent 1260 Infinity II LC system with an Infinity Lab Poroshell 120 EC-C18 column (3.0 × 150 mm, 2.7 μm) and an Infinity Lab Poroshell 120 EC-C18 guard column (3.0 × 5 mm, 2.7 μm). Separation was achieved with a 1 mL min-1 flow rate at 35.0°C with a gradient of water:acetonitrile 50:50 to 37.5:62.5 over 10 min, then held at 37.5:62.5 for 3 min, gradient 37.5:62.5 to 50:50 over 3 min and held at 50:50 for 4 min. The time courses were performed as experimental duplicates for each mutant of DPS. Data were analyzed in base R version 4.2.1.
2.7 NanoDifferential scanning fluorimetry
Protein was expressed and purified as per the protocol for the activity assays. DPS and mutant solutions were made up to 250 μL at 1 mg mL-1 using MOPS Buffer B. The samples were loaded into nano-differential scanning fluorimetry (nanoDSF) grade Standard Capillaries and analyzed using a Prometheus NT.48 nanoDSF machine. Protein unfolding was monitored using a linear thermal ramp of 20–95°C at 1°C min-1 at an excitation of 40%. Data were analyzed in base R version 4.2.1.
2.8 Halogenation assays
Cell free extract (CFE) in MOPS buffer C from an expression of D186G (as for the protocol for wild-type DPS) was prepared for an activity assay. The assay comprised 40% v/v CFE, 2 mM yatein in DMSO, 3 mM 2-OG, 3.2 mM sodium ascorbate, 1.0 mM FeSO4 and 160 uL of MOPS buffer C (200 mM MOPS, 100 mM NaCl at pH 7.4). The reactions were shaken at 500 rpm at 25°C and a 100 µL sample taken after 1 h and quenched with an equal volume of acetonitrile. This was centrifuged at 16,000 × g for 5 min and the supernatant transferred to a HPLC vial. The HPLC-MS analysis was performed on an UltiMate 3000 HPLC system connected to HCTultra ETD II mass spectrometer with an Infinity Lab with a Poroshell 120 EC-C18 column (3.0 × 150 mm, 2.7 μm) and an Infinity Lab Poroshell 120 EC-C18 guard column (3.0 × 5 mm, 2.7 μm). Separation was achieved with a 1 mL min-1 flow rate at 35.0°C with a gradient of water:acetonitrile 50:50 to 37.5:62.5 over 10 min, then held at 37.5:62.5 for 5 min, and returned to 50:50 to equilibrate.
3 Results and discussion
3.1 Crystal structure of DPS
The structure of DPS was determined by X-ray crystallography and refined to a resolution of 1.41 Å (Figure 3A). Crystals were obtained in space group P21 and featured one molecule in the asymmetric unit. The structure was refined to give Rcryst and Rfree values of 18.8% and 22.0% respectively (Table 1). Electron density for the backbone of the model was complete throughout the length of the chain, although poorer density for K17 and N18 did not permit their side chains to be modelled. A search on the DALI server (Holm, 2022) revealed the three closest structural homologs, apart from the extant DPS structure, to be an auxin dioxygenase from rice (6KUN; Takehara et al., 2020, 28% sequence i. d.; rmsd 2.7 Å over 294 Cα atoms); a second auxin dioxygenase, catalyzing the conversion of indole acetic acid into 2-oxoindole-3-acetic acid from Arabidopsis thaliana (6 KWA; Jin et al., 2020, 26%; 2.6 over 267 Cαs) and a hyoscyamine 6-hydroxylase (tH6H) from Datura metel (6TTO; Kluza et al., 2020, 20%; 2.8 over 311 Cαs), which catalyzes the last two steps in the biosynthesis of scopolamine.
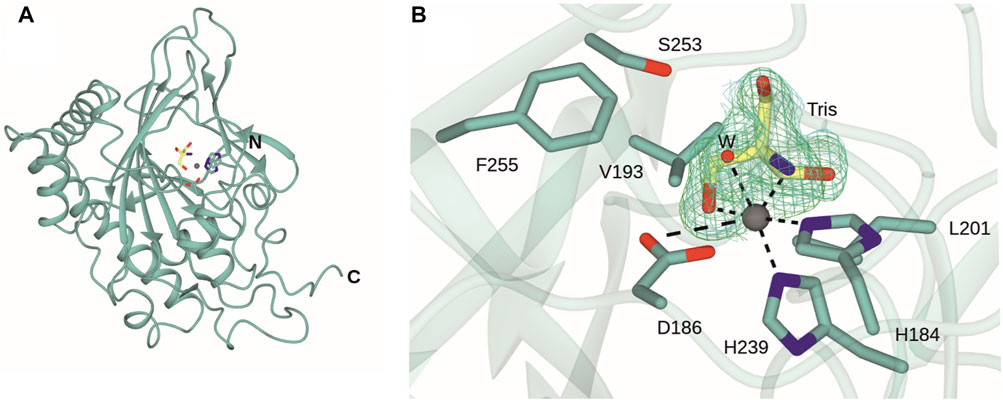
FIGURE 3. Structure of DPS. (A) Overall structure of DPS. (B) Active site of DPS highlighting coordination of the iron atom (grey sphere) to side chains of H184, D186 and H239, one water molecule (W) and an oxygen and nitrogen atom of the Tris ligand (carbon atoms in yellow). Electron density corresponds to the Fo-Fc and 2 Fo - Fc maps (each obtained prior to ligand refinement) in green and blue and at levels of 3σ and 1σ respectively.
The active site of DPS is located towards the top of the double-stranded β-helix (DSBH) fold. The iron atom is coordinated by the “facial triad” side chains of H184, D186 and H239, each of which is conserved in the closest structural homologues. Additional conserved residues in the substrate binding pockets of DPS and 6KUN include Y169 (Y158); L181 (L170); R249 (R242); L201 (L191); F255 (F248) and S251 (S244). Y158 and R242 are each involved in binding the gamma carboxylate of 2OG in the 6KUN complex (Takehara et al., 2010).
A comparison of the new structure with both 7E37 and 7E38 showed that this dataset revealed electron density for residues 292–299 absent in 7E37 but present in 7E38 and that the r.m.s.d. for the new structure with the ‘A’ chain of 7E38 was 0.43 Å over 305 Cα atoms. Significant differences in side chain orientations were observed for R8 and also the region E150/K151/H152 at the periphery. These are likely due to changes in crystal contacts between the older and new structures, which have two and one molecules in the asymmetric unit respectively. The major difference in the new structure is at the iron center. Following building of the protein chain’s atoms and the active site metal, there was persistent residual density in the omit map around the iron atom (Figure 3B). This was modelled with a molecule of Tris, with one hydroxymethyl and the amine group in the two equatorial coordination positions that are occupied by the chelating carboxylate of succinate in the 7E38 complex structure. The distal axial coordination position is occupied by a water molecule. Tris was a buffer constituent and there were two other clearly defined Tris molecules in the maps at different locations. There is also precedent for the coordination of Tris to transition metal ions such as cobalt as part of an octahedral coordination sphere in, for example, hydroxypropyl phosphonic acid epoxidase (PDB ID:1ZZC; Higgins et al., 2005). This coordination of Tris to the catalytic iron in DPS has possible implications for its use as a buffer in the further study and application of the enzyme.
In the interests of investigating the structural determinants of activity and substrate specificity in DPS, the (−)-yatein coordinate positions from 7E38 were superimposed with the new structure (Figure 4). Chang and co-workers showed that the (−)-yatein molecule adopts a U-shaped conformation in the active site that brings the C6 and C7’ atoms into close proximity for the ring closing reaction to occur (Tang et al., 2022). The benzodioxole moiety (rings A and B) was observed to be bound in a pocket formed by T86, M167 and H165; ring E in a pocket formed by L181, H184, V298, and F290. In addition, S182 was observed as a possible gatekeeper residue to the active site in this region.
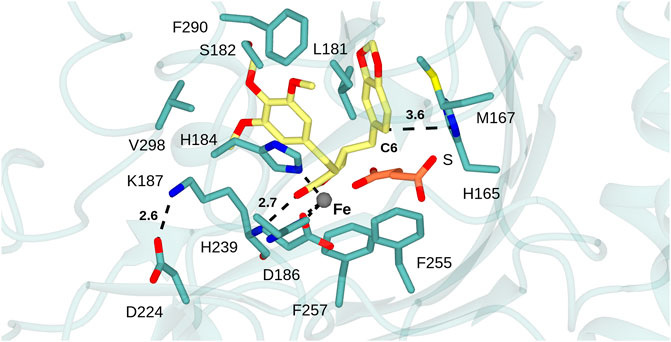
FIGURE 4. DPS substrate binding mode. Active site of DPS superimposed with atom coordinates of (−)-yatein (carbon atoms in yellow) and succinate (S, carbon atoms in coral) derived from 7E38. Black dashed lines indicate interactions between: the iron atom and the side chains of D186, H239 and H184; the salt-bridge between the side-chains of K187 and D224; the H-bond between the lactone carbonyl of the ligand and backbone N-H of K187. Distances are given in Ångstroms.
In the case of each ligand, there were no H-bonding interactions with active site side-chains that would suggest that either moiety would be crucial to substrate recognition. This suggestion is borne out by the substrate promiscuity of DPS revealed by both Fuchs and Renata and co-workers (Lazzarotto et al., 2019; Li et al., 2019). The C6 atom of the benzodioxole, which is thought to be deprotonated as the final step in ring closure by DPS, is 3.6 Å from the side chain of H165, which is therefore a candidate residue for the base-catalyzed reaction (Figure 5). A further bonding interaction of note is the interaction of the lactone carbonyl (ring D) with the K187 main chain amide (2.8 Å). The conformation of K187 is maintained via a salt bridge formed between the side-chain of residues D224 and K187 (2.6 Å) (Figure 4). Kroutil et al. have demonstrated that the lactone moiety in (−)-yatein is necessary for DPS activity (Lazzarotto et al., 2019).
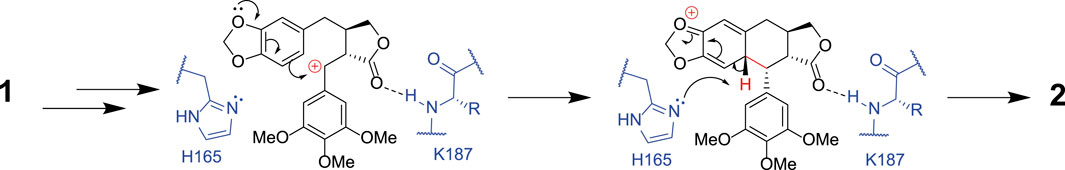
FIGURE 5. The proposed mechanism for formation of deoxypodophyllotoxin 2 from (−)-yatein 1. This shows how H165 may act as a base, abstracting a proton from the C6 atom in the final stages of the mechanism. The interaction between the lactone ring (ring D) of the substrate and the amide backbone of K187 is also highlighted.
3.2 Mutational analysis of DPS
The structural analysis of DPS was used to inform the selection of mutants, in order to investigate different aspects of DPS activity and stability. The substitution H239A was selected to investigate if H239 was a necessary part of the facial triad. M167A and H165A were chosen to study the effect on substrate binding within the benzodioxole pocket and the catalytic role of the histidine. L181A and S182A probed the region of ring E binding, again to observe the effects of changes in this region to substrate recognition. The substitution D224A was selected to break the D224-K187 salt bridge, to examine the effect on activity or enzyme stability of the interaction with K187. In addition, we prepared the mutant D186G, as previous work had shown that mutation of this conserved aspartate to glycine can enable or increase halogenation activity of 2-OG dependent dioxygenases, as observed for the D157A mutant of the N-acyl amino acid hydroxylase SadA (Mitchell et al., 2017). Each mutant was assayed for activity using (−)-yatein as substrate and their catalytic performance analyzed using HPLC (Figures 6A, B).
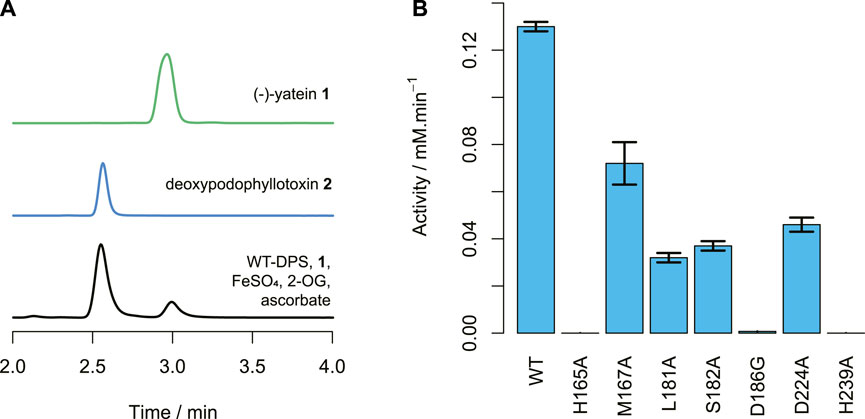
FIGURE 6. DPS activity. (A) HPLC traces of (−)-yatein 1 chemical standard (0.5 mM), deoxypodophyllotoxin 2 chemical standard (0.1 mM), and the wild-type DPS (1.2 μM) catalyzed conversion of (−)-yatein (0.2 mM) to deoxypodophyllotoxin (2 min). Chromatogram intensities were adjusted to compensate for the differences in the concentration of the standards. (B) The average activities of the DPS mutants with (−)-yatein 1, calculated by formation of deoxypodophyllotoxin 2 when the concentration of DPS is 1.2 µM. Error bars show standard error of the mean calculated from the experimental duplicates.
All the mutants showed reduced activities in comparison to the wild-type (Figure 6B). Neither H165A nor H239A demonstrated detectable activity. This is unsurprising for H239, as this residue coordinates to the central ion atom in the active site. However, it suggests that H165 plays a crucial role in DPS catalysis. Nano-scale differential scanning fluorimetry (nano-DSF) data (Figure 7) shows that this mutation results in a slightly reduced thermostability in comparison to the wild-type but not to a degree that would be expected to render a mutant inactive. Instead, its proximity to the C6 atom of the benzodioxole ring suggests it may be a plausible candidate for the base that is proposed to catalyze the final step in ring closure: the abstraction of a proton from the C6 atom.
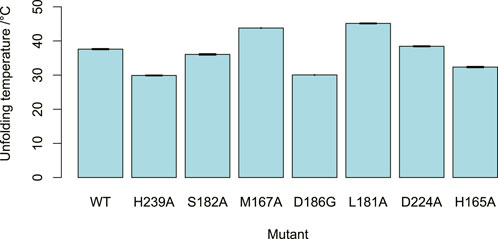
FIGURE 7. DPS mutant stability. The unfolding temperatures of the DPS mutants, as determined by nano-DSF. At least three measurements were taken for each mutant. Error bars showing standard error of the mean depicted but barely visible due to high repeatability.
Mutation of the residue M167, which is in the benzodioxole-binding region, to alanine, resulted in a reduction in the enzyme’s activity and little change to its thermostability. Reduced activities were also seen for the mutants S182A and L181A, despite little significant change in thermostability. This supports the contention that these residues may be involved in substrate binding in the region of ring E, as predicted by the structural analysis. The mutation of D224 to alanine breaks an ionic interaction with the side chain of K187, the amide nitrogen of which is proposed to bind to the lactone carbonyl. This resulted in a mutant with reduced activity. The nano-DSF data suggest that the D224-K187 interaction only has a minor impact on stability in comparison to the wild-type and the lower activity caused by D224A indicates the D224-K187 interaction may be more related to the catalytic activity of the enzyme. This observation fits with the proposed role of the K187 main chain amide interaction with the substrate lactone carbonyl, with the orientation of the amide maintained by the D224-K187 salt bridge.
Halogenase activity is of particular relevance to biocatalysis as bioactive compounds often contain halogens and these can be difficult to install selectively through traditional synthetic routes. Halogens can also be used as handles for compound derivatization via cross-coupling catalysis. Halogenated derivatives of yatein and deoxypodophyllotoxin would allow greater exploration of chemical and bioactivity space. Previous work with SaDAH and SadA (21% and 11% amino acid identity to DPS respectively) showed that substitution of the typical metal-binding aspartic acid with glycine can act as a switch for halogenation activity (Mitchell et al., 2017; Kim et al., 2020). The equivalent mutation of D186 to glycine was performed in DPS in order to investigate the possibility of switching the activity, at least partially, to halogenation. However, the activity assays of D186G showed it had extremely reduced native C-C bond forming activity (<1% of WT activity) and no halogenase activity. Work performed since the creation of this mutant has further investigated the determinants of switching activity for 2-ODDs and shown that the exact positioning of the substrate is an important factor in enabling the halogenation reaction, as demonstrated by engineering the activity of the hydroxylase MBT76 to chlorinate lysine (Neugebauer et al., 2022; Kastner et al., 2023). It is possible that rational site directed mutagenesis based on these studied enzymes could still permit the creation of a halogenation activity for DPS (Mitchell et al., 2017).
4 Conclusion
We set out to increase our understanding of the residues responsible for controlling catalysis in DPS using structurally informed amino acid substitutions. In doing so, we determined a new, high-resolution structure of DPS in complex with tris buffer, which complements existing structures that were released as we progressed this project. The structures informed the first mutational analysis of DPS. We generated six mutants, testing their activity and stability. Most significantly, we found that H165 is necessary for catalysis, potentially acting as a base in the reaction mechanism (Figure 5). Activity of mutants also implied that the salt bridge between D224 and K187 may be important for mediating substrate interactions. This work provides important mechanistic information about an unusual, C-C bond-forming member of the 2-ODD family. As an enzyme with high potential for biocatalytic use, the improved knowledge of structural determinants of the catalytic mechanism provide an excellent starting point for future work including directed evolution experiments to expand substrate scope.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: www.rcsb.org, 8CI9. Other reported data is available from authors at reasonable request.
Author contributions
ZI conducted the experimental work. ZI, GG, and BL interpreted the results and jointly wrote this manuscript. All authors contributed to the article and approved the submitted version.
Funding
ZI was funded by a grant (BB/T007222/1) from the BBSRC White Rose Doctoral Training Program (DTP) in Mechanistic Biology. BL is funded by a UKRI Future Leaders Fellowship (MR/S01862X/1).
Acknowledgments
We thank Sam Hart and Dr. Johan P. Turkenburg for assistance with X-ray data collection. We acknowledge the Diamond Light Source Didcot UK for access to beamline I04 under grant number mx24948. We thank the Technology Facility at the University of York for the use of their facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2023.1178345/full#supplementary-material
References
Alizadeh, B. H., Emami, S., Dehghan, G., Foroumadi, A., and Shafiee, A. (2017). Synthesis of cytotoxic isodeoxypodophyllotoxin analogs. J. Heterocycl. Chem. 54 (1), 539–545. doi:10.1002/jhet.2618
Chang, W. C., Yang, Z. J., Tu, Y. H., and Chien, T. C. (2019). Reaction mechanism of a nonheme iron enzyme catalyzed oxidative cyclization via C–C bond formation. Org. Lett. 21, 228–232. doi:10.1021/acs.orglett.8b03670
Decembrino, D., Raffaele, A., Knöfel, R., Girhard, M., and Urlacher, V. B. (2021). Synthesis of (−)−deoxypodophyllotoxin and (−)−epipodophyllotoxin via a multi-enzyme cascade in E. coli. Microb. Cell Factories 20, 183. doi:10.1186/s12934-021-01673-5
Decembrino, D., Ricklefs, E., Wohlgemuth, S., Marco, G., Schulleher, K., Jach, G., et al. (2020). Assembly of plant enzymes in E. coli for the production of the valuable (−)-Podophyllotoxin precursor (−)-Pluviatolide. ACS Synth. Biol. 9 (11), 3091–3103. doi:10.1021/acssynbio.0c00354
Eljounaidi, K., and Lichman, B. R. (2020). Nature's chemists: The discovery and engineering of phytochemical biosynthesis. Front. Chem. 9 (8), 596479. doi:10.3389/fchem.2020.596479
Emsley, P., and Cowtan, K. (2004). Coot: Model building tools for molecular graphics. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60, 2126–2132. doi:10.1107/S0907444904019158
Evans, P. (2006). Scaling and assessment of data quality. Acta Crystallogr. Sect. D. Biol. Crystallogr. 62, 72–82. doi:10.1107/S0907444905036693
Hagel, J. M., and Facchini, P. J. (2018). Expanding the roles for 2-oxoglutarate-dependent oxygenases in plant metabolism. Nat. Prod. Rep. 35, 721–734. doi:10.1039/C7NP00060J
Higgins, L. J., Yan, F., Liu, P., Liu, H. W., and Drennan, C. L. (2005). Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature 437, 838–844. doi:10.1038/nature03924
Holm, L. (2022). Dali server: Structural unification of protein families. Nucleic Acids Res. 50 (W1), W210–W215. doi:10.1093/nar/gkac387
Islam, M. S., Leissing, T. M., Chowdhury, R., Hopkinson, R. J., and Schofield, C. J. (2018). 2-Oxoglutarate-Dependent oxygenases. Ann. Rev.Biochem. 87, 585–620. doi:10.1146/annurev-biochem-061516-044724
Jin, S. H., Lee, H., Shin, Y., Kim, J. H., and Rhee, S. (2020). Crystal structure of the indole-3-acetic acid-catabolizing enzyme DAO1 from Arabidopsis thaliana. J. Struct. Biol. 212, 107632. doi:10.1016/j.jsb.2020.107632
Kabsch, W. X. D. S. (2010). Xds. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 125–132. doi:10.1107/S0907444909047337
Kastner, D. W., Nandy, A., Mehmood, R., and Kulik, H. J. (2023). Mechanistic insights into substrate positioning that distinguish non-heme Fe(II)/α-Ketoglutarate-Dependent halogenases and hydroxylases. ACS Catal. 13, 2489–2501. doi:10.1021/acscatal.2c06241
Kim, C. Y., Mitchell, A. J., Glinkerman, C. M., Li, F. S., Pluskal, T., and Weng, J. K. (2020). The chloroalkaloid (−)-acutumine is biosynthesized via a Fe(II)- and 2-oxoglutarate-dependent halogenase in Menispermaceae plants. Nat. Comms. 11, 1867. doi:10.1038/s41467-020-15777-w
Kim, S. S., Wengier, D. L., Ragland, C. J., and Sattely, E. S. (2022). Transcriptional reactivation of lignin biosynthesis for the heterologous production of etoposide aglycone in Nicotiana benthamiana. ACS Synth. Biol. 11 (10), 3379–3387. doi:10.1021/acssynbio.2c00289
Kluza, A., Wojdyla, Z., Mrugala, B., Kurpiewska, K., Porebski, P. J., Niedzialkwska, E., et al. (2020). Regioselectivity of hyoscyamine 6β-hydroxylase-catalysed hydroxylation as revealed by high-resolution structural information and QM/MM calculations. Dalton Trans. 49, 4454–4469. doi:10.1039/D0DT00302F
Lau, W., and Sattely, E. S. (2015). Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science 349 (6253), 1224–1228. doi:10.1126/science.aac7202
Lazzarotto, M., Hammerer, L., Hetmann, M., Borg, A., Schmermund, L., Steiner, L., et al. (2019). Chemoenzymatic total synthesis of deoxy-, epi-, and podophyllotoxin and a biocatalytic kinetic resolution of dibenzylbutyrolactones. Angew. Chem. Int. Ed. 58 (24), 8226–8230. doi:10.1002/anie.201900926
Li, J., Zhang, X., and Renata, H. (2019). Asymmetric chemoenzymatic synthesis of (−)-Podophyllotoxin and related aryltetralin lignans. Angew. Chem. Int. Ed. 58 (34), 11657–11660. doi:10.1002/anie.201904102
Mitchell, A. J., Dunham, N. P., Bergman, J. A., Wang, B., Zhu, Q., Chang, W. C., et al. (2017). Structure-guided reprogramming of a hydroxylase to halogenate its small molecule substrate. Biochemistry 56, 441–444. doi:10.1021/acs.biochem.6b01173
Mitchell, A. J., and Weng, J. K. (2019). Unleashing the synthetic power of plant oxygenases: From mechanism to application. Plant Physiol. 179, 813–829. doi:10.1104/pp.18.01223
Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D. Biol. Crystallogr. 53, 240–255. doi:10.1107/S0907444996012255
Nett, R. S., Dho, Y., Low, Y. Y., and Sattely, E. S. (2021). A metabolic regulon reveals early and late acting enzymes in neuroactive Lycopodium alkaloid biosynthesis. PNAS 118, e2102949118. doi:10.1073/pnas.2102949118
Neugebauer, M. E., Kissman, E. N., Marchand, J. A., Pelton, J. G., Sambold, N. A., Millar, D. C., et al. (2022). Reaction pathway engineering converts a radical hydroxylase into a halogenase. Nat. Chem. Biol. 18, 171–179. doi:10.1038/s41589-021-00944-x
Peters, C., and Buller, R. M. (2019). Industrial application of 2-oxoglutarate-dependent oxygenases. Catalysts 9 (3), 221. doi:10.3390/catal9030221
Schultz, B. J., Kim, S. Y., Lau, W., and Sattely, E. S. (2019). Total biosynthesis for milligram-scale production of etoposide intermediates in a plant chassis. J.Am. Chem. Soc. 141 (49), 19231–19235. doi:10.1021/jacs.9b10717
Takehara, S., Sakuraba, S., Mikami, B., Yoshida, H., Itoh, A., Endo, M., et al. (2010). A common allosteric mechanism regulates homeostatic inactivation of auxin and gibberellin. Nat. Commun. 11, 2143. doi:10.1038/s41467-020-16068-0
Takehara, S., Sakuraba, S., Mikami, B., Yoshida, H., Yoshimura, H., Itoh, A., et al. (2020). A common allosteric mechanism regulates homeostatic inactivation of auxin and gibberellin. Nat. Comms 11, 2143. doi:10.1038/s41467-020-16068-0
Tang, H., Wu, M. H., Lin, H. Y., Han, M. R., Yang, Z. J., Chien, T. C., et al. (2022). Mechanistic analysis of carbon–carbon bond formation by deoxypodophyllotoxin synthase. PNAS 119 (1), e2113770119. doi:10.1073/pnas.2113770119
Vagin, A., and Teplyakov, A. M. (1997). Molrep: An automated Program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. doi:10.1107/S0021889897006766
Keywords: biocatalysis, crystallography, deoxypodophyllotoxin, ring-formation, oxygenase, natural product
Citation: Ingold Z, Grogan G and Lichman BR (2023) Structure and mutation of deoxypodophyllotoxin synthase (DPS) from Podophyllum hexandrum. Front. Catal. 3:1178345. doi: 10.3389/fctls.2023.1178345
Received: 02 March 2023; Accepted: 25 April 2023;
Published: 24 May 2023.
Edited by:
Mohamed Nagia, Leibniz Institute of Plant Biochemistry, GermanyReviewed by:
Nicole Leferink, The University of Manchester, United KingdomJordan Froese, Ball State University, United States
Jan Muschiol, Helmholtz Association of German Research Centres (HZ), Germany
Florian Rudroff, Vienna University of Technology, Austria
Vlada B. Urlacher, Heinrich Heine University of Düsseldorf, Germany
Copyright © 2023 Ingold, Grogan and Lichman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gideon Grogan, Z2lkZW9uLmdyb2dhbkB5b3JrLmFjLnVr; Benjamin R. Lichman, YmVuamFtaW4ubGljaG1hbkB5b3JrLmFjLnVr