 Wenhong Jiang
Wenhong Jiang Zhanman Zhang
Zhanman Zhang Xiao Qin
Xiao Qin- Department of Vascular Surgery, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
Arterial calcification refers to the abnormal deposition of calcium salts in the arterial wall, which results in vessel lumen stenosis and vascular remodeling. Studies increasingly show that arterial calcification is a cell mediated, reversible and active regulated process similar to physiological bone mineralization. The osteoblasts and chondrocytes-like cells are present in large numbers in the calcified lesions, and express osteogenic transcription factor and bone matrix proteins that are known to initiate and promote arterial calcification. In addition, osteoclast-like cells have also been detected in calcified arterial walls wherein they possibly inhibit vascular calcification, similar to the catabolic process of bone mineral resorption. Therefore, tilting the balance between osteoblast-like and osteoclast-like cells to the latter maybe a promising therapeutic strategy against vascular calcification. In this review, we have summarized the current findings on the origin and functions of osteoblast-like and osteoclast-like cells in the development and progression of vascular progression, and explored novel therapeutic possibilities.
Introduction
Vascular calcification, or the pathological accumulation of calcium phosphate crystals in the intimal and medial layers of vessel walls, is the pathological basis of many cardiovascular diseases (1, 2). It decreases blood vessel compliance and leads to lumen stenosis, eventually aggravating the cardiovascular symptoms. More than 60% of the middle-aged and elderly individuals in the 40–75 years age group, and more than 70% of patients undergoing chronic dialysis exhibit aortic calcification (3, 4). Furthermore, the prevalence of vascular calcification reaches as high as 80% in the patients with atherosclerosis, aged 80 and above (5). Although the mechanisms underlying vascular calcification have been elucidated to a large extent, there is still a dearth of effective pharmacological therapies. Thus, there is an urgent need to further clarify its molecular and cellular basis.
Vascular calcification was initially recognized as a passive and degenerative pathological process. However, recent findings indicate a cell mediated, active and reversible process that is similar to physiological bone mineralization (6). For instance, chondroblast-like cells, osteoblast-like cells, and even complete lamellar bone and regenerated bone marrow have been observed in the walls of calcified arteries (7–9). Furthermore, several bone-specific transcription factors and bone matrix proteins are overexpressed during vascular calcification, and their inhibition blocks osteoblast-like transdifferentiation and calcification (10, 11). In addition, calcium salts released from osteoporotic bones are deposited in the vascular lesions, which further points to a crosstalk between the osteogenic and vascular systems (12–15). Therefore, researchers are increasingly recognizing a novel bone-vascular axis in the pathogenesis of vascular calcification (16, 17), wherein osteoblast-like cells play a central role. Although there are no osteoblast-like cells in the normal arterial walls, studies show that vascular smooth muscle cells (VSMCs) can differentiate into chondroblast/osteoblast-like cells in the presence of high calcium levels (18, 19). In addition, endothelial cells, fibroblasts, pericytes, mesenchymal stem cells, and progenitor cells can also transdifferentiate into osteoblast-like cells (20–24). Therefore, the exact origin of osteoblast-like cells in the calcified arterial walls and the subsequent phenotypic remodeling are largely unknown.
Interestingly, vascular calcification may be reversible. For instance, calcification scores of both coronary and carotid artery are significantly declined in several patients with end-stage renal disease after they undergo subtotal parathyroidectomy (25). Additionally, aortic calcification induced in uremic rats with calcitriol and high phosphorus diet can be partially resolved when reducing phosphorus intake or using calcimimetics or phosphate binders, which may be involved in increase of mineral phagocytes and urinary calcium excretion (26–29). Rat medial elastocalcinosis that is induced by warfarin-rich diet feeding would also be inhibited when they are feeding with vitamin K-rich diet, with content in the aorta decreased by 50% (30). Interesting, aortic mineral loss of rat with medial elastocalcinosis is associated with activation of carbonic anhydrase IV that plays an important role in bone resorption, suggesting vascular mineral loss has a similar mechanism with bone resorption (31). Medial artery calcification is induced with calcitriol. It will regress rapidly, displaying a 75% decline of aortic calcium and phosphorus in 9 weeks. Furthermore, the reversibility of calcitriol induced vascular calcification may be mediated by macrophages (32). Apart from these, the phenotype of osteoblast-like VSMCs can be also reversed. It is reported that the initial deposition of hydroxyapatite-like mineral in medical arterial calcification occurs on degraded elastin first and that causes VSMCs transdifferentiation into osteogenic phenotype expressing bone related proteins and contribute to VSMCs calcification in vitro, but these VSMCs will return to original phenotype of VSMC after calcified conditions are removed (33). These researches indicate vascular calcification is reversible that is possibly involved in mineral loss similar to bone resorption, but the reversal process is still largely unknown.
Under physiological conditions, the osteoblasts and osteoclasts maintain the balance between bone calcium absorption and resorption, which is essential for proper bone mineralization (34, 35). There is evidence showing the involvement of osteoclast-like cells in vascular calcification (36). Ge et al. reported the increased expression of TRAP, which is a osteoclast phenotypic marker in the calcific vessels from hypertensive patients (37). Qiao et al. also identify osteoclast-like cells are present in carotid artery atheromatous lesions containing calcified plaque based on cell morphology, TRAP-positivity, and osteoclast-associated marker genes. Furthermore, they think osteoclast-like cells absorb deposited minerals preventing vascular calcification and counteract the effects of osteoblasts (38). In addition, osteoclasts cultured in vitro with calcified elastin reduced the extent of calcification and restored the structural integrity of elastin, indicating that these cells have an inhibitory role in vascular calcification as well (39, 40). Therefore, enhancing the proportion of osteoclast-like cells in the calcified arteries might be a suitable therapeutic intervention. However, the origin, phenotypic transformation and functions of the osteoclast-like cells during vascular calcification also remain to be elucidated.
Therefore, we have summarized the current knowledge regarding the osteoblast/osteoclast-like cells in vascular calcification, and discussed their potential as therapeutic targets.
Molecular Mechanism Underlying Osteoblast-Like Cells-Mediated Vascular Calcification
Studies show that the initiation and progression of vascular calcification likely involve both active and passive mechanisms (41–43). Passive calcification depends on the natural affinity of elastin and collagen in the extracellular matrix (ECM) for calcium ions. These proteins bind to calcium ions and increase their deposition in the ECM, resulting in extracellular calcium and phosphorus imbalance. The positively charged calcium ions adsorb the negatively charged phosphoric acid or carbonate ions to form amorphous calcium salt, which continuously adsorbs more mineral ions to form large mineral deposits and eventually lead to vascular calcification (41, 44, 45). Active vascular calcification resembles physiological bone mineralization. The cells in the vessel walls differentiate or transdifferentiate into chondroblast/osteoblast-like cells in the pro-calcific milieu of diabetes, chronic kidney disease, aging, and atherosclerosis. The osteoblast-like cells express osteogenic transcription factors and genes, such as runt-related transcription factor 2 (Runx2), osterix, Msh homeobox (Msx), SRY-box transcription factor 9 (SOX9), and alkaline phosphatase (ALP), bone sialoprotein (BSP) and osteocalcin (OC), all of which play a central role in bone mineralization. Knocking down RUNX2, osterix, SOX9, MSX1, or MSX2 suppressed osteogenic differentiation and subsequent vascular calcification (10, 46–49). The osteoblast/chondroblast-like cells initiate vascular calcification by releasing matrix vesicles (MV) (50) that are loaded with mineral crystals and are deposited in the ECM. Mineralized MVs have been detected in the calcified lesions of chronic kidney disease, atherosclerosis and diabetes. Furthermore, VSMCs cultured with high levels of phosphorus, calcium synthesize and secrete MVs harboring calcium, and phosphorus crystal-like structures (51, 52). MVs from osteoclast-like VSMCs and osteoblasts and hypertrophic chondrocytes have similar morphology and proteomics, which bind to collagen and induce apatite nucleation, resulting in endochondral and membranous osteogenesis (53–55). A recent study showed that hydroxyapatite nanocrystals reversibly induced osteogenic differentiation and calcification of VSMCs in vitro, and removal of the nanocrystals restored the original phenotype of the VSMCs and prevented calcification (33, 56). In a CKD rat model, inhibition of hydroxyapatite growth by sevelamer or pyrophosphate also slowed the progression of moderate to severe aortic calcification (57). Thus, osteoblast-like cells may also initiate vascular calcification through hydroxyapatite. Interestingly, the MVs from osteoblast-like cells express lower levels of calcification inhibitors like matrix gla protein (MGP) and fetuin A compared to normal VSMCs-derived MVs, whereas ALP, biomineralization-associated TNAP and phosphatidylserine (PS) are overexpressed in the former. TNAP hydrolyzes pyrophosphate, an inhibitor of vascular calcification, to produce inorganic phosphorus and PS that adhere to MV surface and adsorb calcium and phosphorus ions. Furthermore, the MVs secreted by osteoblast-like cells also express the annexins 2, 5, and 6, which function as calcium channels and allow ion influx (51, 58). Since MVs and exosomes aid intercellular communication, osteoblast-like cells may promote vascular calcification by secreting MVs that express aberrantly high levels of pro-calcification factors. Indeed, normal VSMCs co-cultured with osteoblast-like cell-derived MVs internalize the latter and undergo phenotypic transition characterized by the loss of contractile markers like smooth muscle 22 alpha (SM22a), and acquisition of the osteogenic markers (59). Microarray and bioinformatics analysis have identified significantly dysregulated non-coding RNAs in these MVs, which are enriched in signaling pathways closely related to osteogenic differentiation and calcification (60). Taken together, osteoblast-like cells in the vascular wall synthesize and secrete calcified MVs, which not only provide a site for calcium salt deposition but also initiate the phenotypic transition and calcification of adjacent cells in a paracrine manner.
Origin of Osteoblast-Like Cells in Vascular Calcification
Although there are no osteoblast-like cells in the normal artery wall, several resident cells like VSMCs, vascular endothelial cells (VECs), pericytes, fibroblasts, calcified vascular cells (CVCs), mesenchymal stem cells (MSCs), or progenitor cells have the potential to differentiate or transdifferentiate into osteoblast-like cells in response to pathological stimuli (61).
VSMCs and Osteoblast-Like Cells in Vascular Calcification
VSMCs are the main cell type in the arterial wall, and maintain vascular elasticity and contractility. Although VSMCs are terminally differentiated cells, they exhibit highly plastic phenotypes (62). The contractile VSMCs in the normal arterial wall are characterized by low growth, migration and protein synthesis, and express contractile markers such as SM22a, SMA, and SMM-HC. They can transdifferentiate into the synthetic phenotype that exhibits higher growth and migrations rates, produces more proteins, and expresses osteoblast and chondroblast markers (63). The calcified arterial lesions from humans and animal models also overexpress osteogenic and chondrogenic genes as a function of calcification degree, and have low levels of contractile markers. Furthermore, the osteogenic or chondrogenic markers are co-expressed with the VSMC contractile proteins, which indicates that VSMC can differentiate into osteogenic and chondrogenic cells as well (64–66). Consistent with this, osteoblast genes were upregulated and VSMC contractile genes were downregulated in human and animal VSMCs exposed to high levels calcium or phosphorus, which also formed massive alizarin red-staining calcium nodules. Likewise, hydroxyapatite and calcified elastin induced cultured VSMCs to lose the contractile phenotype and transdifferentiate into osteogenic cells, and removal of both restored the contractile phenotype and down-regulated bone-related genes, indicating that transdifferentiation of VSMCs is a reversible process (33). VSMCs expressing SM22α-Cre recombinase and Rosa26-LacZ Cre reporter alleles transdifferentiated into osteochondrogenic precursors and chondrocytes in MGP–/– or LDLr–/– and ApoE–/– mice, resulting in atherosclerotic medial and intimal calcification (67). Furthermore, calcium deposition and vascular calcification occur prior to the osteogenic phenotypic switch of VSMCs (42), and blocking the latter can inhibit or even reverse vascular calcification both in vitro and in vivo. There is evidence indicating that the different phenotypes of VSMCs have distinct embryonic origins (68, 69). It is possible that the contractile VSMCs first transdifferentiate into stem cells or other intermediate phenotypes prior to the osteoblast-like stage rather than differentiate directly. Thus, phenotypic reprogramming of VSMCs is a complex process and the regulatory pathways are largely unknown.
VECs and Osteoblast-Like Cells in Vascular Calcification
VECs are derived from the mesoderm, and are the main cell type in the vascular intima. Under physiological conditions, VECs form a vascular barrier through tight junctions with endothelial-specific proteins. However, pathological stimuli can trigger endothelial-mesenchymal transition (EndMT) in the VECs, resulting in the acquisition of MSC-like multipotent differentiation capacity. Human aortic ECs cultured in hyperglycemic conditions express low levels of EC marker genes like CD31, and gain MSC markers like CD44 and CD10. Furthermore, the chondrocyte genes SOX9 and type II collagen, cartilage proteoglycan and calcium nodules are induced in VECs cultured in osteogenic media (70). Calcium nodules are also formed in MGP–/– human aortic ECs induced with high glucose and bone morphogenetic protein (BMP), as well as in human and bovine aortic ECs stimulated with BMP6 and oxidized low-density lipoprotein (LDL), or tumor necrosis factor-a (TNF-a) and interleukin-β (IL-β) (22, 71, 72). Furtherly, the research of Malhotra et al. shows EndMT and vascular calcification in vitro and in vivo are dependent upon BMP signaling in MGP-deficient mice, while the other hand, also suggests that activation of BMP signaling inhibits atherosclerosis in MGP-deficient mice fed a standard diet when compared to LDLR–/– mice fed a high fat diet, a murine model of atherosclerosis (73). These results indicate that VECs can transdifferentiate into osteoblast-like cells and promote vascular calcification. Mechanistically, BMP signal activation induces the phenotypic switch of VECs via EndMT. Consistent with this, calcified arteries from Mgp–/– and Ins2Akita/+ mice express CD31, multipotent markers like SOX2, Nanog and Oct3/4, and Runx2 and Osterix (74, 75). Interestingly, MGP–/– human VECs cultured with osteogenic medium or BMP2 show upregulation of SOX2, Nanog and Oct3/4 prior to that of Runx2 and Osterix, indicating that multipotent marker appear earlier in the endothelium. Lineage tracing in the Mgp–/– and Tie2-Gfp transgenic mice confirmed the localization of GFP-labeled ECs in calcified lesions, along with increased expression of Runx2 and Osterix, which further demonstrated that osteogenic cells can originate from VECs (74). In addition, VECs can also regulate vascular calcification in a paracrine manner by activating the VSMCs, MSCs, and fibroblasts. VSMCs and MSCs co-cultured with RANKL-expressing ECs or their conditioned medium transdifferentiated into osteoblast-like cells expressing Runx2 and ALP (76). Likewise, fibroblasts co-cultured with mechanically stimulated ECs or the conditioned medium also underwent calcification (77). Furthermore, the conditioned medium from inorganic phosphorus or indoxyl sulfate-induced umbilical vein ECs inhibited expression of osteopontin in MSCs and induced calcification in the presence of IL-8 (78). Finally, the MVs and exosomes secreted by aging or inflammatory factor-stimulated ECs express high amounts of calcium, BMP2, annexin and Notch3, and promote calcification of recipient cells following internalization (79).
Pericytes and Osteoblast-Like Cells in Vascular Calcification
Pericytes are part of the microvascular system and closely related to endothelial cells. Recent studies show that pericytes or pericyte-like cells are abundant in the intima, media and adventitia of large, medium and small arteries, and can differentiate into osteoblasts, chondroblast-like cells and adipocytes. Furthermore, atherosclerotic calcified plaques of carotid and femoral arteries have significantly more pericytes compared to the non-calcified lesions, suggesting that these cells are involved in the formation of calcified plaques (80, 81). Consistent with the above, pericytes cultured with high content of advanced glycation end products, beta- glycerophosphate or glucocorticoid differentiate into osteoblast-like cells expressing Runx2, ALP, OC and other bone-related genes, and form calcium nodules. The latter may block MGP, an inhibitor of BMP4 and OPN, and activate the Axl signaling pathway (82–84). In addition, collagen glycosaminoglycan scaffolds loaded with pericytes gave rise to bone-like tissues in mice at the site of implantation, indicating that pericytes have spontaneous osteogenic potential (85, 86). Indeed, pericytes grown in normal medium can form multicellular nodules with a mineralized matrix containing extensive calcium salt deposits, and express high levels of Runx2 and the other osteogenic genes (21, 85). Therefore, pericytes have the potential to differentiate into osteoblast-like cells both spontaneously and in a stimuli-responsive manner, and contribute to mineralization.
Fibroblasts and Osteoblast-Like Cells in Vascular Calcification
Fibroblasts are the major cell type located in the outer arterial membrane, and a potential source of osteoblast-like cells during vascular calcification. This is supported by the significant calcification seen in the aorta adventitia of high fat diet-fed ApoE–/– mice and individuals older than 60 years of age (87). Shao et al. found that transgenic mice expressing the osteoblast gene Msx2 in the adventitial cells developed extensive vascular calcification (88). Furthermore, primary fibroblasts cultured in the presence of recombinant transforming growth factor-1 (TGF-β) and beta-glycerophosphoric acid transformed into myofibroblasts and formed calcium nodules (89, 90). Lai et al. also confirmed that MSX2 expression levels increased in the myofibroblasts and subsequently contributed to calcification (91). Simionescu et al. found rat fibroblasts induced by elastin degradation products and/or TGF-β expressed high levels of Runx2, ALP, OC, and other bone-related genes, and formed calcium deposits that stained with the von Kossa dye (89). Likewise, Runx2 and OC were upregulated and calcium deposition was increased in the three dimensional cultures of Wistar rat fibroblasts after 2 weeks of exposure to high levels of inorganic phosphate or β-glycerophosphate (92). Therefore, adventitial fibroblasts are likely to transform into myofibroblasts and osteoblast-like cells to regulate vascular calcification.
MSCs and Osteoblast-Like Cells in Vascular Calcification
MSCs are adult stem cells derived from the early mesoderm, and are characterized by high proliferation rates, self-renewal ability, multipotent differentiation potential, and immunosuppressive ability. Therefore, MSCs are the ideal biological tool for tissue repair and regeneration. The bone marrow-derived MSCs (BMSCs) differentiate into multiple cell types that are involved in cardiovascular pathologies, including vascular calcification. BMSCs transplanted into high cholesterol diet-fed rats with balloon injury in the abdominal aorta migrated to the BMP2-overexpressing arterial intima, and resulted in extensive calcification. Thus, MSCs can promote vascular calcification via the BMP2 signaling pathway (93). Kramann et al. implanted MSC-loaded collagen gels in the peritoneal cavity of rats with 5/6th nephrectomy or HFD-induced CKD, and detected Runx2, ALP, and sclerostin in the circulating MSCs, indicating osteogenic differentiation. Furthermore, a similar degree of calcification was observed after 8 weeks in the MSC-loaded gels as well as the arteries, both in terms of X-ray images and calcium content, indicating that MSCs contributed to vascular calcification after phenotypic conversion to osteoblast-like cells (94). Leszczynska et al. also confirmed the subcutaneous implantation of MSCs pretreated with chondrogenic medium gave rise to bone tissue in ApoE-/- and wild-type mice, and the extent of calcification was greater in the former (95). In addition, MSCs cultured in osteogenesis medium express osteogenic transcription factors and form calcium deposits in vitro. The MSCs isolated from ApoE–/– mice have greater osteogenic and chondrogenic capacity compared to wild-type cells, and is likely associated with the activation of uPAR-C5aR/Erk1/2/NF-kB axis (23). Wang et al. found that fluorescently labeled MSCs migrated to the calcified lesions in the HFD-fed LDLr–/– mice, and expressed osteogenic genes via TGF-β/SMAD2/4 signal activation (96). Interestingly, several groups have reported an inhibitory effect of MSCs on vascular calcification. For instance, intravenously injected MSCs significantly inhibited calcium and phosphorus deposition in the aortic walls, and also decreased serum phosphorus levels in the adenine-fed rat model of CKD (97). Similarly, conditioned medium from MSCs inhibited the osteogenic phenotypic switch and calcification of VSMCs even in the presence of β-glycerophosphate, and this effect was reversed by recombinant BMP2 via activation of the SMAD1/5/8 pathway. In addition, MSCs reduced calcification of co-cultured VSMCs in the osteogenic medium by inactivating the Wnt5a/β-catenin pathway (98). Therefore, the role of MSCs in vascular calcification is highly complex, and may depend on the environmental cues.
Progenitor Cells and Osteoblast-Like Cells in Vascular Calcification
Progenitor cells are precursors with multipotent differentiation potential. Studies increasingly show that both endothelial and VSMC progenitors are closely related to arterial calcification. For instance, circulating endothelial progenitor cells expressing the late osteoblast differentiation marker osteocalcin (OC) are abundant in patients with diabetes, CKD and coronary atherosclerosis. Furthermore, the number of OC+ circulating endothelial progenitor cells correlates positively with the calcium score and spotty calcification in coronary artery disease and hemodialysis patients (99, 100). Endothelial progenitors isolated from the peripheral blood of hemodialysis patients showed significantly increased calcium deposition in vitro. However, adding vitamin D receptor activator into medium of serum from uremia patients would inhibit calcification of endothelial progenitor cells (99). Similarly, the number of circulating OC+ endothelial progenitor cells in postmenopausal women and hemodialysis patients decreased significantly by administering bisphosphonates and vitamin D receptor activator, which also reduced vascular calcification (20). Taken together, OC+ endothelial progenitor cells can differentiate into osteoblast-like cells and promote vascular calcification. The CD10+ human perivascular progenitor cells, also known as adventitial cells due to their anatomical location, also show osteogenic potential. CD10 is significantly upregulated in the calcified vs. normal arteries. The CD10+ progenitor cells isolated from human abdominal white adipose tissue and placenta expressed osteoblastic transcription factors and other proteins in an NF-κB-dependent manner when cultured in osteogenic medium for 28 days, and formed mineralized nodules. Silencing the CD10 gene significantly decreased the bone forming ability of these progenitors (101). Kramann et al. detected abundant Gli1+ VSMC progenitors in the arterial adventitia, media and intima of mice with acute femoral artery injury or CKD, which promoted tissue repair (102). Furthermore, genetic fate tracing indicated that Gli1+ progenitor cells migrated from the adventitia into the intima and media of ApoE–/– mice with CKD, and expressed osteogenic genes that promoted arterial calcification. Furthermore, knocking down Gli1 inhibited arterial calcification (102). Cho et al. discovered Sca-1+/PDGFRα- progenitor cells in the bone marrow and blood vessel walls that differentiated into both osteoblasts and osteoclasts. Subcutaneous transplantation of Sca-1+/PDGFRα- progenitor cells and bone matrix in C57 mice resulted in a highly mineralized bone-like structure after 8 weeks. However, simultaneous injection of PPARγ significantly decreased calcification volume and calcium scores by inducing the differentiation of Sca-1+/PDGFRα- progenitor cells into osteoclasts. Consistent with the above, Sca-1+/PDGFRα- progenitor cells also aggravated arterial calcification in atherosclerotic ApoE–/– mice, which was reversed by PPARγ (39, 103).
CVCs and Osteoblast-Like Cells in Vascular Calcification
CVCs are a subset of VSMCs that characteristically express a modified form of ganglioside sialyllactose ceramide, along with osteogenic and chondrogenic markers. The CVCs can form calcium nodules following stimulation with transforming growth factor-β or 25-hydroxycholesterol. Similarly, mechanical stimulation or cAMP treatment also promotes osteogenic differentiation and mineralization of the CVCs. Overexpression of liver X receptor enhanced CVC mineralization by increasing fatty acid synthesis and lipid accumulation. In addition, subcutaneously transplanted CVCs formed calcium nodules akin to calcified atherosclerotic plaques in the Apoe–/– mice (104). Mechanistically, osteogenic differentiation and calcification of CVCs is mediated by the heat shock protein (HSP) 70, which binds to MGP and activates the BMP2/P-SMAD1/3/5 pathway (105). In addition, macrophages stimulated with ultrafine particles or their conditioned media induced osteogenic differentiation and mineralization of CVCs by partially activating the NF-κB signaling pathway (106). Conversely, the spontaneous differentiation and calcification of CVCs were inhibited by the protein kinase A-specific inhibitor KT5720, or insulin-like growth factor I, which are known to activate the ERK and PI3K pathways (107). Abedin et al. found that activation of p38-MAPK and PPARγ pathways decreased both spontaneous and IL-6-mediated osteogenesis of CVCs (108). These results indicate that CVCs can not only form calcium nodules spontaneously, but also be induced to exacerbate vascular calcification.
Origin and Function of Osteoclast-Like Cells in Vascular Calcification
Bone formation depends on the balance between the osteoblasts and osteoclasts. The osteoclasts mediate bone mineral matrix resorption and prevent excessive bone mineralization by producing various enzymes. While excessive bone resorption by osteoclast activity leads to osteoporosis, aberrant bone formation by hyperactive osteoblasts result in osteopetrosis. Osteoblast-like cells are abundant in calcified blood vessels and promotes vascular calcification, although the role of osteoclast-like cells are poorly understood.
Osteoclast-like cells have been detected in both atherosclerotic plaques and calcified medial layer, although the number is small and mainly concentrated in the heavily calcified lesions. Qiao et al. found osteoclast-like cells in human carotid plaques (38). Han et al. analyzed the calcified media and intima of 282 lower extremity artery samples, and detected bone tissue and osteoclasts in some heavily calcified regions (109). Similarly, tartrate-resistant acid phosphatase (TRAP), an osteoclast-specific marker gene, was also detected in the calcified vessels of hypertensive patients (37). Carbonic anhydrase, the key enzyme involved in osteoclast-mediated bone resorption, is significantly up-regulated in the atherosclerotic plaques of carotid, femoral and aortic vessels, and co-expressed with TRAP (110). These findings indicate that osteoclast-like cells regulate atherosclerosis and vascular calcification, although the mechanisms remain unknown. Monocytes and macrophages can differentiate into osteoclasts in the presence of inflammatory cytokines such as IL-6, IL-1, and TNF-α via activation of the RANKL/Akt signaling pathway. Osteoclastogenic differentiation of these cells is inhibited by oxidized low density lipoprotein or high levels of inorganic phosphate (111, 112). In the atherogenic ApoE–/– mice as well, macrophages transdifferentiate into osteoclasts via the Runx2/RANKL axis. Consistent with this, macrophages derived from human peripheral blood mononuclear cells differentiate into osteoclasts expressing cathepsin K and TRAP, and their calcium resorption activity is controlled by N-acetylglucosamine-1-phosphate transferase alpha and beta subunits (113). In addition, macrophages can effectively decalcify ectopic deposits by overexpressing carbonic anhydrase 2, which is silenced in mice with medial calcification in small arteries (114). Vascular progenitor cells also differentiate into osteoclast-like cells in the calcified lesions. The Sca-1+/PDGFRa- progenitor cells from mouse aorta differentiate into functional osteoclast-like cells in vitro in the presence of suitable factors, indicating their bipotent differentiation potential. In addition, Sca-1+/PDGFRa- cells implanted subcutaneously in C57 mice and atherogenic ApoE–/– mice differentiated into osteoclast-like cells in the presence of PPARγ, resulting in reduced calcium scores and calcification volume, suggesting a suitable therapeutic strategy for vascular calcification (39). Co-culture of calcified elastin and bone-marrow-derived osteoclast-like cells decreased significantly calcium content of the calcified elastin without elastin degradation and subcutaneous transplantation in rat of the mixture containing pure aortic elastin and osteoclast-like cells limited elastin calcification compared to control group (40). Bas et al. and Qiao et al. also found that osteoclast-like cells and macrophages inhibited and reversed calcification through mineral resorption (32, 38). However, the exact role of osteoclast-like cells in vascular calcification is largely ambiguous. RNA-seq analysis of rabbit atherosclerotic plaques showed enrichment of genes and KEGG pathways related to osteoclast differentiation, which have a bidirectional impact on osteoblast differentiation (115). In another study, knocking out Runx2 in VSMCs decreased RANKL expression, osteoclast numbers and the severity of calcification. Furthermore, VSMCs co-cultured with RANKL-overexpressing bone marrow-derived macrophages differentiated into osteoclast-like cells and increased vascular calcification (116). Thus, the role of osteoclast-like cells in vascular calcification needs further clarification.
The Potential of Cell-Based Therapeutic Strategies for Vascular Calcification
Over the years, great progress has been obtained about the pathogenesis of vascular calcification, but the treatment strategies for vascular calcification are limited. As discussion above, osteoblast-like cells play an important role in vascular calcification. Owning the role of osteoblast-like cells, many researches showed inhibition of osteogenic differentiation of VSMCs or recovery of contractile phenotype of VSMCs ameliorated vascular calcification both in vitro and in vivo (117–119). Lin et al. also reported SMC-specific Runx2 knockout significantly reduced vascular osteochondrogenesis and calcification in mouse (10). These results suggest it is possible to treat vascular calcification at cellular level by regulating phenotype changes of VSMCs. In addition, owing mineral resorption capacity of osteoclast-like cells, osteoclasts efficiently remove deposited minerals from calcified elastin both in vitro and in vivo (40). Co-culture of macrophages and VSMCs, inhibition of macrophages differentiated into osteoclasts contributed to VSMCs calcification. Furthermore, Barinda et al. found also macrophages reversed ectopic calcification in extracellular matrix through overexpressing carbonic anhydrase 2, which mediates hydrogen ions formation inducing mineral dissolution (114). In a mouse model of ectopic calcification, promoting the differentiation of Sca-1(+)/PDGFRα (-) progenitor cells with a PPARγ agonist into osteoclast-like cells attenuated ectopic calcification severity (39). These studies indicate osteoclast-like cells—and macrophage-based vascular calcification cell therapy has potential. However, Gbaguidi et al. reported macrophages near calcium deposits were deficient in RANKL-RANK axis and cathepsin K hindering their ability to reabsorb minerals in human atherosclerotic plaques (120). Recently, MSC therapy has been developed in many areas, which is a new strategy for treatment of vascular calcification. Calcified VSMCs induced with β-GP were cultured with conditioned media from MSCs and mineral deposition in VSMCs was inhibited, which was associated with inactivation of the BMP2–Smad1/5/8 signaling pathway and suppression of osteogenic phenotype, inflammation, and apoptosis (97, 98). Exosomes from MSCs were used to culture VSMCs and inhibited high phosphorus-induced VSMCs calcification (121). To similar these studies, indirect co-culture of VSMCs and MSCs in a transwell system reduced also VSMCs calcification involving in inhibiting of wnt signaling pathways (122). These researches suggest MSCs regulate vascular calcification in a paracrine manner. In vivo, adipose-derived MSCs were injected into a rat model of chronic kidney disease, which inhibited the progression of vascular calcification (123). However, some studies also reported MSCs have the potential of osteogenic differentiation (124). BMSCs transplantation promoted vascular calcification in hyperlipidemic rats subjected balloon injury and MSCs were implanted in c57BL/6 mice and Apoe–/– mice can form bone (93, 95). Taken together, cell treatment for vascular calcification based on osteoclast-like cells or macrophages and even MSCs is possible, but there are still some controversies and more research is needed.
Future Research
Although the potential sources of osteoblast-like cells in calcified lesions have been identified, the transdifferentiation of vascular cells into osteoblast-like cells is more complex and needs further research. Different subsets of VSMCs and ECs had been identified in vascular walls, and the VSMCs located in different segments of the aorta originate from different embryo layers. The obvious question herein is whether specific VSMC and ECs subsets are wired to differentiate into osteoblast-like cells. Both VSMCs and ECs undergo a phenotypic trajectory during transdifferentiation into osteoblast-like cells. The underlying molecular mechanisms remain to be elucidated, especially during early phenotypic transition. Limited number of osteoclast-like cells have also been identified in calcified arteries, especially in areas of severe calcification. Recent studies show that osteoclast-like cells promote atherosclerotic calcification in Apoe–/– mice and rabbits, although others have correlated increased number of osteoclast-like cells with atherosclerotic decalcification. Therefore, the exact role and origin of these cells are ambiguous, and have to be elucidated. Osteoclasts have bone resorptive activity, and an imbalance between osteoblastogenesis and osteoclastogenesis is present in vascular calcification. It remains to be seen whether osteoclasts or osteoclast-like cells are a promising therapeutic strategy for vascular calcification, and the most pressing question in this regard is to optimize the differentiation of vascular wall cells into osteoclast-like cells.
Conclusions
Vascular calcification is a cell-mediated, reversible and active regulated process resembled to bone mineralization. Osteoblast-like cells and limited number of osteoclast-like cells have been identified in calcified lesions (Figure 1). While the former promote calcification of the arterial walls, the latter have a demineralizing effect. Therefore, tracing the origin of these cells and the trajectory of their phenotypic remodeling, along with identification of core regulatory molecules, can further elucidate the mechanism of vascular calcification and reveal new therapeutic targets.
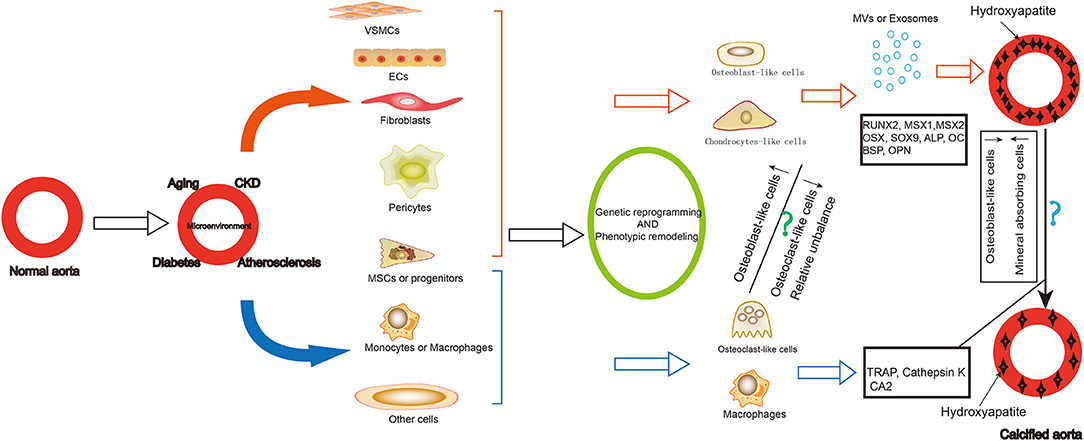
Figure 1. Summary of the origin and role of osteoblast-like cells and osteoclast-like-cells in vascular calcification. Vascular calcification is common in older adults, patients with chronic kidney disease, diabetes, and atherosclerosis. During the formation of vascular calcification, many cells differentiated into osteoblast-like cells or chondrocytes-like cells, including VSMCs, ECs, fibroblasts, pericytes, MSCs and progenitors. They express osteoblastic differentiation transcription factors and bone matrix proteins as well as secrete exosomes and matrix vesicles contributing to the development and progression of vascular calcification. Some cells can also differentiate into osteoclast—like cells, including some MSCs or progenitors, macrophages. They play a role in demineralization of the vascular calcification. The reduction of osteogenic phenotype cells and the increase of mineral reabsorption cells may be a strategy for vascular calcification cell therapy. VSMCs, vascular smooth muscle cells; ECs, endothelial cells; MSCs, mesenchymal stem cells; MV, matrix vesicles; RUNX2, RUNX family transcription factor 2; MSX, msh homeobox; SOX9, SRY-related high mobility group-box gene9; OSX, Sp7 transcription factor; ALP, alkaline phosphatase; OC, osteocalcin; BSP, bone sialoprotein; OPN, osteopontin; TRAP, acid phosphatase 5, tartrate resistant; CA2, carbonic anhydrase 2.
Author Contributions
WJ, ZZ, YL, CC, HY, QL, and MH: literature search and analysis. WJ: writing the manuscript. XQ and WJ: review and revise the manuscript. XQ: the funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the National Nature Science Foundation of China (no. 81960091).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Strauss HW, Nakahara T, Narula N, Narula J. Vascular calcification: the evolving relationship of vascular calcification to major acute coronary events. J Nucl Med. (2019) 60:1207–12. doi: 10.2967/jnumed.119.230276
2. Joshi FR, Rajani NK, Abt M, Woodward M, Bucerius J, Mani V, et al. Does vascular calcification accelerate inflammation?: a substudy of the dal-PLAQUE trial. J Am Coll Cardiol. (2016) 67:69–78. doi: 10.1016/j.jacc.2015.10.050
3. Kalsch H, Lehmann N, Mohlenkamp S, Hammer C, Mahabadi AA, Moebus S, et al. Prevalence of thoracic aortic calcification and its relationship to cardiovascular risk factors and coronary calcification in an unselected population-based cohort: the Heinz Nixdorf Recall Study. Int J Cardiovasc Imaging. (2013) 29:207–16. doi: 10.1007/s10554-012-0051-3
4. Liu ZH, Yu XQ, Yang JW, Jiang AL, Liu BC, Xing CY, et al. Prevalence and risk factors for vascular calcification in Chinese patients receiving dialysis: baseline results from a prospective cohort study. Curr Med Res Opin. (2018) 34:1491–1500. doi: 10.1080/03007995.2018.1467886
5. Wong ND, Sciammarella M, Arad Y, Miranda-Peats R, Polk D, Hachamovich R, et al. Relation of thoracic aortic and aortic valve calcium to coronary artery calcium and risk assessment. Am J Cardiol. (2003) 92:951–5. doi: 10.1016/S0002-9149(03)00976-7
6. Chen Y, Zhao X, Wu H. Arterial stiffness: a focus on vascular calcification and its link to bone mineralization. Arterioscler Thromb Vasc Biol. (2020) 40:1078–93. doi: 10.1161/ATVBAHA.120.313131
7. Neven E, Dauwe S, De Broe ME, D'Haese PC, Persy V. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int. (2007) 72:574–81. doi: 10.1038/sj.ki.5002353
8. Neven E, Persy V, Dauwe S, De Schutter T, De Broe ME, D'Haese PC. Chondrocyte rather than osteoblast conversion of vascular cells underlies medial calcification in uremic rats. Arterioscler Thromb Vasc Biol. (2010) 30:1741–50. doi: 10.1161/ATVBAHA.110.204834
9. Schmidt N, Brandsch C, Schutkowski A, Hirche F, Stangl GI. Dietary vitamin D inadequacy accelerates calcification and osteoblast-like cell formation in the vascular system of LDL receptor knockout and wild-type mice. J Nutr. (2014) 144:638–46. doi: 10.3945/jn.113.189118
10. Lin ME, Chen TM, Wallingford MC, Nguyen NB, Yamada S, Sawangmake C, et al. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc Res. (2016) 112:606–16. doi: 10.1093/cvr/cvw205
11. Rashdan NA, Sim AM, Cui L, Phadwal K, Roberts FL, Carter R, et al. Osteocalcin regulates arterial calcification via altered Wnt signaling and glucose metabolism. J Bone Miner Res. (2020) 35:357–67. doi: 10.1002/jbmr.3888
12. Lampropoulos CE, Kalamara P, Konsta M, Papaioannou I, Papadima E, Antoniou Z, et al. Osteoporosis and vascular calcification in postmenopausal women: a cross-sectional study. Climacteric. (2016) 19:303–7. doi: 10.3109/13697137.2016.1164134
13. De Schutter TM, Neven E, Persy VP, Behets GJ, Postnov AA, De Clerck NM, et al. Vascular calcification is associated with cortical bone loss in chronic renal failure rats with and without ovariectomy: the calcification paradox. Am J Nephrol. (2011) 34:356–66. doi: 10.1159/000331056
14. Kiel DP, Kauppila LI, Cupples LA, Hannan MT, O'Donnell CJ, Wilson PW. Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham Heart Study. Calcif Tissue Int. (2001) 68:271–6. doi: 10.1007/BF02390833
15. Choi SH, An JH, Lim S, Koo BK, Park SE, Chang HJ, et al. Lower bone mineral density is associated with higher coronary calcification and coronary plaque burdens by multidetector row coronary computed tomography in pre- and postmenopausal women. Clin Endocrinol. (2009) 71:644–51. doi: 10.1111/j.1365-2265.2009.03535.x
16. Sotomayor CG, Benjamens S, Gomes-Neto AW, Pol RA, Groothof D, Te Velde-Keyzer CA, et al. Bone mineral density and aortic calcification: evidence for a bone-vascular axis after kidney transplantation. Transplantation. (2020) 105:231–9. doi: 10.1097/TP.0000000000003226
17. De Mare A, Maudsley S, Azmi A, Hendrickx JO, Opdebeeck B, Neven E, et al. Sclerostin as regulatory molecule in vascular media calcification and the bone-vascular axis. Toxins. (2019) 11:70428. doi: 10.3390/toxins11070428
18. Fakhry M, Roszkowska M, Briolay A, Bougault C, Guignandon A, Diaz-Hernandez JI, et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochimica et Biophysica Acta. (2017) 1863:643–53. doi: 10.1016/j.bbadis.2016.12.003
19. Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone. (2019) 124:53–61. doi: 10.1016/j.bone.2019.04.006
20. Peris P, Atkinson EJ, Gossl M, Kane TL, McCready LK, Lerman A, et al. Effects of bisphosphonate treatment on circulating osteogenic endothelial progenitor cells in postmenopausal women. Mayo Clin Proc. (2013) 88:46–55. doi: 10.1016/j.mayocp.2012.08.019
21. Doherty MJ, Ashton BA, Walsh S, Beresford JN, Grant ME, Canfield AE. Vascular pericytes express osteogenic potential in vitro and in vivo. J Bone Miner Res. (1998) 13:828–38. doi: 10.1359/jbmr.1998.13.5.828
22. Sánchez-Duffhues G, García de Vinuesa A, van de Pol V, Geerts ME, de Vries MR, Janson SGT, et al. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. (2019) 247:333–46. doi: 10.1002/path.5193
23. Kalbasi Anaraki P, Patecki M, Larmann J, Tkachuk S, Jurk K, Haller H, et al. Urokinase receptor mediates osteogenic differentiation of mesenchymal stem cells and vascular calcification via the complement C5a receptor. Stem Cells Dev. (2014) 23:352–62. doi: 10.1089/scd.2013.0318
24. Lu Z, Chiu J, Lee LR, Schindeler A, Jackson M, Ramaswamy Y, et al. Reprogramming of human fibroblasts into osteoblasts by insulin-like growth factor-binding protein 7. Stem Cells Transl Med. (2020) 9:403–15. doi: 10.1002/sctm.19-0281
25. Bleyer AJ, Burkart J, Piazza M, Russell G, Rohr M, Carr JJ. Changes in cardiovascular calcification after parathyroidectomy in patients with ESRD. Am J Kidney Dis. (2005) 46:464–9. doi: 10.1053/j.ajkd.2005.04.035
26. Lopez I, Mendoza FJ, Guerrero F, Almaden Y, Henley C, Aguilera-Tejero E, et al. The calcimimetic AMG 641 accelerates regression of extraosseous calcification in uremic rats. Am J Physiol Renal Physiol. (2009) 296:F1376-85. doi: 10.1152/ajprenal.90737.2008
27. De Schutter TM, Behets GJ, Geryl H, Peter ME, Steppan S, Gundlach K, et al. Effect of a magnesium-based phosphate binder on medial calcification in a rat model of uremia. Kidney Int. (2013) 83:1109–17. doi: 10.1038/ki.2013.34
28. Finch JL, Lee DH, Liapis H, Ritter C, Zhang S, Suarez E, et al. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney Int. (2013) 84:1145–53. doi: 10.1038/ki.2013.213
29. Yamada S, Tatsumoto N, Tokumoto M, Noguchi H, Ooboshi H, Kitazono T, et al. Phosphate binders prevent phosphate-induced cellular senescence of vascular smooth muscle cells and vascular calcification in a modified, adenine-based uremic rat model. Calcif Tissue Int. (2015) 96:347–58. doi: 10.1007/s00223-014-9929-5
30. Schurgers LJ, Spronk HM, Soute BA, Schiffers PM, DeMey JG, Vermeer C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood. (2007) 109:2823–31. doi: 10.1182/blood-2006-07-035345
31. Essalihi R, Dao HH, Gilbert LA, Bouvet C, Semerjian Y, McKee MD, et al. Regression of medial elastocalcinosis in rat aorta: a new vascular function for carbonic anhydrase. Circulation. (2005) 112:1628–35. doi: 10.1161/CIRCULATIONAHA.104.528984
32. Bas A, Lopez I, Perez J, Rodriguez M, Aguilera-Tejero E. Reversibility of calcitriol-induced medial artery calcification in rats with intact renal function. J Bone Mineral Res. (2005) 21:484–90. doi: 10.1359/JBMR.051211
33. Lei Y, Sinha A, Nosoudi N, Grover A, Vyavahare N. Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp Cell Res. (2014) 323:198–208. doi: 10.1016/j.yexcr.2014.01.011
34. Arthur A, Nguyen TM, Paton S, Klisuric A, Zannettino ACW, Gronthos S. The osteoprogenitor-specific loss of ephrinB1 results in an osteoporotic phenotype affecting the balance between bone formation and resorption. Sci Rep. (2018) 8:12756. doi: 10.1038/s41598-018-31190-2
35. Wang X, Wu J, Shidoji Y, Muto Y, Ohishi N, Yagi K, et al. Effects of geranylgeranoic acid in bone: induction of osteoblast differentiation and inhibition of osteoclast formation. J Bone Miner Res. (2002) 17:91–100. doi: 10.1359/jbmr.2002.17.1.91
36. Li J, Xing G, Zhang L, Shang J, Li Y, Li C, et al. Satb1 promotes osteoclastogenesis by recruiting CBP to upregulate miR-223 expression in chronic kidney disease-mineral and bone disorder. Pharmazie. (2017) 72:680–6. doi: 10.1691/ph.2017.7606
37. Ge Q, Ruan CC, Ma Y, Tang XF, Wu QH, Wang JG, et al. Osteopontin regulates macrophage activation and osteoclast formation in hypertensive patients with vascular calcification. Sci Rep. (2017) 7:40253. doi: 10.1038/srep40253
38. Qiao JH, Mishra V, Fishbein MC, Sinha SK, Rajavashisth TB. Multinucleated giant cells in atherosclerotic plaques of human carotid arteries: identification of osteoclast-like cells and their specific proteins in artery wall. Exp Mol Pathol. (2015) 99:654–62. doi: 10.1016/j.yexmp.2015.11.010
39. Cho HJ, Cho HJ, Lee HJ, Song MK, Seo JY, Bae YH, et al. Vascular calcifying progenitor cells possess bidirectional differentiation potentials. PLoS Biol. (2013) 11:e1001534. doi: 10.1371/journal.pbio.1001534
40. Simpson CL, Lindley S, Eisenberg C, Basalyga DM, Starcher BC, Simionescu DT, et al. Toward cell therapy for vascular calcification: osteoclast-mediated demineralization of calcified elastin. Cardiovasc Pathol. (2007) 16:29–37. doi: 10.1016/j.carpath.2006.07.001
41. Urry DW. Neutral sites for calcium ion binding to elastin and collagen: a charge neutralization theory for calcification and its relationship to atherosclerosis. Proc Natl Acad Sci USA. (1971) 68:810–4. doi: 10.1073/pnas.68.4.810
42. Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. (2009) 104:733–41. doi: 10.1161/CIRCRESAHA.108.183053
43. Montezano AC, Zimmerman D, Yusuf H, Burger D, Chignalia AZ, Wadhera V, et al. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension. (2010) 56:453–62. doi: 10.1161/HYPERTENSIONAHA.110.152058
44. Silver FH, Landis WJ. Deposition of apatite in mineralizing vertebrate extracellular matrices: a model of possible nucleation sites on type I collagen. Connect Tissue Res. (2011) 52:242–54. doi: 10.3109/03008207.2010.551567
45. Kuzan A, Chwilkowska A, Pezowicz C, Witkiewicz W, Gamian A, Maksymowicz K, et al. The content of collagen type II in human arteries is correlated with the stage of atherosclerosis and calcification foci. Cardiovasc Pathol. (2017) 28:21–7. doi: 10.1016/j.carpath.2017.02.003
46. Taylor J, Butcher M, Zeadin M, Politano A, Shaughnessy SG. Oxidized low-density lipoprotein promotes osteoblast differentiation in primary cultures of vascular smooth muscle cells by up-regulating Osterix expression in an Msx2-dependent manner. J Cell Biochem. (2011) 112:581–8. doi: 10.1002/jcb.22948
47. Xu Z, Ji G, Shen J, Wang X, Zhou J, Li L. SOX9 and myocardin counteract each other in regulating vascular smooth muscle cell differentiation. Biochem Biophys Res Commun. (2012) 422:285–90. doi: 10.1016/j.bbrc.2012.04.149
48. Hayashi K, Nakamura S, Nishida W, Sobue K. Bone morphogenetic protein-induced MSX1 and MSX2 inhibit myocardin-dependent smooth muscle gene transcription. Mol Cell Biol. (2006) 26:9456–70. doi: 10.1128/MCB.00759-06
49. Zhao YG, Meng FX, Li BW, Sheng YM, Liu MM, Wang B, et al. Gelatinases promote calcification of vascular smooth muscle cells by up-regulating bone morphogenetic protein-2. Biochem Biophys Res Commun. (2016) 470:287–93. doi: 10.1016/j.bbrc.2016.01.067
50. Zazzeroni L, Faggioli G, Pasquinelli G. Mechanisms of arterial calcification: the role of matrix vesicles. Eur J Vasc Endovasc Surg. (2018) 55:425–32. doi: 10.1016/j.ejvs.2017.12.009
51. Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. (2011) 109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808
52. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. (2015) 116:1312–23. doi: 10.1161/CIRCRESAHA.116.305012
53. Chaudhary SC, Khalid S, Smethurst V, Monier D, Mobley J, Huet A, et al. Proteomic profiling of extracellular vesicles released from vascular smooth muscle cells during initiation of phosphate-induced mineralization. Connect Tissue Res. (2018) 59(supp.1):55–61. doi: 10.1080/03008207.2018.1444759
54. Balcerzak M, Malinowska A, Thouverey C, Sekrecka A, Dadlez M, Buchet R, et al. Proteome analysis of matrix vesicles isolated from femurs of chicken embryo. Proteomics. (2008) 8:192–205. doi: 10.1002/pmic.200700612
55. Cui L, Houston DA, Farquharson C, MacRae VE. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone. (2016) 87:147–58. doi: 10.1016/j.bone.2016.04.007
56. Nahar-Gohad P, Gohad N, Tsai CC, Bordia R, Vyavahare N. Rat aortic smooth muscle cells cultured on hydroxyapatite differentiate into osteoblast-like cells via BMP-2-SMAD-5 pathway. Calcif Tissue Int. (2015) 96:359–69. doi: 10.1007/s00223-015-9962-z
57. Neven E, Opdebeeck B, De Mare A, Bashir-Dar R, Dams G, Marynissen R, et al. Can intestinal phosphate binding or inhibition of hydroxyapatite growth in the vascular wall halt the progression of established aortic calcification in chronic kidney disease? Calcif Tissue Int. (2016) 99:525–34. doi: 10.1007/s00223-016-0178-7
58. Kirsch T, Harrison G, Golub EE, Nah HD. The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J Biol Chem. (2000) 275:35577–83. doi: 10.1074/jbc.M005648200
59. Chen NX, O'Neill KD, Moe SM. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. (2017) 93:343–54. doi: 10.1016/j.kint.2017.07.019
60. Chaturvedi P, Chen NX, O'Neill K, McClintick JN, Moe SM, Janga SC. Differential miRNA expression in cells and matrix vesicles in vascular smooth muscle cells from rats with kidney disease. PLoS ONE. (2015) 10:e0131589. doi: 10.1371/journal.pone.0131589
61. Hortells L, Sur S, St Hilaire C. Cell phenotype transitions in cardiovascular calcification. Front Cardiovasc Med. (2018) 5:27. doi: 10.3389/fcvm.2018.00027
62. Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp. (2007) 283:174–91. doi: 10.1002/9780470319413.ch14
63. Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. (2018) 52:48–64. doi: 10.1016/j.cellsig.2018.08.019
64. Lee GL, Yeh CC, Wu JY, Lin HC, Wang YF, Kuo YY, et al. TLR2 promotes vascular smooth muscle cell chondrogenic differentiation and consequent calcification via the concerted actions of osteoprotegerin suppression and IL-6-mediated RANKL induction. Arterioscler Thromb Vasc Biol. (2019) 39:432–45. doi: 10.1161/ATVBAHA.118.311874
65. Shen J, Yang M, Jiang H, Ju D, Zheng JP, Xu Z, et al. Arterial injury promotes medial chondrogenesis in Sm22 knockout mice. Cardiovasc Res. (2011) 90:28–37. doi: 10.1093/cvr/cvq378
66. McRobb LS, McGrath KCY, Tsatralis T, Liong EC, Tan JTM, Hughes G, et al. Estrogen receptor control of atherosclerotic calcification and smooth muscle cell osteogenic differentiation. Arterioscler Thromb Vasc Biol. (2017) 37:1127–37. doi: 10.1161/ATVBAHA.117.309054
67. Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB, Giachelli CM, et al. Sources of cells that contribute to atherosclerotic intimal calcification: an in vivo genetic fate mapping study. Cardiovasc Res. (2012) 94:545–54. doi: 10.1093/cvr/cvs126
68. Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. (2019) 25:1280–9. doi: 10.1038/s41591-019-0512-5
69. Espitia O, Chatelais M, Steenman M, Charrier C, Maurel B, Georges S, et al. Implication of molecular vascular smooth muscle cell heterogeneity among arterial beds in arterial calcification. PLoS ONE. (2018) 13:e0191976. doi: 10.1371/journal.pone.0191976
70. Tang R, Gao M, Wu M, Liu H, Zhang X, Liu B. High glucose mediates endothelial-to-chondrocyte transition in human aortic endothelial cells. Cardiovasc Diabetol. (2012) 11:113. doi: 10.1186/1475-2840-11-113
71. Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS, et al. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler Thromb Vasc Biol. (2012) 32:613–22. doi: 10.1161/ATVBAHA.111.242594
72. Yung L-M, Sánchez-Duffhues G, ten Dijke P, Yu PB. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res. (2015) 108:278–87. doi: 10.1093/cvr/cvv221
73. Malhotra R, Burke MF, Martyn T, Shakartzi HR, Thayer TE, O'Rourke C, et al. Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix Gla protein deficiency. PLoS ONE. (2015) 10:e0117098. doi: 10.1371/journal.pone.0117098
74. Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Boström KI. A role for the endothelium in vascular calcification. Circulat Res. (2013) 113:495–504. doi: 10.1161/CIRCRESAHA.113.301792
75. Guihard PJ, Yao J, Blazquez-Medela AM, Iruela-Arispe L, Bostrom KI, Yao Y. Endothelial-mesenchymal transition in vascular calcification of Ins2Akita/+ mice. PLoS ONE. (2016) 11:e0167936. doi: 10.1371/journal.pone.0167936
76. Davenport C, Harper E, Forde H, Rochfort KD, Murphy RP, Smith D, et al. RANKL promotes osteoblastic activity in vascular smooth muscle cells by upregulating endothelial BMP-2 release. Int J Biochem Cell Biol. (2016) 77:171–80. doi: 10.1016/j.biocel.2016.06.009
77. Rutkovskiy A, Lund M, Siamansour TS, Reine TM, Kolset SO, Sand KL, et al. Mechanical stress alters the expression of calcification-related genes in vascular interstitial and endothelial cells. Interact Cardiovasc Thorac Surg. (2019) 28:803–11. doi: 10.1093/icvts/ivy339
78. Bouabdallah J, Zibara K, Issa H, Lenglet G, Kchour G, Caus T, et al. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol Dial Transplant. (2019) 34:1125–34. doi: 10.1093/ndt/gfy325
79. Alique M, Ruiz-Torres MP, Bodega G, Noci MV, Troyano N, Bohorquez L, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging. (2017) 9:778–89. doi: 10.18632/aging.101191
80. Davaine JM, Quillard T, Chatelais M, Guilbaud F, Brion R, Guyomarch B, et al. Bone like arterial calcification in femoral atherosclerotic lesions: prevalence and role of osteoprotegerin and pericytes. Eur J Vasc Endovasc Surg. (2016) 51:259–67. doi: 10.1016/j.ejvs.2015.10.004
81. Aikawa E, Davaine J-M, Quillard T, Brion R, Lapérine O, Guyomarch B, et al. Osteoprotegerin, pericytes and bone-like vascular calcification are associated with carotid plaque stability. PLoS ONE. (2014) 9:e107642. doi: 10.1371/journal.pone.0107642
82. Kirton JP, Wilkinson FL, Canfield AE, Alexander MY. Dexamethasone downregulates calcification-inhibitor molecules and accelerates osteogenic differentiation of vascular pericytes: implications for vascular calcification. Circ Res. (2006) 98:1264–72. doi: 10.1161/01.RES.0000223056.68892.8b
83. Collett G, Wood A, Alexander MY, Varnum BC, Boot-Handford RP, Ohanian V, et al. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ Res. (2003) 92:1123–9. doi: 10.1161/01.RES.0000074881.56564.46
84. Yamagishi S, Fujimori H, Yonekura H, Tanaka N, Yamamoto H. Advanced glycation endproducts accelerate calcification in microvascular pericytes. Biochem Biophys Res Commun. (1999) 258:353–7. doi: 10.1006/bbrc.1999.0625
85. Canfield AE, Doherty MJ, Wood AC, Farrington C, Ashton B, Begum N, et al. Role of pericytes in vascular calcification: a review. Z Kardiol. (2000) 89(Suppl.2):20–7. doi: 10.1007/s003920070096
86. Schor AM, Allen TD, Canfield AE, Sloan P, Schor SL. Pericytes derived from the retinal microvasculature undergo calcification in vitro. J Cell Sci. (1990) 97:449–61.
87. Li N, Cheng W, Huang T, Yuan J, Wang X, Song M. Vascular adventitia calcification and its underlying mechanism. PLoS ONE. (2015) 10:e0132506. doi: 10.1371/journal.pone.0132506
88. Shao J-S, Cheng S-L, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Investig. (2005) 115:1210–20. doi: 10.1172/JCI24140
89. Simionescu A, Simionescu DT, Vyavahare NR. Osteogenic responses in fibroblasts activated by elastin degradation products and transforming growth factor-beta1: role of myofibroblasts in vascular calcification. Am J Pathol. (2007) 171:116–23. doi: 10.2353/ajpath.2007.060930
90. Watanabe Y, Suzuki M, Oyama Y, Kusano E, Tamba K, Iimura O, et al. Cellular component of vascular calcification. Fibroblasts are essential for calcium deposition in cultured cells. Nephron. (2002) 92:840–5. doi: 10.1159/000065461
91. Towler DA, Cheng S-L, Krchma K, Behrmann A, Shao J-S, Lai C-F. TNFR1-activated reactive oxidative species signals up-regulate osteogenic Msx2 programs in aortic myofibroblasts. Endocrinology. (2012) 153:3897–910. doi: 10.1210/en.2012-1216
92. Suyama T, Hatta M, Hata S, Ishikawa H, Yamazaki J. Differentiation of rat dermal mesenchymal cells and calcification in three-dimensional cultures. Tissue Eng Regen Med. (2016) 13:527–37. doi: 10.1007/s13770-016-9124-z
93. Liao J, Chen X, Li Y, Ge Z, Duan H, Zou Y, et al. Transfer of bone-marrow-derived mesenchymal stem cells influences vascular remodeling and calcification after balloon injury in hyperlipidemic rats. J Biomed Biotechnol. (2012) 2012:165296. doi: 10.1155/2012/165296
94. Kramann R, Kunter U, Brandenburg VM, Leisten I, Ehling J, Klinkhammer BM, et al. Osteogenesis of heterotopically transplanted mesenchymal stromal cells in rat models of chronic kidney disease. J Bone Miner Res. (2013) 28:2523–34. doi: 10.1002/jbmr.1994
95. Leszczynska A, O'Doherty A, Farrell E, Pindjakova J, O'Brien FJ, O'Brien T, et al. Differentiation of vascular stem cells contributes to ectopic calcification of atherosclerotic plaque. Stem Cells. (2016) 34:913–23. doi: 10.1002/stem.2315
96. Wang W, Li C, Pang L, Shi C, Guo F, Chen A, et al. Mesenchymal stem cells recruited by active TGFβ contribute to osteogenic vascular calcification. Stem Cells Dev. (2014) 23:1392–404. doi: 10.1089/scd.2013.0528
97. Wang S, Tong M, Hu S, Chen X. The bioactive substance secreted by MSC retards mouse aortic vascular smooth muscle cells calcification. Biomed Res Int. (2018) 2018:6053567. doi: 10.1155/2018/6053567
98. Wang S, Hu S, Wang J, Liu Y, Zhao R, Tong M, et al. Conditioned medium from bone marrow-derived mesenchymal stem cells inhibits vascular calcification through blockade of the BMP2-Smad1/5/8 signaling pathway. Stem Cell Res Ther. (2018) 9:160. doi: 10.1186/s13287-018-0894-1
99. Cianciolo G, La Manna G, Della Bella E, Cappuccilli ML, Angelini ML, Dormi A, et al. Effect of vitamin D receptor activator therapy on vitamin D receptor and osteocalcin expression in circulating endothelial progenitor cells of hemodialysis patients. Blood Purif . (2013) 35:187–95. doi: 10.1159/000347102
100. Zhang H, Wang LJ, Si DL, Wang C, Yang JC, Jiang P, et al. Correlation between osteocalcin-positive endothelial progenitor cells and spotty calcification in patients with coronary artery disease. Clin Exp Pharmacol Physiol. (2015) 42:734–9. doi: 10.1111/1440-1681.12366
101. Ding L, Vezzani B, Khan N, Su J, Xu L, Yan G, et al. CD10 expression identifies a subset of human perivascular progenitor cells with high proliferation and calcification potentials. Stem Cells. (2019) 38:261–75. doi: 10.1002/stem.3112
102. Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, et al. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell. (2016) 19:628–42. doi: 10.1016/j.stem.2016.08.001
103. Cho HJ, Lee JW, Cho HJ, Lee CS, Kim HS. Identification of adult mesodermal progenitor cells and hierarchy in atherosclerotic vascular calcification. Stem Cells. (2018) 36:1075–96. doi: 10.1002/stem.2814
104. Li R, Mittelstein D, Lee J, Fang K, Majumdar R, Tintut Y, et al. A dynamic model of calcific nodule destabilization in response to monocyte- and oxidized lipid-induced matrix metalloproteinases. Am J Physiol Cell Physiol. (2012) 302:C658–65. doi: 10.1152/ajpcell.00313.2011
105. Yao Y, Watson AD, Ji S, Boström KI. Heat shock protein 70 enhances vascular bone morphogenetic protein-4 signaling by binding matrix Gla protein. Circulat Res. (2009) 105:575–84. doi: 10.1161/CIRCRESAHA.109.202333
106. Li R, Mittelstein D, Kam W, Pakbin P, Du Y, Tintut Y, et al. Atmospheric ultrafine particles promote vascular calcification via the NF-kappaB signaling pathway. Am J Physiol Cell Physiol. (2013) 304:C362–9. doi: 10.1152/ajpcell.00322.2012
107. Radcliff K, Tang TB, Lim J, Zhang Z, Abedin M, Demer LL, et al. Insulin-like growth factor-I regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circ Res. (2005) 96:398–400. doi: 10.1161/01.RES.0000157671.47477.71
108. Abedin M, Lim J, Tang TB, Park D, Demer LL, Tintut Y. N-3 fatty acids inhibit vascular calcification via the p38-mitogen-activated protein kinase and peroxisome proliferator-activated receptor-gamma pathways. Circ Res. (2006) 98:727–9. doi: 10.1161/01.RES.0000216009.68958.e6
109. Han KH, Hennigar RA, O'Neill WC. The association of bone and osteoclasts with vascular calcification. Vasc Med. (2015) 20:527–33. doi: 10.1177/1358863X15597076
110. Oksala N, Levula M, Pelto-Huikko M, Kytomaki L, Soini JT, Salenius J, et al. Carbonic anhydrases II and XII are up-regulated in osteoclast-like cells in advanced human atherosclerotic plaques-Tampere Vascular Study. Ann Med. (2010) 42:360–70. doi: 10.3109/07853890.2010.486408
111. Itou T, Maldonado N, Yamada I, Goettsch C, Matsumoto J, Aikawa M, et al. Cystathionine gamma-lyase accelerates osteoclast differentiation: identification of a novel regulator of osteoclastogenesis by proteomic analysis. Arterioscler Thromb Vasc Biol. (2014) 34:626–34. doi: 10.1161/ATVBAHA.113.302576
112. Maziere C, Louvet L, Gomila C, Kamel S, Massy Z, Maziere JC. Oxidized low density lipoprotein decreases Rankl-induced differentiation of osteoclasts by inhibition of Rankl signaling. J Cell Physiol. (2009) 221:572–8. doi: 10.1002/jcp.21886
113. Lei Y, Iwashita M, Choi J, Aikawa M, Aikawa E. N-acetylglucosamine-1-phosphate transferase suppresses lysosomal hydrolases in dysfunctional osteoclasts: a potential mechanism for vascular calcification. J Cardiovasc Dev Dis. (2015) 2:31–47. doi: 10.3390/jcdd2020031
114. Barinda AJ, Ikeda K, Hirata KI, Emoto N. Macrophages highly express carbonic anhydrase 2 and play a significant role in demineralization of the ectopic calcification. Kobe J Med Sci. (2017) 63:E45–50.
115. Tan L, Wang Z, Li Y. Rabbit models provide insights into bone formation related biological process in atherosclerotic vascular calcification. Biochem Biophys Res Commun. (2018) 496:1369–75. doi: 10.1016/j.bbrc.2018.02.035
116. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, et al. Runx2-upregulated receptor activator of nuclear factor kappaB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. (2011) 31:1387–96. doi: 10.1161/ATVBAHA.110.222547
117. Hodroge A, Trecherel E, Cornu M, Darwiche W, Mansour A, Ait-Mohand K, et al. Oligogalacturonic acid inhibits vascular calcification by two mechanisms: inhibition of vascular smooth muscle cell osteogenic conversion and interaction with collagen. Arterioscler Thromb Vasc Biol. (2017) 37:1391–401. doi: 10.1161/ATVBAHA.117.309513
118. Shi Y, Lu W, Hou Y, Fu K, Gan F, Liu J. Fibroblast growth factor 21 ameliorates vascular calcification by inhibiting osteogenic transition in vitamin D3 plus nicotine-treated rats. Biochem Biophys Res Commun. (2018) 495:2448–55. doi: 10.1016/j.bbrc.2017.10.115
119. Sapkota M, Shrestha SK, Yang M, Park YR, Soh Y. Aloe-emodin inhibits osteogenic differentiation and calcification of mouse vascular smooth muscle cells. Eur J Pharmacol. (2019) 865:172772. doi: 10.1016/j.ejphar.2019.172772
120. Chinetti-Gbaguidi G, Daoudi M, Rosa M, Vinod M, Louvet L, Copin C, et al. Human alternative macrophages populate calcified areas of atherosclerotic lesions and display impaired RANKL-induced osteoclastic bone resorption activity. Circ Res. (2017) 121:19–30. doi: 10.1161/CIRCRESAHA.116.310262
121. Guo Y, Bao S, Guo W, Diao Z, Wang L, Han X, et al. Bone marrow mesenchymal stem cell-derived exosomes alleviate high phosphorus-induced vascular smooth muscle cells calcification by modifying microRNA profiles. Funct Integr Genomics. (2019) 19:633–43. doi: 10.1007/s10142-019-00669-0
122. Zhu M, Fang X, Zhou S, Li W, Guan S. Indirect coculture of vascular smooth muscle cells with bone marrow mesenchymal stem cells inhibits vascular calcification and downregulates the Wnt signaling pathways. Mol Med Rep. (2016) 13:5141–8. doi: 10.3892/mmr.2016.5182
123. Yokote S, Katsuoka Y, Yamada A, Ohkido I, Yokoo T. Effect of adipose-derived mesenchymal stem cell transplantation on vascular calcification in rats with adenine-induced kidney disease. Sci Rep. (2017) 7:14036. doi: 10.1038/s41598-017-14492-9
Keywords: vascular calcification, osteoblast-like cells, osteoclast-like cells, origin, reversibe, cell therapy
Citation: Jiang W, Zhang Z, Li Y, Chen C, Yang H, Lin Q, Hu M and Qin X (2021) The Cell Origin and Role of Osteoclastogenesis and Osteoblastogenesis in Vascular Calcification. Front. Cardiovasc. Med. 8:639740. doi: 10.3389/fcvm.2021.639740
Received: 09 December 2020; Accepted: 24 March 2021;
Published: 23 April 2021.
Edited by:
Hiroshi Iwata, Juntendo University, JapanReviewed by:
Joshua D. Hutcheson, Florida International University, United StatesDaiju Fukuda, Tokushima University, Japan
Copyright © 2021 Jiang, Zhang, Li, Chen, Yang, Lin, Hu and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Qin, ZHJfcWlueGlhb0Bob3RtYWlsLmNvbQ==