 Graham Chakafana1,2
Graham Chakafana1,2 Timothy F. Spracklen1,2Stephen Kamuli1,2
Timothy F. Spracklen1,2Stephen Kamuli1,2 Tawanda Zininga3
Tawanda Zininga3 Addmore Shonhai4Ntobeko A. B. Ntusi1,2,5Karen Sliwa1,2*
Addmore Shonhai4Ntobeko A. B. Ntusi1,2,5Karen Sliwa1,2*- 1Department of Medicine, Faculty of Health Sciences, Cape Heart Institute, University of Cape Town, Cape Town, South Africa
- 2Division of Cardiology, Department of Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa
- 3Department of Biochemistry, Stellenbosch University, Stellenbosch, South Africa
- 4Department of Biochemistry, University of Venda, Thohoyandou, South Africa
- 5Cape Universities Body Imaging Centre, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa
Peripartum cardiomyopathy (PPCM) is a potentially life-threatening condition in which heart failure and systolic dysfunction occur late in pregnancy or within months following delivery. To date, no reliable biomarkers or therapeutic interventions for the condition exist, thus necessitating an urgent need for identification of novel PPCM drug targets and candidate biomarkers. Leads for novel treatments and biomarkers are therefore being investigated worldwide. Pregnancy is generally accompanied by dramatic hemodynamic changes, including a reduced afterload and a 50% increase in cardiac output. These increased cardiac stresses during pregnancy potentially impair protein folding processes within the cardiac tissue. The accumulation of misfolded proteins results in increased toxicity and cardiac insults that trigger heart failure. Under stress conditions, molecular chaperones such as heat shock proteins (Hsps) play crucial roles in maintaining cellular proteostasis. Here, we critically assess the potential role of Hsps in PPCM. We further predict specific associations between the Hsp types Hsp70, Hsp90 and small Hsps with several proteins implicated in PPCM pathophysiology. Furthermore, we explore the possibility of select Hsps as novel candidate PPCM biomarkers and drug targets. A better understanding of how these Hsps modulate PPCM pathogenesis holds promise in improving treatment, prognosis and management of the condition, and possibly other forms of acute heart failure.
Introduction
Protein folding processes are fundamental in the maintenance of cardiac tissue integrity (1). The metabolic and mechanical demands of the heart, such as its continuous contractile activities, place a burden for robust protein quality control systems (2). Several cardiovascular diseases such as ischemic heart disease and heart failure (HF) are characterised by increased mechanical and oxidative pressures which trigger an accumulation of misfolded proteins in cardiomyocytes. Misfolded proteins are toxic to cardiomyocytes, potentially causing cardiac insults that lead to HF (3). Under ensuing stress conditions, protein folding processes which typically occur in the sarcoplasmic reticulum, sarcomeres and mitochondria, are crucial in maintaining cardiac muscle integrity (4). Molecular chaperones such as heat shock proteins (Hsps) are an important class of proteins involved in the maintenance of proteostasis in various cell types under both normal and stress conditions. Here, we review the potential involvement of Hsps in peripartum cardiomyopathy (PPCM). Notably, the additional cardiac stress associated with pregnancy may further induce unique protein folding pathways in PPCM. Using bioinformatics tools, we further propose novel interactions (between proteins involved in PPCM pathogenesis and Hsps) which can be targeted toward drug interventions. Currently, echocardiography is the principal diagnostic tool for PPCM, as no reliable biomarkers exist. In this review, we also critically assess the potential of Hsps as candidate PPCM biomarkers.
Pathophysiology and Molecular Pathways of PPCM
PPCM is a common and devastating disease that is associated with the unexpected loss of maternal cardiac function in the period surrounding parturition i.e., toward the end of pregnancy or within months following delivery (5). PPCM is characterised by a decreased left ventricular ejection fraction (LVEF) that is <45% in patients without prior cardiac disease (6). Generally, PPCM presents as congestive HF and systolic dysfunction, with typical symptoms including dyspnoea, fatigue, palpitations, oedema and chest pain (5). Although 90% of PPCM cases present after parturition, in rare cases disease onset has also been reported in the second trimester (7, 8). With a reported incidence ranging from 1:100 to 1:10,000 deliveries (9, 10), the frequency of PPCM appears to be influenced by ethnicity, with individuals of African origin at greater risk of developing the disease (10, 11). Other risk factors for PPCM include preeclampsia, multiparity, maternal age and multiple pregnancies.
Although the pathophysiology of PPCM is not entirely understood, several mechanisms of disease have been suggested. These include malnutrition, viral infection, autoimmunity and increased haemodynamic strain, although their roles in PPCM have proved nebulous (12). Current evidence, however, strongly suggests that PPCM may be driven by a pathological imbalance of pro- and anti-angiogenic hormones, as well as genetic factors. Two main pathways underlying disease pathogenesis have accrued from studies of mouse models of PPCM, as well as observations in human patients. These are reviewed in more detail elsewhere (12), but both involve the creation of a profoundly vasculotoxic environment through imbalances in angiogenic hormones.
The first pathway is characterised by increased expression of the pituitary hormone prolactin (PRL) which, in conditions of high oxidative stress, ultimately leads to cardiomyocyte apoptosis and cardiac dysfunction. The post translational processing of PRL is complex and the full length 23 kDa protein may be cleaved by peptidases (such as Cathepsin D) into a smaller 16 kDa variant. The 16 kDa PRL variant is a potent anti-angiogenic factor which acts as a vasoinhibin which can also cause vascular dropout, global systolic dysfunction and cardiac endothelial apoptosis. A murine PPCM model demonstrated that cardiac-specific deletion of the STAT3 gene caused increased oxidative stress through reduced MnSOD expression (13). Further investigation also revealed an increase in cathepsin D activity and a corresponding increase in 16 kDa PRL levels (13). Altogether, this implies a vital role of STAT3 in cardioprotection during pregnancy, suggesting that dysregulation of STAT3 may also underlie PPCM. Recently, inhibition of Notch1/Hes1 has been found to induce PPCM through suppression of STAT3 activation, as well as increasing cathepsin D expression (14). Another protein involved in PPCM pathophysiology, Akt, is highly activated during pregnancy and promotes cardiac hypertrophy, and was shown to be activated by both PRL and interferon-γ (IFNγ) (15).
The second PPCM pathophysiology pathway involves the increased placental secretion of soluble Fms-like tyrosine kinase 1 (sFlt1) into the maternal system. Precisely why sFlt1 is secreted by the placenta is unclear, but both sFlt1 and membrane bound Flt1 are decoy receptors for vascular endothelial growth factors (VEGFs). VEGFA and VEGFB are proangiogenic factors and important mediators of cardiac homeostasis, but the binding of sFlt1 inhibits their activity (16). VEGF expression is driven by PGC-1α, and suppression of this in murine hearts led to PPCM and an increased susceptibility to sFlt1-induced cardiomyopathy (17). In this study, excessively high sFlt1 levels were able to cause cardiomyopathy, even in mice without the PGC1-1α deletion or pregnancy, indicating that excess sFlt1 alone can induce cardiac dysfunction. This emphasises the sensitivity of the heart to angiogenic imbalance as a result of placental sFlt1, that may occur during pregnancy. Synchtiotrophoblasts of the placenta secrete copious amounts of sFlt1 and as such, plasma levels of the protein rise exponentially toward birth (18). Most of the free VEGF in maternal circulation is thus neutralised by sFlt1 during pregnancy. More so, elevated sFlt1 have been described in women with PPCM (17), and have been directly correlated with disease severity and the occurrence of adverse clinical events (19). Notably, higher sFlt1 levels have been reported in twin pregnancies, another risk factor for PPCM (20, 21), possibly as a result of the larger placenta (22).
Cardiac Mechanical Stress During Pregnancy
Pregnancy is accompanied by dramatic hemodynamic changes, including reduced resistance during systole (afterload) and a 50% increase in cardiac output and blood volume (23). Furthermore, foetal microchimeric cells may reduce cardiac function resulting in increased cardiac mechanical stress during pregnancy (24, 25). It is however worth noting that most of these changes typically occur early in gestation, many months before PPCM typically presents (Figure 1). These changes trigger homeostatic and structural remodelling of cardiovascular tissues. Whereas, hemodynamic changes of pregnancy peak in the second trimester, hormonal changes of pregnancy are most drastic in the third trimester and early postpartum (Figure 1) (26). These changes also coincide with the presentation of PPCM. As such, the vasculotoxic hormonal changes that occur during and after parturition act as a trigger for PPCM. Indeed, several studies have demonstrated that PPCM is triggered by the rapidly changing environment of late gestation thus inducing vasculopathy in susceptible women (13, 27, 28). This is supported by the fact that hormones that likely trigger PPCM (PRL and sFlt1) are mostly at their peak in late pregnancy and postpartum. In addition, unlike other forms of cardiomyopathy, cardiac function is usually restored upon a drop in these hormones which comes with delivery. Apart from sFlt1, the placenta also secretes several other hormones which may result in maternal stress during pregnancy. Despite these cardiac demands associated with pregnancy, there is a need for protein quality control to be maintained in the cardiomyocytes.

Figure 1. Stress associated with PPCM development. Approximately 10% of PPCM cases present within the third trimester, while 90% of PPCM cases present postpartum. Hemodynamic changes associated with pregnancy begin in the second trimester and persist to approximately 6 months postpartum. Several other stresses, such as hormonal changes are also associated with pregnancy.
Cardiomyocyte Proteostasis and Potential Roles of Hsps in PPCM
The robust maintenance of proteostasis in cardiomyocytes is crucial in ensuring the integrity of cardiac tissue. An accumulation of misfolded and unfolded proteins results in the formation of aggregates which are usually cytotoxic. The heart is constantly exposed to mechanical stresses associated with its continuous contractile activities, as well as chemical stresses induced by free radicals and hormones. In PPCM, pregnancy further burdens cardiac tissue, as the heart readjust to the needs of the developing foetus. Stress supresses the cell's capacity to maintain proteostasis thus compromising the ability of proteins to attain native conformation. Cellular stress can lead to protein misfolding or unfolding, leading to proteins that are unable to carry out their normal functions (29). Such stresses typically impair protein folding, potentially resulting in the formation of functionally impaired and toxic protein aggregates that trigger cardiac insults. Under the ensuing stress conditions, molecular chaperones such as Hsps likely facilitate cardiomyocyte proteostasis. Generally, Hsps perform a myriad of housekeeping and stress-protective roles in cells to maintain proteostasis (Figure 2) (30). We propose that Hsps are particularly important in PPCM since the heart is further burdened by pregnancy related stresses that may impair optimal protein folding processes in the cardiomyocytes.
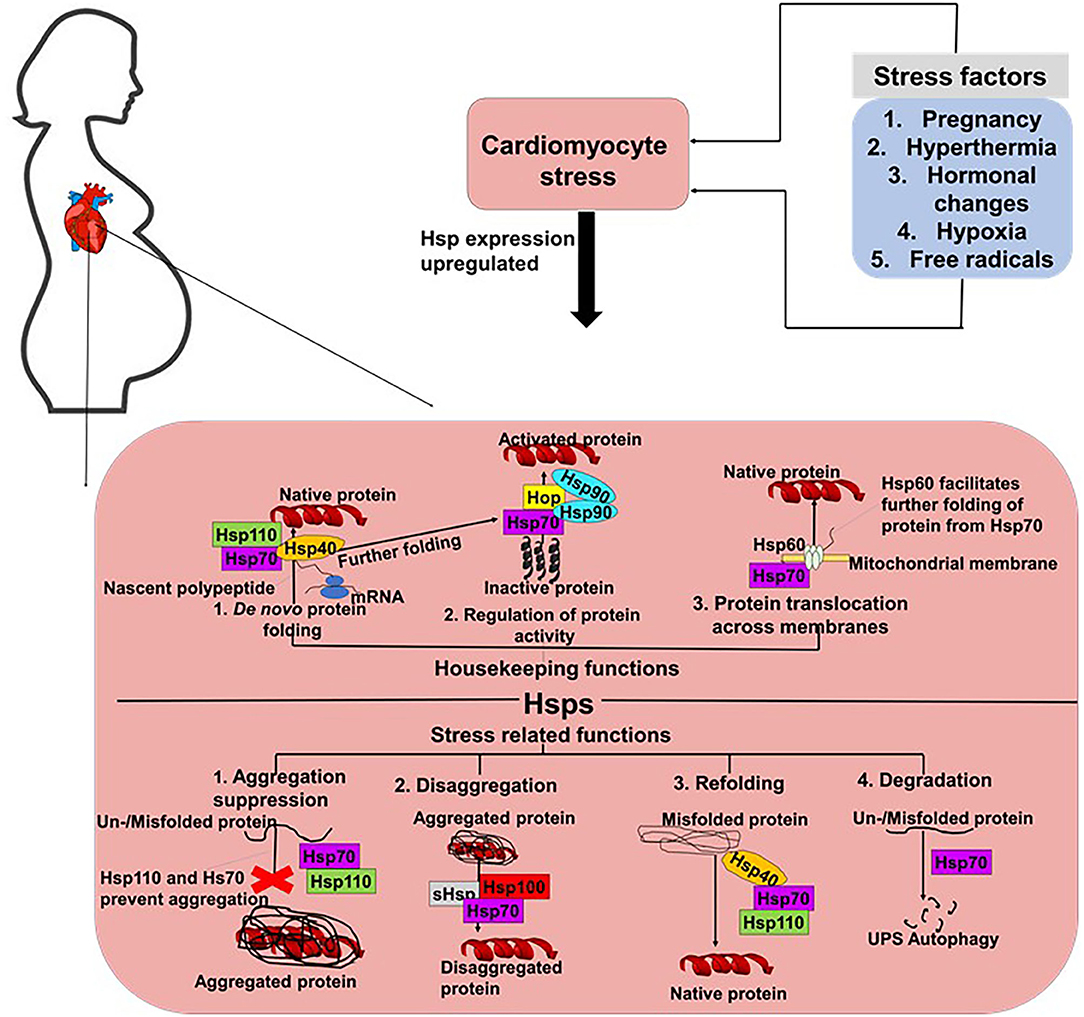
Figure 2. Proposed roles of Hsps in PPCM. In cells, Hsp70s perform both housekeeping and stress response related roles. Hsp70s co-operate with various co-chaperones such as Hsp40 and Hsp110 to facilitate the de novo folding of polypeptides from the ribosome to facilitate their folding into their native conformations. Hsp70s also co-operate with Hsp90 and Hop to activate proteins. Hsp70s also form partnerships with Hsp60 to facilitate the translocation of proteins across membranes. Under stress conditions, Hsp70s prevent the formation of aggregates and also facilitate the degradation of misfolded/unfolded proteins.
Hsps can generally be classified into seven families, based on structural and functional features (Table 1). Broadly, Hsps function to facilitate the correct folding and assembly of polypeptides, thus preventing the formation of misfolded or incorrectly assembled proteins (44). Hsps also play an important role in the suppression or inhibition of polypeptide aggregation in cells (45). When a nascent polypeptide chain exits the ribosome or an organellar import pore, or when a labile native protein becomes transiently heat denatured, it may transiently unfold and expose hydrophobic segments to the aqueous environment (31). Depending on the intensity and duration of the stress as well as the degree of hydrophobic exposure, the misfolded monomers may clamp together through intermolecular hydrophobic associations to form aggregates (46). Hsps of the “holdase” class (such as small Hsps (sHsps), Hsp40, Hsp70, and Hsp110) can bind to the exposed hydrophobic residues on the surface of misfolded polypeptides to prevent the formation of aggregates (47).
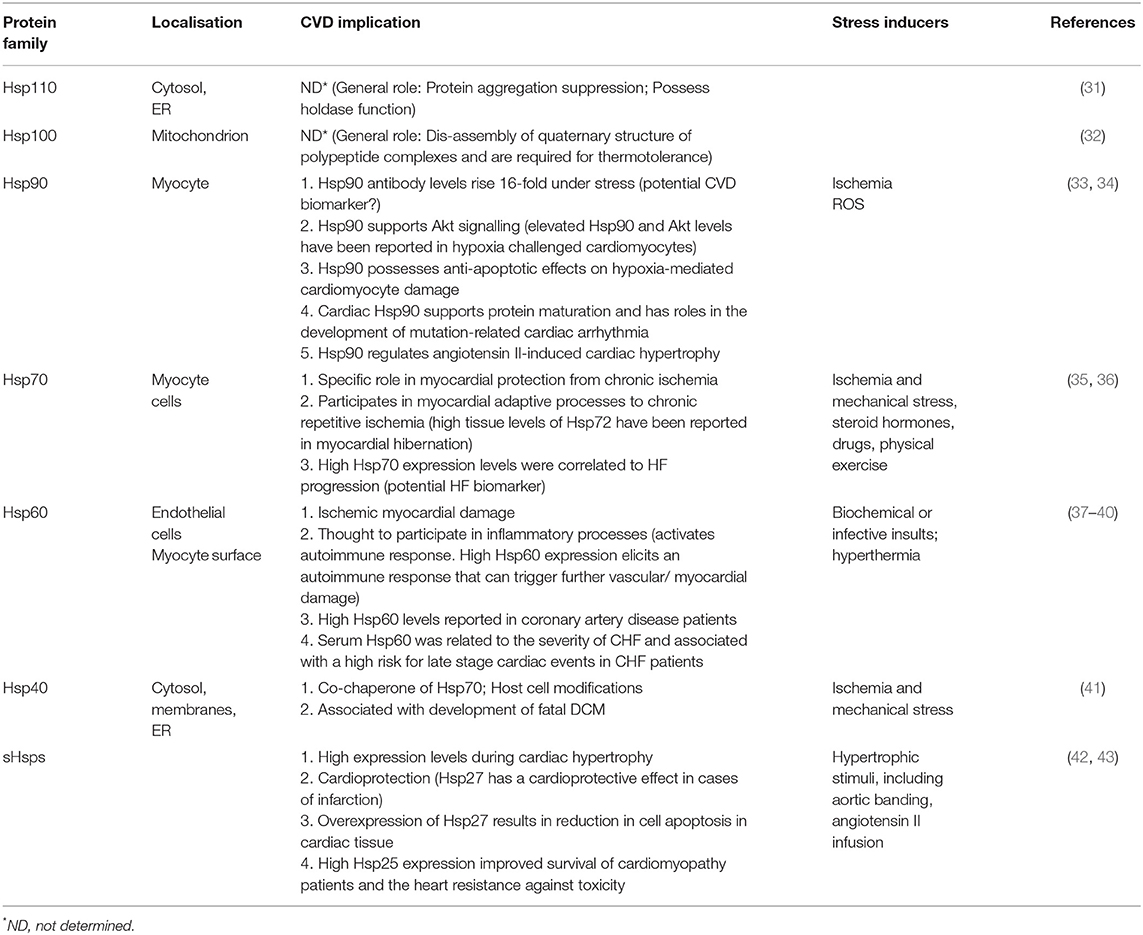
Table 1. Major Hsp families and cardiovascular roles.
The roles of Hsps in several cardiovascular conditions such as HF and dilated cardiomyopathy (DCM) have been reported (Table 1). The Hsp70 family of molecular chaperones is a central hub for the maintenance of proteostasis in cells (48). Hsp70s are actively involved in almost every stage of a protein's life course (Figure 2). Thus, Hsp70s facilitate folding of nascent peptides emerging at the ribosomes (49), protein trafficking and translocation across membranes (50). In addition, Hsp70s facilitate the refolding of misfolded protein (31, 51), and also channelling misfolded proteins which are beyond repair toward degradation (52, 53). In order for Hsp70s to function efficiently, they depend on assistance from functional networks formed with members of several co-chaperones which include Carboxyl terminus of HSC70-interacting protein (CHIP), Bcl-2 associated athanogene 3 (BAG3) and Hsp70-Hsp90 organising protein (Hop) (2). Additionally, Hsp70 also forms functional networks with Hsp40 (54), Hsp90 (55), and sHsps (56). Protein folding by Hsp70 is tightly controlled by J-domain proteins (Hsp40) and nucleotide exchange factors (NEFs) such as BAG3. The co-ordinated action of Hsp70 and Hsp90 facilitates the folding of most structural and signalling proteins (Figure 2). In fact, Hsp70 has been described as a promiscuous chaperone that is capable of binding virtually any peptide sequence (57).
Hsps also play crucial roles in the ubiquitin proteasome system (UPS) which is the main proteolytic system in eukaryotic cells facilitating the degradation of misfolded proteins. It has previously been established that the constitutively expressed Hsp70 (Hsc70) is required for ubiquitylation of several proteasome substrates (58). Furthermore, the majority of E3 ligase complexes of the UPS pathway have been shown to cooperate with Hsps (59). Hsps also function as escort factors that either deliver or dock the Ub-protein conjugates to the proteasome thus preventing the formation of ubiquitylated protein aggregates (Figure 3A) (53). Functional co-operations of Hsp70 could be pivotal in maintaining cardiomyocyte proteostasis during pregnancy-induced stress. The co-chaperone CHIP (a ubiquitin ligase) is ubiquitously expressed, although it is prominently expressed in striated muscle such as cardiac tissue (2). Functionally, CHIP co-operates with Hsp70 to ubiquitinate misfolded proteins that cannot be repaired, targeting them for protein degradation by the proteasome (Figure 3A) (60). Previous studies have implicated CHIP in cardiac disease. In a murine model, genetic knockout of CHIP resulted in exaggerated cardiac hypertrophy, as evidenced by increased heart weights, wall thickness and cardiomyocyte size following exercise or pressure overload (61, 62). Genetic knockout of CHIP was also associated with a dramatic decline in cardiac function in response to pressure overload (63). Due to its ability to regulate Hsp70 chaperone activity, CHIP may also be an important determinant in modulating Hsp70-chaperoned proteins in the cardiomyocyte.
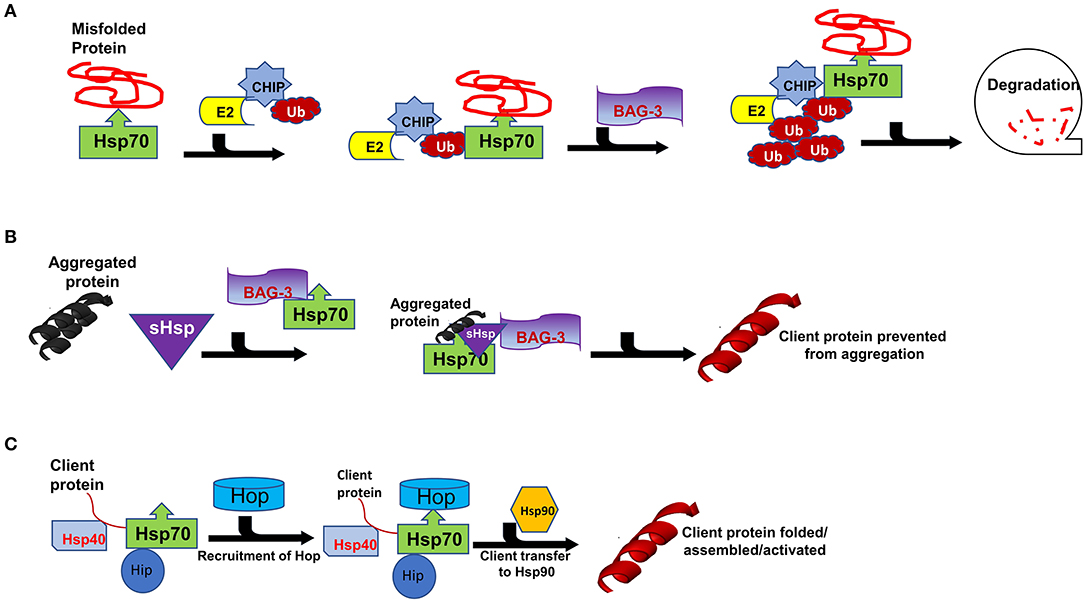
Figure 3. Interaction of Carboxyl terminus of HSC70-interacting protein (CHIP) with binding partners for protein quality control. Hsp70 functions with co-chaperones CHIP, BAG3 and Hop in maintain cellular proteostasis. (A) The CHIP pathway involves the association of CHIP with Ub, E2, BAG3, Bcl-2-associated athanogene 3 and Hsp70 to facilitate degradation. (B) Formation of sHSPs-BAG3-HSP70 complex to prevent aggregation of proteins thus forming insoluble substrates in the cardiomyocytes. (C) Hsp70-Hsp90 Organising Protein (Hop) adaptor properties to link Heat shock protein 70 (Hsp70) with Heat shock protein 90 for client substrates transfer.
The interaction of Hsp70 with another co-chaperone, BAG3, is also critical for cardiac muscle development and vascular disease pathogenesis (64). BAG3 is generally involved in a range of cellular functions which include protein folding, apoptosis, autophagy as well as CMA/chaperone-assisted selective autophagy (CASA) to the UPS (2). BAG3 not only forms functional co-operations with Hsp70, but also with sHsps such as HspB5, HspB6 and HspB8 (65, 66). sHSPs act in concert with Hsp70 to facilitate protein refolding (Figure 3B) (67, 68). Since sHSPS lack ATP-dependent enzymatic activity that is necessary for active protein refolding, BAG3 facilitates the formation of a BAG3-sHsp-Hsp70 complex through which protein refolding can occur. The direct involvement of BAG3 in cardiac disease has previously been reported in ΔBAG3 mice that were observed to develop cardiomyopathy and non-inflammatory myofibrillar myopathy (MFM) (64).
In the heart, Hsp70 is induced by several factors which include steroid hormones (e.g., vasopressin), free radicals, drugs, probiotic derived proteins, physical exercise and environmental changes (69, 70). Elevated Hsp70 expression results in reduced myocardial infarction (71). In addition, Hsp70 levels are also thought to correlate with a timecourse of cardioprotection (36). Furthermore, high Hsp70 levels are linked with a decrease in cardiac apoptosis (33, 72). As such, high Hsp70 levels have been reported to confer improvement in the recovery of post ischemia reperfusion injury (73). Wei et al. (74) demonstrated high Hsp70 expression levels in hearts showing failure due to cardiomyopathy (arrhythmogenic cardiomyopathy, DCM) and ischemia. The degradation of misfolded proteins is important for the constant turnover of sarcomeric proteins required for correct function and regulation of cardiac mass (75). In addition, Hsp70 co-operates with CHIP and BAG1 to control the degradation of myosin and sarcomeric proteins (76). Given these roles of Hsp70 in cardiac function, it is tempting to speculate implications of the chaperone in PPCM. This hypothesis may need experimental validation.
Hsp90 is a unique molecular chaperone which possesses the ability to bind target proteins that are in a near native state in order to mediate the final stages of folding (77). In addition, Hsp90 is specialised to facilitate the folding of specific, defined sets of client proteins such as steroid hormone receptors, TLR innate immunity receptors, RNA polymerases and PI3-kinase-related kinases (PIKKs) (78). As such, Hsp90 modulates cell signalling, genome maintenance and assembly of transcriptional and translational apparatuses in cells (36). Together with Hsp70, Hsp90 plays key roles in protein folding as they facilitate the folding, maturation and activation of virtually all proteins in the cell (50, 57). While Hsp70 generally binds to nascent polypeptide chains at the ribosome, Hsp90 binds a more specialised clientome that includes steroid hormones and kinases. Hop acts as an adaptor protein to enable Hsp70-Hsp90 interaction through their C-terminal sequences (Figure 3C) (79, 80). As such, Hop enables the transfer of client proteins such as kinases, nuclear receptors like the steroid hormone receptors (SHR) and transcription factors between the chaperones Hsp70 and Hsp90 for folding, assembly and activation (Figure 2) (81, 82).
Hsp90 also plays an active role in protein quality control by directing misfolded proteins toward the UPS for degradation (83). Hsp90 possesses anti-apoptotic effects on hypoxia-mediated cardiomyocyte damage (84). An Hsp90 client, ErBB2, was shown to be linked to the development of HF in a murine model (85). Mice lacking cardiac specific ErBB2 developed HF characterised by left ventricular (LV) dilation, wall thinning and decreased systolic function (85). Due to its large clientome, we speculate a likelihood of Hsp90 in chaperoning several other proteins involved in PPCM pathogenesis. We therefore sought to predict the interaction and roles of Hsp90, Hsp70 and sHsps in PPCM using bioinformatics.
Predicted Associations Between Major Hsp Families and the PPCM Proteome
Using STRING analysis, we predicted the associations of several proteins currently known to be involved in PPCM pathophysiology with members of the Hsp70, Hsp90 and sHsp family members (Table 2 and Figure 4). Although the actual implications of such interactions are at present unknown, experimental validation may provide insights into candidate drug design or biomarker discovery. These Hsps and PPCM protein associations are described in the following sections.
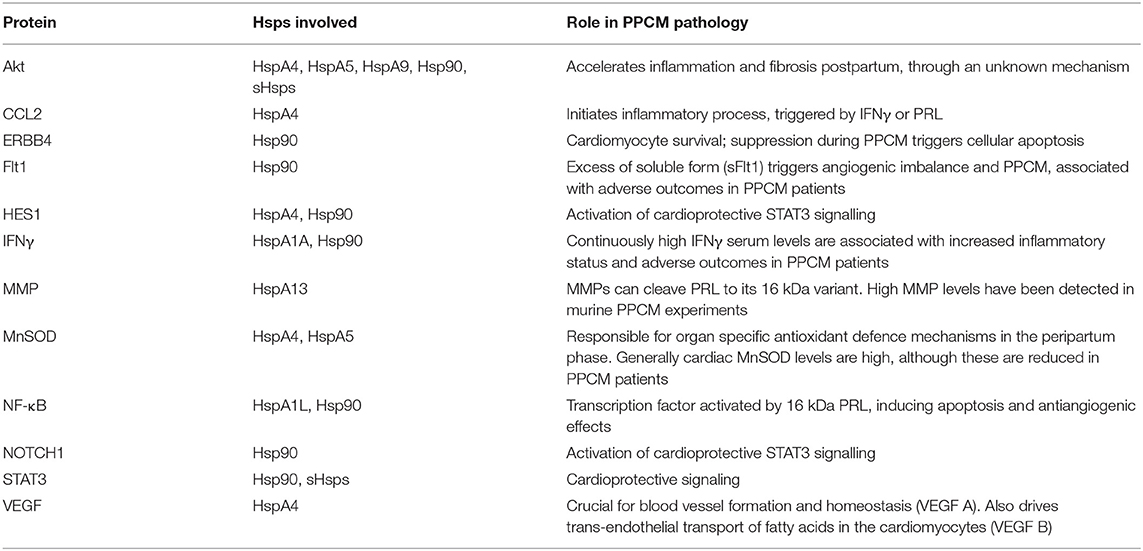
Table 2. STRING interactions between heat shock and PPCM proteins.
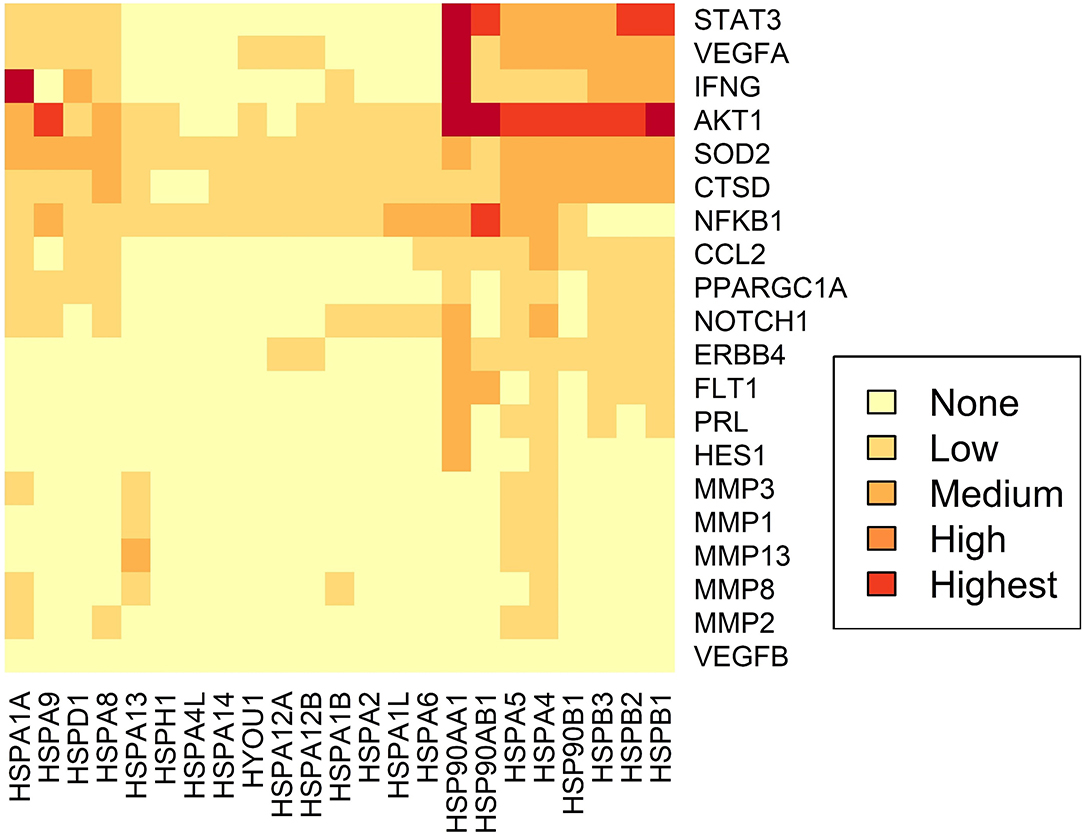
Figure 4. Predicted interactome of several Hsps with PPCM proteins. The heat map shows the interaction of PPCM pathogenesis proteins with Hsp70, Hsp90 and sHsp group members.
Akt
Akt is a protein kinase and a key component of the ubiquitous PI3K/Akt signalling pathway. Activated Akt promotes cellular survival, proliferation and growth. In the heart, Akt signalling induces both pathological hypertrophy and physiological hypertrophy (for example, during pregnancy) (86). However, Akt appears to have an exacerbatory effect in PPCM (15), possibly by aggravating an underlying angiogenic imbalance. Select Hsp70 (HspA4, HspA5, HspA9), Hsp90 (Hsp90AB1), and sHsps (HspB1) are predicted to interact with Akt (Table 2).
HspA4 (a non-canonical Hsp70 member), HspA5 (an Hsp of the endoplasmic reticulum (ER) also referred to as Grp78), and HspA9 (a mitochondrial Hsp also referred to as Grp75) (Figure 4) have been demonstrated to activate Akt in cancer models (87–90). Interestingly, Akt has also been shown to phosphorylate HspA5 (91) and regulate its expression (92), thus suggesting a possible feedback loop in which Akt and Hsp70 proteins positively regulate each other. This may be protective in cellular conditions such as oxidative stress and ER stress, which induce HspA9 and HspA5, respectively (89, 90). Since these Hsp70 isoforms are predicted to bind and possibly activate Akt, it is possible that HspA4, HspA5, and HspA9 play decisive roles in PPCM pathogenesis. HspA5/Akt interaction has been demonstrated as essential for cardiac development, function, and stress response. The loss of HspA5 in cardiomyocytes leaves them vulnerable to apoptosis following oxidative and ER stresses through suppressed Akt signalling (93), while upregulation of HspA5 attenuated ischaemia/reperfusion-induced cardiac damage by stimulating Akt activity (94, 95). Recently, ER stressors and ischaemia were also shown to increase the secretion of HspA5 by cardiomyocytes (96), indicating an extracellular cardioprotective role of this Hsp by activating Akt signalling.
Hsp90 proteins and sHsps are also predicted to interact with Akt (Figure 4). Similar to Hsp70s, these also appear to be involved in the stabilisation and activation of Akt kinase activity (97–100). Hsp90AB1/HspB1 interaction with Akt may have cardioprotective effects against a range of cardiac insults (101–105). While the activation of Akt by Hsps is generally beneficial in response to several cardiac insults, Akt stimulation is detrimental in the case of PPCM (15). As PPCM is characterised by oxidative and other stresses (Figure 2), the induction of Hsp70s, Hsp90s and sHsps during PPCM is highly probable, as is the subsequent stimulation of the Akt signalling pathway, although more research will be needed to confirm this hypothesis.
STAT3
Contrary to Akt, STAT3 activation may inhibit PPCM (13). Although no interactions between Hsp70s and STAT3 were observed, members of the Hsp90 and sHsp families are predicted to interact with STAT3 (Figure 4). The role of Hsp90 and the small HspB1 (Hsp27) in STAT3 signalling has recently been reviewed in detail (106), with these Hsps acting as key chaperones at numerous stages of the STAT3 pathway, including phosphorylation, activation and nuclear localisation of STAT3, as well as limiting its proteasomal degradation. It is therefore plausible that Hsp90 promotes STAT3 activation which may reverse the progression to fulminant PPCM. Therapeutic interventions that promote elevated Hsp90 expression levels in the cardiomyocytes may therefore prove beneficiual to PPCM patients. Further research is however needed to ascertain the functions of Hsps in modulating these pathways.
Other Interactions
Several other interactions were observed between Hsps and proteins in the PPCM pathway (Table 2), although the functional and biochemical roles of these remain to be determined. For instance, excess sFlt1 and reduced VEGF levels are key components of the angiogenic imbalance that characterises PPCM. While an interaction between Hsp90 and Flt1 was observed, current knowledge has only implicated Hsp90 in the maintenance of membrane-bound Flt1 in endothelial cells (107); interaction with the soluble form of the protein is unknown. However, because inhibition of Hsp90 could reduce levels of Flt1 (107), the role of Hsps in the stabilisation or folding of Flt1 may be worth exploring, especially in the context of placentally-derived sFlt1. We also predicted association of HspA4 with VEGFA. It has previously been demonstrated that HspA4 has a stabilising effect on VEGFA mRNA in cancer cells (108). It is conceivable that HspA4 suppresses aggregation of VEGFA under stress conditions, keeping the protein in a folding competent form, as has been demonstrated with other Hsp70 isoforms (56).
The Role of Hsps in Buffering Mutations
PPCM has been demonstrated to have a genetic basis, in at least a subset of patients (109–111). It is notable that molecular chaperones play a key role in the translation of genetic variation, by silencing or potentiating mutations (112). Hsp90, in particular, may potentiate mutations by assisting the folding and function of the mutant proteins, allowing them to have immediate phenotypic consequences (113). Alternatively, Hsp90 can silence mutations so that they have no phenotypic manifestation, although these buffering effects can be overwhelmed by environmental stresses (113). The implications of this are that Hsp90, or other Hsps, can buffer genetic mutations in a manner that is dependent on the environmental conditions. This may explain, in part, why the same mutations can cause DCM in some individuals, and PPCM in others.
The modulation of mutations by Hsps has also been described in Fanconi Anaemia, where Hsp70 was found to bind to inactive mutant proteins with severe phenotypic effects (114). The roles of Hsps in the buffering of cardiomyopathy-causing mutations is largely unknown, although heritable cardiomyopathies including DCM and hypertrophic cardiomyopathy (HCM) may be characterised by dysfunction of the UPS and other protein quality control mechanisms (115, 116). DCM- and HCM-causing mutations typically occur as truncations of genes encoding the sarcomeric proteins titin and cMyBP-C, respectively, and mutations in both have been described in PPCM patients, as well as other sarcomeric gene mutations (109–111, 117). Notably, these truncated protein products are not incorporated into the sarcomeres of mutation carriers (118, 119), although in the case of titin this haploinsufficiency has been attributed to mRNA degradation by nonsense-mediated decay (120). The role of Hsps in the buffering of truncating PPCM-causing mutations remains to be determined.
The Role of Hsps in the PPCM Inflammasome
In addition to their proteostatic roles, Hsps may also act as “chaperokines” which present antigens to the immune system. Hsp70, Hsp90 and sHps are secreted under stress conditions, where they can have pro- or anti-inflammatory effects [reviewed in (121)]. As myocardial inflammation is thought to be a key contributor to PPCM pathogenesis (12), the immunomodulatory effects of Hsps may be of interest. Indeed, several studies have demonstrated potential roles of Hsps in cardiac inflammatory pathways in response to myocarditis (122, 123) and myocardial injury (discussed below).
Extracellular Hsp70 can induce cardiomyocyte inflammation and cell death, in contrast with the pro-survival role of intracellular Hsp70 (124). Increased levels of circulating Hsp70 have been reported in models of acute myocardial infarction, autoimmune myocarditis and left ventricular dysfunction, all in association with elevated inflammatory markers and worse outcomes (37, 125, 126). Up-regulation of Hsp70 by treatment with Melusin in a mouse model of myocardial infarction was shown to reduce inflammatory cell infiltrates in the myocardium and improve cardiac function (127). The roles of Hsp90, Hsp60 and sHsps in cardiac inflammatory responses has also been demonstrated. Hsp90 has been shown to have cardioprotective effects in ischaemic pre- and post-conditioning by suppression of immune responses (128, 129). On the other hand, HspB1 may down-regulate leukocyte recruitment and cardiac inflammation (130). Hsp60 appears to induce cardiac inflammation and cytokine production (131, 132). It is therefore plausible that the level of circulating Hsps may promote or suppress cardiac inflammation, although it is unclear at this stage whether Hsp induction in PPCM would be beneficial or detrimental to recovery of cardiac function.
Targeting Hsps Toward Novel PPCM Therapy
To date, no disease-specific interventions for PPCM exist. As is the case with other cardiomyopathies, PPCM management is primarily focused on managing volume status, neutralising neurohormonal maladaptive responses, and preventing complications (133). One of the most used PPCM treatments is bromocriptine which functions by suppressing PRL production postpartum. A small proof-of-concept randomised trial of bromocriptine in 20 women with PPCM in Africa demonstrated improvements in mortality and LVEF at 6 months (134). Those observations were confirmed in a multi-center randomized study (135). Based on these data the 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy states that bromocriptine may be considered in women with newly diagnosed PPCM (5). However, more research in this area is needed. As such, there is an urgent need for the identification of novel PPCM drug targets in the design of novel therapeutic interventions.
Hsps have previously been suggested as druggable candidates in human disease models (136). Several Hsps, such as Hsp70 and Hsp90 members, show great potential as drug targets in several cancers. For instance, the proteasome inhibitor, Bortezomib, has been used in anticancer therapy where it exerts antitumor effects by upregulating Hsp60 and Hsp90 on the surface of cancer cells (137). Attention is currently being drawn toward Hsp-directed therapies as candidates for novel cardiovascular disease therapies. A study recently demonstrated that blocking Hsp70 activity could be therapeutically beneficial in HF treatment (138). Given its ATP-dependent nature, Hsp70 is also amenable to inhibition using ATP-mimicking drugs. Interestingly, predictions from our STRING analysis revealed several stages which can potentially be modulated by Hsp70 (Figure 5A).
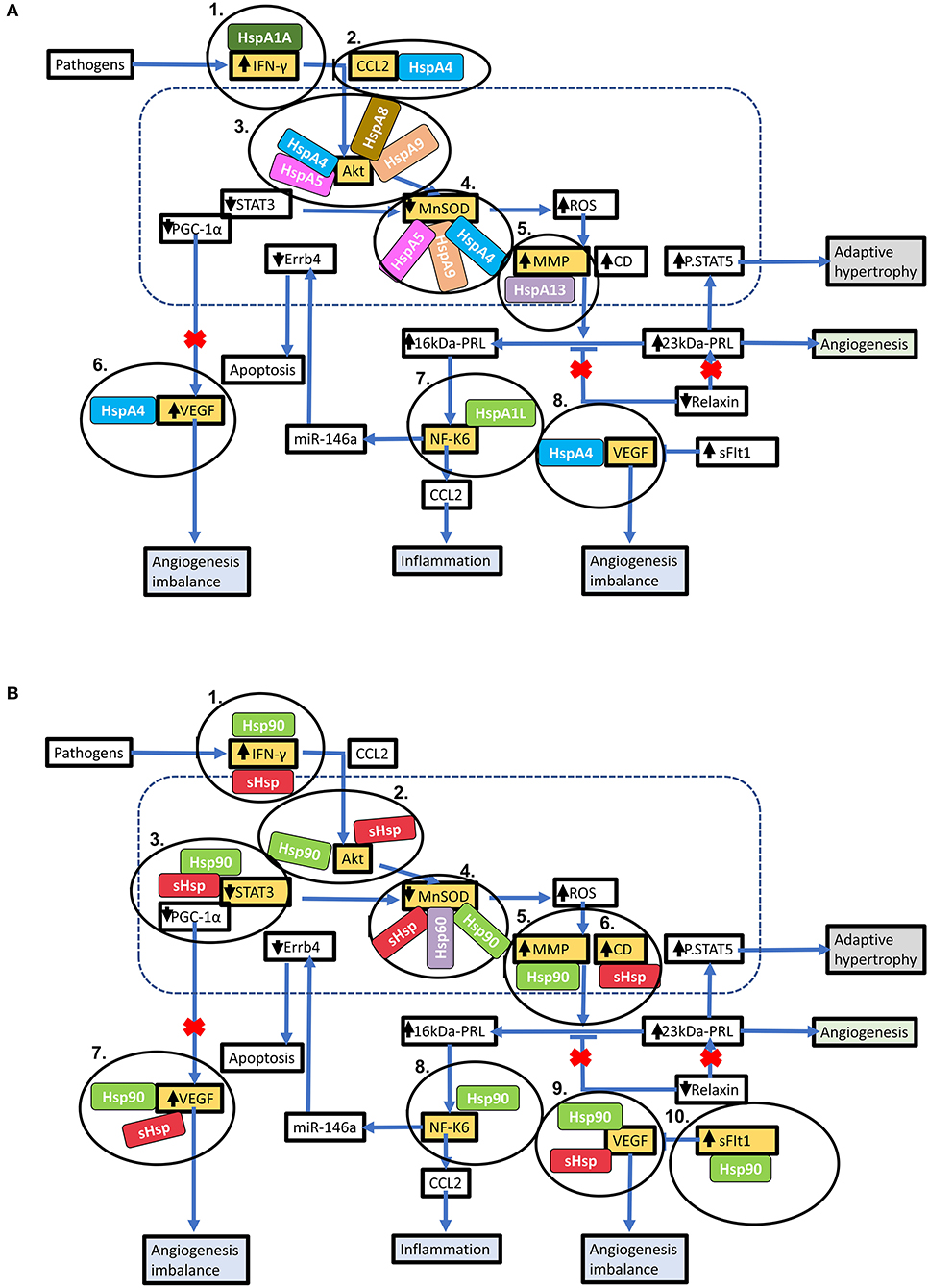
Figure 5. Key stages in the PPCM pathophysiology pathway which are modulated by Hsps. Hsps interact with several proteins involved in the PPCM pathophysiological pathway. (A) Several Hsp70 isoforms interact with various proteins involved in PPCM pathogenesis. (B) The stages within the PPCM pathophysiology pathway where Hsp90 and sHsps interact with PPCM proteins are shown. Targeting these stages may be crucial in novel PPCM interventions.
Hsp-centred drug modulation strategies may be centred around one of the following strategies; (i) boosting Hsp expression and (ii) inhibition of Hsp functionality. Each of these possible strategies is assessed in detail (Table 3). Inhibition would generally involve the development of small molecule inhibitors that target the functional domains of the Hsps thus disrupting their chaperone function. The direct inhibition of Hsps, resulting in the subsequent disruption of key protein-associations can be an efficacious way to modulate protein fate. Small molecule inhibitors such as polymyxin B and colistin sulphate possess great potential in this regard, as they have been successfully used to inhibit the activity Hsp70 in vitro (139). These two compounds are directed toward the nucleotide-binding domain, thus lowering the basal ATPase activity of the Hsp70. Designing domain specific inhibitory compounds may prove useful in Hsp70-targeted PPCM therapy. Since Hsp70 functions in co-operation with several other proteins involved in the pathophysiology of PPCM, selective targeting of cardiac Hsp70 may become a decisive step in inhibiting PPCM pathology. An alternative strategy worth exploring in designing novel PPCM therapy involves targeting heat shock factors (e.g., HSF1) which are responsible for modulation of Hsp expression. Although the generally high sequence and structural conservation of Hsp70 may be a snag in targeted inhibition, specific signature motifs in individual Hsp70 family members may be targeted toward this. Particularly, we predicted the association of several PPCM pathophysiology proteins with HspA4 (Figure 5A). HspA4 belongs to the Hsp110 family of chaperones, which are a specialised subclass of Hsp70s functionally and structurally distinguished from the canonical Hsp70s (56). This makes HspA4 amenable to selective targeting by inhibitors since it is unique from the more conserved canonical Hsp70s.
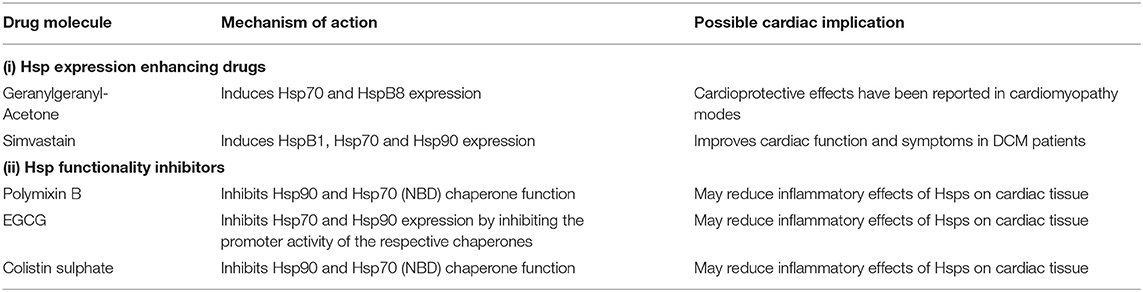
Table 3. Hsp-centred drug modulation strategies in novel PPCM drug design.
Prospects of targeting co-chaperones that are crucial for Hsp70 function also exist. The Hsp70 co-chaperone, CHIP, has been shown to play a crucial role in regulating intracellular protein signalling as evidenced by an increase in Akt phosphorylation which in turn leads to activation of the Akt signalling pathway leading to cardiac hypertrophy (63, 140). CHIP-directed therapies may potentially disrupt Hsp70 activity in turn causing detrimental downstream effects on Akt signalling within cardiomyocytes (Figure 6). Another Hsp70 co-chaperone, BAG3, may also be a crucial PPCM drug target. Disruption of the BAG3-sHsp-Hsp70 complex has previously been shown to be associated with DCM and non-inflammatory MFM (141–143). The specific roles of BAG3 in PPCM pathophysiology are however yet to be validated. BAG3 mutations have however been proposed to be associated with PPCM. Thus, enhancing BAG3 expression in PPCM patients may possibly offer cardioprotection by augmenting cardiomyocyte proteostasis. As such, it may therefore be generally hypothesised that these co-chaperones may act as decisive players in Hsp-modulated proteostasis within the cardiomyocyte and may thus act as PPCM drug targets.
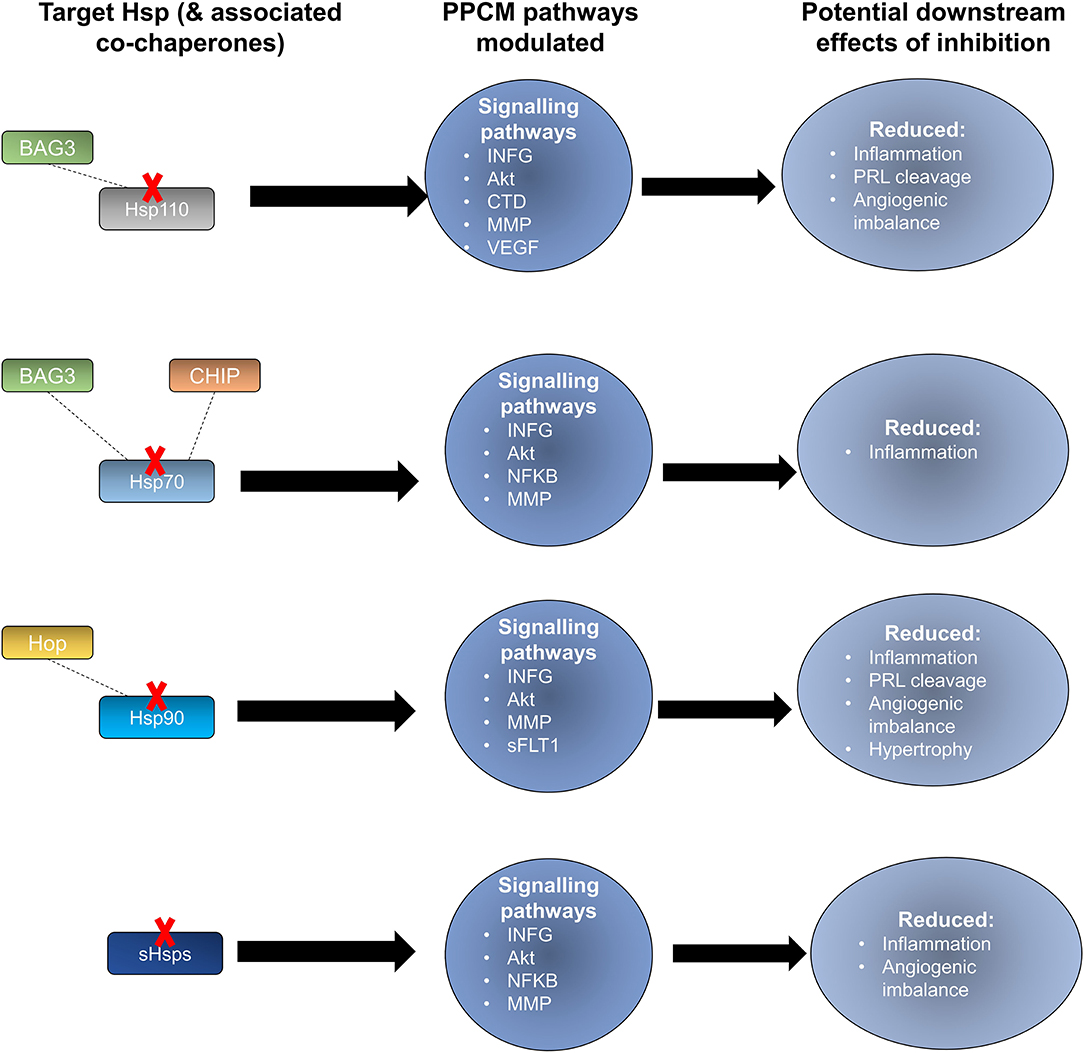
Figure 6. Potential effects of Hsp-directed inhibition in PPCM therapy. Targeting the Hsp types sHsp, Hsp110, Hsp90, and Hsp70 has the potential to disrupt key signalling pathways that are important in PPCM pathophysiology.
Hsp90 also serves as an attractive candidate for novel PPCM drug interventions given that it is predicted to modulate the PPCM pathogenesis pathway (Figure 5B) via its interaction with AKT and NFKB1 (Figure 6). It is well-established that Hsp90 plays important functions in regulating client proteins into their active conformations. As such, Hsp90 possibly activates AKT triggering a cascade of events that leads to PPCM. Furthermore, Hsp90 is predicted to interact with NFKB which is associated with cardiac inflammation in PPCM. Hsp90 thus likely plays decisive roles in PPCM pathophysiology via AKT and NFKB signalling. Consequently, Hsp90-inhibiting directed therapies may prove beneficial in novel PPCM therapies. To date, several classes of Hsp90-targeting small molecule inhibitors have been proposed for disrupting Hsp90 chaperone function. The majority of these compete with ATP for binding onto the N-terminal domain (NTD) ultimately keeping the chaperone in an inhibitor-bound conformation that abrogates its function (144). Currently, the following natural and synthetic Hsp90-targeting drugs, such as ansamycin and derivatives of purine, resorcinol, benzamide, and tricyclic imidapyridines have been described in several disease models such as cancer [reviewed in (145)]. However, the potential of Hsp90-targeting drugs in PPCM is yet to be explored.
Hsps As Candidate PPCM Biomarkers
Despite the importance of early diagnosis for full cardiac recovery in PPCM patients, physicians are often faced with the difficulty of distinguishing between peripartum discomfort in healthy women and the pathological PPCM symptoms. PPCM diagnosis thus relies on a high index of suspicion. A thorough history of the onset of symptoms combined with a comprehensive echocardiography report confirming <45% LVEF are important diagnostic determinants. Currently, the only clinically confirmed PPCM biomarker is the brain natriuretic peptide NT (proBNP) which is however not specific for PPCM diagnosis (12, 146). Since Hsps are triggered by stress, we hypothesise that Hsp expression is upregulated to allow the chaperones to form functional partnerships with markers of inflammation in PPCM patients. It is important to monitor Hsp expression levels through the progression of PPCM as this presents a basis for the potential use of Hsps as biomarkers. Indeed, STRING analysis predicts the interaction of Hsp70, Hsp90, sHsps, and Hsp60 with several PPCM pathophysiology proteins (Figure 7). The prospects of Hsps as PPCM biomarkers either individually or in a panel with the already established proBNP marker is promising.
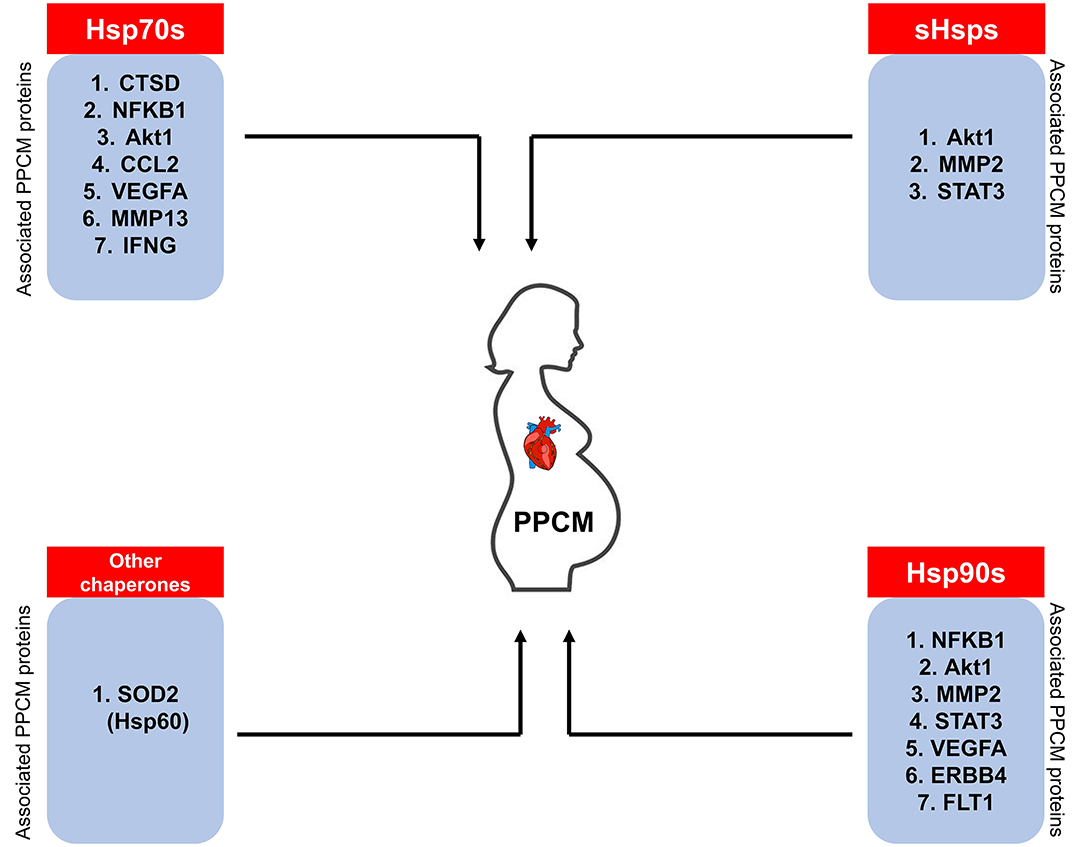
Figure 7. Hsps as potential PPCM biomarkers. The Hsps (sHsps, Hsp60, Hsp70, Hsp90) predicted to associate with PPCM pathogenesis proteins may be used as candidate biomarkers.
At present, some Hsps have been proposed as biomarkers in cardiovascular disease. A study by Giannessi and colleagues (37) demonstrated that circulating Hsp60 and HspA1A levels correlate with LV dysfunction severity. Interestingly, the same study also established that HspA1A protein levels are significantly correlated with BNP levels. Hsp70 and Hsp60 activation and inflammation markers, such as IL6 are also correlated with the extent of cardiac and microvascular dysfunction in patients with angiographically normal coronary arteries (37). Hsps have also been reported to mediate coronary endothelial dysfunction and produce microvascular damage in response to metabolic or infective insults (147). The potential of Hsp60 as a potential diagnostic or prognostic marker of heart disease has also been investigated. Veres et al. (148) noted the increased risk of heart disease when Hsp60 expression is upregulated. Also, elevated Hsp60 concentrations are positively associated with the severity of coronary arterial disease and ischemic heart disease in a dose-dependent fashion (149). Hsp70 levels were found to be significantly higher in HF and myocardial infarction, potentially implicating the protein's role as a CVD biomarker (150, 151). To date, not many proteomics-based studies have been conducted to assess the expression levels of stress proteins in PPCM. Nonetheless, the exacerbated stress associated with PPCM may trigger a unique chaperone response which may be studied toward biomarker design. Therefore, it may also be necessary to validate the role of other Hsp families such as the Hsp110 and Hsp40 members to determine their specific roles in PPCM. Recently, Hsp110 family members have been demonstrated to play important roles in cancers (152, 153). As such, the differential expression of Hsps in PPCM patients may lay a foundation toward novel biomarker identification.
Hsp70 may serve as an important diagnostic or prognostic biomarker of PPCM since Hsp70 expression levels are correlated with traditional injury markers such as AST, ALT, γGT and bilirubin in HF patients (154). Furthermore, a study by Baba et al. (155) predicted that worse outcomes correlate with increased Hsp70 levels after heart transplantation. Although similar studies have not yet been applied to PPCM at present, they are worth exploring. Given that there are 13 different Hsp70 isoforms which are subtly distinguished from each other by unique signature motifs, the Hsp70 family members are promising PPCM biomarkers. sHSPs have also been proposed as biomarkers of congestive HF. It has been demonstrated that Hsp20, Hsp27, and Hsp32 expression correlates to disease (156). The Hsp70 co-chaperone, BAG3 is also an attractive potential PPCM biomarker. Increased BAG3 levels were observed in sera of patients with end stage HF, purporting that BAG3 is released by cardiomyocytes as a stress response (157–159). As such, BAG3 may be a useful biomarker to monitor HF progression. The aggravated stress conditions associated with PPCM potentially trigger upregulated Hsp-expression to maintain proteostasis and alleviate the effects of cardiotoxicity. We therefore hypothesise an Hsp expression profile that is unique from other forms of cardiomyopathy toward the identification of novel PPCM biomarkers of diagnosis or prognosis. Indeed, while in HF several studies have focused on the potential role of Hsp60 and Hsp70, there is need to investigate the role of co-chaperones and other Hsps (e.g., Hsp40 and Hsp110) as PPCM biomarkers.
Conclusion
The involvement of Hsps in protein quality control systems has been reported in several cardiovascular disorders such as ischaemic heart disease and HF. Given the exaggerated cardiac stress associated with pregnancy, Hsps likely play crucial roles in PPCM pathophysiology. Here, we predicted associations between proteins involved in PPCM pathogenesis and sHsp, Hsp70 as well as Hsp90. Furthermore, Hsps and their respective co-chaperones hold promise as novel candidate PPCM biomarkers. However, fundamental research is needed in this regard to experimentally validate the utility of Hsps as PPCM biomarkers. These associations could also pave the way for the development of novel Hsp-targeted therapeutic PPCM drug interventions.
Author Contributions
GC, TS, and SK wrote the draft, the original idea was conceived by GC and KS. Critical feedback was provided by TZ, AS, NN, and KS. All authors contributed to the article and approved the submitted version.
Funding
GC is a recipient of the UCT Faculty of Health Sciences, URC grant. TS acknowledges funding received from the South African Medical Research Council. NN gratefully acknowledges support from the South African Medical Research Council, the NRF and the Lily and Ernst Hausmann Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Willis MS, Schisler JC, Portbury AL, Patterson C. Build it up-Tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. (2009) 81:439–48. doi: 10.1093/cvr/cvn289
2. Ranek MJ, Stachowski MJ, Kirk JA, Willis MS. The role of heat shock proteins and co-chaperones in heart failure. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20160530. doi: 10.1098/rstb.2016.0530
3. McLendon PM, Robbins J. Proteotoxicity and cardiac dysfunction. Circ Res. (2015) 116:1863–82. doi: 10.1161/CIRCRESAHA.116.305372
4. Bennett PM, Ehler E, Wilson AJ. Sarcoplasmic reticulum is an intermediary of mitochondrial and myofibrillar growth at the intercalated disc. J Muscle Res Cell Motil. (2016) 37:55–69. doi: 10.1007/s10974-016-9444-6
5. Bauersachs J, Konig T, van der Meer P, Petrie MC, Hilfiker-Kleiner D, Mbakwem A, et al. Pathophysiology, diagnosis and management of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur J Heart Fail. (2019) 21:827–43. doi: 10.1002/ejhf.1493
6. Sliwa K, Hilfiker-Kleiner D, Petrie MC, Mebazaa A, Pieske B, Buchmann E, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail. (2010) 12:767–78. doi: 10.1093/eurjhf/hfq120
7. Arany Z. Understanding peripartum cardiomyopathy. Annu Rev Med. (2018) 69:165–76. doi: 10.1146/annurev-med-041316-090545
8. Shaikh N. An obstetric emergency called peripartum cardiomyopathy! J Emerg Trauma Shock. (2010) 3:39–42. doi: 10.4103/0974-2700.58664
9. Ersbøll AS, Johansen M, Damm P, Rasmussen S, Vejlstrup NG, Gustafsson F. Peripartum cardiomyopathy in Denmark: a retrospective, population-based study of incidence, management and outcome. Eur J Heart Fail. (2017) 19:1712–20. doi: 10.1002/ejhf.882
10. Karaye KM, Ishaq NA, Sa'idu H, Balarabe SA, Talle MA, Isa MS, et al. Incidence, clinical characteristics, and risk factors of peripartum cardiomyopathy in Nigeria: results from the PEACE Registry. ESC Heart Fail. (2020) 7:235–43. doi: 10.1002/ehf2.12562
11. Kolte D, Khera S, Aronow WS, Palaniswamy C, Mujib M, Ahn C, et al. Temporal trends in incidence and outcomes of peripartum cardiomyopathy in the United States: a nationwide population-based study. J Am Heart Assoc. (2014) 3:e001056. doi: 10.1161/JAHA.114.001056
12. Azibani F, Sliwa K. Peripartum cardiomyopathy: an update. Curr Heart Fail Rep. (2018) 15:297–306. doi: 10.1007/s11897-018-0404-x
13. Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. (2007) 128:589–600. doi: 10.1016/j.cell.2006.12.036
14. Zhu R-R, Chen Q, Liu Z-B, Ruan H-G, Wu Q-C, Zhou X-L. Inhibition of the Notch1 pathway induces peripartum cardiomyopathy. J Cell Mol Med. (2020) 24:7907–14. doi: 10.1111/jcmm.15423
15. Ricke-Hoch M, Bultmann I, Stapel B, Condorelli G, Rinas U, Sliwa K, et al. Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovasc Res. (2014) 101:587–96. doi: 10.1093/cvr/cvu010
16. Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. (2008) 8:924–56. doi: 10.1038/nrc2524
17. Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. (2012) 485:333–8. doi: 10.1038/nature11040
18. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. (2003) 111:649–58. doi: 10.1172/JCI17189
19. Damp J, Givertz MM, Semigran M, Alharethi R, Ewald G, Felker GM, et al. Relaxin-2 and soluble Flt1 levels in peripartum cardiomyopathy: results of the multicenter IPAC study. JACC Heart Fail. (2016) 4:380–8. doi: 10.1016/j.jchf.2016.01.004
20. Ntusi NBA, Badri M, Gumedze F, Sliwa K, Mayosi BM. Pregnancy-associated heart failure: a comparison of clinical presentation and outcome between hypertensive heart failure of pregnancy and idiopathic peripartum cardiomyopathy. PLoS ONE. (2015) 10:e0133466. doi: 10.1371/journal.pone.0133466
21. Barasa A, Rosengren A, Sandström TZ, Ladfors L, Schaufelberger M. Heart failure in late pregnancy and postpartum: incidence and long-term mortality in Sweden from 1997 to 2010. J Card Fail. (2017) 23:370–8. doi: 10.1016/j.cardfail.2016.12.011
22. Faupel-Badger JM, McElrath TF, Lauria M, Houghton LC, Lim KH, Parry S, et al. Maternal circulating angiogenic factors in twin and singleton pregnancies. Am J Obstet Gynecol. (2015) 212:636.e1–8. doi: 10.1016/j.ajog.2014.11.035
23. Hall ME, George EM, Granger JP. The heart during pregnancy. Rev Esp Cardiol. (2011) 64:1045–50. doi: 10.1016/j.rec.2011.07.008
24. Kara RJ, Bolli P, Karakikes I, Matsunaga I, Tripodi J, Tanweer O, et al. Fetal cells traffic to injured maternal myocardium and undergo cardiac differentiation. Circ Res. (2012) 110:82–93. doi: 10.1161/CIRCRESAHA.111.249037
25. Boddy AM, Fortunato A, Wilson Sayres M, Aktipis A. Fetal microchimerism and maternal health: a review and evolutionary analysis of cooperation and conflict beyond the womb. Bioessays. (2015) 37:1106–18. doi: 10.1002/bies.201500059
26. Kodogo V, Azibani F, Sliwa K. Role of pregnancy hormones and hormonal interaction on the maternal cardiovascular system: a literature review. Clin Res Cardiol. (2019) 108:831–46. doi: 10.1007/s00392-019-01441-x
27. Halkein J, Tabruyn SP, Ricke-Hoch M, Haghikia A, Nguyen NQ, Scherr M, et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. (2013) 123:2143–54. doi: 10.1172/JCI64365
28. Bajou K, Herkenne S, Thijssen VL, D'Amico S, Nguyen NQ, Bouché A, et al. PAI-1 mediates the antiangiogenic and profibrolytic effects of 16K prolactin. Nat Med. (2014) 2014:741–7. doi: 10.1038/nm.3552
29. Basaiawmoit RV, Rattan SI. Cellular stress and protein misfolding during aging. Methods Mol Biol. (2010) 648:107–17. doi: 10.1007/978-1-60761-756-3_7
30. Lindquist S. The heat-shock response. Ann Rev Biochem. (1986) 55:1151–91. doi: 10.1146/annurev.bi.55.070186.005443
31. Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J Biol Chem. (2013) 288:21399–411. doi: 10.1074/jbc.M113.479253
32. Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. (1998) 94:73–82. doi: 10.1016/S0092-8674(00)81223-4
33. Zhao Y, Wang W, Qian L. Hsp70 may protect cardiomyocytes from stress-induced injury by inhibiting Fas-mediated apoptosis. Cell Stress Chaperones. (2007) 12:83–95. doi: 10.1379/CSC-231R.1
34. Datta R, Bansal T, Rana S, Datta K, Chaudhuri RD, Chawla-Sarkar M, et al. Myocyte-derived Hsp90 modulates collagen upregulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Mol Biol Cell. (2017) 37:e00611–16. doi: 10.1128/MCB.00611-16
35. Biasucci LM, Liuzzo G, Ciervo A, Petrucca A, Piro M, Angiolillo DJ, et al. Antibody response to chlamydial heat shock protein 60 is strongly associated with acute coronary syndromes. Circulation. (2003) 107:3015–7. doi: 10.1161/01.CIR.0000078632.76653.6C
36. Li Q, Shi M, Li B. Anandamide enhances expression of heat shock protein 72 to protect against ischemia-reperfusion injury in rat heart. J Physiol Sci. (2013) 63:47–53. doi: 10.1007/s12576-012-0228-5
37. Giannessi D, Colotti C, Maltinti M, Del Ry S, Prontera C, Turchi S, et al. Circulating heat shock proteins and inflammatory markers in patients with idiopathic left ventricular dysfunction: their relationships with myocardial and microvascular impairment. Cell Stress Chaperones. (2007) 12:265–74. doi: 10.1379/CSC-272.1
38. Dybdahl B, Slørdahl SA, Waage A, Kierulf P, Espevik T, Sundan A. Myocardial ischaemia and the inflammatory response: release of heat shock protein 70 after myocardial infarction. Heart. (2005) 91:299–304. doi: 10.1136/hrt.2003.028092
39. Kanwar JR, Shen WP, Kanwar RK, Berg RW, Krissansen GW. Effects of survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J Natl Cancer Inst. (2001) 93:1541–52. doi: 10.1093/jnci/93.20.1541
40. Xu Q, Schett G, Perschinka H, Mayr M, Egger G, Oberhollenzer F, et al. Serum soluble heat shock protein 60 is elevated in subjects with atherosclerosis in a general population. Circulation. (2000) 102:14–20. doi: 10.1161/01.CIR.102.1.14
41. Hayashi M, Imanaka-Yoshida K, Yoshida T, Wood M, Fearns C, Tatake RJ, et al. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat Med. (2006) 12:128–32. doi: 10.1038/nm1327
42. Zhu Y, Ma T, Wang X. Gene transfer of heat-shock protein 20 protects against ischemia/reperfusion injury in rat hearts. Acta Pharmacol Sin. (2005) 26:1193–200. doi: 10.1111/j.1745-7254.2005.00139.x
43. Krishnamurthy K, Vedam K, Kanagasabai R, Druhan LJ, Ilangovan G. Heat shock factor-1 knockout induces multidrug resistance gene, MDR1b, and enhances P-glycoprotein (ABCB1)-based drug extrusion in the heart. Proc Natl Acad Sci USA. (2012) 109:9023–8. doi: 10.1073/pnas.1200731109
44. Zügel U, Kaufmann SH. Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin Microbiol Rev. (1999) 12:19–39. doi: 10.1128/CMR.12.1.19
45. Zininga T, Makumire S, Gitau GW, Njunge JM, Pooe OJ, Klimek H, et al. Plasmodium falciparum Hop (PfHop) interacts with the Hsp70 chaperone in a nucleotide-dependent fashion and exhibits ligand selectivity. PLoS ONE. (2015) 10:e0135326. doi: 10.1371/journal.pone.0135326
46. Marinko JT, Huang H, Penn WD, Capra JA, Schlebach JP, Sanders CR. Folding and misfolding of human membrane proteins in health and disease: from single molecules to cellular proteostasis. Chem Rev. (2019) 119:5537–606. doi: 10.1021/acs.chemrev.8b00532
47. Welch WJ. How cells respond to stress. Sci Am. (1993) 268:56–64. doi: 10.1038/scientificamerican0593-56
48. Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, Agostini F, et al. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. (2012) 1:251–64. doi: 10.1016/j.celrep.2011.12.007
49. Szabo A, Langer T, Schröder H, Flanagan J, Bukau B, Hartl FU. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc Natl Acad Sci USA. (1994) 91:10345–9. doi: 10.1073/pnas.91.22.10345
50. Rosenzweig R, Sekhar A, Nagesh J, Kay LE. Promiscuous binding by Hsp70 results in conformational heterogeneity and fuzzy chaperone-substrate ensembles. Elife. (2017) 6:e28030. doi: 10.7554/eLife.28030.025
51. Goloubinoff P, De Los Rios P. The mechanism of Hsp70 chaperones: (entropic) pulling the models together. Trends Biochem Sci. (2007) 32:372–80. doi: 10.1016/j.tibs.2007.06.008
52. Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. (2010) 11:579–92. doi: 10.1038/nrm2941
53. Shiber A, Ravid T. Chaperoning proteins for destruction: diverse roles of Hsp70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome. Biomolecules. (2014) 4:704–24. doi: 10.3390/biom4030704
54. Alderson TR, Kim JH, Markley JL. Dynamical structures of Hsp70 and Hsp70-Hsp40 complexes. Structure. (2016) 24:1014–30. doi: 10.1016/j.str.2016.05.011
55. Genest O, Reidy M, Street TO, Hoskins JR, Camberg JL, Agard DA, et al. Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol Cell. (2013) 49:464–73. doi: 10.1016/j.molcel.2012.11.017
56. Zwirowski S, Kłosowska A, Obuchowski I, Nillegoda NB, Piróg A, Zietkiewicz S, et al. Hsp70 displaces small heat shock proteins from aggregates to initiate protein refolding. EMBO J. (2017) 36:783–96. doi: 10.15252/embj.201593378
57. Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. (2019) 20:665–80. doi: 10.1038/s41580-019-0133-3
58. Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, et al. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem. (1997) 272:9002–10. doi: 10.1074/jbc.272.14.9002
59. Kriegenburg F, Ellgaard L, Hartmann-Petersen R. Molecular chaperones in targeting misfolded proteins for ubiquitin-dependent degradation. FEBS J. (2012) 279:532–42. doi: 10.1111/j.1742-4658.2011.08456.x
60. McLellan AJ, Frydman J. Molecular chaperones and the art of recognizing a lost cause. Nat Cell Biol. (2001) 3:E51–3. doi: 10.1038/35055162
61. Schisler JC, Rubel CE, Zhang C, Lockyer P, Cyr DM, Patterson C. CHIP protects against cardiac pressure overload through regulation of AMPK. J Clin Invest. (2013) 123:3588–99. doi: 10.1172/JCI69080
62. Willis MS, Min JN, Wang S, McDonough H, Lockyer P, Wadosky KM, et al. Carboxyl terminus of Hsp70-interacting protein (CHIP) is required to modulate cardiac hypertrophy and attenuate autophagy during exercise. Cell Biochem Funct. (2013) 31:724–35. doi: 10.1002/cbf.2962
63. McMullen JR, Izumo S. Role of the insulin-like growth factor 1 (IGF1)/phosphoinositide-3-kinase (PI3K) pathway mediating physiological cardiac hypertrophy. Novartis Found Symp. (2006) 274:90–111. doi: 10.1002/0470029331.ch7
64. Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC, Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. (2006) 169:761–73. doi: 10.2353/ajpath.2006.060250
65. Fuchs M, Poirier DJ, Seguin SJ, Lambert H, Carra S, Charette SJ, et al. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem J. (2009) 425:245–55. doi: 10.1042/BJ20090907
66. Rauch JN, Tse E, Freilich R, Mok S-A, Makley LN, Southworth DR, et al. BAG3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J Mol Biol. (2017) 429:128–41. doi: 10.1016/j.jmb.2016.11.013
67. Giffard RG, Macario AJL, de Macario EC. The future of molecular chaperones and beyond. J Clin Invest. (2013) 123:3206–8. doi: 10.1172/JCI70799
68. Mymrikov EV, Daake M, Richter B, Haslbeck M, Buchner J. The chaperone activity and substrate spectrum of human small heat shock proteins. J Biol Chem. (2017) 292:672–84. doi: 10.1074/jbc.M116.760413
69. Moalic JM, Bauters C, Himbert D, Bercovici J, Mouas C, Guicheney P, et al. Phenylephrine, vasopressin and angiotensin II as determinants of proto-oncogene and heat-shock protein gene expression in adult rat heart and aorta. J Hypertens. (1989) 7:195–201. doi: 10.1097/00004872-198903000-00005
70. Kukreja RC, Kontos MC, Loesser KE, Batra SK, Qian YZ, Gbur CJJ, et al. Oxidant stress increases heat shock protein 70 mRNA in isolated perfused rat heart. Am J Physiol. (1994) 267:H2213–9. doi: 10.1152/ajpheart.1994.267.6.H2213
71. Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. (1993) 88:1264–72. doi: 10.1161/01.CIR.88.3.1264
72. Shinohara T, Takahashi N, Ooie T, Ichinose M, Hara M, Yonemochi H, et al. Estrogen inhibits hyperthermia-induced expression of heat-shock protein 72 and cardioprotection against ischemia/reperfusion injury in female rat heart. J Mol Cell Cardiol. (2004) 37:1053–61. doi: 10.1016/j.yjmcc.2004.09.006
73. Qian YZ, Bernardo NL, Nayeem MA, Chelliah J, Kukreja RC. Induction of 72-kDa heat shock protein does not produce second window of ischemic preconditioning in rat heart. Am J Physiol. (1999) 276:H224–34. doi: 10.1152/ajpheart.1999.276.1.H224
74. Wei J, Tong J, Yu L, Zhang J. EMF protects cardiomyocytes against hypoxia-induced injury via heat shock protein 70 activation. Chem Biol Interact. (2016) 248:8–17. doi: 10.1016/j.cbi.2016.02.003
75. Tarone G, Brancaccio M. Keep your heart in shape: molecular chaperone networks for treating heart disease. Cardiovasc Res. (2014) 102:346–61. doi: 10.1093/cvr/cvu049
76. Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. (2003) 92:e87–100. doi: 10.1161/01.RES.0000079028.31393.15
77. Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. (2001) 154:267–73. doi: 10.1083/jcb.200104079
78. Kakihara Y, Houry WA. The R2TP complex: discovery and functions. Biochim Biophys Acta. (2012) 1823:101–7. doi: 10.1016/j.bbamcr.2011.08.016
79. Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. (2000) 101:199–210. doi: 10.1016/S0092-8674(00)80830-2
80. Schmid AB, Lagleder S, Gräwert MA, Röhl A, Hagn F, Wandinger SK, et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. (2012) 31:1506–17. doi: 10.1038/emboj.2011.472
81. Pratt WB, Morishima Y, Osawa Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J Biol Chem. (2008) 283:22885–9. doi: 10.1074/jbc.R800023200
82. Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. (2012) 150:987–1001. doi: 10.1016/j.cell.2012.06.047
83. Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. (2009) 28:899–901. doi: 10.1038/emboj.2009.29
84. Wang W, Peng Y, Wang Y, Zhao X, Yuan Z. Anti-apoptotic effect of heat shock protein 90 on hypoxia-mediated cardiomyocyte damage is mediated via the phosphatidylinositol 3-kinase/AKT pathway. Clin Exp Pharmacol Physiol. (2009) 36:899–903. doi: 10.1111/j.1440-1681.2009.05167.x
85. Negro A, Brar BK, Lee K-F. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog Horm Res. (2004) 59:1–12. doi: 10.1210/rp.59.1.1
86. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. (2016) 97:245–62. doi: 10.1016/j.yjmcc.2016.06.001
87. Choi DW, Lim MS, Lee JW, Chun W, Lee SH, Nam YH, et al. The cytotoxicity of kahweol in HT-29 human colorectal cancer cells is mediated by apoptosis and suppression of Heat Shock Protein 70 expression. Biomol Ther. (2015) 23:128–33. doi: 10.4062/biomolther.2014.133
88. Fu Y, Wey S, Wang M, Ye R, Liao C-P, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA. (2008) 105:19444–9. doi: 10.1073/pnas.0807691105
89. Guo W, Yan L, Yang L, Liu X, E Q, Gao P, et al. Targeting GRP75 improves HSP90 inhibitor efficacy by enhancing p53-mediated apoptosis in hepatocellular carcinoma. PLoS ONE. (2014) 9:e85766. doi: 10.1371/journal.pone.0085766
90. Yung HW, Charnock-Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE. (2011) 6:e17894. doi: 10.1371/journal.pone.0017894
91. Barati MT, Rane MJ, Klein JB, McLeish KR. A proteomic screen identified stress-induced chaperone proteins as targets of Akt phosphorylation in mesangial cells. J Proteome Res. (2006) 5:1636–46. doi: 10.1021/pr0502469
92. Gray MJ, Mhawech-Fauceglia P, Yoo E, Yang W, Wu E, Lee AS, et al. AKT inhibition mitigates GRP78 (glucose-regulated protein) expression and contribution to chemoresistance in endometrial cancers. Int J Cancer. (2013) 133:21–30. doi: 10.1002/ijc.27994
93. Wang X, Bi X, Zhang G, Deng Y, Luo X, Xu L, et al. Glucose-regulated protein 78 is essential for cardiac myocyte survival. Cell Death Differ. (2018) 25:2181–94. doi: 10.1038/s41418-018-0109-4
94. Bi X, Zhang G, Wang X, Nguyen C, May HI, Li X, et al. Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ Res. (2018) 122:1545–54. doi: 10.1161/CIRCRESAHA.117.312641
95. Petrovski G, Das S, Juhasz B, Kertesz A, Tosaki A, Das DK. Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxid Redox Signal. (2011) 14:2191–200. doi: 10.1089/ars.2010.3486
96. Blackwood EA, Thuerauf DJ, Stastna M, Stephens H, Sand Z, Pentoney A, et al. Proteomic analysis of the cardiac myocyte secretome reveals extracellular protective functions for the ER stress response. J Mol Cell Cardiol. (2020) 143:132–44. doi: 10.1016/j.yjmcc.2020.04.012
97. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. (2002) 277:39858–66. doi: 10.1074/jbc.M206322200
98. Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA. (2000) 97:10832–7. doi: 10.1073/pnas.170276797
99. Rane MJ, Pan Y, Singh S, Powell DW, Wu R, Cummins T, et al. Heat shock protein 27 controls apoptosis by regulating Akt activation. J Biol Chem. (2003) 278:27828–35. doi: 10.1074/jbc.M303417200
100. Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem. (2007) 282:21598–608. doi: 10.1074/jbc.M611316200
101. Liu L, Zhang X, Jiang S, Ding Z, Ding G, Huang J, et al. Heat shock protein 27 regulates oxidative stress-induced apoptosis in cardiomyocytes: mechanisms via reactive oxygen species generation and Akt activation. Chin Med J. (2007) 120:2271–7. doi: 10.1097/00029330-200712020-00023
102. You W, Min X, Zhang X, Qian B, Pang S, Ding Z, et al. Cardiac-specific expression of heat shock protein 27 attenuated endotoxin-induced cardiac dysfunction and mortality in mice through a PI3K/Akt-dependent mechanism. Shock. (2009) 32:108–17. doi: 10.1097/SHK.0b013e318199165d
103. Penna C, Brancaccio M, Tullio F, Rubinetto C, Perrelli M-G, Angotti C, et al. Overexpression of the muscle-specific protein, melusin, protects from cardiac ischemia/reperfusion injury. Basic Res Cardiol. (2014) 109:418. doi: 10.1007/s00395-014-0418-9
104. Ke X, Chen J, Peng L, Zhang W, Yang Y, Liao X, et al. Heat shock protein 90/Akt pathway participates in the cardioprotective effect of exogenous hydrogen sulfide against high glucose-induced injury to H9c2 cells. Int J Mol Med. (2017) 39:1001–10. doi: 10.3892/ijmm.2017.2891
105. Zhang X-H, Wu J-X, Sha J-Z, Yang B, Sun J-R, Bao E-D. Heat shock protein 90 relieves heat stress damage of myocardial cells by regulating Akt and PKM2 signaling in vivo. Int J Mol Med. (2020) 45:1888–908. doi: 10.3892/ijmm.2020.4560
106. Jego G, Hermetet F, Girodon F, Garrido C. Chaperoning STAT3/5 by heat shock proteins: interest of their targeting in cancer therapy. Cancers. (2019) 12:21. doi: 10.3390/cancers12010021
107. Sanderson S, Valenti M, Gowan S, Patterson L, Ahmad Z, Workman P, et al. Benzoquinone ansamycin heat shock protein 90 inhibitors modulate multiple functions required for tumor angiogenesis. Mol Cancer Ther. (2006) 5:522–32. doi: 10.1158/1535-7163.MCT-05-0439
108. Kishor A, Tandukar B, Ly YV, Toth EA, Suarez Y, Brewer G, et al. Hsp70 is a novel posttranscriptional regulator of gene expression that binds and stabilizes selected mRNAs containing AU-rich elements. Mol Cell Biol. (2013) 33:71–84. doi: 10.1128/MCB.01275-12
109. Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N, et al. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation. (2010) 121:2176–82. doi: 10.1161/CIRCULATIONAHA.109.931220
110. van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J. (2014) 35:2165–73. doi: 10.1093/eurheartj/ehu050
111. Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. (2016) 374:233–41. doi: 10.1056/NEJMoa1505517
112. Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. (2010) 44:189–216. doi: 10.1146/annurev.genet.40.110405.090412
113. Zabinsky RA, Mason GA, Queitsch C, Jarosz DF. It's not magic - Hsp90 and its effects on genetic and epigenetic variation. Semin Cell Dev Biol. (2019) 88:21–35. doi: 10.1016/j.semcdb.2018.05.015
114. Karras GI, Yi S, Sahni N, Fischer M, Xie J, Vidal M, et al. HSP90 shapes the consequences of human genetic variation. Cell. (2017) 168:856–66. doi: 10.1016/j.cell.2017.01.023
115. Dorsch LM, Schuldt M, dos Remedios CG, Schinkel AFL, de Jong PL, Michels M, et al. Protein quality control activation and microtubule remodeling in hypertrophic cardiomyopathy. Cells. (2019) 8:741. doi: 10.3390/cells8070741
116. Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. (2014) 71:25–31. doi: 10.1016/j.yjmcc.2013.12.016
117. Moller DV, Andersen PS, Hedley P, Ersboll MK, Bundgaard H, Moolman-Smook J, et al. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet. (2009) 17:1241–9. doi: 10.1038/ejhg.2009.34
118. Glazier AA, Hafeez N, Mellacheruvu D, Basrur V, Nesvizhskii AI, Lee LM, et al. HSC70 is a chaperone for wild-type and mutant cardiac myosin binding protein C. JCI Insight. (2018) 3:e99319. doi: 10.1172/jci.insight.99319
119. Parbhudayal RY, Garra AR, Götte MJW, Michels M, Pei J, Harakalova M, et al. Variable cardiac myosin binding protein-C expression in the myofilaments due to MYBPC3 mutations in hypertrophic cardiomyopathy. J Mol Cell Cardiol. (2018) 123:59–63. doi: 10.1016/j.yjmcc.2018.08.023
120. Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L. The giant protein Titin's role in cardiomyopathy: genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front Physiol. (2019) 10:1436. doi: 10.3389/fphys.2019.01436
121. Zininga T, Ramatsui L, Shonhai A. Heat shock proteins as immunomodulants. Molecules. (2018) 23:2846. doi: 10.3390/molecules23112846
122. Qiu Y, Ye X, Hanson PJ, Zhang HM, Zong J, Cho B, et al. Hsp70-1: upregulation via selective phosphorylation of heat shock factor 1 during coxsackieviral infection and promotion of viral replication via the AU-rich element. Cell Mol Life Sci. (2016) 73:1067–84. doi: 10.1007/s00018-015-2036-6
123. Wang Y, Zhao S, Chen Y, Wang T, Dong C, Wo X, et al. The capsid protein VP1 of Coxsackievirus B induces cell cycle arrest by up-regulating Heat shock protein 70. Front Microbiol. (2019) 10:1633. doi: 10.3389/fmicb.2019.01633
124. Mathur S, Walley KR, Wang Y, Indrambarya T, Boyd JH. Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ J. (2011) 75:2445–52. doi: 10.1253/circj.CJ-11-0194
125. Sanzen Y, Ito M, Ohta Y, Yoshida Y, Kawada T, Sato H, et al. Functional proteomic analysis of experimental autoimmune myocarditis-induced chronic heart failure in the rat. Biol Pharm Bull. (2010) 33:477–86. doi: 10.1248/bpb.33.477
126. Satoh M, Shimoda Y, Akatsu T, Ishikawa Y, Minami Y, Nakamura M. Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte Toll signal in patients with heart failure after acute myocardial infarction. Eur J Heart Fail. (2006) 8:810–5. doi: 10.1016/j.ejheart.2006.03.004
127. Unsöld B, Kaul A, Sbroggiò M, Schubert C, Regitz-Zagrosek V, Brancaccio M, et al. Melusin protects from cardiac rupture and improves functional remodelling after myocardial infarction. Cardiovasc Res. (2014) 101:97–107. doi: 10.1093/cvr/cvt235
128. Wang DX, Huang Z, Li QJ, Zhong GQ, He Y, Huang WQ, et al. Involvement of HSP90 in ischemic postconditioning-induced cardioprotection by inhibition of the complement system, JNK and inflammation. Acta Cir Bras. (2020) 35:e202000105. doi: 10.1590/s0102-865020200010000005
129. Zhang XY, Huang Z, Li QJ, Zhong GQ, Meng JJ, Wang DX, et al. Role of HSP90 in suppressing TLR4-mediated inflammation in ischemic postconditioning. Clin Hemorheol Microcirc. (2020) 76:51–62. doi: 10.3233/CH-200840
130. Wang Y, Liu J, Kong Q, Cheng H, Tu F, Yu P, et al. Cardiomyocyte-specific deficiency of HSPB1 worsens cardiac dysfunction by activating NFκB-mediated leucocyte recruitment after myocardial infarction. Cardiovasc Res. (2019) 115:154–67. doi: 10.1093/cvr/cvy163
131. Li Y, Si R, Feng Y, Chen HH, Zou L, Wang E, et al. Myocardial ischemia activates an injurious innate immune signaling via cardiac heat shock protein 60 and toll-like receptor 4. J Biol Chem. (2011) 286:31308–19. doi: 10.1074/jbc.M111.246124
132. Tian J, Guo X, Liu XM, Liu L, Weng QF, Dong SJ, et al. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc Res. (2013) 98:391–401. doi: 10.1093/cvr/cvt047
133. Arany Z, Elkayam U. Peripartum cardiomyopathy. Circulation. (2016) 133:1397–409. doi: 10.1161/CIRCULATIONAHA.115.020491
134. Sliwa K, Blauwet L, Tibazarwa K, Libhaber E, Smedema JP, Becker A, et al. Evaluation of bromocriptine in the treatment of acute severe peripartum cardiomyopathy: a proof-of-concept pilot study. Circulation. (2010) 121:1465–73. doi: 10.1161/CIRCULATIONAHA.109.901496
135. Hilfiker-Kleiner D, Haghikia A, Berliner D, Vogel-Claussen J, Schwab J, Franke A, et al. Bromocriptine for the treatment of peripartum cardiomyopathy: a multicentre randomised study. Eur Heart J. (2017) 38:2671–9. doi: 10.1093/eurheartj/ehx35
136. Zininga T, Shonhai A. Are heat shock proteins druggable candidates? Am J Biochem Biotechnol. (2004) 10:209–10. doi: 10.3844/ajbbsp.2014.208.210
137. Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu T-C, et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol. (2012) 189:3209–20. doi: 10.4049/jimmunol.1103826
138. Liu P, Bao HY, Jin CC, Zhou JC, Hua F, Li K, et al. Targeting extracellular heat shock protein 70 ameliorates doxorubicin-induced heart failure through resolution of Toll-like receptor 2-mediated myocardial inflammation. J Am Heart Assoc. (2019) 8:e012338. doi: 10.1161/JAHA.119.012338
139. Zininga T, Pooe OJ, Makhado PB, Ramatsui L, Prinsloo E, Achilonu I, et al. Polymyxin B inhibits the chaperone activity of Plasmodium falciparum Hsp70. Cell Stress Chaperones. (2017) 22:707–15. doi: 10.1007/s12192-017-0797-6
140. McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. (2004) 279:4782–93. doi: 10.1074/jbc.M310405200
141. Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, et al. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci USA. (1999) 96:6137–42. doi: 10.1073/pnas.96.11.6137
142. Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. (2017) 127:3189–200. doi: 10.1172/JCI94310
143. Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. (2003) 54:804–10. doi: 10.1002/ana.10767
144. Jhaveri K, Ochiana SO, Dunphy MP, Gerecitano JF, Corben AD, Peter RI, et al. Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Exp Opin Investig Drugs. (2014) 23:611–28. doi: 10.1517/13543784.2014.902442
145. Zininga T, Shonhai A. Small molecule inhibitors targeting the heat shock protein system of human obligate protozoan parasites. Int J Mol Sci. (2019) 20:5930. doi: 10.3390/ijms20235930
146. Ricke-Hoch M, Hoes MF, Pfeffer TJ, Schlothauer S, Nonhoff J, Haidari S, et al. In peripartum cardiomyopathy plasminogen activator inhibitor-1 is a potential new biomarker with controversial roles. Cardiovasc Res. (2019) 116:1875–86. doi: 10.1093/cvr/cvz300
147. Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation. (2005) 111:887–93. doi: 10.1161/01.CIR.0000155616.07901.35
148. Veres A, Füst G, Smieja M, McQueen M, Horváth A, Yi Q, et al. Relationship of anti-60 kDa heat shock protein and anti-cholesterol antibodies to cardiovascular events. Circulation. (2002) 106:2775–80. doi: 10.1161/01.CIR.0000038890.39298.8D
149. Zhang X, He M, Cheng L, Chen Y, Zhou L, Zeng H, et al. Elevated heat shock protein 60 levels are associated with higher risk of coronary heart disease in Chinese. Circulation. (2008) 118:2687–93. doi: 10.1161/CIRCULATIONAHA.108.781856
150. Genth-Zotz S, Bolger AP, Kalra PR, von Haehling S, Doehner W, Coats AJ, et al. Heat shock protein 70 in patients with chronic heart failure: relation to disease severity and survival. Int J Cardiol. (2004) 96:397–401. doi: 10.1016/j.ijcard.2003.08.008
151. Comini L, Gaia G, Curello S, Ceconi C, Pasini E, Benigno M, et al. Right heart failure chronically stimulates heat shock protein 72 in heart and liver but not in other tissues. Cardiovasc Res. (1996) 31:882–90. doi: 10.1016/S0008-6363(96)00039-9
152. Berthenet K, Bokhari A, Lagrange A, Marcion G, Boudesco C, Causse S, et al. HSP110 promotes colorectal cancer growth through STAT3 activation. Oncogene. (2017) 36:2328–36. doi: 10.1038/onc.2016.403
153. Gozzi GJ, Gonzalez D, Boudesco C, Dias AMM, Gotthard G, Uyanik B, et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ. (2020) 27:117–29. doi: 10.1038/s41418-019-0343-4
154. Gombos T, Förhécz Z, Pozsonyi Z, Jánoskuti L, Prohászka Z. Interaction of serum 70-kDa heat shock protein levels and HspA1B (+1267) gene polymorphism with disease severity in patients with chronic heart failure. Cell Stress Chaperones. (2008) 13:199–206. doi: 10.1007/s12192-007-0001-5
155. Baba HA, Schmid KW, Schmid C, Blasius S, Heinecke A, Kerber S, et al. Possible relationship between heat shock protein 70, cardiac hemodynamics, and survival in the early period after heart transplantation. Transplantation. (1998) 65:799–804. doi: 10.1097/00007890-199803270-00006
156. Dohke T, Wada A, Isono T, Fujii M, Yamamoto T, Tsutamoto T, et al. Proteomic analysis reveals significant alternations of cardiac small heat shock protein expression in congestive heart failure. J Card Fail. (2006) 12:77–84. doi: 10.1016/j.cardfail.2005.07.006
157. De Marco M, D'Auria R, Rosati A, Vitulano G, Gigantino A, Citro R, et al. BAG3 protein in advanced-stage heart failure. JACC Heart Fail. (2014) 2:673–5. doi: 10.1016/j.jchf.2014.05.012
158. De Marco M, Falco A, Basile A, Rosati A, Festa M, d'Avenia M, et al. Detection of soluble BAG3 and anti-BAG3 antibodies in patients with chronic heart failure. Cell Death Dis. (2013) 4:e495. doi: 10.1038/cddis.2013.8
Keywords: heat shock protein, stress, cardiomyopathy, pregnancy, biomarkers, drug targets
Citation: Chakafana G, Spracklen TF, Kamuli S, Zininga T, Shonhai A, Ntusi NAB and Sliwa K (2021) Heat Shock Proteins: Potential Modulators and Candidate Biomarkers of Peripartum Cardiomyopathy. Front. Cardiovasc. Med. 8:633013. doi: 10.3389/fcvm.2021.633013
Received: 24 November 2020; Accepted: 06 May 2021;
Published: 16 June 2021.
Edited by:
Jun Ren, University of Washington, United StatesReviewed by:
Tom Van Agtmael, University of Glasgow, United KingdomTilman Grune, German Institute of Human Nutrition Potsdam-Rehbruecke (DIfE), Germany
Copyright © 2021 Chakafana, Spracklen, Kamuli, Zininga, Shonhai, Ntusi and Sliwa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen Sliwa, a2FyZW4uc2xpd2EtaGFobmxlQHVjdC5hYy56YQ==