 Yucheng Yao
Yucheng Yao Jiayi Yao1
Jiayi Yao1- 1Division of Cardiology, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
- 2Molecular Biology Institute, UCLA, Los Angeles, CA, United States
The SRY (Sex Determining Region Y)-related HMG box of DNA binding proteins, referred to as SOX transcription factors, were first identified as critical regulators of male sex determination but are now known to play an important role in vascular development and disease. SOX7, 17, and 18 are essential in endothelial differentiation and SOX2 has emerged as an essential mediator of endothelial-mesenchymal transitions (EndMTs), a mechanism that enables the endothelium to contribute cells with abnormal cell differentiation to vascular disease such as calcific vasculopathy. In the following paper, we review published information on the SOX transcription factors in endothelial differentiation and hypothesize that SOX2 acts as a mediator of EndMTs that contribute to vascular calcification.
Introduction
Appropriate endothelial cell (EC) differentiation is essential to support vascularization of tissues and maintain proper vascular homeostasis. In coordination with tissue development, ECs are derived from progenitor cells that undergo endothelial lineage differentiation to form functional vascular networks (1, 2). In fully developed tissues, quiescent endothelium can be converted to active endothelium as needed for tissue regeneration or repair, and mature endothelial lineage is required to return to and maintain normal vasculature (3). Disease often borrows elements from development such as excessive production of morphogenic factors (4), dysregulation of stem cells (5), abnormal angiogenesis (6), and ectopic cell differentiation (7). Albeit a normal process in neural crest development and cardiac valves and neovascularization (8, 9), endothelial-mesenchymal transitions (EndMTs) contribute to vascular disease when the transitions re-emerge in atypical locations (10–16). EndMTs have been revealed as novel sources of calcifying cells for vascular calcification, which is considered to be a form of ectopic bone formation and involves multipotent cells and networks of growth factors and transcription factors (10). The SOX transcription factors have been shown to be essential mediators in vascular development. Here, we review the SOX factors in endothelial differentiation and EndMTs and include some of our results to support previous studies. In addition, we briefly review the SOX factors in EndMTs, and argue that SOX2 induces EndMTs and serves as a novel cellular source in vascular calcification.
Vascular SOX Transcription Factors
The sex-determining region of the Y-chromosome, the SRY gene, was initially discovered as a testis-determining gene in human and mice (17, 18). It led to the discovery of the family of SRY (Sex Determining Region Y)-related HMG box of DNA binding proteins, referred to as SOX transcription factors, which consists of more than 20 Sox genes (19).
The SOX transcription factors are characterized by the evolutionarily conserved high mobility group (HMG) box, a 79-amino-acid DNA-binding motif that binds to a common consensus site with variable efficiency (19) and are regulated by multiple signaling pathways during vascular development (19–21). They are subdivided into the A-J groups based on phylogenetic analysis of the HMG box sequences, protein structure, gene organization, and function within developmental programs (19, 22, 23). The SOX transcription factors that have been associated with the vasculature are SOX7, SOX17, and SOX18, members of the SOXF subgroup that appear in vascular developmental programs (19–21), and SOX2, a member of the SOXB subgroup that primarily has been associated with EndMTs.
SOX7, SOX17, and SOX18
SOX7, SOX17, and SOX18 are active in early vasculogenesis at the onset of endothelial differentiation and function upstream of signaling cascades that regulate cell fate decisions (19–21). Already on embryonic day (E) 7.5 in mice, ETV2+/FLK1+/CD41– cell populations enriched for endothelial progenitor cells show SOX7 expression in 97%, SOX18 expression in 50%, and SOX17 expression in 75% of the cells (21). SOX7 and SOX18 continue to be expressed in the dorsal aorta, cardinal vein and intersomitic vessels by E8.25 and throughout the developing vascular network at later dates (21, 24, 25). SOX17, on the other hand is detected in EphrinB2+ arterial cells by E10.5, suggesting arterial specificity already at this stage (21, 26, 27). Post-natally, SOX7 and SOX18 continue to be expressed in both arterial and venous endothelium in mice, whereas SOX17 expression is restricted to the arterial endothelium (28). Studies in zebrafish have confirmed similar roles of Sox7 and Sox18 in vascular regulation [reviewed in (21)], whereas Sox17 is not expressed in the developing vasculature of zebrafish.
SOX7 haploinsufficiency has been linked to cardiac defects and congenital diaphragmatic hernia, and is characterized by microdeletions at 8p23.1 that include the Sox7 gene (29). Global gene deletion of Sox7 in mice is associated with embryonic lethality due to absence of the major vessels in the yolk sac and cardiovascular failure (29). Global loss of Sox17 results in depletion of the definitive endoderm and early embryonic lethality (30). However, the cardiovascular defects in Sox17−/− mice are more pronounced in mice with combined loss of Sox17 and Sox18 (24), suggesting redundancy between these two factors. Conditional endothelial-specific Sox17 deletion using Tie2-Cre mice results in blocks in the vascular remodeling of the yolk sac, absence of arteries, and fusion between the aorta and the cardinal vein associated with loss of arteriovenous identity (21, 27). It implies a connection between the SOX transcription factors and Notch signaling that recognizes the important role of Notch in arteriovenous differentiation (20). It has also been shown that Notch signaling can suppress endothelial SOX17, and that this repression induces venous genes such as CoupTFII, while suppressing arterial genes such as EphrinB2, Notch4, and Delta-like ligand (Dll)4 (20, 31). SOX17 may also be a mediator of canonical Wnt signaling in arterial differentiation (32). Global Sox18−/− mice on 129/Sv-CD1 mixed genetic background were initially reported to be viable without gross abnormalities in the cardiovascular system (33). However, Sox18−/− mice on pure C57BL/6 background develop subcutaneous edema and embryonic lethality due to interference in the lymphangiogenesis (34, 35), which supports the hypothesis that SOX18 plays an important role in lymphangiogenesis (21, 34, 35).
SOX2
SOX2 is essential for regulation of interactions between the epithelium and the mesenchyme (36), differentiation of multiple cell lineages (37–40) and cell fate transitions (41, 42). SOX2 may be best known as one of the four original pluripotent factors that together with octamer-binding transcription factor 3/4 (Oct3/4), Kruppel-like factor 4 (Klf4), and c-Myc is used for the reprogramming of cells (43) and serves as a marker of neural stem cells (44, 45). SOX2 also enhances the reprogramming capacity of cardiovascular cells, and has been shown to induce endothelial differentiation in isolated adult mesoangioblasts (46) and participate in the reprogramming of corneal endothelial cells (47).
To study the role of SOX2 in the developing endothelium, we used an embryonic stem cells (ESCs) model of endothelial differentiation (48) and examined the temporal expression of SOX2 and endothelial markers. The endothelial markers emerged between day 3 and 6 of endothelial induction, as the expression of SOX2 increased (Figure 1A), which suggested that involvement of SOX2 might be required for EC differentiation. Therefore, we depleted Sox2 transcripts in the ESCs on day 3 using siRNA, and found that the reduction of SOX2 suppressed EC differentiation (Figure 1B). Interestingly, the cells still kept the ability to differentiate into other lineages after depletion of Sox2, including neuronal differentiation (data not shown), suggesting that suppression of Sox2 may alter the direction of endothelial differentiation. Our results supported a role for SOX2 in the endothelial integrity, although it is unknown if SOX2 directly targets or interacts with early drivers of endothelial differentiation.
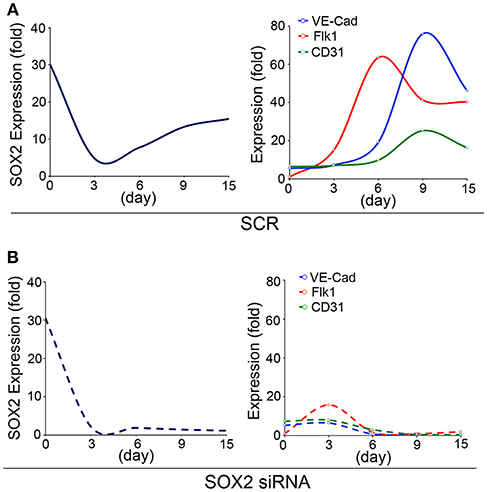
Figure 1. SOX2 plays a role in endothelial differentiation. Time course of expression of (left panels) SOX2, and (right panels) fetal liver kinase 1 (Flk1), VE-cadherin (VE-Cad) and cluster of differentiation 31 (CD31), as determined by real-time PCR during endothelial cell derivation from wild type embryonic stem cells (15 days). (A) Scrambled siRNA (SCR) or (B) specific Sox2 siRNAs were transfected into the embryonic stem cells on day 3. Gene expression is shown as fold change compared to the expression on day 0 (n = 5).
SOX2 is also known to be a key regulator of neuronal differentiation (37). In previous studies, we found ECs that were double positive for the endothelial marker fetal liver kinase 1 (Flk1) and SOX2 adjacent to differentiating brain cells on E10.5 and E14 (49). Flow cytometric analysis of dissected E12.5 embryonic brains confirmed the presence of cerebral ECs that co-expressed the endothelial marker VE-cadherin and SOX2 (49). This suggested that the ECs and the brain cells originated from the same progenitor cells and the endothelial and neuronal differentiation were coordinated. Similar observations were made in other organs, such as the lungs and the liver (49), suggesting the presence of differentiation “forks” involving ECs and organ-specific cells that are aimed at coordinating the developmental progression.
To further examine the potential role of SOX2 in such a coordination, we induced neuronal differentiation in ESCs (50). We found that the Sox2 depletion delayed the endothelial marker induction and changed the temporal sequence of neuronal and endothelial differentiation (Figure 2A). The results showed that expression of SOX2 peaked twice during the neuronal differentiation, on day 3 and day 6 (Figure 2B), which differs from the SOX2 expression in endothelial differentiation (Figure 1). We examined the expression of the neuronal lineage markers SOX1, paired box protein (Pax6), and Nestin, together with the endothelial markers VE-cadherin, Flk1, and cluster of differentiation 31 (CD31) for up to 15 days. The results showed that the endothelial markers were induced during neuronal differentiation even without specific EC induction (Figures 2C–F). The time course suggested that the two types of differentiation were orchestrated, such that expression of endothelial markers was high when expression of neuronal markers was low, and vice versa (Figures 2C–F). The temporal sequence in this cell model appeared to be neuronal-endothelial-neuronal-endothelial (Figure 2A).
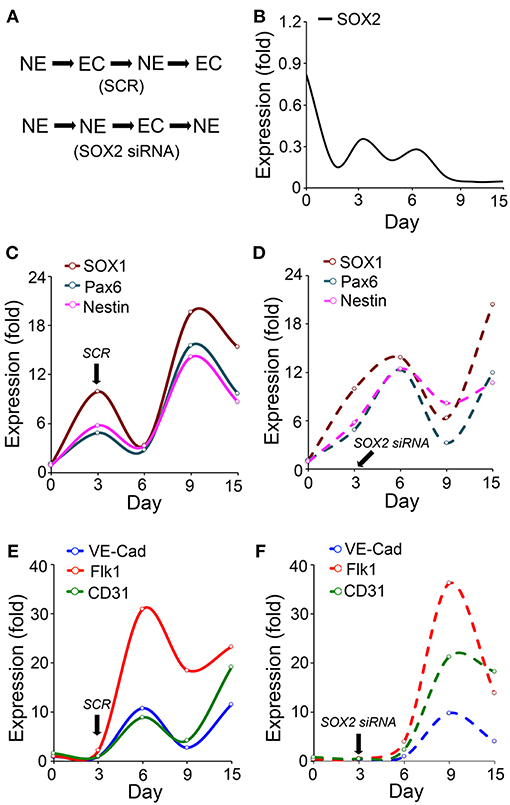
Figure 2. Endothelial differentiation co-exists with neural differentiation. (A) Schematic diagram of the temporal induction patterns of coordinated endothelial (EC) and neuronal (NE) differentiation. (B–F) Marker expression during neuronal differentiation in wild type embryonic stem cells with or without suppression of SOX2 (n = 5). Scrambled siRNA (SCR) or specific Sox2 siRNAs were transfected into the cells on day 3. Expression was determined by real-time PCR and calculated as fold change compared to the expression on day 0. (B–D) Time course of expression of SOX2, and the neural progenitor markers SOX1, paired box protein (Pax6), and nestin. (E,F) Time course expression of the endothelial markers VE-cadherin (VE-Cad), fetal liver kinase 1 (Flk1) and cluster of differentiation 31 (CD31).
SOX2, EndMTs, and Vascular Calcification
Vascular calcification is a frequent complication of vascular disease (7, 51–54) that exhibits multiple patterns of calcification depending on the type of disease, the type of vessel and the vascular layer that is affected (55). Several sources of calcifying cells have been identified, including vascular medial cells such as smooth muscle cells and pericytes, adventitial cells, ECs, various progenitor cells and osteoclast-like cells (55). Reports of endothelial contributions to calcific lesions suggest that EndMTs mediate direct contributions of osteogenic cells from the endothelium, thereby giving the endothelium a direct role in the development of vascular calcification.
EndMTs occur first in development and have been clearly demonstrated during heart valve formation (8, 56, 57) and recur in adult disease processes in the cardiac valves (8, 58), pulmonary artery hypertension (59), atherosclerosis and vascular calcification (59, 60). Several members of the transforming growth factor (TGF)beta superfamily, such as TGFbeta and bone morphogenetic proteins (BMPs) (61) have been shown to be important regulators of EndMTs.
Abnormal TGFbeta signaling induces mesenchymal-like phenotype in a variety of ECs (8, 59, 62–64) and both BMP4 and BMP6 have been implicated in EndMTs (65–67). Mutations in the BMP receptor activin receptor-like kinase 2 (ALK2) are causative in fibrodysplasia ossificans progressiva, where capillary ECs contribute cells to calcific lesions through endothelial transitions (13). Gene deletion of the BMP inhibitor matrix Gla protein (MGP) results in excess BMP signaling and rapidly developing arterial calcification (68) involving extensive EndMTs (10, 65).
Several factors have emerged as important participants in the crosstalk between TGFbeta and BMP signaling in EndMTs. These include Notch signaling, which is essential in EndMTs in heart development and valve formation [reviewed in (59, 69)], hypoxia (70) and fibroblast growth factor (FGF) signaling (71). Another factor is Wnt signaling, which is active in lymphatic ECs responding to Wnt5b (72), valvular ECs responding to matrix stiffness (73), and ECs transitioning to cardiac smooth muscle cells and pericytes under the influence of paracrine Wnt ligands (74). Interestingly, lack of primary cilia in a model of mouse embryonic ECs has been shown to increase the propensity to undergo EndMTs and osteogenesis in response to BMP signaling (75), potentially due to altered responses to mechanical and chemical stimuli.
In our studies using the Mgp−/− and Ins2Akita/+ diabetic mouse models of vascular calcification, we identified SOX2 as a response gene to ectopic BMP activity and a master regulator of EndMTs (10, 65). EndMTs are especially prominent in the aortas of Mgp−/− mice (10–12, 65), where they contribute to the rapid calcification. Observed through phase contrast and transmission electron microscopy, the endothelium was highly abnormal with a mixture of cells largely replacing normal ECs, including chondroblast-like cells (65). EC-like cells were surrounded by abnormal matrix and detached from the internal elastic lamina (IEL). Transmission and scanning electron microscopy showed a marked degradation of the IEL, usually in close contact with endothelium [(65), Figure 3A]. Ultimately, the IEL became undetectable with the EC-like cells positioned as if migrating from the luminal side toward the calcifying lesions (65). Endothelial markers were detected deep in the calcified media, where they co-localized with osteogenic markers (65). Further studies revealed that the degradation of the IEL resulted from the induction of a complex of specific serine proteases including elastase 1 and 2 and kallikrein 1, 5, and 6 (65). The expression of these serine proteases increased dramatically in association with the degradation of both the IEL and the elastic lamellae in the media and induction of endothelial SOX2 as the Mgp−/− aortas calcified (Figures 3B,C). Assessment of the proteolytic activity showed that the proteases are able to degrade collagen I, II, III, and IV, fibronectin, fibrinogen, and laminin (Figure 3D). In addition, the serine proteases were able to induce endothelial SOX2 and activate EndMTs (65). Both serine protease inhibition and Sox2 depletion in the endothelium diminished EndMTs and vascular calcification in vitro and in vivo (65). Similar results for SOX2 as a mediator of vascular calcification were also found in atherosclerotic and diabetic mice, which showed that genetically limiting SOX2 in InsAkita/+ mice or inhibiting SOX2 by siRNA in Apoe−/− mice fed a Western diet reduced vascular calcification (11, 12). We argue that on one hand, the induction of the serine proteases plays an initial role in triggering endothelial SOX2 and activating EndMTs. On other hand, the local milieu with excessive degradation of elastin and cell-matrix allows the transitioning ECs to migrate and contribute to the calcification (Figure 4).
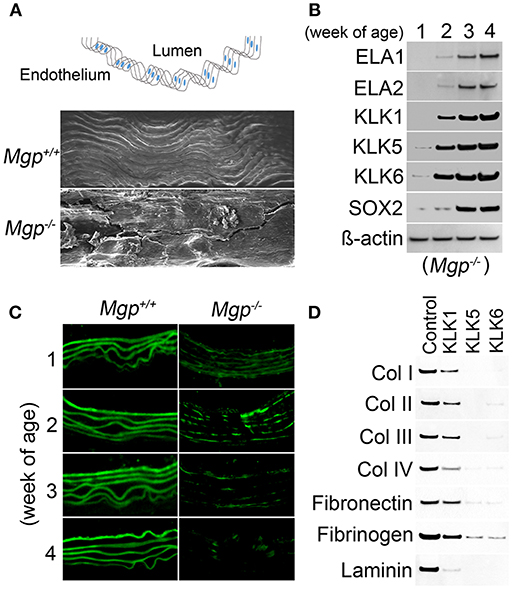
Figure 3. Serine proteases degrade Mgp-/- aortic tissue (A) Wild type (Mgp+/+) and Mgp−/− aortic endothelium at 4 weeks of age was examined by scanning electron microscopy (magnification 5 × 102). The image shows significant disruption of the Mgp−/− endothelium and elastic lamina (n = 3). Top, schematic diagram indicating the locations of lumen and endothelium. (B) Expression of elastase (ELA) 1 and 2 and kallikrein (KLK) 1, 5, and 6 and SOX2 in Mgp−/− aortas at 1–4 weeks of age, as determined by immunoblotting. Beta-actin was used as a loading control (n = 3). (C) Immunofluorescent staining of aortic elastin of wild type (Mgp+/+) and Mgp−/− mice at 1–4 weeks of age. The images reveal the degradation of Mgp−/− aortic elastin starting at 2 weeks of age (n = 5). (D) Immunoblotting demonstrates that kallikrein (KLK) 1, 5, and 6 degrade matrix components of aortic tissues including collagen (Col) 1, II, III, and IV, fibronectin, fibrinogen and laminin in vitro. One mg of substrate protein was mixed with 100 ng of enzyme or control at 37°C for 1 h before the samples were analyzed by immunoblotting (n = 3).
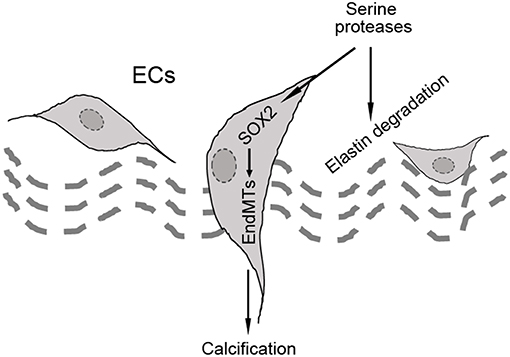
Figure 4. Schematic diagram of the hypothesis that SOX2 acts as a mediator of endothelial-mesenchymal transitions (EndMTs).
Potential roles for other SOX transcription factors in acquired vascular disease have not been well-studied. SOX17 has been identified as a risk locus for intracranial aneurysms (76) and SOX17 deficiency, which affects EC regeneration, may predispose to stress-induced intracranial aneurysms in hypertensive mice (77). SOX18 expression has been reported in advanced coronary atherosclerotic lesions (78), where it is expressed in both ECs and smooth muscle cells and may be involved in cell growth. However, it is unknown if SOX2 interacts with other members of SOX family to induce EndMTs.
Altogether, the SOX transcription factors are emerging as increasingly important players in cellular transitions, endothelial dysfunction and vascular disease. The SOX factors may be useful targets for endothelial modulation in the prevention or treatment of vascular calcification.
Methods
Animals
Mgp+/− (B6.129S7-Mgptm1Kry/KbosJ) on C57BL/6J background were obtained from the Jackson Laboratory. Genotypes were confirmed by PCR (79), and experiments were performed with generations F4-F6. Littermates were used as wild type controls. All mice were fed a standard chow diet (Diet 8604, HarlanTeklad Laboratory). The studies were reviewed and approved by the Institutional Review Board and conducted in accordance with the animal care guidelines set by the University of California, Los Angeles. The investigation conformed to the National Research Council, Guide for the Care and Use of Laboratory Animals, Eighth Edition (Washington, DC: The National Academies Press, 2011).
Tissue Culture and Cell Differentiation
Wild type ESCs (C57BL/6J background) were obtained from American Type Culture Collection (ATCC) (SCRC-1002). Mouse ESCs were cultured and maintained as previously described (48, 80). The derivation of endothelial cell differentiation from ESCs was performed using previously published protocols (48, 81). BMP-4, Activin A, FGF-2, and VEGF (all from R&D Systems) were added to StemPro-34® medium prior to use. The process of derivation lasted 14 days.
Neuronal differentiation in ESCs was performed using previously published protocols (82). Briefly, mouse ESCs without feeder cells were dispersed into a single cell suspension with 0.25% trypsin. Aggregation of ESCs was induced by preparing hanging drops of medium (20 μL) on the lids of petri dishes (2000 cells per drop) on day 0. After 2 days, embryonic bodies were harvested in petri dishes, where they matured for 3 days. The medium was changed every 1–2 days based on the number of dead cells. On day 5, the embryonic bodies were plated on coated cell culture dishes. In order to increase the adherence of embryonic bodies, the dishes were coated with laminin for one day and D-lysine for another day before use. The embryonic bodies were subsequently cultured for two more days. The medium was changed on day 7, and every 1–2 days after that. Mature neurons were observed subsequently to day 7.
Protease Assay
Kallikrein 1, 5, and 6 (all 10 ng/mL; Abnova) were individually added to the purified proteins, collagen I, II, III, and IV, fibronectin, fibrinogen, and laminin, and incubated at 37°C for 1 h. After the incubation, the mixtures were examined by immunoblotting to assess the degradation of each protein using specific antibodies. The carrier was used as a control.
RNA Analysis
Real-time PCR analysis was performed as previously described (65). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control gene (65). Primers and probes for mouse SOX2, Flk1, CD31, VE-cadherin, SOX2, Pax6, and Nestin were obtained from Applied Biosystems as part of Taqman® Gene Expression Assays.
Immunofluorescence
Immunofluorescence was performed as previously described (65) with specific antibodies for elastin (Abcam). The nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich).
Scanning Electron Microscopy
Aortic tissue samples were analyzed by scanning electron microscopy as previously described (65).
Immunoblotting
Immunoblotting and immunoprecipitation were performed as previously described (65). Equal amounts of tissue lysates were analyzed, and the blots were incubated with specific antibodies to elastase 1 (Sigma-Aldrich), elastase 2 (Abgent), kallikrein 1 (Sigma-Aldrich), kallikrein 2 (Abgent), kallikrein 5 (Acris Antibodies), kallikrein 6 (Sigma-Aldrich), and collagen I, II, III, IV, fibronectin, fibrinogen and laminin (all from Abcam) as previously described (65). Beta-Actin (Sigma-Aldrich) was used as a loading control.
Statistical Analysis
Data were analyzed for statistical significance by ANOVA with post-hoc Tukey's analysis. The analyses were performed using GraphPad Instat®, version 3.0 (GraphPad Software). Experiments were repeated a minimum of three times.
Author Contributions
YY and KB supervised the experiments, analyzed data, and wrote the manuscript. JY performed experiments and data analysis.
Funding
Funding for this work was provided in part by NIH grants NS79353 (YY), HL139675 (YY), HL30568 (KB), and HL81397 (KB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Marcelo KL, Goldie LC, Hirschi KK. Regulation of endothelial cell differentiation and specification. Circ Res. (2013) 112:1272–87. doi: 10.1161/CIRCRESAHA.113.300506
2. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. (2016) 529:316–25. doi: 10.1038/nature17040
3. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. (2015) 116:1231–44. doi: 10.1161/CIRCRESAHA.116.302855
4. Yao Y, Jumabay M, Wang A, Bostrom KI. Matrix Gla protein deficiency causes arteriovenous malformations in mice. J Clin Invest. (2011) 121:2993–3004. doi: 10.1172/JCI57567
5. Lee KH. “Sack of marbles” in mature cystic ovarian teratoma. Abdom Radiol. (2017) 42:1616–7. doi: 10.1007/s00261-017-1059-x
6. Marchuk DA, Srinivasan S, Squire TL, Zawistowski JS. Vascular morphogenesis: tales of two syndromes. Hum Mol Genet. (2003) 12:R97–112. doi: 10.1093/hmg/ddg103
7. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. (2010) 7:528–36. doi: 10.1038/nrcardio.2010.115
8. Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. (2018) 15:445–56. doi: 10.1038/s41569-018-0023-y
9. Ubil E, Duan J, Pillai IC, Rosa-Garrido M, Wu Y, Bargiacchi F, et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. (2014) 514:585–90. doi: 10.1038/nature13839
10. Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res. (2013) 113:495–504. doi: 10.1161/CIRCRESAHA.113.301792
11. Bostrom KI, Yao J, Guihard PJ, Blazquez-Medela AM, Yao Y. Endothelial-mesenchymal transition in atherosclerotic lesion calcification. Atherosclerosis. (2016) 253:124–7. doi: 10.1016/j.atherosclerosis.2016.08.046
12. Guihard PJ, Yao J, Blazquez-Medela AM, Iruela-Arispe L, Bostrom KI, Yao Y. Endothelial-Mesenchymal Transition in Vascular Calcification of Ins2Akita/+ Mice. PLoS ONE. (2016) 11:e0167936. doi: 10.1371/journal.pone.0167936
13. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. (2010) 16:1400–6. doi: 10.1038/nm.2252
14. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. (2003) 112:1776–84. doi: 10.1172/JCI200320530
15. Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. (2015) 131:1006–18. doi: 10.1161/CIRCULATIONAHA.114.008750
16. Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. (2008) 13:6991–8. doi: 10.2741/3204
17. Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Munsterberg A, et al. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. (1990) 346:245–50. doi: 10.1038/346245a0
18. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. (1990) 346:240–4. doi: 10.1038/346240a0
19. She ZY, Yang WX. SOX family transcription factors involved in diverse cellular events during development. Eur J Cell Biol. (2015) 94:547–63. doi: 10.1016/j.ejcb.2015.08.002
20. Fish JE, Wythe JD. The molecular regulation of arteriovenous specification and maintenance. Dev Dyn. (2015) 244:391–409. doi: 10.1002/dvdy.24252
21. Lilly AJ, Lacaud G, Kouskoff V. SOXF transcription factors in cardiovascular development. Semin Cell Dev Biol. (2017) 63:50–57. doi: 10.1016/j.semcdb.2016.07.021
22. Schepers GE, Teasdale RD, Koopman P. Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell. (2002) 3:167–70. doi: 10.1016/S1534-5807(02)00223-X
23. Bowles J, Schepers G, Koopman P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol. (2000) 227:239–55. doi: 10.1006/dbio.2000.9883
24. Sakamoto Y, Hara K, Kanai-Azuma M, Matsui T, Miura Y, Tsunekawa N, et al. Redundant roles of Sox17 and Sox18 in early cardiovascular development of mouse embryos. Biochem Biophys Res Commun. (2007) 360:539–44. doi: 10.1016/j.bbrc.2007.06.093
25. Takash W, Canizares J, Bonneaud N, Poulat F, Mattei MG, Jay P, et al. SOX7 transcription factor: sequence, chromosomal localisation, expression, transactivation and interference with Wnt signalling. Nucl Acids Res. (2001) 29:4274–83. doi: 10.1093/nar/29.21.4274
26. Matsui T, Kanai-Azuma M, Hara K, Matoba S, Hiramatsu R, Kawakami H, et al. Redundant roles of Sox17 and Sox18 in postnatal angiogenesis in mice. J Cell Sci. (2006) 119:3513–26. doi: 10.1242/jcs.03081
27. Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, et al. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun. (2013) 4:2609. doi: 10.1038/ncomms3609
28. Zhou Y, Williams J, Smallwood PM, Nathans J. Sox7, Sox17, and Sox18 cooperatively regulate vascular development in the mouse retina. PLoS ONE. (2015) 10:e0143650. doi: 10.1371/journal.pone.0143650
29. Wat MJ, Beck TF, Hernandez-Garcia A, Yu Z, Veenma D, Garcia M, et al. Mouse model reveals the role of SOX7 in the development of congenital diaphragmatic hernia associated with recurrent deletions of 8p23.1. Hum Mol Genet. (2012) 21:4115–25. doi: 10.1093/hmg/dds241
30. Kanai-Azuma M, Kanai Y, Gad JM, Tajima Y, Taya C, Kurohmaru M, et al. Depletion of definitive gut endoderm in Sox17-null mutant mice. Development. (2002) 129:2367–79.
31. Corada M, Morini MF, Dejana E. Signaling pathways in the specification of arteries and veins. Arterioscler Thromb Vasc Biol. (2014) 34:2372–7. doi: 10.1161/ATVBAHA.114.303218
32. Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, et al. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. (2009) 139:285–98. doi: 10.1016/j.cell.2009.07.047
33. Pennisi D, Bowles J, Nagy A, Muscat G, Koopman P. Mice null for sox18 are viable and display a mild coat defect. Mol Cell Biol. (2000) 20:9331–6. doi: 10.1128/MCB.20.24.9331-9336.2000
34. Francois M, Caprini A, Hosking B, Orsenigo F, Wilhelm D, Browne C, et al. Sox18 induces development of the lymphatic vasculature in mice. Nature. (2008) 456:643–7. doi: 10.1038/nature07391
35. Hosking B, Francois M, Wilhelm D, Orsenigo F, Caprini A, Svingen T, et al. Sox7 and Sox17 are strain-specific modifiers of the lymphangiogenic defects caused by Sox18 dysfunction in mice. Development. (2009) 136:2385–91. doi: 10.1242/dev.034827
36. Pispa J, Thesleff I. Mechanisms of ectodermal organogenesis. Dev Biol. (2003) 262:195–205. doi: 10.1016/S0012-1606(03)00325-7
37. Amador-Arjona A, Cimadamore F, Huang CT, Wright R, Lewis S, Gage FH, et al. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proc Natl Acad Sci USA. (2015) 112:E1936–45. doi: 10.1073/pnas.1421480112
38. Ochieng JK, Schilders K, Kool H, Boerema-De Munck A, Buscop-Van Kempen M, et al. Sox2 regulates the emergence of lung basal cells by directly activating the transcription of Trp63. Am J Respir Cell Mol Biol. (2014) 51:311–22. doi: 10.1165/rcmb.2013-0419OC
39. Clavel C, Grisanti L, Zemla R, Rezza A, Barros R, Sennett R, et al. Sox2 in the dermal papilla niche controls hair growth by fine-tuning BMP signaling in differentiating hair shaft progenitors. Dev Cell. (2012) 23:981–94. doi: 10.1016/j.devcel.2012.10.013
40. Basu-Roy U, Ambrosetti D, Favaro R, Nicolis SK, Mansukhani A, Basilico C. The transcription factor Sox2 is required for osteoblast self-renewal. Cell Death Different. (2010) 17:1345–53. doi: 10.1038/cdd.2010.57
41. Mandalos N, Rhinn M, Granchi Z, Karampelas I, Mitsiadis T, Economides AN, et al. Sox2 acts as a rheostat of epithelial to mesenchymal transition during neural crest development. Front Physiol. (2014) 5:345. doi: 10.3389/fphys.2014.00345
42. Luo W, Li S, Peng B, Ye Y, Deng X, Yao K. Embryonic stem cells markers SOX2, OCT4 and Nanog expression and their correlations with epithelial-mesenchymal transition in nasopharyngeal carcinoma. PLoS ONE. (2013) 8:e56324. doi: 10.1371/journal.pone.0056324
43. Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev. (2016) 17:183–93. doi: 10.1038/nrm.2016.8
44. Sarlak G, Vincent B. The roles of the stem cell-controlling sox2 transcription factor: from neuroectoderm development to alzheimer's disease? Mol Neurobiol. (2016) 53:1679–98. doi: 10.1007/s12035-015-9123-4
45. Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. (2004) 131:3805–19. doi: 10.1242/dev.01204
46. Koyanagi M, Iwasaki M, Rupp S, Tedesco FS, Yoon CH, Boeckel JN, et al. Sox2 transduction enhances cardiovascular repair capacity of blood-derived mesoangioblasts. Circ Res. (2010) 106:1290–302. doi: 10.1161/CIRCRESAHA.109.206045
47. Zhu YT, Li F, Han B, Tighe S, Zhang S, Chen SY, et al. Activation of RhoA-ROCK-BMP signaling reprograms adult human corneal endothelial cells. J Cell Biol. (2014) 206:799–811. doi: 10.1083/jcb.201404032
48. Yao J, Guihard PJ, Blazquez-Medela AM, Guo Y, Liu T, Bostrom KI, et al. Matrix Gla protein regulates differentiation of endothelial cells derived from mouse embryonic stem cells. Angiogenesis. (2016) 19:1–7. doi: 10.1007/s10456-015-9484-3
49. Bostrom KI, Yao J, Wu X, Yao Y. Endothelial cells may have tissue-specific origins. J Cell Biol Histol. (2018) 1:104. doi: 10.15744/2638-082x.1.104
50. Kim M, Habiba A, Doherty JM, Mills JC, Mercer RW, Huettner JE. Regulation of mouse embryonic stem cell neural differentiation by retinoic acid. Dev Biol. (2009) 328:456–71. doi: 10.1016/j.ydbio.2009.02.001
51. Ladich E, Yahagi K, Romero ME, Virmani R. Vascular diseases: aortitis, aortic aneurysms, and vascular calcification. Cardiovasc Pathol. (2016) 25:432–41. doi: 10.1016/j.carpath.2016.07.002
52. Shao JS, Cheng SL, Sadhu J, Towler DA. Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension. (2010) 55:579–92. doi: 10.1161/HYPERTENSIONAHA.109.134205
53. Schlieper G, Schurgers L, Brandenburg V, Reutelingsperger C, Floege J. Vascular calcification in chronic kidney disease: an update. Nephrol Dial Transl. (2016) 31:31–9. doi: 10.1093/ndt/gfv111
54. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
55. Bostrom KI. Where do we stand on vascular calcification? Vasc Pharmacol. (2016) 84:8–14. doi: 10.1016/j.vph.2016.05.014
56. Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. (2004) 18:99–115. doi: 10.1101/gad.276304
57. Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res. (2011) 109:183–92. doi: 10.1161/CIRCRESAHA.111.245035
58. Wylie-Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler Thromb Vasc Biol. (2011) 31:598–607. doi: 10.1161/ATVBAHA.110.216184
59. Sanchez-Duffhues G, Garcia de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev Dyn. (2018) 247:492–508. doi: 10.1002/dvdy.24589
60. Hortells L, Sur S, St Hilaire C. Cell Phenotype Transitions in Cardiovascular Calcification. Front Cardiovasc Med. (2018) 5:27. doi: 10.3389/fcvm.2018.00027
61. Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. (2016) 12:203–21. doi: 10.1038/nrendo.2016.12
62. Diez M, Musri MM, Ferrer E, Barbera JA, Peinado VI. Endothelial progenitor cells undergo an endothelial-to-mesenchymal transition-like process mediated by TGFbetaRI. Cardiovasc Res. (2010) 88:502–11. doi: 10.1093/cvr/cvq236
63. Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. (2002) 90:1189–96. doi: 10.1161/01.RES.0000021432.70309.28
64. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. (2008) 121:3317–24. doi: 10.1242/jcs.028282
65. Yao J, Guihard P, Blazquez-Medela AM, Guo Y, Moon JH, Jumabay M, et al. Serine protease activation essential for endothelial-mesenchymal transition in vascular calcification. Circ Res. (2015) 117:758–69. doi: 10.1161/CIRCRESAHA.115.306751
66. Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. (2013) 498:492–6. doi: 10.1038/nature12207
67. Yung LM, Sanchez-Duffhues G, Ten Dijke P, Yu PB. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res. (2015) 108:278–87. doi: 10.1093/cvr/cvv221
68. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. (1997) 386:78–81. doi: 10.1038/386078a0
69. Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res. (2008) 102:1169–81. doi: 10.1161/CIRCRESAHA.108.174318
70. Doerr M, Morrison J, Bergeron L, Coomber BL, Viloria-Petit A. Differential effect of hypoxia on early endothelial-mesenchymal transition response to transforming growth beta isoforms 1 and 2. Microvasc Res. (2016) 108:48–63. doi: 10.1016/j.mvr.2016.08.001
71. Xiao L, Dudley AC. Fine-tuning vascular fate during endothelial-mesenchymal transition. J Pathol. (2017) 241:25–35. doi: 10.1002/path.4814
72. Wang SH, Chang JS, Hsiao JR, Yen YC, Jiang SS, Liu SH, et al. Tumour cell-derived WNT5B modulates in vitro lymphangiogenesis via induction of partial endothelial-mesenchymal transition of lymphatic endothelial cells. Oncogene. (2017) 36:1503–1515. doi: 10.1038/onc.2016.317
73. Zhong A, Mirzaei Z, Simmons CA. The Roles of Matrix Stiffness and ss-Catenin Signaling in Endothelial-to-Mesenchymal Transition of Aortic Valve Endothelial Cells. Cardiovasc Eng Technol. 2018. doi: 10.1007/s13239-018-0363-0
74. Chen Q, Zhang H, Liu Y, Adams S, Eilken H, Stehling M, et al. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat Commun. (2016) 7:12422. doi: 10.1038/ncomms12422
75. Sanchez-Duffhues G, de Vinuesa AG, Lindeman JH, Mulder-Stapel A, DeRuiter MC, Van Munsteren C, et al. SLUG is expressed in endothelial cells lacking primary cilia to promote cellular calcification. Arterioscler Thromb Vasc Biol. (2015) 35:616–27. doi: 10.1161/ATVBAHA.115.305268
76. Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet. (2010) 42:420–5. doi: 10.1038/ng.563
77. Lee S, Kim IK, Ahn JS, Woo DC, Kim ST, Song S, Koh GY, Kim HS, Jeon BH, Kim I. Deficiency of endothelium-specific transcription factor Sox17 induces intracranial aneurysm. Circulation. (2015) 131:995–1005. doi: 10.1161/CIRCULATIONAHA.114.012568
78. Garcia-Ramirez M, Martinez-Gonzalez J, Juan-Babot JO, Rodriguez C, Badimon L. Transcription factor SOX18 is expressed in human coronary atherosclerotic lesions and regulates DNA synthesis and vascular cell growth. Arterioscler Thromb Vasc Biol. (2005) 25:2398–403. doi: 10.1161/01.ATV.0000187464.81959.23
79. Yao Y, Jumabay M, Ly A, Radparvar M, Wang AH, Abdmaulen R, et al. Crossveinless 2 regulates bone morphogenetic protein 9 in human and mouse vascular endothelium. Blood. (2012) 119:5037–47. doi: 10.1182/blood-2011-10-385906
80. Czechanski A, Byers C, Greenstein I, Schrode N, Donahue LR, Hadjantonakis AK, et al. Derivation and characterization of mouse embryonic stem cells from permissive and non-permissive strains. Nat Protoc. (2014) 9:559–74. doi: 10.1038/nprot.2014.030
81. Israely E, Ginsberg M, Nolan D, Ding BS, James D, Elemento O, et al. Akt suppression of TGFbeta signaling contributes to the maintenance of vascular identity in embryonic stem cell-derived endothelial cells. Stem Cells. (2014) 32:177–90. doi: 10.1002/stem.1521
Keywords: vascular calcification, sex determining region y-box, endothelial-mesenchymal transition, endothelium, differentiation
Citation: Yao Y, Yao J and Boström KI (2019) SOX Transcription Factors in Endothelial Differentiation and Endothelial-Mesenchymal Transitions. Front. Cardiovasc. Med. 6:30. doi: 10.3389/fcvm.2019.00030
Received: 31 August 2018; Accepted: 07 March 2019;
Published: 28 March 2019.
Edited by:
Dwight A. Towler, University of Texas Southwestern Medical Center, United StatesReviewed by:
Cécile Oury, University of Liège, BelgiumKathleen Ann Martin, Yale University, United States
Copyright © 2019 Yao, Yao and Boström. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yucheng Yao, eXlhb0BtZWRuZXQudWNsYS5lZHU=
Kristina I. Boström, a2Jvc3Ryb21AbWVkbmV0LnVjbGEuZWR1