 Kelly Stam1
Kelly Stam1 Sebastian Clauss
Sebastian Clauss Yannick J. H. J. Taverne
Yannick J. H. J. Taverne Daphne Merkus
Daphne Merkus- 1Department of Cardiology, Erasmus University Medical Center Rotterdam, Rotterdam, Netherlands
- 2Department of Medicine I, University Hospital Munich, Ludwig-Maximilians University Munich, Munich, Germany
- 3Institute of Surgical Research at the Walter-Brendel-Centre of Experimental Medicine, University Hospital, LMU Munich, Munich, Germany
- 4DZHK (German Centre for Cardiovascular Research), Partner Site Munich, Munich Heart Alliance, Munich, Germany
- 5Department of Cardiothoracic Surgery, Erasmus University Medical Center Rotterdam, Rotterdam, Netherlands
Chronic thrombo-embolic pulmonary hypertension (CTEPH) develops in a subset of patients after acute pulmonary embolism. In CTEPH, pulmonary vascular resistance, which is initially elevated due to the obstructions in the larger pulmonary arteries, is further increased by pulmonary microvascular remodeling. The increased afterload of the right ventricle (RV) leads to RV dilation and hypertrophy. This RV remodeling predisposes to arrhythmogenesis and RV failure. Yet, mechanisms involved in pulmonary microvascular remodeling, processes underlying the RV structural and functional adaptability in CTEPH as well as determinants of the susceptibility to arrhythmias such as atrial fibrillation in the context of CTEPH remain incompletely understood. Several large animal models with critical clinical features of human CTEPH and subsequent RV remodeling have relatively recently been developed in swine, sheep, and dogs. In this review we will discuss the current knowledge on the processes underlying development and progression of CTEPH, and on how animal models can help enlarge understanding of these processes.
Introduction
Chronic thrombo-embolic pulmonary hypertension (CTEPH) develops in a some patients after acute pulmonary embolism (1, 2). In CTEPH, pulmonary vascular resistance, initially increases due to the obstructions in the larger pulmonary arteries but is further elevated by pulmonary microvascular remodeling (1–3). CTEPH is defined as a mean pulmonary artery pressure ≥25 mmHg at rest persisting for at least 6 weeks in patients with previous pulmonary artery embolism (4). In addition to the increase in pressure, flow is redistributed toward the unobstructed parts of the pulmonary vasculature, causing local alterations in shear stress. Altered shear stress combined with systemic risk factors contribute to pathological processes like endothelial dysfunction, inflammation, vasoconstriction, and impaired vasodilation (5). In addition, these processes promote structural remodeling of both the obstructed and unobstructed pulmonary vasculature (6, 7). This remodeling leads to increased pulmonary vascular resistance that in turn augments afterload of the right ventricle (RV), thereby resulting in RV dilation and RV hypertrophy.
The degree of RV structural remodeling and functional adaptability have been demonstrated to be important determinants of functional capacity and survival in patients with CTEPH (8–10). In addition to RV remodeling, CTEPH is a risk factor for development of arrhythmias, especially atrial fibrillation (AF), which adds to the morbidity and mortality of patients with CTEPH (11, 12).
Yet, mechanisms involved in pulmonary microvascular remodeling, processes underlying the RV structural and functional adaptability in CTEPH as well as determinants of the susceptibility to arrhythmias such as AF in the context of CTEPH remain incompletely understood. Gathering knowledge about these processes has proven notoriously difficult, at least in part because (large) animal models with critical clinical features of human CTEPH and subsequent RV remodeling have only been developed relatively recently. In this review we will discuss current knowledge on the processes underlying development and progression of CTEPH, and on how porcine, canine and ovine models can help enlarge understanding of these processes.
Definition, Prevalence, Incidence, and Clinical Phenotype of CTEPH
Pulmonary embolism results in an acute increase in pulmonary vascular resistance and a decrease in pulmonary vascular compliance, both of which contribute to an increase in RV afterload. CTEPH is defined as a persistent pulmonary artery pressure (PAP) above 25 mmHg, with a pulmonary wedge pressure below 15 mmHg, at rest for at least 3 months, despite therapeutic anticoagulation in order to discriminate this condition from “subacute” PE (4, 13–16). It is increasingly recognized that patients that fail to meet the PH cut-off value of 25 mmHg, yet have complaints similar to CTEPH patients, may have a milder form of the disease, so-called chronic thrombo-embolic disease (CTED) (3, 8, 17, 18). Indeed, PAP above 19 mmHg at rest following embolism are already associated with increased mortality at long term (15, 19). Mismatched perfusion defects on a ventilation/perfusion scan and specific diagnostic signs for CTEPH seen by multidetector CT angiography, MR imaging or conventional pulmonary cineangiography, such as ring-like stenoses, webs/slits and chronic total occlusions are imaging hallmarks for CTEPH.
The prevalence of CTEPH is still largely unknown. CTEPH develops in about 3–4% of patients after acute pulmonary embolism and in up to 10% of patients with recurrent pulmonary embolism (3, 20–22). The reported annual incidence of acute pulmonary embolism ranges from 750 to 2,700 per million adults (23–25). According to these numbers, the expected incidence of CTEPH would be 22.5–108 per million adults, while the reported numbers of diagnosed patients with CTEPH are substantially smaller. Three countries assessed the CTEPH incidence through nationwide registries. In the United Kingdom the CTEPH incidence was 1.75 per million (26), in Spain it was 0.9 per million adults (12) and in Germany it was 5.7 per million adults (27). Between ~300 and 400 patients were newly diagnosed with CTEPH per year in France and Germany (28, 29).
The symptoms of CTEPH are similar to those of other forms of PH, being shortness of breath, fatigue, syncope, chest pain, palpitations, and reduced exercise capacity, which (together with unawareness in many physicians' daily practice) contribute to the late diagnosis in a large number of patients. This delayed diagnosis negatively impacts the prognosis (30). Treatment options for CTEPH are limited and, even when treated, the disease often advances to right heart failure and even death. Proximal obstructions can be removed with surgical interventions such as pulmonary endarterectomy or balloon angioplasty (31–33) although these can only be performed in eligible patients. Therapeutic agents to modulate the pulmonary vascular resistance are limited to date. Riociguat, which is a soluble guanylyl cyclase stimulator, that activates the nitric oxide (NO) pathway without endogenous NO, thus acting as a vasodilator, inhibiting pulmonary smooth muscle cell growth and antagonizes platelet inhibition (i.e., preventing clot formation) is the only approved therapeutic agent in CTEPH to date (15, 31, 34–36).
Large Animal Models of CTEPH
Over the past decades, the pathophysiology of CTEPH has been studied in large animal models. Different embolization protocols and materials including air, autologous blood clots, Sephadex beads, and glue have been utilized in swine, sheep and dogs (Table 1). Measurements in these models include measurement of PAP, with either indwelling (chronic), or Swan Ganz catheters. Cardiac output and stroke volume was measured using either the Swan Ganz catheter, a chronically implanted flow probe or via echo, CMR or PV loop catheter (Tables 1, 2). The PV loop catheter also allows measurement of right ventricular- pulmonary arterial (RV-PA) coupling, a measure of how well the right ventricle can cope with the increased afterload. In the absence of a PV-loop catheter, RV-PA coupling can also be assessed using the single beat method (59).
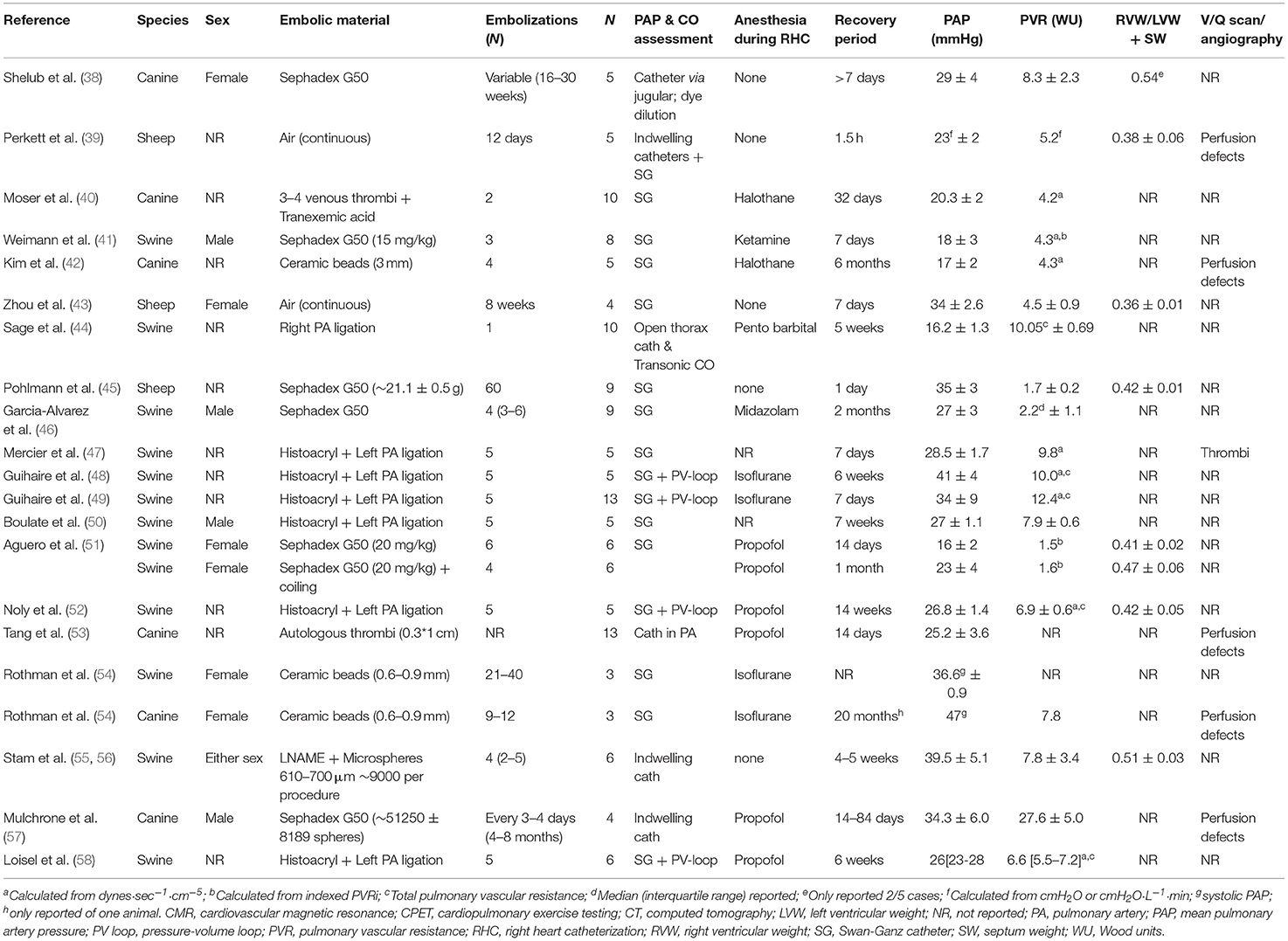
Table 1. Overview of large animal studies utilizing embolization techniques to create CTEPH/CTED, adapted from (37).
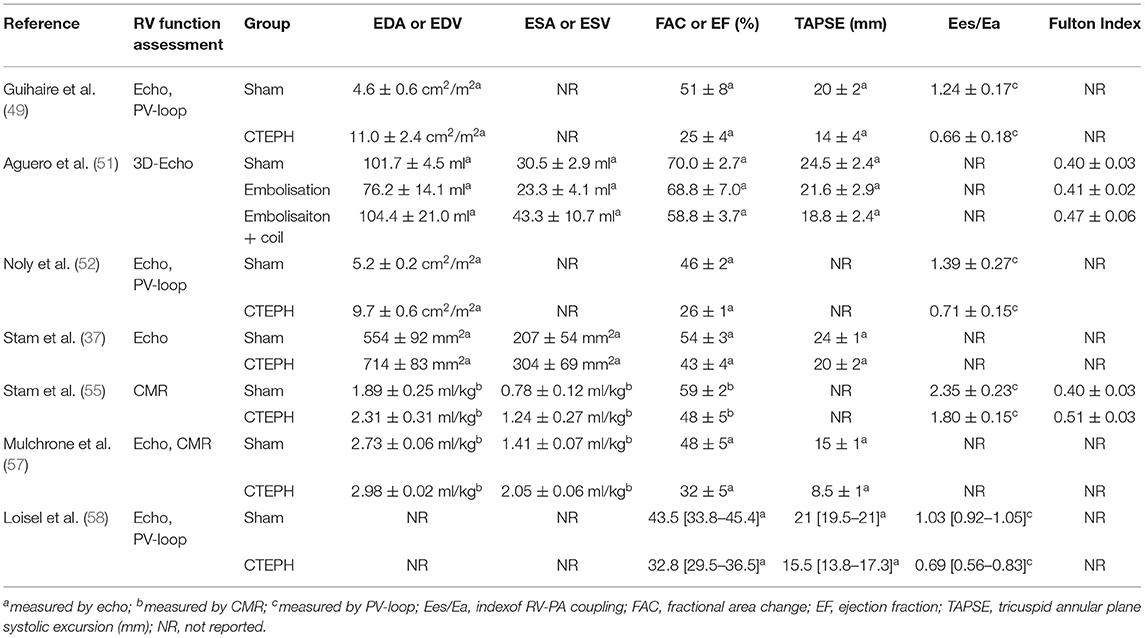
Table 2. Overview of RV-functional measurements in CTEPH/CTED models.
Very few investigators succeeded in establishing CTEPH during prolonged follow-up although the PAP increases acutely upon embolization (51). Ideally, autologous thrombi should be to induce CTEPH, in order to mimic the contribution of potential factors released from and cells interacting with these thrombi. However, studies with autologous thrombi to induce CTEPH have generally failed, which has initially been ascribed to a more active fibrinolytic system in animals as compared to humans (3, 60). Plasminogen activator inbibitor-1 was upregulated in the endothelial cells lining the thrombus in humans (60). Furthermore, a prothrombotic milieu, evidenced by elevated levels of von Willebrand Factor and C-reactive protein is associated with CTEPH in humans (60) and CTEPH severity and prognosis correlate with circulating D-dimer levels (61). These conditions are generally difficult to mimic in animal models. Nevertheless, inhibition of the fibrinolytic system in animals with tranexamic acid did not result in successful establishment of CTEPH in dogs (40). This failure to maintain high PAP during follow-up suggests that many models resemble CTED (i.e., mean PAP between 20 and 25 mmHg) more than CTEPH (mean PAP ≥25 mmHg).
It should be noted however, that PAP is mostly assessed under general anesthesia, which due to its cardiodepressive effects, may have resulted in an underestimation of PAP. Prerequisites for successful induction of CTEPH appear to include repeated embolization procedures (37, 38, 43, 45–50, 54, 56, 57) and obstruction of at least 40–60% of the pulmonary vasculature (62, 63). In these studies, pulmonary artery pressure and pulmonary vascular resistance decreased in between embolization procedures, but showed a slow but progressive increase over time and remained elevated after cessation of the embolisations. Obstruction of a large part of the pulmonary vasculature has been performed in swine, by combined ligation of the left pulmonary artery and progressive embolization of the segmental arteries of the right lower lobe, to mimic the proximal obstructions induced by thrombo-emboli (47–50, 64). An alternative method is to use smaller (micro)spheres to obstruct the pulmonary vasculature. The relative amount of obstruction of the pulmonary vasculature can be estimated by comparing the amount of microspheres infused with the number of vascular branches of this size present in the pulmonary vascular bed. The latter can be estimated from the morphometrical description of the pulmonary vasculature, which, in humans contains 15 Strahler orders, from the capillaries to the main pulmonary artery (65). It has been proposed that the pulmonary vascular bed of dogs (10 kg) and rats (500 g) are 1 and 3 log orders smaller than those of humans (66). Extrapolation of these numbers to swine of 20 kg, the size most commonly used in embolization experiments, would result in a pulmonary vasculature that is ~0.5 log order, or 3-fold, smaller than that of humans. The porcine pulmonary vasculature showed 10 branching orders of vessels larger than 160 μm in swine (67). The number of 3rd order pulmonary small arteries (diameter 430 μm, range 380–570 μ) was estimated to be 2,100, while 590 4th order vessels (diameter 760 μm, range 660–990 μm) were estimated to be present (67). These numbers exclude supernumerary vessels, which are 1.6- (68) to 2.8-fold (69) more than conventional arteries. Taking into account these supernumerary vessels, a total of 5,460–7,980 arteries of order 3, and 1,530–2,240 arteries of order 4 are present in the porcine lung.
In the porcine studies from our laboratory, CTEPH was induced with multiple (up to 5) embolizations using an average of 9,000 microspheres of 600–710 μm in diameter per embolization procedure, hence 45,000 microspheres in total (37, 55, 56). This number is much higher than the number of order 3 and 4 pulmonary small arteries present in the porcine lung and hence should be able to obstruct a sufficiently large part of the pulmonary vasculature. Despite this large number of emboli, sustained CTEPH only developed when embolizations were combined with chronic inhibition of eNOS, suggesting that endothelial dysfunction is required for CTEPH to develop in swine. These findings are in accordance with a study by Rothman and co-workers, who used ceramic beads of similar size but failed to induce sustained CTEPH in swine (54). Conversely, when a similar protocol was used in dogs, CTEPH was successfully induced in the absence of eNOS inhibition (54). Other studies in dogs also show that repeated embolization (Sephadex beads of 100–300 μm in diameter) does result in sustained CTEPH (38, 57), but this requires a large number of embolizations over a period of several months. A total of ~50,000 beads was injected over a period of several months (57), which is similar to the number of beads required to induced sustained CTEPH in our swine model. However, it should be noted that these smaller beads penetrate deeper into the pulmonary vasculature, and that the number of pulmonary small arteries increases with decreasing size. Thus, in the human lung 67,400 vessels of order 5 (diameter 150 ± 20 μm), 285,000 vessels of order 6 (diameter 220 ± 30μm) and 86,000 vessels of order 7 (diameter 350 ± 90μm) 86,000 vessels are present. Particularly when taking into account the 0.5 log-order fold difference between dogs and pigs, the relative number of beads required to induce sustained CTEPH in dogs seems to be smaller as compared to pigs.
Pulmonary Microvasculopathy in CTEPH
CTEPH not only involves the obstructed pulmonary arteries, but causes progressive microvascular remodeling of the non-obstructed distal pulmonary small arteries, which is associated with a further increase in resistance and decrease in compliance (1, 70), and contributes to worsening of CTEPH. The exact time-course of microvascular remodeling is unknown. It is difficult to distinguish between an increase in resistance due to recurrent embolizations and an increase in resistance due to microvascular remodeling even in animal models. Nevertheless, pulmonary vascular resistance continued to increase after cessation of the embolization procedures in our swine model, which suggest persistent remodeling of the distal vasculature (37, 56). Even after cessation of the eNOS inhibitor, which may also have contributed to a higher pulmonary vascular resistance by increasing microvascular tone, the high pulmonary pressure and resistance persisted (37, 56). This microvasculopathy was reversible upon reperfusion of the obstructed left lung vasculature (50).
Increased microvascular wall thickness results in narrowing of the lumen and stiffening of the pulmonary microvasculature. The mechanisms behind the pulmonary microvascular remodeling remain incompletely understood. A shift in balance of angiogenic factors, with decreased VEGF and high levels of angiopoietin 1 and 2 have been reported (1). Furthermore, chronic inflammation, evidenced by increased C-Reactive protein (CRP), as well as interleukin (IL)-10, monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1α (MIP-1α) and matrix metalloproteinase (MMP)-9 was present in patients with CTEPH (71–73), indicating that chronic inflammatory processes may play a role in the microvasculopathy. In accordance with this concept, studies in swine showed an elevation in IL-6 expression in the unobstructed territory (50), while no change in IL-6 was found in obstructed territories (50) and a trend toward an increase in mixed territories (56). However, neither mRNA expression of interferon-γ, TNF-α, nor expression of TGF-β were altered in the lungs of swine with CTEPH (56). It is however likely that a general systemic inflammatory phenotype in patients with CTEPH induces endothelial dysfunction, thereby predisposing to pulmonary microvasculopathy.
The structural changes in the pulmonary microvasculature are accompanied by functional changes. These functional changes can be assessed using isolated pulmonary small arteries in animal models of CTEPH. These thicker wall of the non-obstructed pulmonary small arteries results in exaggerated vasoconstriction to both KCl and the thromboxane analog U46619 (56). In addition, microvascular remodeling induced alterations in two major endothelial signaling pathways, i.e., endothelin (ET) and NO. NO sensitivity was increased in pulmonary small arteries from swine with CTEPH. This increase in NO sensitivity was evidenced by an enhanced vasodilator response to the NO-donor SNP. Furthermore, while the vasodilator response to bradykinin was reduced, there was a similar attenuation of bradykinin-induced vasodilation by eNOS-inhibition. The increased NO sensitivity was therefore likely accompanied by and/or a consequence of a decrease in NO production (56). Loss of NO is in accordance with data from CTEPH patients showing reduced circulating NO levels and increased levels of the endogenous eNOS-inhibitor asymmetric dimethyl arginine (ADMA) (74). In addition, the vasodilator response to PDE5 inhibition with Sildenafil was reduced in isolated pulmonary small arteries from swine with CTEPH as compared to healthy controls (56). This reduced PDE5 activity likely reduced the rate of cGMP breakdown and therefore may have contributed to the increased NO-sensitivity. Furthermore, a reduction in endogenous PDE5 activity may explain that PDE5 -inhibition is less effective for treatment of CTEPH and underlines the importance of intervening more upstream in the NO-pathway either by administering NO (75) or stimulating soluable guanylyl cyclase (sGC) by Riociguat, which has recently been approved for therapeutic use in CTEPH (35, 76–78).
NO not only impacts the vasculature directly, but also indirectly by inhibiting the production of the potent vasoconstrictor ET. ET can induce vasoconstriction and vascular remodeling and plasma levels of ET are higher in both animal models (42, 56) and patients with CTEPH (50) and correlate with clinical severity (79, 80). Although beneficial effects of ET receptor antagonism (ERA) in preventing vasculopathy were shown in dogs (42) conflicting results of ERA therapy have been reported in CTEPH patients with some studies showing positive effects (81–83), whereas, the randomized, double-blind, placebo-controlled BENEFIT study, which investigated dual ERA therapy with Bosentan, showed no statistically significant effects of Bosentan in CTEPH patients (84).
The lack of clinical effect of ERA in CTEPH may be due to a decreased sensitivity of the pulmonary microvasculature to ET (56). This decreased sensitivity to ET was accompanied by an increased ETA but decreased ETB contribution to ET-induced vasoconstriction. These data were consistent with an increased ETA/ETB gene expression ratio in lung tissue (56) as well as with the increased ETA receptor expression in human pulmonary endarterectomy tissues (85) and in the lungs of swine with CTEPH (47, 50). These data from large animal models and humans suggest that ET signaling is altered in the pulmonary microvasculature in CTEPH. The loss of ETB mediated vasoactive effects in isolated pulmonary small arteries (56) suggests that specific ETA blockade alone would be more therapeutically efficacious. Although the AMBER trial, which investigated the ETA specific ERA Ambrisentan was terminated early (ClinicalTrials.gov NCT02021292, NCT02060721), there are ongoing studies to ETA specific ERAs such as Macitentan (MERIT) which is an ERA with high specificity for ETA (86).
ET can signal via two pathways, the first one being calcium-dependent and involving phospholipase C (PLC)-mediated activation of myosin-light chain kinase. The second pathway is calcium-independent and encompasses RhoA-Rho-kinase mediated inactivation of myosin phosphatase (87). Although the Rho-kinase inhibitor Fasudil is therapeutically effective in some patients with PAH (88), knowledge about this pathway in the pathogenesis of CTEPH is limited. ET-induced vasoconstriction was mediated in part through Rho-kinase in isolated pulmonary small arteries from healthy swine, whereas, Rho-kinase inhibition had no effect in pulmonary small arteries from swine with CTEPH (56), implying that ET-induced vasoconstriction in CTEPH is mediated through the PLC-pathway. These data imply that microvascular constriction and remodeling occur via different pathways in CTEPH than in PAH in which the contribution of Rho-kinase was found to be augmented (89). Hence, using Rho-kinase inhibitors to alleviate pulmonary vasoconstriction and remodeling may not be efficacious in CTEPH patients.
Characterization of Cardiac Remodeling in CTEPH
Cardiac Dimensions and Function
The afterload of the RV increases during development and progression of CTEPH. To cope with this increased afterload, the RV exhibits structural and functional changes to augment contractility (Figure 1). RV dilation and remodeling are dependent on the magnitude of the increase in afterload (51). The effects of CTEPH on cardiac structure and function have been examined using a variety of non-invasive, including ultrasound, CMR, CT, and invasive (pressure volume relation, assessment of RV-PA coupling) methods (for overview see Table 2). These studies show RV dilation (increase in enddiastolic area or volume) and a reduced contractile function, as evidenced by a decrease in fractional area change or ejection fraction, a decrease in TAPSE and a progessive decrease in RV-PA coupling in animals with CTED and CTEPH (Figure 1, for results of individual studies see Table 2).
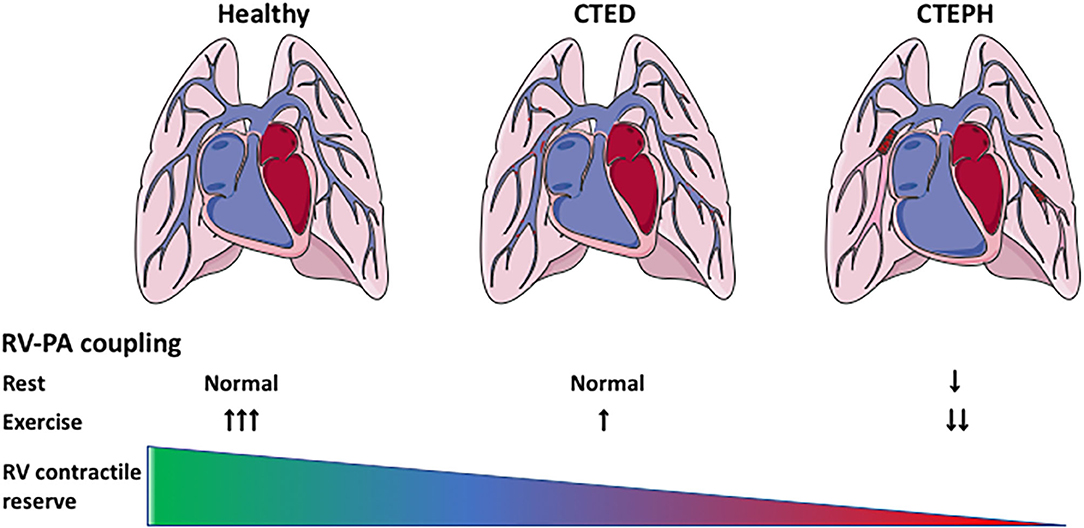
Figure 1. Cardiac remodeling and reserve in CTEPH. Cardiac remodeling in CTEPH, and the effects of exercise on the cardiac reserve. CTED, chronic thrombo-embolic disease; CTEPH, chronic thrombo-embolic pulmonary hypertension; RV-PA coupling: right ventricular-pulmonary artery coupling.
RV remodeling is associated with cardiomyocyte hypertrophy, interstitial fibrosis and changes in capillary density (48, 55). RV hypertrophy, both in terms of RV weight and RV cardiomyocyte size was accompanied by activation of both pro- and antiapoptotic gene expression (upregulation of Caspase-3 and BCL2 mRNA, respectively). RV resting function is generally preserved, but BNP expression is increased (48, 49, 55), suggestive of an increased wall stress. The increased BNP showed a negative correlation with stroke volume and a positive correlation with global RV hypertrophy (48). RV structural and functional adaptability are important determinants of functional capacity and survival in patients with CTEPH (8–10).
Thus, reduced RV – PA coupling, which denotes the ratio of RV contractility and RV afterload, is associated with a lower exercise capacity (8) and patients with a dilated RV have a worse prognosis compared to patients in which RV function and geometry are preserved (90, 91).
It is increasingly recognized that in addition to RV systolic dysfunction, RV diastolic dysfunction is associated with a worse prognosis in patients with PAH (92). Diastolic dysfunction, evidenced by an increase in stiffness, is also present in pigs with type II pulmonary hypertension (93). Diastolic stiffness is determined by passive myocyte stiffness as well as interstitial fibrosis. Consistent with the findings in a rat model of pulmonary artery banding (94), the mild RV dysfunction in swine with CTEPH was characterized by an increase in the stiff titin isoform N2BA but not with changes in myocardial fibrosis as measured histologically. Furthermore, there was a change in the ratio between Col1 and Col3 in the RV, suggesting relatively more expression of the stiff Col1 isoform (55), which is consistent with data from rats with PAH (94), and may have contributed to a stiffer RV. Although, neither SERCA nor phospholamban gene expression were changed in our CTEPH swine model, it is possible that changes in their phosphorylation may play a role in altered Ca2+ handling, which is in turn implied to play a role in the development of RV dysfunction (93). Hence, future studies in relevant large animal models should investigate contractile function of individual cardiomyocytes as well as expression and phosphorylation of the contractile and calcium handling proteins SERCA, phospholamban, smooth muscle actin, titin, and troponins.
Cardiac Inflammation, Oxidative Stress, Apoptosis, and Angiogenesis
A key factor that distinguishes compensated RV remodeling from RV failure is adequate myocardial perfusion (95). Angiogenesis is required to enhance RV perfusion commensurate with the increase in RV mass. Capillary density is preserved or even increased in adaptive RV remodeling, whereas, it is reduced in RV failure (95). Furthermore, RV maladaptive remodeling in CTEPH and PAH is accomanied by a reduction in myocardial perfusion reserve (96, 97). Conversely, capillary density was either unaltered (52) or increased in swine with CTEPH (55, 58), which is beneficial for myocardial perfusion and oxygenation, and suggests a state of adaptive RV remodeling. Nevertheless, HIF1α (52) and VEGFA-expression were higher in swine with CTEPH (52, 55, 58), suggesting that there was still a need for additional perfusion. Furthermore, VEGFA expression corelated inversely with RV-PA coupling (55) and further increasing capillary density using a therapeutic intervention with infusion of endothelial progenitor cells into the right coronary artery improved right ventricular function independent of changes in PVR (58). Hence, future studies should address myocardial perfusion and coronary flow reserve in different stages of RV (mal) adaptation and assess whether RV adaptations is different in CTED vs. CTEPH.
The transition from compensated RV remodeling to RV failure is also associated with inflammation and activation of the immune system (98–100). Although, the mRNA of genes involved in immune modulation (TNF-α, IL-6, IFN-γ) was not altered in the RV of swine with CTEPH, expression of TGF-β was higher (52, 55). Activation of the TGF-β pathway is known to play a pivotal role in the development of PH (101, 102). TGF-β pathway activation was further supported by the higher expression PAI, a downstream target of TGF-β signaling in these animals. Just like endothelin, activation of the TGF-β pathway can result in activation of the Rho-kinase pathway (103–105). Indeed, ROCK2 expression was upregulated in the RV of swine with CTEPH. Importantly, ROCK2 activation is involved in cardiac hypertrophy, oxidative stress, angiogenesis, apoptosis, and fibrosis and may therefore present a major deleterious factor in RV-remodeling (106–108) (Figure 2). In addition, ROCK2 activation increases phosphorylation of protein phosphatase 1 (PP1), which in turn regulates Ca2+ handling and Ca2+ sensitivity in the cardiomyocytes (108).
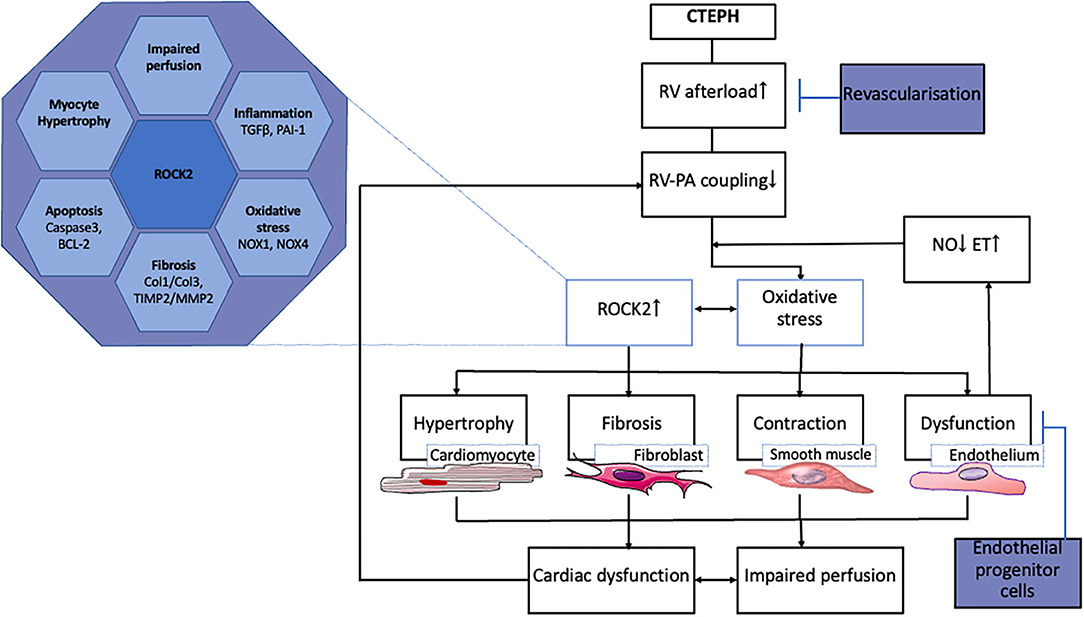
Figure 2. Processes involved in RV remodeling in CTEPH and therapeutic interventions tested in animal models.
ROCK2 is also expressed in the coronary vasculature, where it is associated with oxidative stress and NOX-expression (109). NOX1, NOX2, and NOX4 expression was elevated in the right coronary artery of swine with RV pressure overload due to pulmonary artery banding. The increased expression of these NOX- isoforms was accompanied by oxidative stress and endothelial dysfunction, notwithstanding unaltered eNOS expression (110). Furthermore, an increase in circulating NOX4 has been shown in patients with PAH (111). The upregulation of NOX1 and NOX4, and the unaltered eNOS expression in the RV of swine with CTEPH are consistent with these data (55). Interestingly, expression of NOX1 and NOX4 as well as ROCK2 correlated inversely with RV-PA coupling, suggesting that oxidative stress in the myocardium may contribute to deterioration of RV-function (55).
Hence, it remains to be investigated whether Rho-kinase inhibition may prevent adverse cardiac remodeling despite observations that it has no beneficial effects on pulmonary vascular tone and remodeling. Furthermore, animal models may be well-suited to further establish markers of RV dysfunction.
Cardiac Arrhythmias in the Context of CTEPH
Epidemiology and Clinical Implications
Although arrhythmias have been identified as relevant contributors for morbidity and mortality in patients with CTEPH, only little is known about the incidence and prevalence of arrhythmias in this patient cohort (112–115). Besides the very limited number of studies performed in general, these studies also included patients with other etiologies leading to pulmonary hypertension (in most cases PAH type 1) which makes it challenging to provide data specifically in patients with CTEPH. In general, supraventricular arrhythmias are more common in patients with CTEPH than in the general population ranging from 11.7 to 22% in retrospective studies (115, 116) and from 13.4 to 25.1% in prospective studies (11). The most common arrhythmias observed in these studies were atrial fibrillation (AF) and atrial flutter (AFlut). Kanemoto studied 101 patients with PAH and observed arrhythmias in 17.8% (113). The most frequent arrhythmias in this cohort were (i) sinus tachycardia (38%), (ii) sinus bradycardia (18%), and (iii) first degree AV block (15%) with sinus tachycardia being significantly associated with mortality. Ventricular arrhythmias such as ventricular tachycardia (VT) or ventricular fibrillation (VF) have been demonstrated only in about 8% of patients with PAH and sudden cardiac death (SCD) (117). Due to the pathophysiologic alterations in PAH and the observation of VF and VT in a rat model of PAH (118, 119) one would expect a higher incidence of VT/VF in PAH patients potentially indicating difficulties in diagnosing ventricular arrhythmias due to a lack of permanent ECG monitoring (e.g., implantable loop recorders) in such patients (12). Even more frequent than specific arrhythmias are ECG abnormalities in PAH patients. Tonelli and colleagues evaluated ECGs over time in 50 patients with PAH and could see a significantly increased heart rate as well as prolonged PR interval, QRS duration and QTc duration in terminally diseased patients (120) which was further confirmed by Rich et al. (121). They also observed that none of the patients had a “normal” ECG before death and demonstrated a correlation between QRS/QTc prolongation and impaired RV function, increased RV mass and poor prognosis with a QTC > 480 ms being an independent predictor for mortality (120). Occurrence of arrhythmias is associated with clinical decompensation, although, it is incompletely understood whether arrhythmias occur as a cause or consequence of clinical worsening or both. However, conversion to sinus rhythm improved clinical status and in most cases reverses cardiac decompensation, whereas, the presence of sustained supraventricular arrhythmias is associated with a worse prognosis (11, 12). Overall, supraventricular arrhythmias are associated with increased mortality in PAH patients (3.81-fold increased hazard of death) (122).
Arrhythmia Mechanisms
The increased vulnerability to arrhythmias in CTEPH is incompletely understood. So far, our knowledge on CTEPH-associated arrhythmogenesis is based on clinical findings in patients. So far, no specific animal model for CTEPH has been investigated with respect to arrhythmias, only in the monocrotaline induced PAH models in rat and dog arrhythmogenesis has been studied. Thus, only extrapolated mechanistic data from non-CTEPH animal models or patients/animal models with non-CTEPH RV dysfunction or cardiac remodeling leading to the clinical phenotype seen in CTEPH patients are currently available and a specific mechanistic understanding of CTEPH-mediated electrophysiologic alterations and arrhythmogenesis is lacking.
Having this major limitation in mind several arrhythmia mechanisms have been suggested in CTEPH, including autonomic, electrical, or structural remodeling (12). Autonomic remodeling in PAH results in decreased heart rate variability, an overall increased sympathetic activity and an adrenergic remodeling in the RV including downregulation of β1-, α-, or dopaminergic receptors. This autonomic dysregulation is clinically reflected by arrhythmias (123) but also by sinus tachycardia which may be a reactive mechanism to maintain RV output (124). The influence of the autonomic system on arrhythmogenesis has been further demonstrated in a dog model of PAH (125). Eight weeks after dehydromonocrotaline treatment beagles develop PAH and show an increased vulnerability to AF/AFlut. They could confirm a downregulation of β1 receptors in RV, but in RA they found an upregulation and an increased density of sympathetic nerves. Finally, they ablated the ganglionated plexi which resulted in reduced AF/AFlut inducibility and further confirms the important role of the autonomic nervous system in PAH-associated arrhythmogenesis.
In patients with PAH increased RV stretch, hypertrophy and fibrosis have been demonstrated – well-known proarrhythmic mechanisms summarized under the term “structural remodeling.” These structural alterations result in reduced conduction velocity and the occurrence of re-entry (126). In addition to that, both in patients and a rat model for PAH electrical alterations (so called electrical remodeling), can be observed including downregulation of potassium channels, connexin-43, or changes in calcium handling leading to conduction slowing, QTc and action potential duration prolongation, or afterdepolarizations and ectopic activity (119–129). All these mechanisms finally result in an increased susceptibility of arrhythmias (126) but their relevance in CTEPH remains to be established.
Cardiopulmonary Stress Testing
Exercise testing after pulmonary embolism is predictive of development of PH and/or patient outcome in established CTEPH (130–133). Both patients with CTED and CTEPH show impaired exercise tolerance (8, 18, 134, 135). In accordance with these human data, swine with CTEPH also showed evidence of exercise intolerance (37, 55). This impaired exercise capacity is principally caused by an exacerbated increase in PAP and pulmonary vascular resistance during exercise, that further increases RV afterload and the V/Q-mismatch in the lungs (Figure 3) (90, 134, 135).
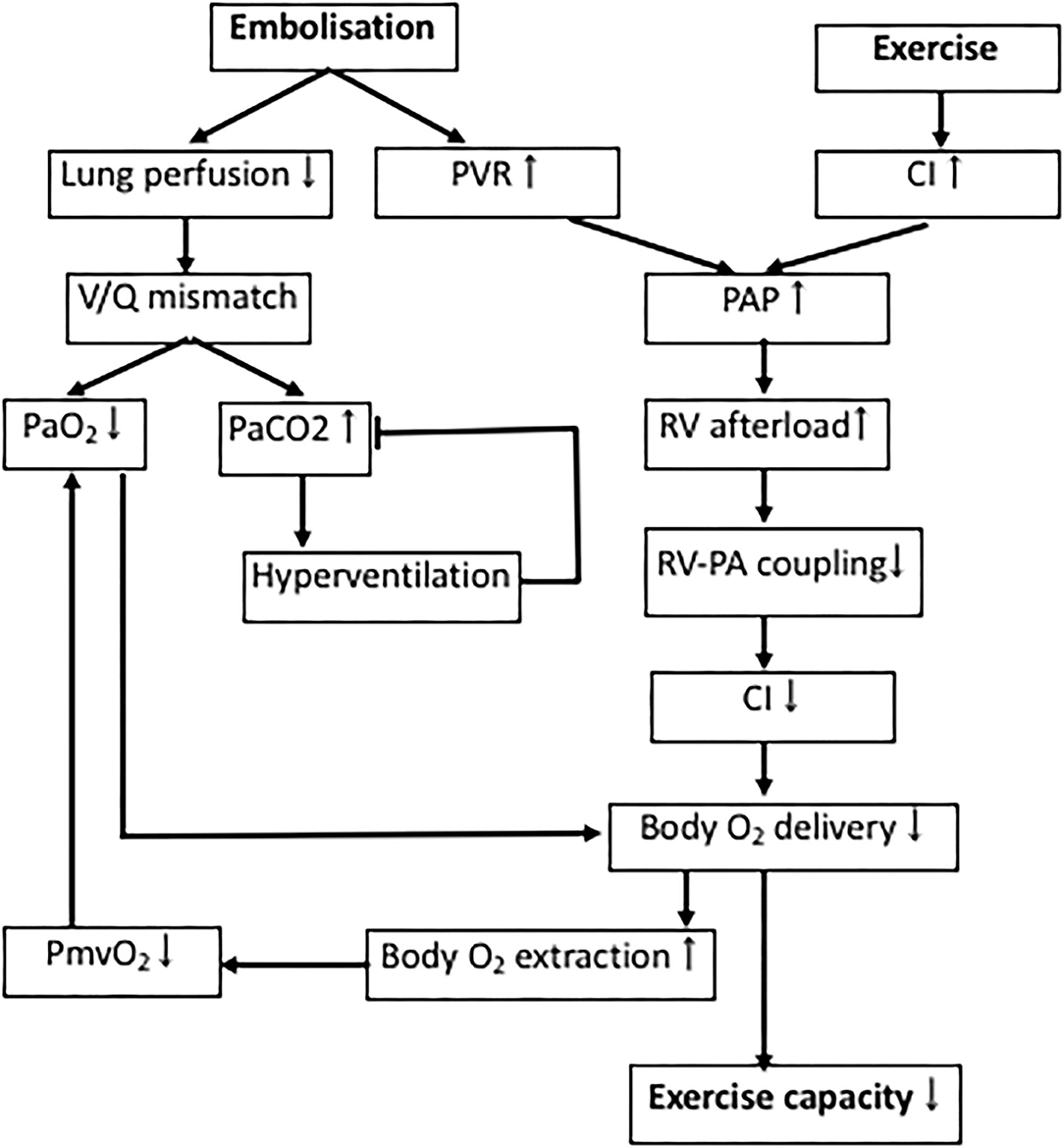
Figure 3. Determinants of exercise intolerance in CTEPH. Processes involved in decreased exercise tolerance in CTEPH. PAP, pulmonary artery pressure; PVR, pulmonary vascular resistance; RV, right ventricle; V/Q-mismatch, ventilation/perfusion-mismatch; PmvO2, mixed venous oxygen pressure; PaO2, arterial oxygen pressure; PaCO2, arterial carbon dioxide pressure.
Physiological dead space is increased in proportion to the increase in PVR in CTEPH, resulting in a lower PaO2, which results in a reduction in PaCO2, due to compensatory hyperventilation (136). Similarly, the slightly lower PaO2 in swine with CTEPH is consistent with dead space ventilation, a mild V/Q mismatch, and a subsequent decrease in capillary transit time at rest. However, PaCO2 was not altered in the swine with CTEPH neither at rest, nor during exercise (37). This discrepancy in CO2 response may in part be due to the observation that swine lack collateral ventilation and therefore cannot equalize intraregional V/Q differences between alveoli. Furthermore, healthy quadrupeds already ventilate and perfuse their entire lungs at rest (137), and hence cannot further recruit hypoventilated lung areas and improve V/Q mismatch by increasing ventilation. Thus, hyperventilation may not be capable of reducing PaCO2 levels below normal as seen in humans (37, 56). The lower PmvO2 observed in CTEPH is partly a consequence of the lower PaO2 due to the V/Q mismatch, and in part reflects the decreased blood flow due to a lower cardiac index (CI), forcing the body to extract more oxygen.
In accordance with the studies in CTED and CTEPH patients in 2015 (8, 18), the RV of swine with CTEPH was not able to cope with this increased afterload evidenced by the limited exercise-induced increase in stroke volume and CI (Figure 1). Furthermore, RV-PA coupling was reduced in swine with CTEPH (49, 55, 64) and a correlation was found between reduced coupling and a reduced SV reserve with dobutamine in swine (49) as well as between afterload and RV-PA coupling (55). Importantly, recent studies in patients with CTEPH show that RV-PA coupling correlates with exercise capacity (8), which in turn is a strong predictor of clinical outcome (90).
During exercise at 4 km/h, the anaerobic threshold was reached in swine with CTEPH and maximal body oxygen consumption was reduced which is consistent with a reduced exercise tolerance in CTEPH patients (18, 111, 132, 138). This reduction in maximal oxygen consumption was principally due to a decreased CI (27% lower as compared to healthy) and to a lesser extent to a decreased arterial oxygen content (10% lower as compared to healthy). Hence, exercise intolerance in CTEPH is principally caused by the increased pulmonary vascular resistance, which augments afterload of the RV, and thereby limits the exercise induced increase in CI. The relative contributions of cardiac and pulmonary dysfunction to the exercise intolerance in patients with more severe RV dysfunction remains to be established.
Added Value of Animal Models
CTEPH is associated with increased mortality and therefore a clinically highly relevant disease. The underlying pathophysiology is complex and currently poorly understood. Animal models are invaluable in this regard to study causal mechanisms and to identify potential targets for drug development or establishment of diagnostic/prognostic biomarkers.
In this context, CTEPH large animal models are of special interest since chronic instrumentation or repeated measurements are possible and they better resemble pathophysiologic hallmarks of the human situation such as microvascular remodeling, as well as remodeling and/or electrophysiologic alterations of the RV in CTED as well as CTEPH (37, 55, 56, 139–141).
Thus, those close-to-human large animal models (37, 55, 56, 139) allow to identify and to validate biomarkers (e.g., circulating biomarkers in the blood, hemodynamic biomarkers or ECG traits) to facilitate early detection of disease and/or disease progression in patients which could lead to the development of preventive strategies in the future as well. Also, in large animal models interventions that interfere with microvascular and/or RV remodeling and have direct clinical applicability (e.g., catheter-based approaches) are possible to get more insight in the pathways involved in this remodeling and get more specific targeted therapy for inoperable CTEPH patients or patients with residual PH after surgery. An example of such treatment to be tested for improvement of RV function based on swine data is ROCK2 inhibition. Thus far, few studies in large animal models have been performed to study the effect of therapeutic interventions. One study investigated lung revascularization (50) and one study investigated the effect of endothelial progenitor cells to improve RV perfusion (58) (see also above).
In addition, animal models may also be used to delineate sex-differences that are known to exist in development, progression and possibly interventions of CTEPH (142) and shed more light on the importance and implications of cardiopulmonary exercise testing in CTED and CTEPH patients. Finally, electrophysiologic evaluation beyond pure mechanistic concepts (which can be obtained in rodents or small animals) and especially studying novel anti-arrhythmic therapeutic approaches are more feasible in larger animals since the electrical properties of the heart are more similar to humans (compared to rodents) (140). Some innovative agents targeting pulmonary vasculature are indeed currently under clinical investigation (ClinicalTrials.gov NCT03689244, NCT01416636, NCT03809650, NCT03273257, NCT02634203, NCT00910429) but have not been tested in large animal models of CTEPH.
Author Contributions
KS drafted the manuscript. SC, YT, and DM revised the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by the German Center for Cardiovascular Research (DZHK; 81X2600210, 81X2600204, 81X3600208, 81X2600249 to SC, 81Z0600207 to DM), the LMU Munich's Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (to SC), the Förderprogramm für Forschung und Lehre (FöFöLe; 962 to SC), the Heinrich-and-Lotte-Mühlfenzl Stiftung (to SC), the ERA-NET on Cardiovascular Diseases (ERA-CVD; 01KL1910 to SC), the Netherlands Cardiovascular Research Initiative, an initiative with financial support from the Dutch Heart Foundation (CVON2014-11, RECONNECT to DM), and the Corona-Foundation (S199/10079/2019 to SC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Simonneau G, Torbicki A, Dorfmuller P, Kim N. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. (2017) 26:160112. doi: 10.1183/16000617.0112-2016
2. Lang IM, Dorfmuller P, Vonk Noordegraaf A. The pathobiology of chronic thromboembolic pulmonary hypertension. Ann Am Thorac Soc. (2016) 13(Suppl. 3):S215–21. doi: 10.1513/AnnalsATS.201509-620AS
3. Lang IM, Campean IA, Sadushi-Kolici R, Badr-Eslam R, Gerges C, Skoro-Sajer N. Chronic thromboembolic disease and chronic thromboembolic pulmonary hypertension. Clin Chest Med. (2021) 42:81–90. doi: 10.1016/j.ccm.2020.11.014
4. Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. (2013) 62:D42–50. doi: 10.1016/j.jacc.2013.10.032
5. Matthews DT, Hemnes AR. Current concepts in the pathogenesis of chronic thromboembolic pulmonary hypertension. Pulm Circ. (2016) 6:145–54. doi: 10.1086/686011
6. Galie N, Kim NH. Pulmonary microvascular disease in chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc. (2006) 3:571–6. doi: 10.1513/pats.200605-113LR
7. Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest. (1993) 103:685–92. doi: 10.1378/chest.103.3.685
8. Claeys M, Claessen G, La Gerche A, Petit T, Belge C, Meyns B, et al. Impaired cardiac reserve and abnormal vascular load limit exercise capacity in chronic thromboembolic disease. JACC Cardiovasc Imaging. (2018) 12(8 Pt. 1):1444–56. doi: 10.1016/j.jcmg.2018.07.021
9. Hardziyenka M, Campian ME, Reesink HJ, Surie S, Bouma BJ, Groenink M, et al. Right ventricular failure following chronic pressure overload is associated with reduction in left ventricular mass: evidence for atrophic remodeling. J Am Coll Cardiol. (2011) 57:921–8. doi: 10.1016/j.jacc.2010.08.648
10. van de Veerdonk MC, Bogaard HJ, Voelkel NF. The right ventricle and pulmonary hypertension. Heart Fail Rev. (2016) 21:259–71. doi: 10.1007/s10741-016-9526-y
11. Olsson KM, Nickel NP, Tongers J, Hoeper MM. Atrial flutter and fibrillation in patients with pulmonary hypertension. Int J Cardiol. (2013) 167:2300–5. doi: 10.1016/j.ijcard.2012.06.024
12. Cirulis MM, Ryan JJ, Archer SL. Pathophysiology, incidence, management, and consequences of cardiac arrhythmia in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Pulm Circ. (2019) 9:1–15. doi: 10.1177/2045894019834890
13. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 37:67–119. doi: 10.1093/eurheartj/ehv317
14. Hoeper MM, Madani MM, Nakanishi N, Meyer B, Cebotari S, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Lancet Respir Med. (2014) 2:573–82. doi: 10.1016/S2213-2600(14)70089-X
15. Kim NH, Delcroix M, Jais X, Madani MM, Matsubara H, Mayer E, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J. (2019) 53:1801915. doi: 10.1183/13993003.01915-2018
16. Lang IM, Madani M. Update on chronic thromboembolic pulmonary hypertension. Circulation. (2014) 130:508–18. doi: 10.1161/CIRCULATIONAHA.114.009309
17. Axell RG, Messer SJ, White PA, McCabe C, Priest A, Statopoulou T, et al. Ventriculo-arterial coupling detects occult RV dysfunction in chronic thromboembolic pulmonary vascular disease. Physiol Rep. (2017) 5:e13227. doi: 10.14814/phy2.13227
18. Held M, Kolb P, Grun M, Jany B, Hubner G, Grgic A, et al. Functional characterization of patients with chronic thromboembolic disease. Respiration. (2016) 91:503–9. doi: 10.1159/000447247
19. Torbicki A. Hypertension: definition of pulmonary hypertension challenged? Nat Rev Cardiol. (2016) 13:250–1. doi: 10.1038/nrcardio.2016.44
20. Ende-Verhaar YM, Cannegieter SC, Vonk Noordegraaf A, Delcroix M, Pruszczyk P, Mairuhu AT, et al. Incidence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: a contemporary view of the published literature. Eur Respir J. (2017) 49:1601792. doi: 10.1183/13993003.01792-2016
21. Vavera Z, Vojacek J, Pudil R, Maly J, Elias P. Chronic thromboembolic pulmonary hypertension after the first episode of pulmonary embolism? How often? Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2016) 160:125–9. doi: 10.5507/bp.2015.021
22. Yang S, Yang Y, Zhai Z, Kuang T, Gong J, Zhang S, et al. Incidence and risk factors of chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. J Thorac Dis. (2015) 7:1927–38. doi: 10.21037/jtd.2018.07.106
23. Konstantinides SV, Barco S, Lankeit M, Meyer G. Management of pulmonary embolism: an update. J Am Coll Cardiol. (2016) 67:976–90. doi: 10.1016/j.jacc.2015.11.061
24. Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, et al. Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol. (2014) 34:2363–71. doi: 10.1161/ATVBAHA.114.304488
25. Vonk Noordegraaf Boonstra A, Konings TC, Marques KM, Bogaard HJ. [Diagnosis and treatment of pulmonary hypertension]. Diagnostiek en behandeling van pulmonale hypertensie. Ned Tijdschr Geneeskd. (2014) 158:A7315.
26. Condliffe R, Kiely DG, Gibbs JS, Corris PA, Peacock AJ, Jenkins DP, et al. Improved outcomes in medically and surgically treated chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med. (2008) 177:1122–7. doi: 10.1164/rccm.200712-1841OC
27. Escribano-Subias P, Blanco I, Lopez-Meseguer M, Lopez-Guarch CJ, Roman A, Morales P, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J. (2012) 40:596–603. doi: 10.1183/09031936.00101211
28. Kramm T, Wilkens H, Fuge J, Schafers HJ, Guth S, Wiedenroth CB, et al. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clin Res Cardiol. (2018) 107:548–53. doi: 10.1007/s00392-018-1215-5
29. Simonneau G, Hoeper MM. Evaluation of the incidence of rare diseases: difficulties and uncertainties, the example of chronic thromboembolic pulmonary hypertension. Eur Respir J. (2017) 49:1602522. doi: 10.1183/13993003.02522-2016
30. Klok FA, Barco S, Konstantinides SV, Dartevelle P, Fadel E, Jenkins D, et al. Determinants of diagnostic delay in chronic thromboembolic pulmonary hypertension: results from the European CTEPH registry. Eur Respir J. (2018) 52:1801687. doi: 10.1183/13993003.01687-2018
31. Edward JA, Mandras S. An update on the management of chronic thromboembolic pulmonary hypertension. Curr Probl Cardiol. (2017) 42:7–38. doi: 10.1016/j.cpcardiol.2016.11.001
32. Gall H, Preston IR, Hinzmann B, Heinz S, Jenkins D, Kim NH, et al. An international physician survey of chronic thromboembolic pulmonary hypertension management. Pulm Circ. (2016) 6:472–82. doi: 10.1086/688084
33. Haythe J. Chronic thromboembolic pulmonary hypertension: a review of current practice. Prog Cardiovasc Dis. (2012) 55:134–43. doi: 10.1016/j.pcad.2012.07.005
34. Albani S, Biondi F, Stolfo D, Lo Giudice F, Sinagra G. Chronic thromboembolic pulmonary hypertension (CTEPH): what do we know about it? A comprehensive review of the literature. J Cardiovasc Med. (2019) 20:159–68. doi: 10.2459/JCM.0000000000000774
35. Halank M, Hoeper MM, Ghofrani HA, Meyer FJ, Stahler G, Behr J, et al. Riociguat for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: results from a phase II long-term extension study. Respir Med. (2017) 128:50–6. doi: 10.1016/j.rmed.2017.05.008
36. Pepke-Zaba J, Ghofrani HA, Hoeper MM. Medical management of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. (2017) 26:160107. doi: 10.1183/16000617.0107-2016
37. Stam K, van Duin RWB, Uitterdijk A, Cai Z, Duncker DJ, Merkus D. Exercise facilitates early recognition of cardiac and vascular remodeling in chronic thromboembolic pulmonary hypertension in swine. Am J Physiol Heart Circ Physiol. (2018) 314:H627–42. doi: 10.1152/ajpheart.00380.2017
38. Shelub, van Grondelle A, McCullough R, Hofmeister S, Reeves JT. A model of embolic chronic pulmonary hypertension in the dog. J Appl Physiol Respir Environ Exerc Physiol. (1984) 56:810–5. doi: 10.1152/jappl.1984.56.3.810
39. Perkett EA, Brigham KL, Meyrick B. Continuous air embolization into sheep causes sustained pulmonary hypertension and increased pulmonary vasoreactivity. Am J Pathol. (1988) 132:444–54.
40. Moser KM, Cantor JP, Olman M, Villespin I, Graif JL, Konopka R, et al. Chronic pulmonary thromboembolism in dogs treated with tranexamic acid. Circulation. (1991) 83:1371–9. doi: 10.1161/01.CIR.83.4.1371
41. Weimann J, Zink W, Schnabel PA, Jakob H, Gebhard MM, Martin E, et al. Selective vasodilation by nitric oxide inhalation during sustained pulmonary hypertension following recurrent microembolism in pigs. J Crit Care. (1999) 14:133–40. doi: 10.1016/S0883-9441(99)90026-6
42. Kim H, Yung GL, Marsh JJ, Konopka RG, Pedersen CA, Chiles PG, et al. Endothelin mediates pulmonary vascular remodelling in a canine model of chronic embolic pulmonary hypertension. Eur Respir J. (2000) 15:640–8. doi: 10.1034/j.1399-3003.2000.15d04.x
43. Zhou X, Wang D, Castro CY, Hawkins H, Lynch JE, Liu X, et al. A pulmonary hypertension model induced by continuous pulmonary air embolization. J Surg Res. (2011) 170:e11–6. doi: 10.1016/j.jss.2011.04.030
44. Sage E, Mercier O, Herve P, Tu L, Dartevelle P, Eddahibi S, et al. Right lung ischemia induces contralateral pulmonary vasculopathy in an animal model. J Thorac Cardiovasc Surg. (2012) 143:967–73. doi: 10.1016/j.jtcvs.2011.12.052
45. Pohlmann JR, Akay B, Camboni D, Koch KL, Mervak BM, Cook KE. A low mortality model of chronic pulmonary hypertension in sheep. J Surg Res. (2012) 175:44–8. doi: 10.1016/j.jss.2011.02.049
46. Garcia-Alvarez A, Fernandez-Friera L, Garcia-Ruiz JM, Nuno-Ayala M, Pereda D, Fernandez-Jimenez R, et al. Non-invasive monitoring of serial changes in pulmonary vascular resistance and acute vasodilator testing using cardiac magnetic resonance. J Am Coll Cardiol. (2013) 62:1621–31. doi: 10.1016/j.jacc.2013.07.037
47. Mercier O, Tivane A, Dorfmuller P, de Perrot M, Raoux F, Decante B, et al. Piglet model of chronic pulmonary hypertension. Pulm Circ. (2013) 3:908–15. doi: 10.1086/674757
48. Guihaire J, Haddad F, Boulate D, Capderou A, Decante B, Flecher E, et al. Right ventricular plasticity in a porcine model of chronic pressure overload. J Heart Lung Transplant. (2014) 33:194–202. doi: 10.1016/j.healun.2013.10.026
49. Guihaire J, Haddad F, Noly PE, Boulate D, Decante B, Dartevelle P, et al. Right ventricular reserve in a piglet model of chronic pulmonary hypertension. Eur Respir J. (2015) 45:709–17. doi: 10.1183/09031936.00081314
50. Boulate D, Perros F, Dorfmuller P, Arthur-Ataam J, Guihaire J, Lamrani L, et al. Pulmonary microvascular lesions regress in reperfused chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant. (2015) 34:457–67. doi: 10.1016/j.healun.2014.07.005
51. Aguero J, Ishikawa K, Fish KM, Hammoudi N, Hadri L, Garcia-Alvarez A, et al. Combination proximal pulmonary artery coiling and distal embolization induces chronic elevations in pulmonary artery pressure in Swine. PLoS ONE. (2015) 10:e0124526. doi: 10.1371/journal.pone.0124526
52. Noly PE, Haddad F, Arthur-Ataam J, Langer N, Dorfmuller P, Loisel F, et al. The importance of capillary density-stroke work mismatch for right ventricular adaptation to chronic pressure overload. J Thorac Cardiovasc Surg. (2017) 154:2070–9. doi: 10.1016/j.jtcvs.2017.05.102
53. Tang CX, Yang GF, Schoepf UJ, Han ZH, Qi L, Zhao YE, et al. Chronic thromboembolic pulmonary hypertension: comparison of dual-energy computed tomography and single photon emission computed tomography in canines. Eur J Radiol. (2016) 85:498–506. doi: 10.1016/j.ejrad.2015.11.035
54. Rothman Wiencek RG, Davidson S, Evans WN, Restrepo H, Sarukhanov V, Mann D. Challenges in the development of chronic pulmonary hypertension models in large animals. Pulm Circ. (2017) 7:156–66. doi: 10.1086/690099
55. Stam K, Cai Z, van der Velde N, van Duin R, Lam E, van der Velden J. Cardiac remodeling in a swine model of chronic thrombo-embolic pulmonary hypertension- comparison of right vs. left ventricle. J Physiol. (2019) 597:4465–80. doi: 10.1113/JP277896
56. Stam K, van Duin RW, Uitterdijk A, Krabbendam-Peters I, Sorop O, Danser AHJ, et al. Pulmonary microvascular remodeling in chronic thrombo-embolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L951–64. doi: 10.1152/ajplung.00043.2018
57. Mulchrone Kellihan HB, Forouzan O, Hacker TA, Bates ML, Francois CJ, Chesler NC. A large animal model of right ventricular failure due to chronic thromboembolic pulmonary hypertension: a focus on function. Front Cardiovasc Med. (2019) 5:189. doi: 10.3389/fcvm.2018.00189
58. Loisel F, Provost B, Guihaire J, Boulate D, Arouche N, Amsallem M, et al. Autologous endothelial progenitor cell therapy improves right ventricular function in a model of chronic thromboembolic pulmonary hypertension. J Thorac Cardiovasc Surg. (2019) 157:655–66.e7. doi: 10.1016/j.jtcvs.2018.08.083
59. Brimioulle S, Wauthy P, Naeije R. Single-beat evaluation of right ventricular contractility. Crit Care Med. (2005) 33:917–8. doi: 10.1097/01.CCM.0000156236.49988.31
60. Toshner M, Pepke-Zaba J. Chronic thromboembolic pulmonary hypertension: time for research in pathophysiology to catch up with developments in treatment. F1000Prime Rep. (2014) 6:38. doi: 10.12703/P6-38
61. Skoro-Sajer N, Gerges C, Gerges M, Panzenbock A, Jakowitsch J, Kurz A, et al. Usefulness of thrombosis and inflammation biomarkers in chronic thromboembolic pulmonary hypertension-sampling plasma and surgical specimens. J Heart Lung Transplant. (2018) 37:1067–74. doi: 10.1016/j.healun.2018.04.003
62. Azarian R, Wartski M, Collignon MA, Parent F, Herve P, Sors H, et al. Lung perfusion scans and hemodynamics in acute and chronic pulmonary embolism. J Nucl Med. (1997) 38:980–3.
63. Fedullo PF, Auger WR, Kerr KM, Rubin LJ. Chronic thromboembolic pulmonary hypertension. N Engl J Med. (2001) 345:1465–72. doi: 10.1056/NEJMra010902
64. Guihaire J, Haddad F, Boulate D, Decante B, Denault AY, Wu J, et al. Non-invasive indices of right ventricular function are markers of ventricular-arterial coupling rather than ventricular contractility: insights from a porcine model of chronic pressure overload. Eur Heart J Cardiovasc Imaging. (2013) 14:1140–9. doi: 10.1093/ehjci/jet092
65. Huang W, Yen RT, McLaurine M, Bledsoe G. Morphometry of the human pulmonary vasculature. J Appl Physiol. (1996) 81:2123–33. doi: 10.1152/jappl.1996.81.5.2123
66. Townsley MI. Structure and composition of pulmonary arteries, capillaries, and veins. Compr Physiol. (2012) 2:675–709. doi: 10.1002/cphy.c100081
67. Lee YC, Clark AR, Fuld MK, Haynes S, Divekar AA, Hoffman EA, et al. MDCT-based quantification of porcine pulmonary arterial morphometry and self-similarity of arterial branching geometry. J Appl Physiol. (2013) 114:1191–201. doi: 10.1152/japplphysiol.00868.2012
68. Rendas Branthwaite M, Reid L. Growth of pulmonary circulation in normal pig–structural analysis and cardiopulmonary function. J Appl Physiol Respir Environ Exerc Physiol. (1978) 45:806–17. doi: 10.1152/jappl.1978.45.5.806
69. Burrowes KS, Hunter PJ, Tawhai MH. Anatomically based finite element models of the human pulmonary arterial and venous trees including supernumerary vessels. J Appl Physiol. (2005) 99:731–8. doi: 10.1152/japplphysiol.01033.2004
70. Delcroix M, Vonk Noordegraaf A, Fadel E, Lang I, Simonneau G, Naeije R. Vascular and right ventricular remodelling in chronic thromboembolic pulmonary hypertension. Eur Respir J. (2013) 41:224–32. doi: 10.1183/09031936.00047712
71. Langer F, Schramm R, Bauer M, Tscholl D, Kunihara T, Schafers HJ. Cytokine response to pulmonary thromboendarterectomy. Chest. (2004) 126:135–41. doi: 10.1378/chest.126.1.135
72. Quarck R, Wynants M, Ronisz A, Sepulveda MR, Wuytack F, Van Raemdonck D, et al. Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients. Respir Res. (2012) 13:27. doi: 10.1186/1465-9921-13-27
73. Zabini D, Heinemann A, Foris V, Nagaraj C, Nierlich P, Balint Z, et al. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J. (2014) 44:951–62. doi: 10.1183/09031936.00145013
74. Skoro-Sajer N, Mittermayer F, Panzenboeck A, Bonderman D, Sadushi R, Hitsch R, et al. Asymmetric dimethylarginine is increased in chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med. (2007) 176:1154–60. doi: 10.1164/rccm.200702-278OC
75. Abe S, Ishida K, Masuda M, Ueda H, Kohno H, Matsuura K, et al. A prospective, randomized study of inhaled prostacyclin versus nitric oxide in patients with residual pulmonary hypertension after pulmonary endarterectomy. Gen Thorac Cardiovasc Surg. (2017) 65:153–9. doi: 10.1007/s11748-016-0724-2
76. Ghofrani HA, D'Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. (2013) 369:319–29. doi: 10.1056/NEJMoa1209657
77. Koress C, Swan K, Kadowitz P. Soluble guanylate cyclase stimulators and activators: novel therapies for pulmonary vascular disease or a different method of increasing cGMP? Curr Hypertens Rep. (2016) 18:42. doi: 10.1007/s11906-016-0645-6
78. Lian TY, Jiang X, Jing ZC. Riociguat: a soluble guanylate cyclase stimulator for the treatment of pulmonary hypertension. Drug Des Devel Ther. (2017) 11:1195–207. doi: 10.2147/DDDT.S117277
79. Guo L, Yang Y, Liu J, Wang L, Li J, Wang Y, et al. Differentially expressed plasma microRNAs and the potential regulatory function of Let-7b in chronic thromboembolic pulmonary hypertension. PLoS ONE. (2014) 9:e101055. doi: 10.1371/journal.pone.0101055
80. Reesink HJ, Meijer RC, Lutter R, Boomsma F, Jansen HM, Kloek JJ, et al. Hemodynamic and clinical correlates of endothelin-1 in chronic thromboembolic pulmonary hypertension. Circ J. (2006) 70:1058–63. doi: 10.1253/circj.70.1058
81. Hirashiki Adachi S, Nakano Y, Kamimura Y, Shimokata S, Takeshita K, Murohara T, et al. Effects of bosentan on peripheral endothelial function in patients with pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension. Pulm Circ. (2016) 6:168–73. doi: 10.1086/685715
82. Hirashiki Adachi S, Nakano Y, Kono Y, Shimazu S, Shimizu S, Morimoto R, et al. Cardiopulmonary exercise testing to evaluate the exercise capacity of patients with inoperable chronic thromboembolic pulmonary hypertension: an endothelin receptor antagonist improves the peak PETCO2. Life Sci. (2014) 118:397–403. doi: 10.1016/j.lfs.2014.03.009
83. Nishikawa-Takahashi M, Ueno S, Kario K. Long-term advanced therapy with bosentan improves symptoms and prevents deterioration of inoperable chronic thromboembolic pulmonary hypertension. Life Sci. (2014) 118:410–3. doi: 10.1016/j.lfs.2014.03.024
84. Jais X, D'Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, et al. Bosentan effects in iNopErable forms of chronIc thromboembolic pulmonary hypertension study, bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable forms of chronIc thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol. (2008) 52:2127–34. doi: 10.1016/j.jacc.2008.08.059
85. Southwood M, MacKenzie Ross RV, Kuc RE, Hagan G, Sheares KK, Jenkins DP, et al. Endothelin ETA receptors predominate in chronic thromboembolic pulmonary hypertension. Life Sci. (2016) 159:104–10. doi: 10.1016/j.lfs.2016.02.036
86. Ghofrani HA, Simonneau G, D'Armini AM, Fedullo P, Howard LS, Jais X, et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med. (2017) 5:785–94. doi: 10.1183/1393003.congress-2017.OA1984
87. Horinouchi T, Terada K, Higashi T, Miwa S. Endothelin receptor signaling: new insight into its regulatory mechanisms. J Pharmacol Sci. (2013) 123:85–101. doi: 10.1254/jphs.13R02CR
88. Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. (2013) 77:2619–25. doi: 10.1253/circj.CJ-13-0443
89. Doe Z, Fukumoto Y, Takaki A, Tawara S, Ohashi J, Nakano M, et al. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ J. (2009) 73:1731–9. doi: 10.1253/circj.CJ-09-0135
90. Blumberg FC, Arzt M, Lange T, Schroll S, Pfeifer M, Wensel R. Impact of right ventricular reserve on exercise capacity and survival in patients with pulmonary hypertension. Eur J Heart Fail. (2013) 15:771–5. doi: 10.1093/eurjhf/hft044
91. Grosse Grosse C, Lang I. Evaluation of the CT imaging findings in patients newly diagnosed with chronic thromboembolic pulmonary hypertension. PLoS ONE. (2018) 13:e0201468. doi: 10.1371/journal.pone.0201468
92. Trip P, Rain S, Handoko ML, van der Bruggen C, Bogaard HJ, Marcus JT, et al. Clinical relevance of right ventricular diastolic stiffness in pulmonary hypertension. Eur Respir J. (2015) 45:1603–12. doi: 10.1183/09031936.00156714
93. Aguero J, Ishikawa K, Hadri L, Santos-Gallego C, Fish K, Hammoudi N, et al. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am J Physiol Heart Circ Physiol. (2014) 307:H1204–15. doi: 10.1152/ajpheart.00246.2014
94. Rain S, Andersen S, Najafi A, Gammelgaard Schultz J, da Silva Goncalves Bos D, Handoko ML, et al. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension: relative contribution of fibrosis and myofibril stiffness. Circ Heart Fail. (2016) 9:e002636. doi: 10.1161/CIRCHEARTFAILURE.115.002636
95. Frump AL, Bonnet S, de Jesus Perez VA, Lahm T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L443–60. doi: 10.1152/ajplung.00374.2017
96. van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. (2008) 29:120–7. doi: 10.1093/eurheartj/ehm567
97. Vogel-Claussen J, Skrok J, Shehata ML, Singh S, Sibley CT, Boyce DM, et al. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology. (2011) 258:119–27. doi: 10.1148/radiol.10100725
98. Dewachter L, Dewachter C. Inflammation in right ventricular failure: does it matter? Front Physiol. (2018) 9:1056. doi: 10.3389/fphys.2018.01056
99. Sun XQ, Abbate A, Bogaard HJ. Role of cardiac inflammation in right ventricular failure. Cardiovasc Res. (2017) 113:1441–52. doi: 10.1093/cvr/cvx159
100. Frangogiannis NG. Fibroblasts and the extracellular matrix in right ventricular disease. Cardiovasc Res. (2017) 113:1453–64. doi: 10.1093/cvr/cvx146
101. Opitz Kirschner MB. Molecular research in chronic thromboembolic pulmonary hypertension. Int J Mol Sci. (2019) 20:784. doi: 10.3390/ijms20030784
102. Rol N, Kurakula KB, Happe C, Bogaard HJ, Goumans MJ. TGF-beta and BMPR2 signaling in PAH: two black sheep in one family. Int J Mol Sci. (2018) 19:2585. doi: 10.3390/ijms19092585
103. Shimizu T, Liao JK. Rho kinases and cardiac remodeling. Circ J. (2016) 80:1491–8. doi: 10.1253/circj.CJ-16-0433
104. Tsai SH, Lu G, Xu X, Ren Y, Hein TW, Kuo L. Enhanced endothelin-1/Rho-kinase signalling and coronary microvascular dysfunction in hypertensive myocardial hypertrophy. Cardiovasc Res. (2017) 113:1329–37. doi: 10.1093/cvr/cvx103
105. Zeidan Gan XT, Thomas A, Karmazyn M. Prevention of RhoA activation and cofilin-mediated actin polymerization mediates the antihypertrophic effect of adenosine receptor agonists in angiotensin II- and endothelin-1-treated cardiomyocytes. Mol Cell Biochem. (2014) 385:239–48. doi: 10.1007/s11010-013-1832-2
106. Sunamura S, Satoh K, Kurosawa R, Ohtsuki T, Kikuchi N, Elias-Al-Mamun M, et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc Natl Acad Sci U S A. (2018) 115:E7129–38. doi: 10.1073/pnas.1721298115
107. Ikeda S, Satoh K, Kikuchi N, Miyata S, Suzuki K, Omura J, et al. Crucial role of rho-kinase in pressure overload-induced right ventricular hypertrophy and dysfunction in mice. Arterioscler Thromb Vasc Biol. (2014) 34:1260–71. doi: 10.1161/ATVBAHA.114.303320
108. Hartmann S, Ridley AJ, Lutz S. The function of Rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front Pharmacol. (2015) 6:276. doi: 10.3389/fphar.2015.00276
109. Chen IC, Tan MS, Wu BN, Chai CY, Yeh JL, Chou SH, et al. Statins ameliorate pulmonary hypertension secondary to left ventricular dysfunction through the Rho-kinase pathway and NADPH oxidase. Pediatr Pulmonol. (2017) 52:443–57. doi: 10.1002/ppul.23610
110. Lu X, Dang CQ, Guo X, Molloi S, Wassall CD, Kemple MD, et al. Elevated oxidative stress and endothelial dysfunction in right coronary artery of right ventricular hypertrophy. J Appl Physiol. (2011) 110:1674–81. doi: 10.1152/japplphysiol.00744.2009
111. Godinas L, Sattler C, Lau EM, Jais X, Taniguchi Y, Jevnikar M, et al. Dead-space ventilation is linked to exercise capacity and survival in distal chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant. (2017) 36:1234–42. doi: 10.1016/j.healun.2017.05.024
112. Anand V, Roy SS, Archer SL, Weir EK, Garg SK, Duval S, et al. Trends and outcomes of pulmonary arterial hypertension-related hospitalizations in the United States: analysis of the nationwide inpatient sample database from 2001 through 2012. JAMA Cardiol. (2016) 1:1021–9. doi: 10.1001/jamacardio.2016.3591
113. Kanemoto N, Sasamoto H. Arrhythmias in primary pulmonary hypertension. Jpn Heart J. (1979) 20:765–75. doi: 10.1536/ihj.20.765
114. Rajdev Garan H, Biviano A. Arrhythmias in pulmonary arterial hypertension. Prog Cardiovasc Dis. (2012) 55:180–6. doi: 10.1016/j.pcad.2012.06.002
115. Tongers J, Schwerdtfeger B, Klein G, Kempf T, Schaefer A, Knapp JM, et al. Incidence and clinical relevance of supraventricular tachyarrhythmias in pulmonary hypertension. Am Heart J. (2007) 153:127–32. doi: 10.1016/j.ahj.2006.09.008
116. Cannillo M, Grosso Marra W, Gili S, D'Ascenzo F, Morello M, Mercante L, et al. Supraventricular arrhythmias in patients with pulmonary arterial hypertension. Am J Cardiol. (2015) 116:1883–9. doi: 10.1016/j.amjcard.2015.09.039
117. Hoeper MM, Galie N, Murali S, Olschewski H, Rubenfire M, Robbins IM, et al. Outcome after cardiopulmonary resuscitation in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. (2002) 165:341–4. doi: 10.1164/ajrccm.165.3.200109-0130c
118. Strauss Sassi Y, Bueno-Beti C, Ilkan Z, Raad N, Cacheux M, Bisserier M, et al. Intra-tracheal gene delivery of aerosolized SERCA2a to the lung suppresses ventricular arrhythmias in a model of pulmonary arterial hypertension. J Mol Cell Cardiol. (2019) 127:20–30. doi: 10.1016/j.yjmcc.2018.11.017
119. Umar S, Lee JH, de Lange E, Iorga A, Partow-Navid R, Bapat A, et al. Spontaneous ventricular fibrillation in right ventricular failure secondary to chronic pulmonary hypertension. Circ Arrhythm Electrophysiol. (2012) 5:181–90. doi: 10.1161/CIRCEP.111.967265
120. Tonelli AR, Baumgartner M, Alkukhun L, Minai OA, Dweik RA. Electrocardiography at diagnosis and close to the time of death in pulmonary arterial hypertension. Ann Noninvasive Electrocardiol. (2014) 19:258–65. doi: 10.1111/anec.12125
121. Rich JD, Thenappan T, Freed B, Patel AR, Thisted RA, Childers R, et al. QTc prolongation is associated with impaired right ventricular function and predicts mortality in pulmonary hypertension. Int J Cardiol. (2013) 167:669–76. doi: 10.1016/j.ijcard.2012.03.071
122. Smith Genuardi MV, Koczo A, Zou RH, Thoma FW, Handen A, Craig E, et al. Atrial arrhythmias are associated with increased mortality in pulmonary arterial hypertension. Pulm Circ. (2018) 8:1–9. doi: 10.1177/2045894018790316
123. Folino AF, Bobbo F, Schiraldi C, Tona F, Romano S, Buja G, et al. Ventricular arrhythmias and autonomic profile in patients with primary pulmonary hypertension. Lung. (2003) 181:321–8. doi: 10.1007/s00408-003-1034-x
124. Ciarka Doan V, Velez-Roa SR. Naeije, van de Borne P. Prognostic significance of sympathetic nervous system activation in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2010) 181:1269–75. doi: 10.1164/rccm.200912-1856OC
125. Zhao Q, Deng H, Jiang X, Dai Z, Wang X, Wang X, et al. Effects of intrinsic and extrinsic cardiac nerves on atrial arrhythmia in experimental pulmonary artery hypertension. Hypertension. (2015) 66:1042–9. doi: 10.1161/HYPERTENSIONAHA.115.05846
126. Medi Kalman JM, Ling LH, Teh AW, Lee G, Lee G, Spence SJ, et al. Atrial electrical and structural remodeling associated with longstanding pulmonary hypertension and right ventricular hypertrophy in humans. J Cardiovasc Electrophysiol. (2012) 23:614–20. doi: 10.1111/j.1540-8167.2011.02255.x
127. Benoist Stones R, Drinkhill M, Bernus O, White E. Arrhythmogenic substrate in hearts of rats with monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Am J Physiol Heart Circ Physiol. (2011) 300:H2230–7. doi: 10.1152/ajpheart.01226.2010
128. Benoist D, Stones R, Drinkhill MJ, Benson AP, Yang Z, Cassan C, et al. Cardiac arrhythmia mechanisms in rats with heart failure induced by pulmonary hypertension. Am J Physiol Heart Circ Physiol. (2012) 302:H2381–95. doi: 10.1152/ajpheart.01084.2011
129. Tanaka Y, Takase B, Yao T, Ishihara M. Right ventricular electrical remodeling and arrhythmogenic substrate in rat pulmonary hypertension. Am J Respir Cell Mol Biol. (2013) 49:426–36. doi: 10.1165/rcmb.2012-0089OC
130. Held M, Grun M, Holl R, Hubner G, Kaiser R, Karl S, et al. Cardiopulmonary exercise testing to detect chronic thromboembolic pulmonary hypertension in patients with normal echocardiography. Respiration. (2014) 87:379–87. doi: 10.1159/000358565
131. Held M, Hesse A, Gott F, Holl R, Hubner G, Kolb P, et al. A symptom-related monitoring program following pulmonary embolism for the early detection of CTEPH: a prospective observational registry study. BMC Pulm Med. (2014) 14:141. doi: 10.1186/1471-2466-14-141
132. Richter MJ, Pader P, Gall H, Reichenberger F, Seeger W, Mayer E, et al. The prognostic relevance of oxygen uptake in inoperable chronic thromboembolic pulmonary hypertension. Clin Respir J. (2015) 11:682–90. doi: 10.1111/crj.12399
133. Hasler ED, Muller-Mottet S, Furian M, Saxer S, Huber LC, Maggiorini M, et al. Pressure-flow during exercise catheterization predicts survival in pulmonary hypertension. Chest. (2016) 150:57–67. doi: 10.1016/j.chest.2016.02.634
134. Claessen La Gerche A, Dymarkowski S, Claus P, Delcroix M, Heidbuchel H. Pulmonary vascular and right ventricular reserve in patients with normalized resting hemodynamics after pulmonary endarterectomy. J Am Heart Assoc. (2015) 4:e001602. doi: 10.1161/JAHA.114.001602
135. Claessen La Gerche A, Wielandts JY, Bogaert J, Van Cleemput J, Wuyts W, Claus P, et al. Exercise pathophysiology and sildenafil effects in chronic thromboembolic pulmonary hypertension. Heart. (2015) 101:637–44. doi: 10.1136/heartjnl-2014-306851
136. Melot C, Naeije R. Pulmonary vascular diseases. Compr Physiol. (2011) 1:593–619. doi: 10.1002/cphy.c090014
137. Merkus D, de Beer VJ, Houweling B, Duncker DJ. Control of pulmonary vascular tone during exercise in health and pulmonary hypertension. Pharmacol Ther. (2008) 119:242–63. doi: 10.1016/j.pharmthera.2008.04.003
138. Iwase T, Nagaya N, Ando M, Satoh T, Sakamaki F, Kyotani S, et al. Acute and chronic effects of surgical thromboendarterectomy on exercise capacity and ventilatory efficiency in patients with chronic thromboembolic pulmonary hypertension. Heart. (2001) 86:188–92. doi: 10.1136/heart.86.2.188
139. Cai Z, Klein T, Geenen LW, Tu L, Tian S, van den Bosch AE, et al. Lower plasma melatonin levels predict worse long-term survival in pulmonary arterial hypertension. J Clin Med. (2020) 9:1248. doi: 10.3390/jcm9051248
140. Clauss S, Bleyer C, Schuttler D, Tomsits P, Renner S, Klymiuk N, et al. Animal models of arrhythmia: classic electrophysiology to genetically modified large animals. Nat Rev Cardiol. (2019) 16:457–75. doi: 10.1038/s41569-019-0179-0
141. Clauss S, Schuttler D, Bleyer C, Vlcek J, Shakarami M, Tomsits P, et al. Characterization of a porcine model of atrial arrhythmogenicity in the context of ischaemic heart failure. PLoS ONE. (2020) 15:e0232374. doi: 10.1371/journal.pone.0232374
Keywords: CTEPH, pulmonary hypertension, pulmonary vasculature, vascular resistence, large animal models, swine models, arrhythmogenesis, cardiac remodeling
Citation: Stam K, Clauss S, Taverne YJHJ and Merkus D (2021) Chronic Thromboembolic Pulmonary Hypertension – What Have We Learned From Large Animal Models. Front. Cardiovasc. Med. 8:574360. doi: 10.3389/fcvm.2021.574360
Received: 19 June 2020; Accepted: 08 March 2021;
Published: 16 April 2021.
Edited by:
Nicola Montano, University of Milan, ItalyReviewed by:
Gaetano Ruocco, Regina Montis Regalis Hospital, ItalyAbraham Rothman, Children's Heart Center Nevada, United States
Copyright © 2021 Stam, Clauss, Taverne and Merkus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daphne Merkus, ZGFwaG5lLm1lcmt1c0BtZWQudW5pLW11ZW5jaGVuLmRl; ZC5tZXJrdXNAZXJhc211c21jLm5s