 Zhuang-Zhuang Yuan
Zhuang-Zhuang Yuan Liang-Liang Fan
Liang-Liang Fan Zi-Chen Jiang4
Zi-Chen Jiang4 Zhi-Ping Tan
Zhi-Ping Tan- 1Clinical Center for Gene Diagnosis and Therapy, Department of Cardiovascular Surgery, The Second Xiangya Hospital of Central South University, Changsha, China
- 2Department of Cell Biology, School of Life Sciences, Central South University, Changsha, China
- 3Hunan Key Laboratory of Animal Models for Human Diseases, School of Life Sciences, Central South University, Changsha, China
- 4University of California, San Diego, San Diego, CA, United States
The position and morphology of human internal organs are asymmetrically distributed along the left–right axis. Aberrant left–right patterning in the developing embryo can lead to a series of congenital laterality defects, such as dextrocardia and heterotaxy syndrome. Laterality defects are a genetic condition; however, pathogenic genetic lesions are found in only one-fifth of patients. In this study, whole-exome sequencing was conducted for 78 patients with laterality defects. We identified a novel stopgain variant in MMP21 (c.G496T; p.G166*) in a Chinese patient with mirror-image dextrocardia. This variant caused a truncated MMP21 mRNA containing only the signal peptide and propeptide, while the coding sequence of matrix metalloproteinase-21 was almost entirely absent. To the best of our knowledge, this novel variant is the first homozygous stopgain variant identified in dextrocardia patients, and the first MMP21 variant found in East Asia. Our findings expand the spectrum of MMP21 variants and provide support for the critical role of MMP21 during left–right patterning in the Han Chinese population.
Introduction
In mammals, the proper left–right (L–R) patterning of internal organs is extremely complex and highly precise. Disordered L–R patterning can lead to a broad spectrum of laterality defects, including situs inversus totalis (SIT), a mirror image of situs solitus (SS), and heterotaxy (HTX), in which at least one organ is discordant along the left–right axis (1, 2). Dextrocardia, a rare congenital heart anomaly and HTX, is caused by the failure of normal L–R asymmetry patterning during heart development. As one of the first congenital heart malformations to be recognized, dextrocardia was mentioned in the early 17th century (3). SIT is rarely associated with congenital malformations, while HTX is highly associated with a series of congenital malformations. Most dextrocardia patients have other defects of the heart and abdominal organs (4).
Matrix metallopeptidase 21 (MMP21) encodes a member of the matrix metalloproteinase (Mmps) family that is known to hydrolyze extracellular matrix (ECM) components and is crucial for morphogenesis (5, 6). Vertebrates Mmps and their inhibitors are expressed in cardiomyocytes during the early stages of cardiac development, which are required for early heart tube assembly and modulate cardiac morphogenesis events, such as heart tube formation, heart directional looping, and differentiation of ostial cells (5).
Using N-ethyl-N-nitrosourea (ENU)-induced mutagenesis and whole-exome sequencing (WES), Li et al. (7) and Akawi et al. (8) identified Mmp21 mutations in mice with congenital heart disease (CHD) and laterality defects, respectively (7). Additionally, MMP21 mutations in several HTX families and sporadic cases were identified by WES (7–11). These mutations and laterality phenotypes in humans and mice indicated that MMP21 plays a critical role in the L–R patterning of visceral organs.
Here, we identified a novel MMP21 variant in one Han individual from among 78 patients with dextrocardia by WES.
Case Presentation
Seventy-eight patients were recruited in this study from Second Xiangya Hospital after providing written informed consent, and family history was extensively investigated. After strict clinical and radiographic examination, all patients were diagnosed with dextrocardia with or without primary ciliary dyskinesia (PCD) and/or CHD. This study was approved by the Ethics Committee of the Second Xiangya Hospital of Central South University.
The patient with MMP21 variation, a 7-year-old boy, was born at full term and liable to catch colds but developed normally. He was diagnosed with complex CHD, mirror-image dextrocardia (Figure 1A), single-ventricle, pulmonary stenosis, and transposition of the great arteries, without PCD. According to the parents of the proband, there are no other people with cardiovascular diseases in his family. A total cavopulmonary connection operation was conducted for the patient and achieved a good outcome. Written informed consent was obtained from the legal guardians of the patient for the publication of any potentially identifiable images or data included in this article.
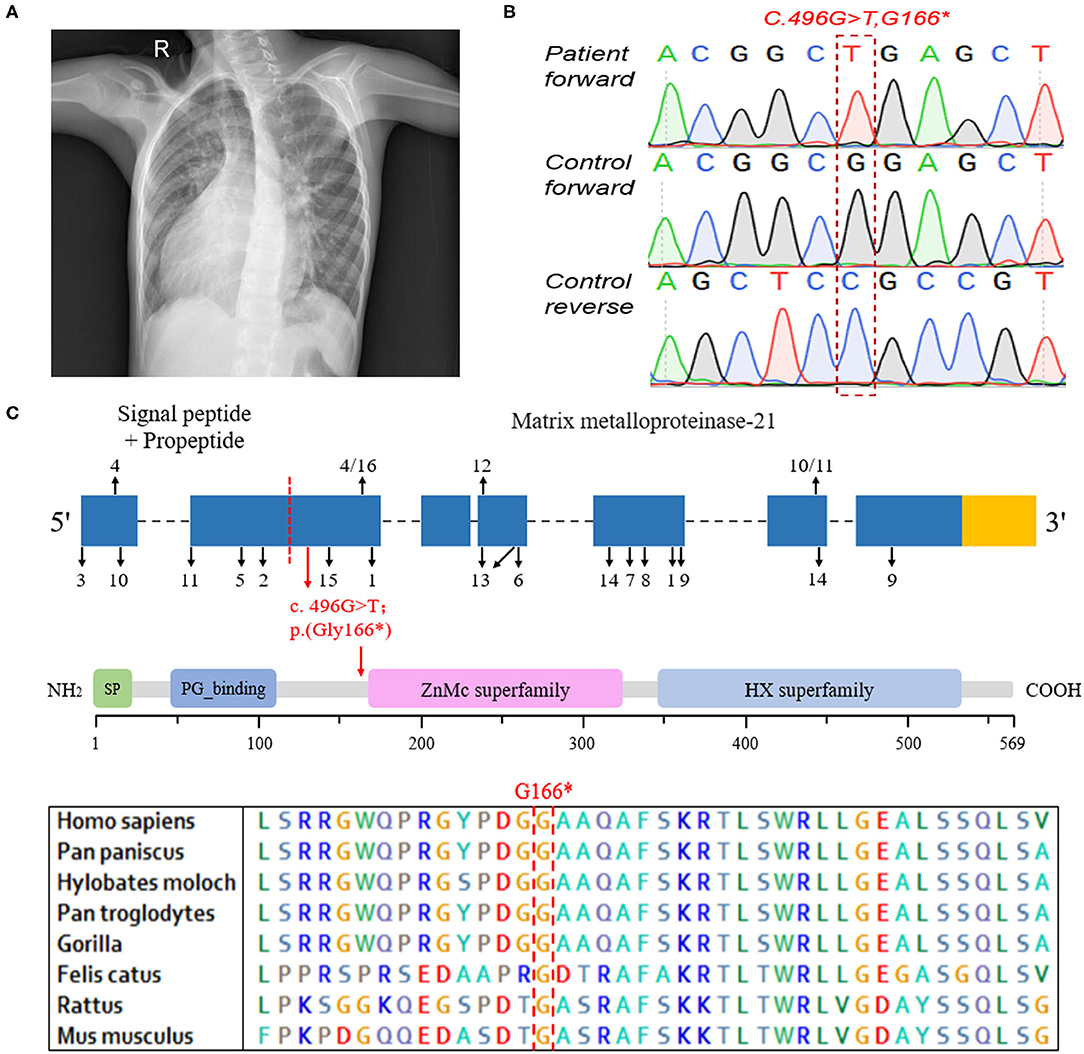
Figure 1. The clinical data and genetic analysis of the patient with MMP21 variant. (A) Chest X-ray of the patient indicating dextrocardia. (B) Sanger DNA sequencing chromatogram demonstrating the homozygosity for an MMP21 stopgain variant (c.G496T; p.G166*). (C) The sequence encoding the signal peptide and propeptide is before the red dotted line, the subsequent sequence encodes matrix metalloproteinase-21, the red arrow indicates the variant identified in this study, and the numbers and black arrows indicate the family in Table 2 and position of MMP21 variants, respectively. Domain structure of MMP21. SP, signal peptide; PG_binding, putative peptidoglycan-binding domain; ZnMc superfamily, zinc-dependent metalloprotease; HX superfamily, hemopexin-like repeats.
Genetic Analysis
Whole peripheral blood samples of the patients were obtained and stored in EDTA tubes. Genomic DNA was extracted by QIAamp DNA Blood Mini Kit (250) (Qiagen, Valencia, CA, USA).
The WES was mainly performed in the Novogene Bioinformatics Institute (Beijing, China). The exomes were captured by means of Agilent SureSelect Human All Exon V6 kits and sequenced on Illumina NovaSeq6000 (Illumina Inc, San Diego, USA). The WES data was filtered using three criteria: (1) Variations outside the coding regions (e.g., intergenic, intronic, and untranslated regions) and synonymous variants were excluded; (2) high allele frequency (>0.01%) compared to population-based databases (e.g., 1,000 Genomes Project, ESP, ExAC, and gnomAD) were excluded; and (3) prediction of a deleterious functional effect using bioinformatics programs (e.g., MutationTaster, CADD, SIFT, and Polyphen2), loss of function, and damaging variations were reserved.
Sanger sequencing was performed to validate the identified variant, and the primers were designed by PrimerQuest Tool (F: 5′-TTTCTCCCTGTCTGTvCTCTCT, R: 5′-GTGCTCTTACCTCTCCCAAAG). PCR conditions consisted of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, for a total of 35 cycles using 2 × Power Taq PCR MasterMix (BioTeke, Beijing, China). PCR products were electrophoresed on 1% agarose gels. The PCR fragment was subsequently cut, and purified fragments were sequenced on 3730XL sequencer (Applied Biosystems).
Identification of MMP21 Variation
WES was conducted and generated 12 Gb data with 99% coverage and a depth of >100 ×. After filtration of the WES data (Table 1), we finally identified a biallelic stopgain variant (c.G496T; p.G166*) in MMP21 and further confirmed it by Sanger sequencing (Figure 1B). This variant caused a truncated MMP21 mRNA containing only the signal peptide and propeptide, while the coding sequence of matrix metalloproteinase-21 was almost entirely absent (Figure 1C). This variant was predicted to be “disease causing” by MutationTaster and not found in the 1,000 Genome Browser, the ExAC Browser, the Exome Variant Server, or 200 unrelated ethnically matched healthy controls. The controls were individuals presenting for routine health checkups or volunteers without similar symptoms or any positive family history of cardiovascular disorders (male/female: 100/100, mean age 36.1 ± 4.3 years). In addition, the variant had the highest CADD score of 35 suggesting that it is deleterious. Multiple alignment of mmp21 orthologs in other animal species showed that the amino acid sequence after position 166 is highly conserved. According to ACMG standards and guidelines, this variant was categorized as pathogenic (PVS1, PM2, PM4, PM6, and PP3) and identified as the genetic lesion of the patient.
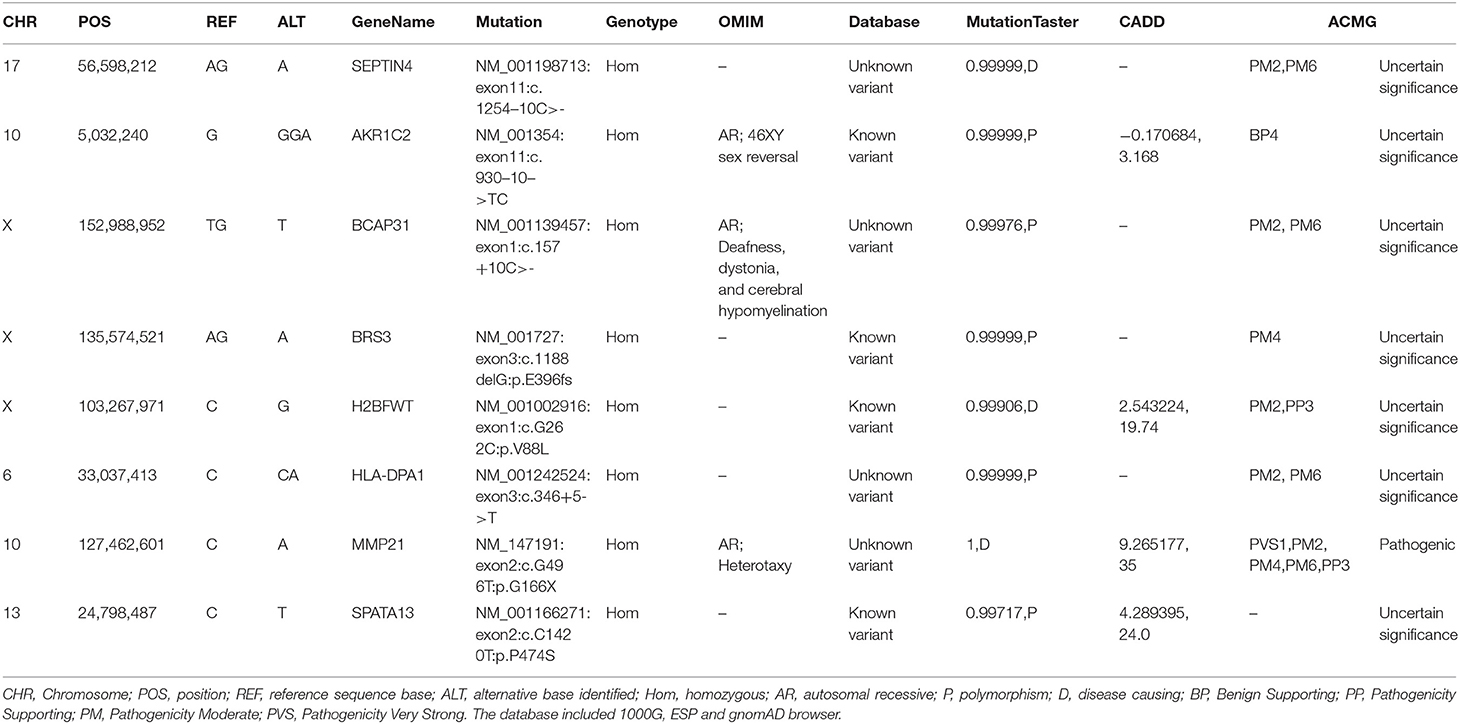
Table 1. The gene list after WES data filtration of the patient.
Discussion
Our group aimed to identify laterality defect genes in a cohort of 78 patients with dextrocardia and CHD. Approximately 10% had variations in genes, i.e., NODAL, ZIC3, NKX2-5, and CCDC151. WES was conducted to identify the causative genes in these patients. A novel biallelic stopgain MMP21 variant (c.G496T; p.G166*) was identified and predicted to be a disease-causing variant by MutationTaster and Combined Annotation Dependent Depletion (CADD). This variant was also not present in the current 1,000 Genome Browser, the ExAC Browser, or the Exome Variant Server. This variant caused a truncated MMP21 mRNA containing only a signal peptide and propeptide, while two functional domains (ZnMc superfamily; HX superfamily) were entirely absent, which may induce the decay of MMP21 mRNA in the patient according to nonsense-mediated mRNA decay theory (12, 13). MMP21 variants that caused laterality defects and complex CHD in humans are summarized in Table 2. To our knowledge, this novel variant (c.G496T; p.G166*) was the first homozygous stopgain variant identified in dextrocardia patients and the first MMP21 variant found in mainland China, suggesting the critical role of MMP21 during left–right patterning in the Han Chinese population.
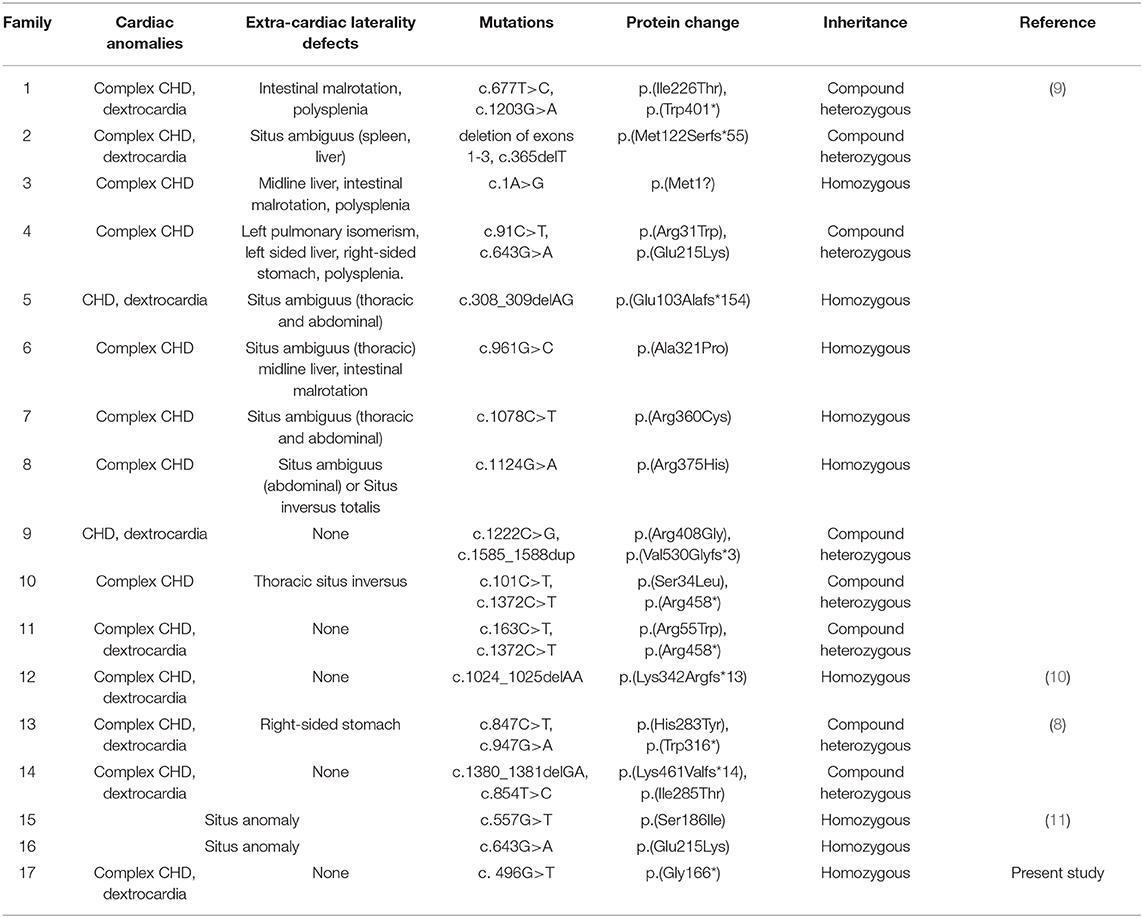
Table 2. The summary of variants in MMP21.
The known pathogenic genes of laterality defects account for only 15–20% of cases and are mainly related to the NODAL/TGFβ signaling pathway, SHH signaling pathway, and ciliary functions (11). Nodal cilia play a role in the origination of L–R asymmetry patterning (14–17). The observation that more than half of patients with primary ciliary dyskinesia (PCD) accompanied by SIT or HTX further reinforces the connection (2). In a prospective study involving 767 participants, Shapiro et al. (2) found that at least 12.1% of patients with classical PCD have HTX ranging from classic to subtle. In our previous studies, we reported a novel CCDC151 mutation in a patient with PCD and SIT and computed tomographic scanning of the sinus and lungs showing diffuse bronchiectasis and chronic sinusitis, respectively. His nasal nitric oxide concentrations (nNO) were far below (2 ppb) the PCD-specific nNO cutoff value (287 ppb). These results indicated that the CCDC151 mutation damaged cilia and further affected L–R patterning (18). However, we did not find PCD or other clinical phenotypes related to cilia in the MMP21 variant patient, suggesting that MMP21 was unrelated to cilia and involved in L–R patterning in another way. In addition, some known variants of NODAL, ZIC3, and NKX2-5 were identified in this cohort.
Knockdown or genome editing of the MMP21 ortholog in zebrafish resulted in heart-looping defects in a dose-dependent manner (9, 10). Mutant zebrafish embryos also showed concomitant disruption of the laterality marker southpaw in the lateral plate mesoderm and disrupted notch signaling in vitro and in vivo, suggesting that the heart-looping defect is related to abnormal L–R patterning and that MMP21 is a negative regulator of notch signaling (10). Consistent with the findings from zebrafish, mutant mouse embryos generated by Mmp21 genome editing and N-ethyl-N-nitrosourea (ENU)-induced mice that have a homozygous missense mutation in Mmp21 both exhibit CHD and laterality defects (7). The findings suggested that MMP21 plays an important role in the establishment of asymmetric organ development.
The Drosophila genome encodes two Mmps, Mmp1 and Mmp2. Experimental results from Drosophila demonstrated that Mmps play essential roles in promoting ECM remodeling, cell polarization, and lumen formation during Drosophila cardiogenesis. In humans, up to 25 Mmps have been identified with overlapping functions, and only MMP21 has been confirmed to be involved in L–R patterning (5, 19). It is very significant to identify the deleterious variants affecting L–R patterning in other members of the MMP family in both human and animal models.
In conclusion, our finding expands the spectrum of MMP21 variants and provides extra support that MMP21 play important roles in L–R patterning.
Data Availability Statement
The datasets generated for this study can be found in NCBI SRA, NCBI Accession No. PRJNA668249.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Second Xiangya Hospital of the Central South University. A signed written informed consent was obtained from the legal guardians of the patient for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Z-PT designed the overall study and performed data analysis. Z-ZY and L-LF processed the WES data, validated the mutation, and drafted the manuscript. Z-CJ contributed to the study as summer intern. Y-FY enrolled the patients. All authors read and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
National Science and Technology Major Project of the Ministry of Science and Technology of China, Grant/Award Number: 2017ZX10103005-006; National Natural Science Foundation of China, Grant/Award Number: 81970268, 81470445, and 81570288; the Fundamental Research Funds of Central South University, Grant/Award Number: 2018zzts394; the Open Sharing Fund for the Large-scale Instruments and Equipments of Central South University, Grant/Award Number: CSUZC201940.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all patients and their family for their participation in this study.
References
1. Sutherland MJ, Ware SM. Disorders of left-right asymmetry: heterotaxy and situs inversus. Am J Med Genet C. (2009) 151C:307–17. doi: 10.1002/ajmg.c.30228
2. Shapiro AJ, Davis SD, Ferkol T, Dell SD, Rosenfeld M, Olivier KN, et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest. (2014) 146:1176–86. doi: 10.1378/chest.13-1704
3. Perloff JK. The cardiac malpositions. Am J Cardiol. (2011) 108:1352–61. doi: 10.1016/j.amjcard.2011.06.055
4. Offen S, Jackson D, Canniffe C, Choudhary P, Celermajer DS. Dextrocardia in adults with congenital heart disease. Heart Lung Circ. (2016) 25:352–7. doi: 10.1016/j.hlc.2015.09.003
5. Raza QS, Vanderploeg JL, Jacobs JR. Matrix metalloproteinases are required for membrane motility and lumenogenesis during drosophila heart development. PLoS. One. (2017) 12:e171905. doi: 10.1371/journal.pone.0171905
6. Marchenko GN, Marchenko ND, Strongin AY. The structure and regulation of the human and mouse matrix metalloproteinase-21 gene and protein. Biochem J. (2003) 372:503–15. doi: 10.1042/bj20030174
7. Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. (2015) 521:520–4. doi: 10.1038/nature14269
8. Akawi N, McRae J, Ansari M, Balasubramanian M, Blyth M, Brady AF, et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families. Nat Genet. (2015) 47:1363–9. doi: 10.1038/ng.3410
9. Guimier A, Gabriel GC, Bajolle F, Tsang M, Liu H, Noll A, et al. Mmp21 is mutated in human heterotaxy and is required for normal left-right asymmetry in vertebrates. Nat Genet. (2015) 47:1260–3. doi: 10.1038/ng.3376
10. Perles Z, Moon S, Ta-Shma A, Yaacov B, Francescatto L, Edvardson S, et al. A human laterality disorder caused by a homozygous deleterious mutation inmmp21. J Med Genet. (2015) 52:840–7. doi: 10.1136/jmedgenet-2015-103336
11. Li AH, Hanchard NA, Azamian M, D'Alessandro L, Coban-Akdemir Z, Lopez KN, et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur J Hum Genet. (2019) 27:563–73. doi: 10.1038/s41431-018-0307-z
12. Fan L, Chen Y, Huang H, Yuan Z, Jin J, Hu M, et al. Exome sequencing identifies a novel nonsense mutation of ring finger protein 207 in a chinese family with long qt syndrome and syncope. J Hum Genet. (2019) 64:233–8. doi: 10.1038/s10038-018-0549-1
13. Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mrna decay. Nat Rev Mol Cell Biol. (2019) 20:406–20. doi: 10.1038/s41580-019-0126-2
14. Loges NT, Antony D, Maver A, Deardorff MA, Güleç EY, Gezdirici A, et al. Recessive dnah9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am J Hum Genet. (2018) 103:995–1008. doi: 10.1016/j.ajhg.2018.10.020
15. Becker-Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan-Brown J, et al. The coiled-coil domain containing protein ccdc40 is essential for motile cilia function and left-right axis formation. Nat Genet. (2011) 43:79–84. doi: 10.1038/ng.727
16. Wallmeier J, Shiratori H, Dougherty GW, Edelbusch C, Hjeij R, Loges NT, et al. Ttc25 deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization. Am J Hum Genet. (2016) 99:460–9. doi: 10.1016/j.ajhg.2016.06.014
17. Fassad MR, Shoemark A, Legendre M, Hirst RA, Koll F, le Borgne P, et al. Mutations in outer dynein arm heavy chain dnah9 cause motile cilia defects and situs inversus. Am J Hum Genet. (2018) 103:984–94. doi: 10.1016/j.ajhg.2018.10.016
18. Zhang W, Li D, Wei S, Guo T, Wang J, Luo H, et al. Whole-exome sequencing identifies a novel ccdc151 mutation, c.325g>t (p.e109x), in a patient with primary ciliary dyskinesia and situs inversus. J Hum Genet. (2019) 64:249–52. doi: 10.1038/s10038-018-0540-x
Keywords: congenital heart defect (CHD), whole exome sequencing, stopgain variant, dextrocardia, MMP21
Citation: Yuan Z-Z, Fan L-L, Jiang Z-C, Yang Y-F and Tan Z-P (2020) A Novel Nonsense MMP21 Variant Causes Dextrocardia and Congenital Heart Disease in a Han Chinese Patient. Front. Cardiovasc. Med. 7:582350. doi: 10.3389/fcvm.2020.582350
Received: 11 July 2020; Accepted: 12 October 2020;
Published: 09 November 2020.
Edited by:
George W. Booz, University of Mississippi Medical Center School of Dentistry, United StatesReviewed by:
Steven Clive Greenway, University of Calgary, CanadaPatrick G. Burgon, Qatar University, Qatar
Copyright © 2020 Yuan, Fan, Jiang, Yang and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Ping Tan, emhpcGluZ3RhbkBjc3UuZWR1LmNu