 Y. T. Candace Ho1,2,3,4Thierry Izoré1,2Joe A. Kaczmarski5,6Edward Marschall1,2,3Minuri S. Ratnayake1,2,3
Y. T. Candace Ho1,2,3,4Thierry Izoré1,2Joe A. Kaczmarski5,6Edward Marschall1,2,3Minuri S. Ratnayake1,2,3 Julien Tailhades1,2,3David L. Steer1,7Ralf B. Schittenhelm1,7
Julien Tailhades1,2,3David L. Steer1,7Ralf B. Schittenhelm1,7 Manuela Tosin4Colin J. Jackson3,5,6,8
Manuela Tosin4Colin J. Jackson3,5,6,8 Max J. Cryle1,2,3*
Max J. Cryle1,2,3*- 1Department of Biochemistry and Molecular Biology, The Monash Biomedicine Discovery Institute Monash University, Clayton, VIC, Australia
- 2EMBL Australia, Monash University, Clayton, VIC, Australia
- 3ARC Centre of Excellence for Innovations in Peptide and Protein Science, Canberra, ACT, Australia
- 4Department of Chemistry, University of Warwick, Coventry, United Kingdom
- 5ARC Centre of Excellence in Synthetic Biology, Australian National University, Canberra, ACT, Australia
- 6Research School of Biology, Australian National University, Acton, ACT, Australia
- 7Monash Proteomics and Metabolomics Facility, Monash University, Clayton, VIC, Australia
- 8Research School of Chemistry, Australian National University, Acton, ACT, Australia
In nonribosomal peptide synthesis, condensation (C) domains are key catalytic domains that most commonly link carrier protein bound substrates to form peptides or depsipeptides. While adenylation domains have been well characterized due to their role in the selection of monomers and hence as gate keepers in nonribosomal peptide biosynthesis, C-domains have been the subject of debate as they do not have apparent “A-domain like” side chain selectivity for their acceptor substrates. To probe the selectivity and specificity of C-domains, here we report our biochemical and structural characterization of the C3-domain from the biosynthesis of the siderophore fusachelin. Our results show that this C-domain is not broadly flexible for monomers bearing significantly alternated side chains or backbones, which suggests there can be a need to consider C-domain specificity for acceptor substrates when undertaking NRPS engineering.
1 Introduction
Nonribosomal peptides (NRPs) are a large group of peptide secondary metabolites that include many medically relevant peptides (e.g., penicillin and vancomycin) and are biosynthesized by nonribosomal peptide synthetases (NRPSs) (Felnagle et al., 2008). The diversity of NRPs is significant and is not restricted to medical applications, with NRPs also identified for use in bioremediation, insect control and metal chelation (e.g., fusachelin, the system under investigation in this work) (Dimise et al., 2008). NRPSs are multi-modular enzymes that act as peptide assembly lines, with each module containing different enzymatic domains working together to join monomers into the “growing” peptide. An NRPS module typically consists of the three domains that are essential for the elongation of a peptide: an adenylation (A) domain, a peptidyl carrier protein (PCP) domain and a condensation (C) domain (Weber and Marahiel, 2001). A-domains function as the key selectivity determinants in NRPS biosynthesis by binding to a specific substrate (typically an amino acid) and activating this via consumption of ATP to form an intermediate mixed anhydride. Next, this activated residue is transferred onto the post-translationally modified 4′-phosphopantetheinyl (PPant) arm of the adjacent PCP-domain, where it is bound as a thioester (Strieker et al., 2010). The PCP-domain then serves as a shuttle to deliver covalently tethered intermediates to different domain active sites. C-domains, which feature donor and acceptor binding sites for upstream and downstream PCP-bound substrates, typically catalyze the formation of peptide bonds. Attack of the downstream (acceptor) amine moiety upon the thioester linkage of the upstream (donor) substrate result in an extended PCP-bound peptide, which become the donor substrate for further peptide chain growth and modification. Beyond these core domains, other additional tailoring domains, such as epimerization (E) (Stachelhaus and Walsh, 2000), methylation (Mt) (Ansari et al., 2008) and heterocylization (Cy) (Bloudoff et al., 2017) domains can be present within NRPS modules where they function to introduce additional complexity into the NRPs produced by various assembly lines. Terminal modules also include domains dedicated to the release of the final peptide, such as thioesterase (TE) domains, which can release the peptide in either a cyclized (via amide or ester formation) or hydrolyzed linear form (Horsman et al., 2016). Depsipeptide NRPs are a broad class of pharmacologically active natural products containing both amide and ester linkages (Alonzo and Schmeing, 2020), such as daptomycin (Baltz et al., 2005) and teixobactin (Tan et al., 2020). The ester bonds found in these compounds are typically installed either in an elongation step by condensation of an α-hydroxy acid with a standard amino acid or peptide (catalysed by C-domains) or in the termination step when a hydroxyl group functions as a nucleophile to initiate cyclization and release of the peptide (TE-catalyzed).
Attempts to modify the NRPS machinery and produce novel compounds have focused on engineering the A-domain as it is the entry point for monomers into the machinery. Reprogramming an A-domain by targeted mutagenesis represents a potentially simple strategy that preserves catalytically relevant conformational changes and maintains protein-protein interactions with adjacent domains and modules of the NRPS (Stanisic and Kries, 2019). A recent and impressive example of this approach is the successful reprogramming of a conventional amino acid-specific A-domain to recognise an alternate α-hydroxy acid substrate, which was achieved using high-throughput yeast display (Camus et al., 2022). While A-domains generally possess high selectivity, C-domains have not broadly displayed signs of “A-domain like” sidechain selectivity in their acceptor sites. Nonetheless, C-domains are the key domains within the NRPS assembly line that link different monomers through amide bond formation. The first structures of a C-domain (VibH from the vibriobactin NRPS) demonstrated that these domains comprise a pseudo-dimer of chloramphenicol acetyltransferase-like domains, possess binding sites for each of their donor and acceptor peptidyl carrier protein-bound ligands (albeit a free amine substrate in the case of VibH). C-domains also contain a conserved motif HHxxxDG located between the two lobes in the active site of the domain (Keating et al., 2002). C-domains can perform diverse functions beyond peptide and ester bond formation, including dehydration (Wang et al., 2021) and β-lactam formation (Gaudelli et al., 2015), whilst structurally related domains (Miller and Gulick, 2016; Bloudoff and Schmeing, 2017) can perform epimerization (E-domains) (Stachelhaus and Walsh, 2000), synthesize ox-/thiazoline moieties (Cy-domains) (Konz et al., 1997; Katsuyama et al., 2021), and recruit trans-acting enzymes (Patteson et al., 2018; Reitz et al., 2019), including X-domains from GPA biosynthesis that recruit a multiple cytochrome P450 enzymes as part of a complex cyclization cascade (Haslinger et al., 2015; Ho et al., 2022). Despite the importance of these reactions for NRPS-catalyzed biosynthesis, the specificity enforced by C-type domains remains unclear and is the subject of ongoing debate. Early studies implicated C-domain selectivity in acceptor substrate selection (albeit with soluble substrate mimics) (Belshaw et al., 1999), and have more recently C-domains have been shown to gate the modification of PCP-bound amino acids by enzymes in trans (Kaniusaite et al., 2019). Thus, it is important to investigate the substrate selectivity of C-domains to ensure the success of future NRPS biosynthetic engineering efforts, which includes the acceptance of substrates with alternate nucleophiles. Here, we report our analysis of the selectivity of the C3-domain (the third C-domain from the NRPS, Figure 1) from the NRPS assembly line of the siderophore fuscachelin. The results of our combined biochemical and structural experiments show that this domain is not broadly flexible for monomers at the acceptor site, a result that highlights the need to consider C-domain specificity when engineering neighboring A-domains to accept different monomers for NRPS biosynthesis.
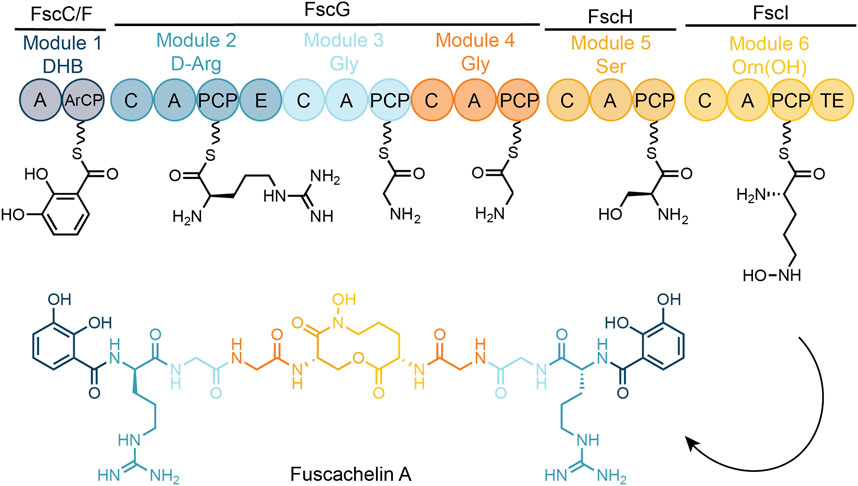
FIGURE 1. Schematic representation of fuscachelin biosynthesis. A—adenylation domain, DHB—2,3-dihydroxybenzoic acid, ArCP—acyl carrier protein, C—condensation domain, PCP—peptidyl carrier protein, E—epimerization domain, TE—thioesterase domain, PPant moieties shown as undulated lines.
2 Materials and methods
2.1 Construct cloning
PCP2-C3 constructs. A wild-type gene encoding the desired PCP2-C3 region of FscG (UniProt ID Q47NR9) and the PCP2-C3 R2577G mutant were amplified and cloned into the pOPINS vector as reported in a previous study (Izore et al., 2021).
PCP2-C3 SpyCatcher and PCP3 SpyTag constructs. The PCP2-C3 SpyCatcher construct (pOPINS vector) and PCP3 SpyTag construct (pHIS17 vector) were obtained in a previous study (Izore et al., 2021). PCP2-C3 SpyCatcher mutants were generated using standard Quick-Change site-directed mutagenesis using the primers listed in Supplementary Table S1 (#1 and #2 for R2577G, and #3 and #4 for E2702S). Five synthetic, codon-optimized genes (optimized for E. coli expression) encoding PCP2-C3 didomain mutants (pOPINS vector) were obtained from Twist Bioscience (Supplementary Table S2).
2.2 Protein expression and purification
All proteins were expressed and purified as reported in a previous study (Izore et al., 2021) with the exception of pOPIN-S derived proteins. For these proteins, the SUMO tag was cleaved with Cth SUMO protease (Cth) (Lau et al., 2018) overnight while being dialyzed in buffer (50 mM Tris–HCl, pH 8.0; 300 mM NaCl, 1 mM DTT) at 4°C. The uncleaved protein, the cleaved tag and Cth then remained bound to the Ni-NTA beads following elution of the cleaved protein.
2.3 Chemical synthesis
Unless specified otherwise, chemicals purchased from Sigma Aldrich, Iris Biotech, Chem-Impex International, GL Biochem and Fisher Scientific were used without further purification. Reagent grade dichloromethane (DCM), N,N-dimethylformamide (DMF), methanol (MeOH), acetonitrile (ACN), diethyl ether and water were purchased from Fisher Scientific.
1H NMR spectra were recorded in D2O and CD3CN using a BACS-400 400 MHz Bruker Avance instrument. HPLC-MS spectra were obtained using a Shimadzu system (LCMS-2020, ESI operating in positive and negative mode) equipped with a Waters XBridge®Peptide BEH C18 column (300 Å, 3.5 μm, 4.6 mm × 250 mm) and employing a gradient of 5%-5%-45% ACN +0.1% formic acid (FA) over 35 min.
2.3.1 Tripeptidyl-CoA synthesis
Tripeptidyl-CoA (benzoic acid (BA)-D-Arg-Gly-CoA, 1) was synthesized, purified and characterized according to a previous study (Izore et al., 2021).
2.3.2 Aminoacyl-CoAs synthesis
All aminoacyl-CoAs were synthesized, purified and characterized as previously reported (Koetsier et al., 2011; Izore et al., 2019; Izore et al., 2021).
2.3.3 Thioether stabilized glycolyl-CoA (Glycostab-CoA) synthesis
CoA (1 eq.) was dissolved in 10 mL of a buffer comprising 0.02 M ammonium bicarbonate and 6.5 mM EDTA, pH 8. Tris (2-carboxyethyl)phosphine (TCEP, 1.2 eq.) was subsequently added and the reaction was stirred for 30 min 2-Bromoethanol (3 eq.) was dissolved in 2 mL of ACN and added to the solution, which was then stirred at room temperature (RT) overnight. The desired compound 3 was concentrated and purified by preparative RP-HPLC purification (ACN gradient 0%–40% over 30 min). Yield: 30.3%. For characterization see Supplementary Figures S1, S2.
2.3.4 Lactyl-CoA synthesis
Lactic acid (2 eq.), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 2 eq.) and Oxyma pure (2 eq.) were dissolved in DMF and stirred in an ice bath for 30 min before a solution of CoA (1 eq.) in DMF was added over a period of 10 min. The mixture was then stirred overnight at RT. The crude product was precipitated by addition of ice cold Et2O and the pellet was collected using a flame-resistant centrifuge, with this process repeated three times to wash the sample. The crude product was then purified by preparative RP-HPLC (ACN gradient 0%–40% over 30 min). Yield: 21.5%. For characterization see Supplementary Figures S3, S4.
2.4 Enzyme activity assays
Assays were performed in three steps: (1) peptide loading of the PCP-containing proteins; (2) in vitro reconstitution of C-domain activity; and (3) cleavage of the elongated peptides from the PCP2-C3-SpyCatcher-SpyTag-PCP3 construct according to the methods reported previously (Izore et al., 2021).
(1) The loading reaction utilized a 1: 2: 0.1 M ratio of the PCP2-C3 construct, peptidyl-CoA and Sfp (R4-4 mutant) (Sunbul et al., 2009), respectively. Peptidyl-CoA (200 μM) was loaded onto PCP-containing construct (100 μM) for 1 h at 30°C through the activity of Sfp (R4-4 mutant, 10 μM) in PCP-loading buffer (50 mM HEPES, pH 7.0; 50 mM NaCl; 10 mM MgCl2). Following the loading reaction, the remaining peptidyl-CoA was removed in three sequential concentration/dilution steps (Amicon Ultra-0.5 mL centrifugal filters, 50 kDa MWCO, Merck-Millipore) with dilution in PCP-loading buffer.
(2) Peptide loaded PCP2-C3 SpyCatcher constructs were incubated with the unloaded PCP3 SpyTag construct (both 100 µM) for 10 min at 30°C, followed by loading of the desired aminoacyl-CoA on the PCP3 as described above. The reaction was then incubated for an additional 1 h at 30°C to allow for the condensation reaction to occur.
(3) For thioether tethered amino acid loaded PCP3 substrates, reaction mixtures were directly analyzed using nano LC-ESI-MS. For thioester tethered amino acid loaded PCP3 substrates, substrates were chemically cleaved by the addition of a 40% methylamine solution in water (0.5 M) to liberate the methylamide peptides; reaction mixtures were incubated for 15 min at RT. For thioester tethered hydroxy acid loaded PCP3 substrates, 15 μL each of 1 M sodium mercaptoethylsulphonate (MESNa) and 1 M cysteamine. HCl solutions were added to offload the substrates; the mixtures were incubated for 20 min at RT. After chemical cleavage, 850 μL of 0.1% FA in water was added into the reaction to adjust the pH to ∼7. Purification was performed using solid phase extraction (SPE) columns (Bond Elut Plexa 30 mg/mL, Agilent Technologies) that had been activated with 0.1% FA in MeOH (1 mL) and equilibrated with 0.1% FA in water (1 mL). The neutralized reaction mixture was applied to the equilibrated SPE column via gravity flow, washed with 0.1% FA in water (1 mL) and the samples eluted with 1 mL of 0.1% FA in ACN/H2O (50:50). These samples were then dried by freeze dryer at −50°C and analyzed by HRMS.
2.5 Liquid chromatography–mass spectrometry (LC-MS) purification and analysis
All RP-HPLC purifications were performed using a Shimadzu high performance liquid chromatography system equipped with an SPD-M20A Prominence photo diode array detector and two LC-20AP pumps. Preparative separations were performed using a Zorbax SB-C18 column from Agilent (7 μm, 21.2 × 250 mm) with a flow rate of 10 mL/min. The solvents used were water +0.1% TFA (solvent A) and HPLC-grade ACN +0.1% TFA (solvent B). And initial assessment of enzyme activity was conducted on an HPLC-MS system from Shimadzu (LCMS-2020, ESI operating in positive and negative mode) using a Waters XBridge®Peptide BEH C18 column (300 Å, 3.5 μm, 4.6 mm × 250 mm) employing a gradient of 5%–45% ACN +0.1% FA over 35 min. The level of peptide extension in the assays calculated using the following formula: percentage conversion = peak area (product)/(peak area (donor) + peak area (product)) × 100.
2.6 Liquid chromatography-high resolution mass spectrometry (LC-HRMS) and MS2 measurements
LC-HRMS measurements were performed on an Orbitrap Fusion mass spectrometer (Thermo Scientific) coupled online to a nano-LC (Ultimate 3,000 RSLCnano; Thermo Scientific) via a nanospray source. Peptides were separated on a 50 cm reverse-phase column (Acclaim PepMap RSLC, 75 μm × 50 cm, nanoViper, C18, 2 μm, 100 Å; Thermo Scientific) after binding to a trap column (Acclaim PepMap 100, 100 μm × 2 cm, nanoViper, C18, 5 μm, 100 Å; Thermo Scientific). Elution was performed on-line with a gradient from 6% MeCN to 30% MeCN in 0.1% formic acid over 30 min at 250 nL min−1. Full scan MS was performed at 60,000 nominal resolutions, with targeted MS2 scans of peptides of interest acquired at 15,000 nominal resolutions using HCD with stepped collision energy (24% ± 5% NCE). Raw data was manually analysed in XCalibur QualBrowser (Thermo Scientific), with extracted ion chromatograms of the predicted species generated with 6 ppm mass tolerance. Predicted MS2 fragments were generated with MS-Product (ProteinProspector v5.22.1, UCSF) and manually assigned to spectra. See Supplementary Figures S6–S12, Supplementary Figures S24–S36.
2.7 PPant ejection
Mass spectrometry measurements were performed on a MicroTOFq mass spectrometer (Bruker Daltonics) coupled online to a 1,200 series capillary/nano-LC (Agilent Technologies) via a Bruker nano ESI source. Proteins were separated on a 150 mm reverse-phase column (ZORBAX 300SB-C18, 3.5 µm, 0.075 × 150 mm; Agilent Technologies) after binding to a trap column (ZORBAX 300SB-C18, 5 μm, 0.30 × 5 mm cartridges; Agilent Technologies). Elution was performed on-line with a gradient from 4% MeCN to 60% MeCN in 0.1% FA over 30 min at 300 nL/min. Proteins greater than 20 kDa were separated on a MabPac SEC-1 5 µm 300 Å 50 × 4 mm (Thermo Scientific) column with an isocratic gradient of 50% MeCN, 0.05% TFA and 0.05% FA at a flow rate of 50 μL/min. The protein is eluted over a 20-min run-time monitored by UV detection at 254 nm. After 20 min the flow path was switched to infuse low concentration tune mix (Agilent Technologies, Santa Clara, CA, United States) to calibrate the spectrum post acquisition. The eluent was nebulized and ionized using the Bruker electrospray source with a capillary voltage of 4,500 V dry gas at 180°C, flow rate of 4 L/min and nebulizer gas pressure at 0.6 bar MS2 spectra were acquired by manual selection of isolation mass and isolation width with a collision energy of 32. The spectra were extracted and deconvoluted using Data explorer software version 3.4 build 192 (Bruker Daltonics, Bremen, Germany). For analysis see Supplementary Figures S13, S14.
2.8 Crystallization of PCP2-C3 proteins
Substrate 3 was loaded onto PCP2-C3 affording the holo form of PCP2-C3 and the protein concentrated to a final concentration of 30 mg/mL in gel-filtration buffer. Crystals of PCP2-C3 grew overnight at RT at a protein concentration of 30 mg/mL in a 1:1 ratio (v/v) with a crystallization solution (2 μL drops) comprising 18%–22% v/v PEG 3350 and 0.17–0.3 M magnesium formate. Crystals were cryoprotected by transferring the crystals to a drop composed of reservoir solution supplemented with glycerol (to a final concertation of 30% v/v). Crystals (WT PCP2-C3 Glycostab and R2577G PCP2-C3 Glycostab) were collected in cryoloops and flash frozen in liquid nitrogen.
2.9 Data collection and structure determination
All data sets were collected using either the MX1 or MX2 beamline (Aragao et al., 2018) at the Australian Synchrotron (Clayton, Victoria, Australia) equipped with an Eiger detector (Dectris) at 100 K (McPhillips et al., 2002). Data processing was performed using XDS (Kabsch, 2010) and AIMLESS as implemented in CCP4 (Winn et al., 2011). Phases were obtained via molecular replacement using the PHENIX in-built Phaser module (Adams et al., 2010) and the starting model PDB 7KW0. The crystals belonged to the P212121 space group, with the unit cell comprising 2 highly similar copies of the PCP2-C3 construct (Supplementary Table S3). Structural models were built in COOT (Emsley and Cowtan, 2004) and refined using PHENIX-refine (Adams et al., 2010). All graphics were generated using Pymol (Schrödinger LLC).
2.10 Modelling and docking
A homology model of the NapL C-domain (from naphthyridinomycin biosynthesis, accession no. JQ996389.1) was prepared using the Robetta server using the RoseTTAFold option and default options. Computational docking of the glycolic acid substrate was performed using the induced fit docking protocol in the Schrodinger Suite (Schrödinger Release 2022-1: Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2021; Prime, Schrödinger, LLC, New York, NY, 2021). Briefly, the model of NapL was prepared using default settings in the protein preparation wizard (pH 7.4, including a minimization step) and the ligand (Glycostab-PPant) was prepared using the LigPrep wizard and default settings (retaining stereochemistry of the substrate). The ligand was then docked into the NapL C-domain using the induced fit protocol with default settings, while placing positional restraints on the terminal phosphate group to keep it within a 5 Å sphere of its location in the equivalent crystal structure of FscG-R2577G bound to Glycostab. The top-scoring pose was compared with the crystal structure of FscG-R2577G bound to Glycostab.
3 Results and discussion
3.1 Acceptance of amino acid substrates by the fusachelin C3-domain
In nature, the FscG C3-domain accepts Gly as the acceptor substrate and 2,3-dihydroxybenzoic acid (DHB)-D-Arg-Gly tripeptide as the donor substrate (Dimise et al., 2008). This suggested that the acceptor pocket in this C-domain could be small and without H-bond donors/acceptors. Given this, we were interested in how tolerant this C-domain acceptor pocket would be in accepting different side chains on potential acceptor substrates. To test the selectivity of this C-domain, we used our established condensation assay that exploits the SpyCatcher-SpyTag system (Zakeri et al., 2012), and which is based on an FscG PCP2-C3 construct loaded with a simplified BA-D-Arg-Gly donor substrate and different acceptor substrates loaded onto the PCP3-domain (Izore et al., 2021). Following reassembly of the NRPS using the SpyCatcher-SpyTag system, the PCP-bound substrates were chemically cleaved and initially analyzed by LC-MS. The presence and structure of the cleaved tetrapeptide was then furthered confirmed by high-resolution LC-MS/MS (Figure 2). The percentage conversion for each acceptor substrate was determined by comparing the peak areas of the tetrapeptide product with any remaining tripeptide starting material based on high-resolution LC-MS analysis.
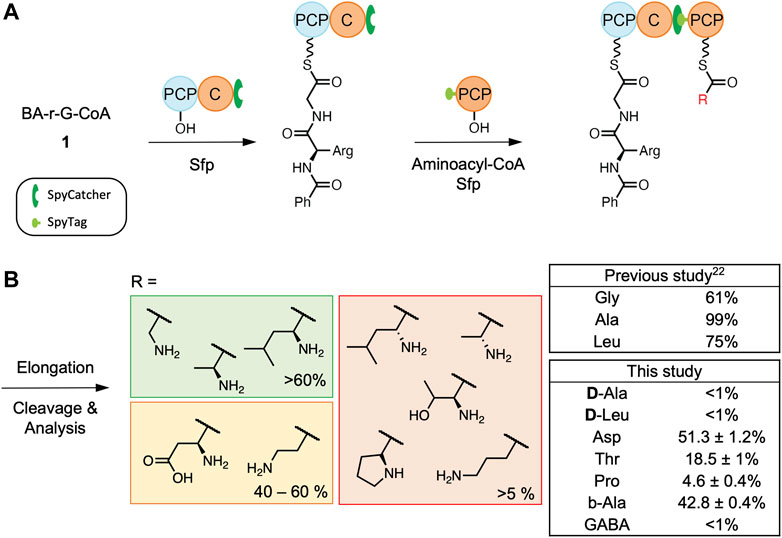
FIGURE 2. C3-domain condensation assays. Scheme of the complete workflow using PCP2-C3 SpyCatcher and SpyTag PCP3 constructs (A) together with tetrapeptide formation by the C3-domain using BA-D-Arg-Gly as a donor substrate and different aminoacyl acceptor substrates (B). All reactions were performed in triplicate. See Supplementary Figures S5–S12 for traces and characterization of the cleaved tetrapeptide. BA- benzoic acid.
As our previous study had showed that the fusachelin C3-domain accepts L-Ala (with 99% conversion) and L-Leu (with 75% conversion) (Izore et al., 2021), we initially decided to test if this domain could utilize acceptor substrates that displayed different sidechain stereochemistry (D-Ala and D-Leu, Figure 2B). Based on the LC-MS results, we determined that this C-domain generally has a strong preference for L-amino acids. This agrees with the general NRPS paradigm of L-configured acceptor substrates being converted into their D-form during or after peptide bond formation (catalyzed by dual C/E or E-domains, respectively) (Linne et al., 2001). Secondly, as we had determined previously that this C-domain has low activity for acceptor substrates with bulky side chains (e.g., phenylalanine), we therefore chose to explore the acceptance of relatively small amino acids as acceptor substrates, including those bearing negatively charged side chains (aspartic acid), polar and uncharged sidechains (threonine), as well as the secondary amino acid proline (Figure 2B). Based on our results, Asp appears to be as well accepted as the control (Gly), whereas there was a significant decrease in peptide extension with Thr and Pro, demonstrating that this C3-domain has limited tolerance for hindered or secondary amino acids. This indicates that whilst the FscG C3-domain can tolerate a range of acceptor substrates with non-polar sidechains (such as Leu), changes beyond relatively “simple” sidechains can lead to unexpected changes in selectivity, possibly proving a hinderance to minimal A-domain engineering approaches for NRPS redesign.
Additionally, we investigated whether the length of the PPant-linked substrate would affect the activity of the FscG C3-domain, as β-Ala and γ-aminobutyric acid (GABA) have been shown to be accepted by C-domains from other systems (Izore et al., 2019; Wei et al., 2023). To this end, we explored the acceptance of β-Ala and GABA to mimic an extension of the PPant-Gly arm by one and two CH2 groups, respectively. The results from these experiments show that extra carbons within the backbone of the amino acid lead to a marked decrease in C3 elongation activity. This is particularly notable when 2 extra carbon atoms were present for the substrate GABA, which afforded only minimal conversion (less than 1%, Figure 2B). This reveals a high degree of the control in terms of acceptor substrate length by the FscG C3-domain and suggests that the PCP interaction with the C-domain is unable to relax sufficiently to allow longer acceptor substrates to be effectively oriented in the C-domain active site.
3.2 Probing C-domain acceptance of altered nucleophile acceptor substrates
Since depsipeptides have a variety of biological activities that are of medical and industrial relevance (Alonzo and Schmeing, 2020), there is great interest in exploring whether archetypal C-domains can catalyze the formation of esters. In this regard, the acceptor substrate glycolic acid is of particular interest as it is widespread in nature (Salusjarvi et al., 2019), raising questions concerning how specificity is maintained in the presence of glycine. To investigate the possibility of ester bond formation by the C3-domain, the thioether stabilized compound 3 was first synthesized as an acceptor substrate and tested using in vitro reconstitution assays. This required analysis by intact protein mass spectrometry coupled with PPant ejection due to the stability of the thioether linkage (Figure 3A). We did not observe the mass of the tetradepsipeptide in these experiments, although the starting reagents were detected (Figure 3B). To probe the causes of the failed elongation reaction, we next turned to X-ray crystallography to gain possible structural insights into the lack of acceptance of this hydroxy acid acceptor by the fusachelin C3-domain.
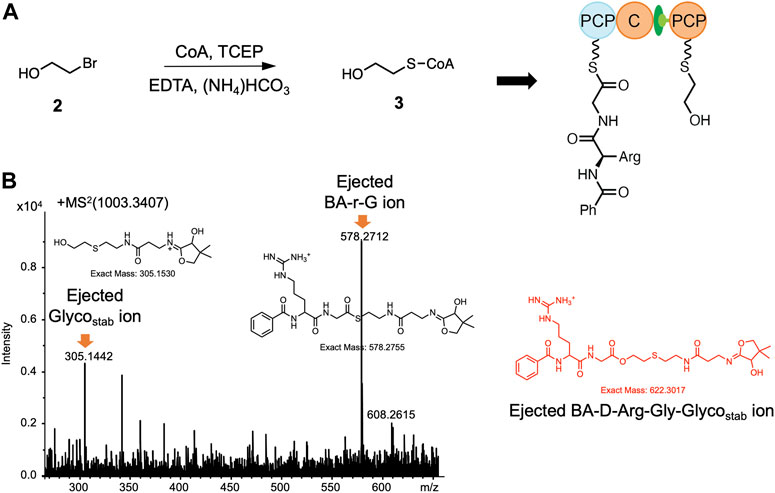
FIGURE 3. The synthesis of 3 and nanoLC-MS analysis of the reconstitution of PCP2-C3:PCP3 WT using 3 as the acceptor substrate. (A) Preparation of 3: 2-bromoethanol (3 eq), CoA (1 eq), TCEP (1.2 eq), EDTA (6.5 mM) (NH4)HCO (20 mM), pH 8, RT, overnight. (B) MS2 spectrum of the 80+ charged ion m/z 1,003.3407, from which the two starting materials were detected but not the anticipated tetrapeptide ester product (m/z = 622.3017). See Supplementary Figures S13 for traces of WT PPant ejection.
3.2.1 Structural characterization of PCP2-C3 didomain loaded with compound 3
Compound 3 was loaded onto the PCP2-C3 didomain and the didomain crystallized as previously described (Izore et al., 2021). Crystals were grown overnight at RT before being harvested, cryoprotected in glycerol and diffraction data collected on the MX1/2 beamlines (Aragao et al., 2018) at the Australian Synchrotron. Phasing was accomplished using molecular replacement (search model: PDB ID 7KW0) and the structure of the complex was determined to a resolution of 3.0 Å in the same space group as the molecular replacement model. We found that the stabilized substrate 3 did not extend into the catalytic channel of C-domain but curled back towards the outer surface of the C-domain (Figure 4A). This has also been previously observed for this didomain with an unloaded PPant arm (PDB ID 7KVW) and could explain why there was no elongation observed in vitro. In the 7KVW structure, the R2577 residue was observed to play a role in preventing the entry of the PPant arm into the acceptor channel of the C-domain, which was rationalized as being caused by a lack of charge-based repulsion for R2577 by the unloaded PPant arm (Izore et al., 2021). As we observed the R2577 residue in the same orientation for the didomain loaded with 3, we suspected that this could be the cause of the lack of elongation in this case. Thus, we next tried to crystallize the PCP2-C3 didomain R2577G mutant loaded with 3. In the structure of this complex, the Glycostab substrate 3 was now fully extended into the catalytic site of the C-domain compared with the WT (Figures 4B,C). Despite this, the hydroxyl group of 3 was not in a favourable orientation for ester formation, being oriented away from the crucial active site histidine residue (Figure 4D), which is also consistent with the lack of elongation seen in the in vitro assays (Supplementary Figure S14).
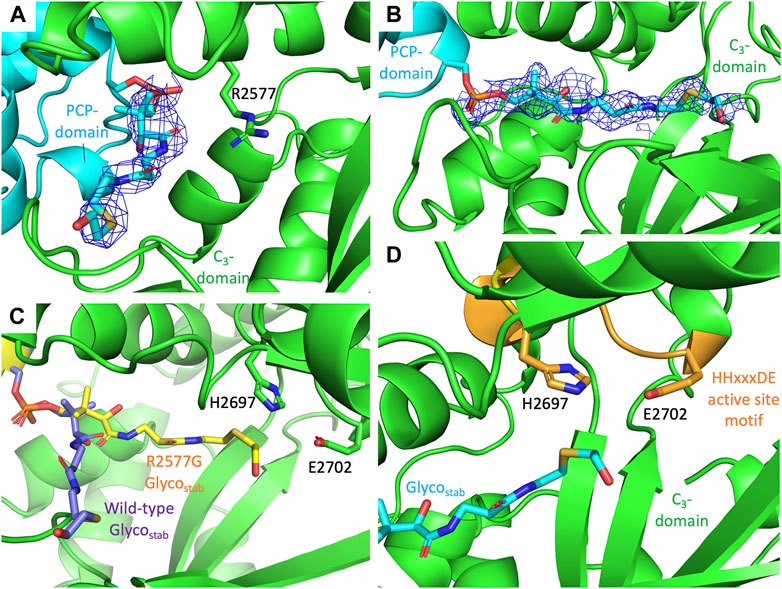
FIGURE 4. PCP2-C3 interactions together with views of the C-domain active site shown together with the position of PPant-Glycostab. (A) Structure of WT C3-domain loaded with Glycostab 3 (PDB: 8G3I), showing the substrate not extending into the C-domain, with R2577 potentially preventing the substrate accessing the C-domain active site. (B) Structure of R2577G C3-domain loaded with Glycostab 3 (PDB: 8G3J), showing the PPant fully extended into the C-domain catalytic channel. (C) Comparison of the positioning of the Glycostab 3 in WT (purple) and R2577G mutant (yellow) variants of the C3-domain. (D) The PPant-Glycostab 3 substrate extends into the C3-domain active site (orange), although the hydroxyl group of is oriented away from the catalytic His residue within the active site HHxxxDE motif (H2696 to E2702). All densities are shown as 2Fo-Fc maps, contoured at 1σ and using a carve value of 1.5 Å.
3.2.2 Comparison of fusachelin C3-domain with the ester-forming C-domain from NapL
To gain insights from other ester forming C-domains, the sequence of the FscG C3-domain was compared with a C-domain that uses a glycolic acid acceptor substrate (from the biosynthesis of naphthyridinomycin, NapL, Figure 5A) (Zhang et al., 2018) and that subsequently forms an ester bond (Supplementary Figure S15). We generated a homology model of the glycolic acid-accepting NapL C-domain and computationally docked the glycolic acid acceptor to identify positions that differed between NapL and FscG and that may account for the differences in their abilities to catalyze the formation of ester bonds (Figure 5B). By combining sequence analysis and docking experiments, we identified several possible active site substitutions that appeared to help position the terminal hydroxyl near the catalytic H2697 residue in NapL: (1) E2702S, allowing the terminal alcohol to orient towards H2697; (2) L2942R, interacting with the terminal hydroxyl group of the substrate; (3) E2950W, preventing the terminal OH from moving away from the catalytic residues; and finally (4) with residues 2,943–2,944 benefiting from shortened sidechains (D2943T, T2944A) when introduced in combination with the L2942R substitution. Taken together with the crystal structure, we designed six different combinations of mutants with a common acceptor-channel mutation (R2577G) and tested their condensation activity: the R2577G E2702S double mutant (GS), the R2577G E2702S L2942R triple mutant (GSR), the R2577G E2702S L2942R D2943T T2944A quintuple mutant (GSRTA), the R2577G E2702S L2942R D2943T T2944A E2950W sextuple mutant (GSRTAW), the R2577G E2702S L2942R E2950W quadruple mutant (GSRW), and the R2577G E2702S E2950W triple mutant (GSW). Firstly, we wanted to test if these 6 mutants would retain the ability to accept Gly as an acceptor substrate. Our results demonstrated that these mutations do indeed alter the C-domain active site as the glycine acceptor was no longer well-accepted by these C-domains (Figure 5C). The result of GS double mutant indicated that changing the E2702 residue in the catalytic motif did not impact the acceptance of Gly, however all other mutants showed a reduction in conversion from 61% to <32% when compared to WT (Supplementary Figures S16–S22).
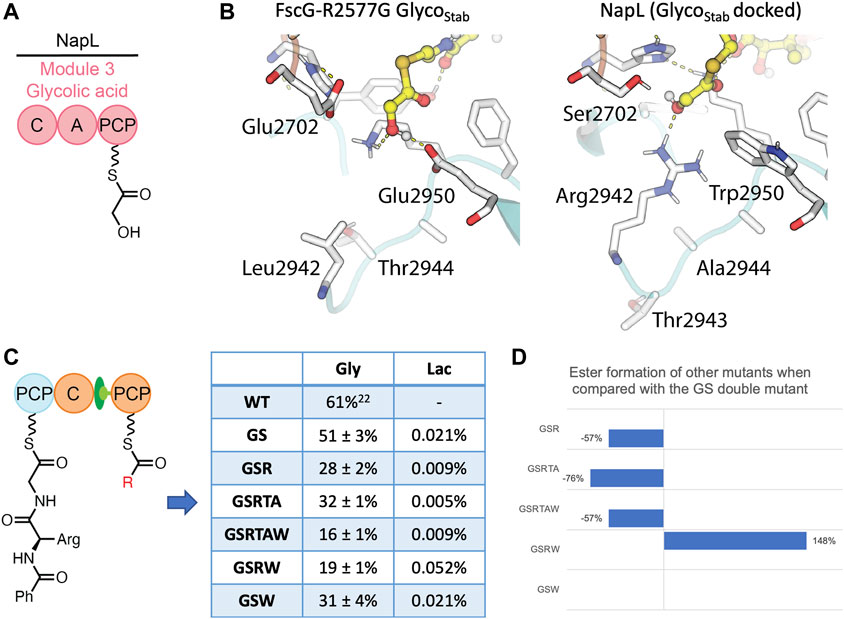
FIGURE 5. Comparison of FscG and NapL C-domains with the results of condensation assays from mutated FscG C3-domain constructs. (A) The NapL C-domain accepts glycolic acid as its acceptor substrate. (B) Comparison of the interactions between the glycolic acid acceptor substrate (yellow) and the active site residues of NapL (left) and FscG (right). Polar interactions shown as yellow dashes. Key residues that differ between NapL and FscG are labelled (using residue numbering for FscG). (C) Level of tetrapeptide formation by designed FscG C-domain mutants using BA-D-Arg-Gly as a donor substrate and Gly/Lac acceptor substrates. (D) The improvement of ester formation level by mutants in comparison with the GS double mutant. All reactions were performed in duplicate. See Supplementary Figures S6–S12, Supplementary Figures S24–S36 for traces and characterization of the cleaved tetrapeptide product.
Next, we explored if these mutants would be active for glycolic acid as an acceptor substrate. As LC-HRMS is a more sensitive and potent tool compared with intact protein MS, we decided to modify our probe from a stabilized glycolyl substrate to a hydrolysable lactyl substrate to simplify both the chemical synthesis of the probe and to enable HRMS analysis. In moving from a system with only one hydrolytically unstable center (PCP-bound thioester) to a depsipeptide system with multiple hydrolytically unstable centers, we first sought to validate our chemical cleavage and analysis protocol. In earlier work, we used a methylamine cleavage protocol to offload the PCP-bound thioester peptides via nucleophilic attack of the amine moiety. To test if these conditions were suitable for recovering ester containing species, we subjected a methionine-methyl ester to these cleavage conditions. We observed complete ester hydrolysis in these experiments, thus indicating a new cleavage strategy was required. The methionine-methyl ester (Supplementary Figures S22) and a PCP-bound tripeptide (Supplementary Figures S23) were then used as substrates for reductive, nucleophilic and thioester exchange conditions, which showed that a MESNa/cysteine-based transthioesterification protocol was able to selectively offload thioesters in the presence of esters (Supplementary Table S4). This protocol was further modified to MESNa/cysteamine to simplify the analytical process. We then applied this optimized cleavage protocol to explore the acceptance of lactic acid by the modified FscG C-domain mutants. All turnovers were initially analyzed by LC-MS, and further by LC-HRMS. LCMS showed that there was limited elongation by any of the six mutants, with LC-HRMS indicating <1% ester formation for all five mutants. Whilst these are a significant improvement in conversion when compared to the lack of ester formation in the wildtype enzyme, these results demonstrate that the control mechanisms governing hydroxy acid acceptance in the FscG C3-domain remain both significant and unclear.
4 Conclusion
In summary, the fuscachelin C3-domain demonstrates selectivity for its acceptor substrates, with those bearing large side chains or D-configured side chains not accepted. Furthermore, reduced activity is observed for acceptor substrates containing secondary amines (Pro) or γ-amino acids. Structural characterization of the PCP-C complex suggests that R2577 can act as a gatekeeper for hydroxy acid acceptor substrates and prevents these from entering the catalytic channel. Although recent studies have shown that engineered A-domains can recognize alternate α-hydroxy acid substrates and that C-domains in some systems are able to accept the hydroxyl substrate and successfully form a depsipeptide (Camus et al., 2022), the FscG C3-domain does not demonstrate this ability. Hence, the mechanisms leading to hydroxy acid acceptance in the FscG C3-domain remain unclear. Our experimental results suggest that C-domains can vary dramatically in their substrate specificity and are not always broadly flexible in accepting different monomers. Taken together, this indicates that future studies using A-domain engineering to introduce different monomers into NRPS biosynthesis may need to consider the specificity of neighboring C-domains. If these domains exhibit unwanted selectivity toward the newly activated substrates this would prevent successful NRPS engineering, and demonstrated that caution is needed when undertaking future NRPS engineering efforts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/pride/archive/, PXD040334; RCSB Protein Data Bank: https://www.rcsb.org/structure/8G3I, https://www.rcsb.org/structure/8G3J.
Author contributions
The study was designed by MC. All cloning and protein purification was performed by YH and TI. Chemical synthesis was performed by YH and JT, structural analysis was performed by YH, TI, and MC with insightful contributions from JK. Condensation assay was performed by YH, MR, and EM and turnovers results were analyzed by YH and DS, with MT assisting with analysis of HRMS experiments. HRMS and protein MS measurements were performed by DS and RS. Computational docking and molecular dynamics simulations were performed and analyzed by JK and CJ. The manuscript was written by YH and MC with input from the other authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Monash University, EMBL Australia, the Australian Research Council (Discovery Project DP210101752 to MC). This research was conducted by the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012) and funded by the Australian Government. This research was undertaken in part using the MX1 and MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.
Acknowledgments
We would like to thank Monash Warwick Alliance for YH studentship; J. Yin (University of Chicago) for the R4-4 Sfp expression plasmid; G. Kong (MMCF, Monash) for assistance with crystal screening experiments. We would also like to thank the beamline scientists at the Australian Synchrotron for their support during data collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2023.1184959/full#supplementary-material
References
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66 (2), 213–221. doi:10.1107/S0907444909052925
Alonzo, D. A., and Schmeing, T. M. (2020). Biosynthesis of depsipeptides, or Depsi: The peptides with varied generations. Protein Sci. 29 (12), 2316–2347. doi:10.1002/pro.3979
Ansari, M. Z., Sharma, J., Gokhale, R. S., and Mohanty, D. (2008). In silico analysis of methyltransferase domains involved in biosynthesis of secondary metabolites. BMC Bioinforma. 9, 454. doi:10.1186/1471-2105-9-454
Aragao, D., Aishima, J., Cherukuvada, H., Clarken, R., Clift, M., Cowieson, N. P., et al. (2018). MX2: A high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 25 (3), 885–891. doi:10.1107/S1600577518003120
Baltz, R. H., Miao, V., and Wrigley, S. K. (2005). Natural products to drugs: Daptomycin and related lipopeptide antibiotics. Nat. Prod. Rep. 22 (6), 717–741. doi:10.1039/b416648p
Belshaw, P. J., Walsh, C. T., and Stachelhaus, T. (1999). Aminoacyl-CoAs as probes of condensation domain selectivity in nonribosomal peptide synthesis. Science 284 (5413), 486–489. doi:10.1126/science.284.5413.486
Bloudoff, K., Fage, C. D., Marahiel, M. A., and Schmeing, T. M. (2017). Structural and mutational analysis of the nonribosomal peptide synthetase heterocyclization domain provides insight into catalysis. Proc. Natl. Acad. Sci. U. S. A. 114 (1), 95–100. doi:10.1073/pnas.1614191114
Bloudoff, K., and Schmeing, T. M. (2017). Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim. Biophys. Acta Proteins Proteom 1865 (11), 1587–1604. doi:10.1016/j.bbapap.2017.05.010
Camus, A., Truong, G., Mittl, P. R. E., Markert, G., and Hilvert, D. (2022). Reprogramming nonribosomal peptide synthetases for site-specific insertion of alpha-hydroxy acids. J. Am. Chem. Soc. 144 (38), 17567–17575. doi:10.1021/jacs.2c07013
Dimise, E. J., Widboom, P. F., and Bruner, S. D. (2008). Structure elucidation and biosynthesis of fuscachelins, peptide siderophores from the moderate thermophile Thermobifida fusca. Proc. Natl. Acad. Sci. U. S. A. 105 (40), 15311–15316. doi:10.1073/pnas.0805451105
Emsley, P., and Cowtan, K. (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132. doi:10.1107/S0907444904019158
Felnagle, E. A., Jackson, E. E., Chan, Y. A., Podevels, A. M., Berti, A. D., McMahon, M. D., et al. (2008). Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 5 (2), 191–211. doi:10.1021/mp700137g
Gaudelli, N. M., Long, D. H., and Townsend, C. A. (2015). beta-Lactam formation by a non-ribosomal peptide synthetase during antibiotic biosynthesis. Nature 520 (7547), 383–387. doi:10.1038/nature14100
Haslinger, K., Peschke, M., Brieke, C., Maximowitsch, E., and Cryle, M. J. (2015). X-domain of peptide synthetases recruits oxygenases crucial for glycopeptide biosynthesis. Nature 521 (7550), 105–109. doi:10.1038/nature14141
Ho, Y. T. C., Schittenhelm, R. B., Iftime, D., Stegmann, E., Tailhades, J., and Cryle, M. J. (2022). Exploring the flexibility of the glycopeptide antibiotic crosslinking cascade for extended peptide backbones. Chembiochem 2022, 00686. doi:10.1002/cbic.202200686
Horsman, M. E., Hari, T. P., and Boddy, C. N. (2016). Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: Logic gate or a victim of fate? Nat. Prod. Rep. 33 (2), 183–202. doi:10.1039/c4np00148f
Izore, T., Candace Ho, Y. T., Kaczmarski, J. A., Gavriilidou, A., Chow, K. H., Steer, D. L., et al. (2021). Structures of a non-ribosomal peptide synthetase condensation domain suggest the basis of substrate selectivity. Nat. Commun. 12 (1), 2511. doi:10.1038/s41467-021-22623-0
Izore, T., Tailhades, J., Hansen, M. H., Kaczmarski, J. A., Jackson, C. J., and Cryle, M. J. (2019). Drosophila melanogaster nonribosomal peptide synthetase Ebony encodes an atypical condensation domain. Proc. Natl. Acad. Sci. U. S. A. 116 (8), 2913–2918. doi:10.1073/pnas.1811194116
Kabsch, W. (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D. Biol. Crystallogr. 66 (2), 133–144. doi:10.1107/S0907444909047374
Kaniusaite, M., Tailhades, J., Marschall, E. A., Goode, R. J. A., Schittenhelm, R. B., and Cryle, M. J. (2019). A proof-reading mechanism for non-proteinogenic amino acid incorporation into glycopeptide antibiotics. Chem. Sci. 10 (41), 9466–9482. doi:10.1039/c9sc03678d
Katsuyama, Y., Sone, K., Harada, A., Kawai, S., Urano, N., Adachi, N., et al. (2021). Structural and functional analyses of the tridomain-nonribosomal peptide synthetase FmoA3 for 4-methyloxazoline ring formation. Angew. Chem. Int. Ed. Engl. 60 (26), 14554–14562. doi:10.1002/anie.202102760
Keating, T. A., Marshall, C. G., Walsh, C. T., and Keating, A. E. (2002). The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat. Struct. Biol. 9 (7), 522–526. doi:10.1038/nsb810
Koetsier, M. J., Jekel, P. A., Wijma, H. J., Bovenberg, R. A., and Janssen, D. B. (2011). Aminoacyl-coenzyme A synthesis catalyzed by a CoA ligase from Penicillium chrysogenum. FEBS Lett. 585 (6), 893–898. doi:10.1016/j.febslet.2011.02.018
Konz, D., Klens, A., Schorgendorfer, K., and Marahiel, M. A. (1997). The bacitracin biosynthesis operon of Bacillus licheniformis ATCC 10716: Molecular characterization of three multi-modular peptide synthetases. Chem. Biol. 4 (12), 927–937. doi:10.1016/s1074-5521(97)90301-x
Lau, Y. K., Baytshtok, V., Howard, T. A., Fiala, B. M., Johnson, J. M., Carter, L. P., et al. (2018). Discovery and engineering of enhanced SUMO protease enzymes. J. Biol. Chem. 293 (34), 13224–13233. doi:10.1074/jbc.RA118.004146
Linne, U., Doekel, S., and Marahiel, M. A. (2001). Portability of epimerization domain and role of peptidyl carrier protein on epimerization activity in nonribosomal peptide synthetases. Biochemistry 40 (51), 15824–15834. doi:10.1021/bi011595t
McPhillips, T. M., McPhillips, S. E., Chiu, H. J., Cohen, A. E., Deacon, A. M., Ellis, P. J., et al. (2002). Blu-ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9 (6), 401–406. doi:10.1107/s0909049502015170
Miller, B. R., and Gulick, A. M. (2016). Structural biology of nonribosomal peptide synthetases. Methods Mol. Biol. 1401, 3–29. doi:10.1007/978-1-4939-3375-4_1
Patteson, J. B., Dunn, Z. D., and Li, B. (2018). In vitro biosynthesis of the nonproteinogenic amino acid methoxyvinylglycine. Angew. Chem. Int. Ed. Engl. 57 (23), 6780–6785. doi:10.1002/anie.201713419
Perez-Riverol, Y., Bai, J., Bandla, C., Garcia-Seisdedos, D., Hewapathirana, S., Kamatchinathan, S., et al. (2022). The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50 (D1), D543–D552. doi:10.1093/nar/gkab1038
Reitz, Z. L., Hardy, C. D., Suk, J., Bouvet, J., and Butler, A. (2019). Genomic analysis of siderophore beta-hydroxylases reveals divergent stereocontrol and expands the condensation domain family. Proc. Natl. Acad. Sci. U. S. A. 116 (40), 19805–19814. doi:10.1073/pnas.1903161116
Salusjarvi, L., Havukainen, S., Koivistoinen, O., and Toivari, M. (2019). Biotechnological production of glycolic acid and ethylene glycol: Current state and perspectives. Appl. Microbiol. Biotechnol. 103 (6), 2525–2535. doi:10.1007/s00253-019-09640-2
Stachelhaus, T., and Walsh, C. T. (2000). Mutational analysis of the epimerization domain in the initiation module PheATE of gramicidin S synthetase. Biochemistry 39 (19), 5775–5787. doi:10.1021/bi9929002
Stanisic, A., and Kries, H. (2019). Adenylation domains in nonribosomal peptide engineering. Chembiochem 20 (11), 1347–1356. doi:10.1002/cbic.201800750
Strieker, M., Tanovic, A., and Marahiel, M. A. (2010). Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 20 (2), 234–240. doi:10.1016/j.sbi.2010.01.009
Sunbul, M., Marshall, N. J., Zou, Y., Zhang, K., and Yin, J. (2009). Catalytic turnover-based phage selection for engineering the substrate specificity of Sfp phosphopantetheinyl transferase. J. Mol. Biol. 387 (4), 883–898. doi:10.1016/j.jmb.2009.02.010
Tan, K., Zhou, M., Jedrzejczak, R. P., Wu, R., Higuera, R. A., Borek, D., et al. (2020). Structures of teixobactin-producing nonribosomal peptide synthetase condensation and adenylation domains. Curr. Res. Struct. Biol. 2, 14–24. doi:10.1016/j.crstbi.2020.01.002
Wang, S., Fang, Q., Lu, Z., Gao, Y., Trembleau, L., Ebel, R., et al. (2021). Discovery and biosynthetic investigation of a new antibacterial dehydrated non-ribosomal tripeptide. Angew. Chem. Int. Ed. Engl. 60 (6), 3229–3237. doi:10.1002/anie.202012902
Weber, T., and Marahiel, M. A. (2001). Exploring the domain structure of modular nonribosomal peptide synthetases. Structure 9 (1), R3–R9. doi:10.1016/s0969-2126(00)00560-8
Wei, X., Chan, T. K., Kong, C. T. D., and Matsuda, Y. (2023). Biosynthetic characterization, heterologous production, and genomics-guided discovery of GABA-containing fungal heptapeptides. J. Nat. Prod. 2023. doi:10.1021/acs.jnatprod.2c01065
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67 (4), 235–242. doi:10.1107/S0907444910045749
Zakeri, B., Fierer, J. O., Celik, E., Chittock, E. C., Schwarz-Linek, U., Moy, V. T., et al. (2012). Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. U. S. A. 109 (12), E690–E697. doi:10.1073/pnas.1115485109
Zhang, Y., Wen, W. H., Pu, J. Y., Tang, M. C., Zhang, L., Peng, C., et al. (2018). Extracellularly oxidative activation and inactivation of matured prodrug for cryptic self-resistance in naphthyridinomycin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 115 (44), 11232–11237. doi:10.1073/pnas.1800502115
Keywords: nonribosomal peptide (NRP), biosynthesis, condensation domain, peptidyl carrier protein, biocatalysis, depsipeptide
Citation: Ho YTC, Izoré T, Kaczmarski JA, Marschall E, Ratnayake MS, Tailhades J, Steer DL, Schittenhelm RB, Tosin M, Jackson CJ and Cryle MJ (2023) Exploring the selectivity and engineering potential of an NRPS condensation domain involved in the biosynthesis of the thermophilic siderophore fuscachelin. Front. Catal. 3:1184959. doi: 10.3389/fctls.2023.1184959
Received: 13 March 2023; Accepted: 23 May 2023;
Published: 16 June 2023.
Edited by:
Mohammed Nabil Ahmed Khalil, Cairo University, EgyptReviewed by:
Steven Gary Van Lanen, University of Kentucky, United StatesKhalid Abdallah Hussein, Assiut University, Egypt
Copyright © 2023 Ho, Izoré, Kaczmarski, Marschall, Ratnayake, Tailhades, Steer, Schittenhelm, Tosin, Jackson and Cryle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Max J. Cryle, TWF4LkNyeWxlQG1vbmFzaC5lZHU=