 Francesca Graziani
Francesca Graziani Rosa Lillo1
Rosa Lillo1- 1Department of Cardiovascular and Thoracic Sciences, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
- 2Catholic University of the Sacred Heart, Rome, Italy
Heart failure with preserved ejection fraction (HFpEF) is a major public health problem with growing prevalence and poor outcomes, mainly due to the lack of an effective treatment. HFpEF pathophysiology is heterogeneous and complex. Recently a “new paradigm” has been proposed, suggesting that cardiovascular and non-cardiovascular coexisting comorbidities lead to a systemic inflammatory state, perturbing the physiology of the endothelium and the perivascular environment and engaging molecular pathways that ultimately converge to myocardial fibrosis. If inflammation and fibrosis are the “fil rouge” in the heterogeneous spectrum of HFpEF, anti-fibrotic and anti-inflammatory drugs may have a role in its treatment. Pirfenidone is an orally bioavailable drug with antifibrotic and anti-inflammatory properties already approved for the treatment of idiopathic pulmonary fibrosis. Pirfenidone has been recently tested in animal models of myocardial fibrosis with promising results. Here we will review the rationale underlying the potential therapeutic effect of Pirfenidone in HFpEF.
Introduction
Heart failure with preserved ejection fraction (HFpEF) is a clinical syndrome characterized by typical symptoms and signs of heart failure (HF) with normal or near-normal left ventricular ejection fraction (LV EF ≥ 50%), echocardiographic features of diastolic dysfunction and/or structural heart disease and elevation of natriuretic peptides (1). The prevalence of HFpEF has grown worldwide and it now represents the dominant form of HF, affecting roughly 5% of the general population aged > 60 years. Contributing factors to this phenomenon are the improvement in diagnostic tools together with the greater clinical awareness and the increase in life expectancy (2). HFpEF patients experience rates of hospitalization, functional decline, and mortality similar to patients with HF and reduced ejection fraction (HFrEF) (3), imposing major economic health care burden. Unlike the established efficacy of several drugs in HFrEF, no specific therapy has yet proven to significantly impact on morbidity and mortality in HFpEF and the current treatment remains “empiric” and mostly symptomatic (4). This unmet need is at least partially explained by the complex and heterogeneous pathophysiology underlying the clinical spectrum of HFpEF. Recently, cardiac fibrosis and microvascular inflammation have emerged as the “fil rouge” in the conundrum of HFpEF. Myocardial fibrosis precedes the clinical diagnosis of HFpEF and is strongly associated with disease severity and adverse outcomes (5, 6).
Based on these evidences, a mechanistic overlap between HFpEF and other fibrotic diseases, such as idiopathic pulmonary fibrosis (IPF) seems likely (7). Pirfenidone is an oral anti-fibrotic drug (with also anti-oxidant and anti-inflammatory effects) approved for clinical use in IPF and it can lead to regression of myocardial fibrosis in animal models. We will discuss the rationale underlying the potential therapeutic effect of Pirfenidone in HFpEF.
Pathophysiology of HFpEF
HFpEF is a systemic syndrome, driven by accumulated risk factors and comorbidities, which, in vulnerable subjects, trigger pathways leading to increased ventricular stiffness, diastolic dysfunction and abnormal ventricular-arterial coupling (8, 9). The underlying mechanisms of diastolic dysfunction are impaired cardiomyocyte relaxation and increased extracellular stiffness, leading to preservation of LV stroke volume at cost of an increase of LV filling pressure (10). Neuro-hormonal system then activates and promotes salt and water retention in the kidney. Over time, the increased circulating volume and high levels of Angiotensin II and aldosterone trigger a maladaptive vicious circle, increasing ventricular stretch, oncostatic pressure in the lungs and peripheries and exerting a pro-hypertrophic and pro-fibrotic effect within the myocardium (11). The original description of HFpEF relied on the relation between arterial hypertension and diastolic dysfunction, with high afterload as trigger of ventricular remodelling and diastolic failure (2). Nowadays it is well-known that HFpEF is a complex disease, and the paradigm of “increased afterload model” is no longer valid.
Comorbidity-Driven Microvascular Inflammation Theory in HFpEF
The classic risk factors for developing HFpEF include age, female gender, hypertension, diabetes, overweight/obesity, renal dysfunction, metabolic syndrome (present in the 85% of patients) and physical inactivity (12, 13). According to the “new paradigm” for HFpEF (9) comorbidities induce a systemic pro-inflammatory state, which causes endothelial dysfunction and coronary microvascular dysfunction (CMD) (Figure 1). Under the effect of persistent pro-inflammatory stimuli (suggested by the elevation of circulating inflammatory biomarkers such as IL-1RL1, C-reactive protein, GDF15, TNF-α, sST-2, pentraxin-3, etc.), coronary microvascular endothelium recruits monocytes and Th1 cells through the production of adhesion molecules (14). These inflammatory cells express transforming growth factor β (TGF-β), interferon-ɤ, Galectin-3 (Gal-3), connective tissue growth factor and angiotensin-converting enzymes, promoting the conversion of fibroblasts to myofibroblasts and collagen deposition (9), with TGF-β playing a pivotal role. Moreover, the microvascular endothelial inflammation enhances oxidative stress. This leads to increased reactive oxygen species (ROS) production which perturb nitric oxide (NO) metabolism decreasing its bioavailability, reducing cyclic guanosine monophosphate (cGMP) content and protein kinase G (PKG) activity in adjacent cardiomyocytes, altering the phosphorylation state of sarcomeric proteins and the calcium handling, thus adversely affecting the cardiomyocyte and inducing hypertrophy. This results in increased myocardial stiffness, impaired energetic metabolism and a pro-fibrotic, pro-inflammatory secretome, which contributes to and perpetuates the haemodynamic changes of HFpEF. Histological studies on LV endomyocardial biopsy samples from HFpEF patients showed high level of expression of inflammatory endothelial adhesion molecules including VCAM1, high numbers of CD3, CD11, and CD45-positive leucocytes in the myocardium, increased expression of TGF-β in inflammatory cells and increased levels of collagen I and III (14). Hage et al. (15) found that myeloperoxidase-dependent oxidative stress, reflected by uric acid and calprotectin, is increased in HFpEF patients, suggesting microvascular neutrophil involvement mirroring endothelial dysfunction as a central component of the HFpEF syndrome. Moreover, Pentraxin 3, a biomarker of inflammation, was found to be significantly elevated in HFpEF patients and its levels at the coronary sinus significantly higher than at the aortic root, suggesting a production in the coronary circulation in patients with LV diastolic dysfunction (16). Interestingly, the systemic inflammatory state seems not only to play a pivotal pathophysiological role, but also to have prognostic implications. Levels of Gal-3, a marker of myocardial fibrosis, inversely correlate with functional capacity and its increase over time is associated with a higher risk of death or hospitalization (17). Shah et al. demonstrated that soluble ST2 (member of the IL-1 receptor family) is a strong predictor of mortality in patients presenting with acute dyspnoea and preserved EF (18). Plasma levels of Neopterin, a molecule mainly secreted by activated macrophages, are significantly increased in HFpEF and correlate with the severity of HF and with future cardiovascular events (19).
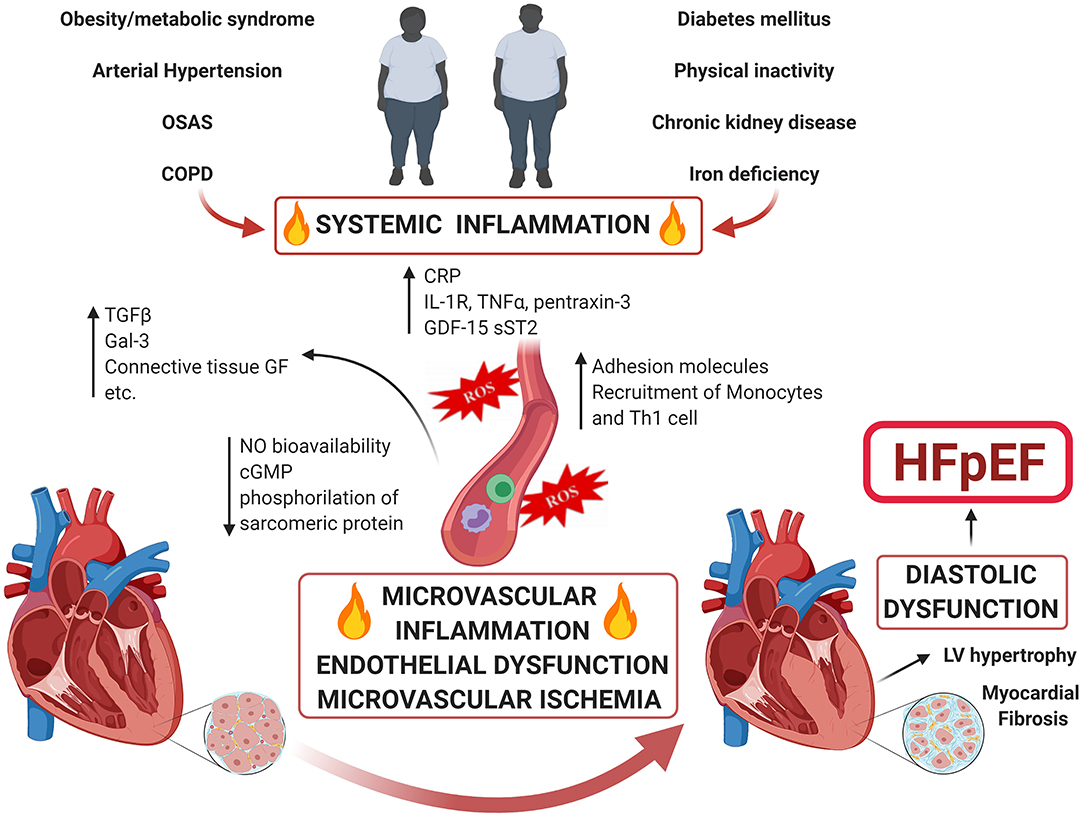
Figure 1. Comorbidity-Driven Microvascular Inflammation Theory in HFpEF. Accumulated risk factors as well as cardiac and non-cardiac comorbidities lead to a systemic inflammatory state and coronary microvascular inflammation. The endothelial dysfunction and perturbation of the physiology of the perivascular environment engage molecular pathways that ultimately converge to microvascular dysfunction and myocardial fibrosis causing HFpEF.
In acute HFpEF, changes in levels of inflammatory markers such as pentraxin-3, TNF- receptor1a, myeloperoxidase, and lymphotoxin β receptor are correlated to outcome (20). The “new HFpEF paradigm theory” found support also from the results of a proteomic analysis (21): Sanders-van Wijk et al. demonstrated that, across two independent cohorts of HFpEF patients, comorbidity burden was associated with abnormal cardiac structure and function and with increased systemic inflammation, which was associated with worse cardiac function and was upregulated in HFpEF as compared to non-HF controls with comorbidities; importantly, inflammation also appeared to mediate the association between comorbidity burden and worse cardiac haemodynamic.
Coronary Microvascular Dysfunction and HFpEF
The role of CMD at the basis of HFpEF pathogenesis and evolution has gained growing consent over years (2). CMD is determined by a variable combination of endothelial dysfunction, vascular smooth muscle cell hyper-reactivity, vascular remodelling, fibrosis and rarefaction, and increased extravascular pressure (22). A cross-talk between the endothelium and the surrounding vascular tissue and architecture, as well as the myocardium, seems to play a key role in the pathogenesis of HFpEF. We proposed an innovative theory that identifies CMD as the “common soil” for the occurrence of both microvascular angina (MVA) and HFpEF. Possible modulating factors may determine an effect in one direction or the other. Beyond molecular mediators, an additional potential mechanism, as suggested by Pepine et al. (23) involves recurrent cycles of ischaemia-reperfusion that affect myocyte relaxation leading to diastolic dysfunction and HFpEF (24). In turn, the increased intra-myocardial pressure can enhance myocardial ischemia increasing myocardial oxygen consumption: this can explain the coexistence of a “vicious circle” with subclinical ischaemia directly contributing to the pathogenesis of HFpEF. In support of these considerations, an autoptic analysis of 124 hearts of HFpEF patients demonstrated an inverse relationship between microvascular density and myocardial fibrosis (25). Moreover, CMD is associated with higher left-sided cardiac filling pressures at rest, with this relationship even more pronounced during exercise.
In the multi-national PRevalence Of MIcrovascular dySfunction in Heart Failure with Preserved Ejection Fraction (PROMIS-HFpEF) study (26), CMD has been documented in 75% of HFpEF patients. In the exploratory assessment of the prognosis at 1 year of the PROMIS study population, Hage et al. interestingly found that coexistence of CMD with HFpEF has prognostic implications, being CMD associated with higher incidence rates of CV death/recurrent HF hospitalizations, all-cause death/first HF, and recurrent but not first all-cause hospitalization (27). Taqueti et al. (28), in a study on symptomatic patients without flow-limiting epicardial coronary artery disease, found that impaired coronary flow reserve (CFR) was independently associated with diastolic dysfunction and adverse events, especially HFpEF events: patients with both diastolic dysfunction and impaired CFR demonstrated a > 5-fold increased risk of HFpEF hospitalization, providing evidence that CMD, alongside myocardial stiffness, may play an important role in the pathophysiology of HFpEF.
Myocardial Fibrosis and HFpEF
Myocardial fibrosis is an endogenous response to different cardiac insults that may become maladaptive over time and contribute to the onset and progression of HF. ECM expansion, secondary to excess collagen accumulation, is considered a key pathophysiological mechanism of HFpEF, a common pathway that exists regardless of aetiology. There is strong evidence demonstrating both the primary aetiological role of myocardial fibrosis in HFpEF, but also the adverse impact that ECM expansion has on myocardial mechanical, electrical and microvascular function, confirming that the myocardial fibrotic burden is strongly and independently associated with adverse outcome (29–33). However, there are some conflicting data on the topic, with histological and imaging studies showing that approximately one-third to one-half of HFpEF patients can have normal measures of myocardial fibrosis (34).
Interestingly, myocardial and pulmonary fibrosis share some characteristics and molecular mediators, with TGF-β and Ang II being the major regulating factors. The activation of the AT1 receptor in fibroblasts by Ang II leads to the secretion of TGF-β, which stimulates fibroblast proliferation and ECM synthesis (fibronectin, proteoglycans, and type I–III collagen) in an autocrine manner and induces cardiomyocytes hypertrophy in a paracrine manner (35). TGF-β then activates downstream effectors including smad-dependent and independent signalling pathways. Several studies have also demonstrated the role of other molecular mediators of both cardiac and pulmonary fibrosis, such as sirtuins (Sirt1, Sirt3, Sirt6, Sirt7), MMPs (MMP-9, MMP-13), microRNAs (miRNA 26, miRNA 29) and others (36–47). Interestingly, Cunningham et al. found that circulating biomarkers reflecting mechanisms of ECM homeostasis (sST2), collagen synthesis (PINP, PIIINP), and collagen degradation and turnover (TIMP-1, CITP) are abnormal in patients with HFpEF (48). In such a scenario, it is reasonable to encourage studies aiming to test the potential beneficial effect of anti-fibrotic therapeutic approach in HFpEF patients, learning from the solid experience of treatment for IPF (7).
Treatment of HFpEF: Knowledge Gaps and Future Perspectives
To date, unlike HFrEF, HFpEF is still orphan of treatments proven to significantly reduce major CV events. Beta-blockers, angiotensin-converting enzyme inhibitors, Ang-II receptor blockers, mineralocorticoid receptor antagonists and angiotensin receptor-neprilysin inhibitors have all failed to reach the pre-specified primary endpoints in trials testing their effects on CV outcomes (Table 1) (49–71), although some have shown improvements in their secondary endpoints.
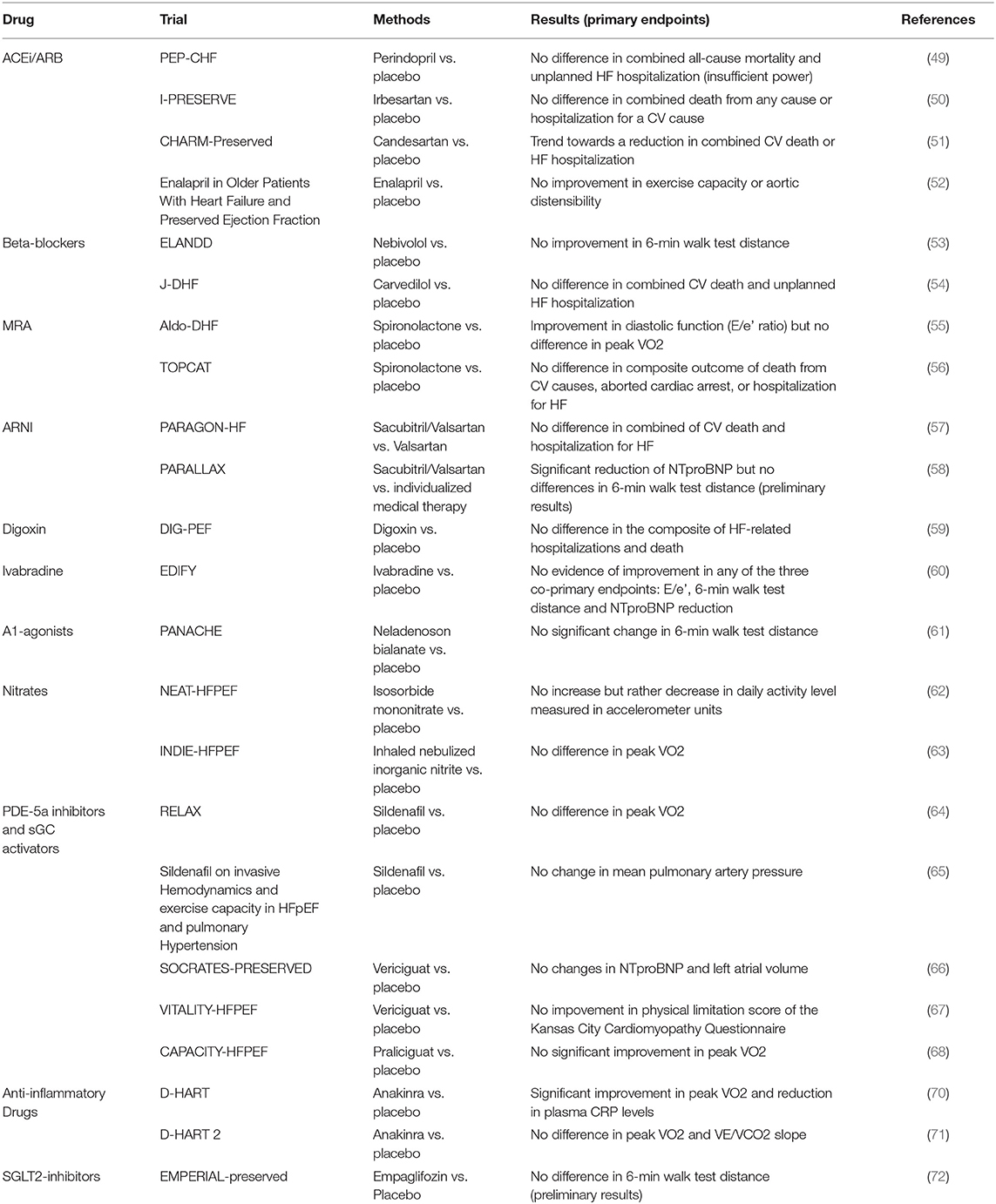
Table 1. Clinical trials of pharmacological therapies for heart failure with preserved ejection fraction.
Over the last years, the new insights on HFpEF pathophysiology have increased the interest in testing new drugs specifically targeting the molecular mediators involved in HFpEF. Given the role of the oxidative stress and NO metabolism perturbation, it seemed reasonable to propose organic NO donors as potentially useful therapeutic tools. Unfortunately, results from the early studies are at best inconclusive and in some patients, paradoxically, a tendency to reduce the total physical activity was observed (62, 63). However, new trials of oral nitrite and nitrate are currently ongoing. Conflicting data were also reported about Phosphodiesterases-5a (PDE-5a) inhibitors (64, 65). A possible explanation of these unsatisfactory results lies on an inadequate production of endogenous cGMP rather than excessive breakdown by PDE-5 (active synthesis of NO is required). This has led to therapeutic strategies specifically targeting the soluble guanylate cyclase (sGC) using direct sGC stimulators that can increase cGMP production through NO-independent pathways. Vericiguat is a stimulator of sGC tested in the SOCRATES-HFpEF trial (66), the pre-specified primary end-point was the change of NT-proBNP levels or left atrium volume over a 12-week treatment period and the trial failed. Recent data from the VITALITY-HFpEF randomized placebo-controlled trial showed that 24-week treatment with Vericiguat compared with placebo did not improve the physical limitation score (67). Similarly, Praliciguat did not improve significantly the Peak Rate of Oxygen Consumption, thus not supporting its use in patients with HFpEF (CAPACITY HFpEF trial) (68).
Drugs specifically targeting inflammation have been tested in HFpEF: in animal models chemokine antagonists (antiMCP1, MCP3) and immuno-modulatory cytokines (interleukin IL-10, pentraxins, and IL-1b blockade) showed promising results (69). Anakinra, a recombinant IL-1 receptor antagonist, reduced C-reactive protein levels and improved exercise capacity in a crossover trial of 12 patients with HFpEF and elevated C-reactive protein (70). Data from the DHART2 trial, however, showed that Anakinra failed to improve aerobic exercise capacity or ventilatory efficiency in patients with HFpEF, even if high-sensitivity C-reactive protein and NT-proBNP levels were lower after treatment compared with baseline (71).
The sodium–glucose cotransporter 2 (SGLT2) inhibitors (dapaglifozin, empaglifozin, and canaglifozin) were found to reduce hospitalizations as well as mortality in HF (73, 74), but nowadays not conclusive data are available on the effect of these drugs in HFpEF (72) and current evidence does not support a widespread use of these drugs in non-diabetic subjects. In a preclinical study performed in a novel co-culture system combined with a high-throughput analysis of cardiomyocyte function (75) cardiac microvascular endothelial cells exerted a direct positive effect on cardiomyocyte contraction and relaxation, mainly mediated by endothelial-derived NO. This effect is lost after pre-incubation of cardiac microvascular endothelial cells with TNF-α and can be restored with empagliflozin, which leads to restoration of endothelial NO bioavailability. In a study on non-diabetic rat models with HFpEF treated with dapagliflozin this drug ameliorated diastolic function, reversed endothelial activation and endothelial nitric oxide synthase deficit, reducing cardiac inflammation and attenuating pro-fibrotic signalling pathways. The potential involvement of coronary endothelium was supported by the endothelial upregulation of Na+/H+ exchanger 1 in vivo and direct effects on dapagliflozin on the activity of this exchanger in endothelial cells demonstrated in vitro (76). These data are promising for future investigations.
Due to the key pathophysiological role of myocardial fibrosis in the development and progression of HFpEF, there is a growing interest about the potential beneficial effects of anti-fibrotic drugs, commonly used in other fibrotic disease such as IPF, also in HFpEF such as Pirfenidone.
Pirfenidone: Pharmacokinetics, Safety Profile, Mechanism of Action
Pirfenidone is an orally bioavailable small synthetic molecule, with proven anti-inflammatory and anti-fibrotic properties and it is authorized by the European Commission for the treatment of adults with IPF (77).
Pirfenidone is rapidly absorbed in the gastrointestinal tract and its half-life is about 3 h (78, 79). It is metabolized in the liver (mainly by CYP1A2) and is mostly excreted as the metabolite 5-carboxy-pirfenidone, by 80% through the urine and by 20% through intestinal elimination. This explains why creatinine clearance <50 mL/min and mild-to-moderate liver dysfunction are relative contraindications to Pirfenidone (78, 79). In the Pirfenidone safety study (PASSPORT) (80) the most frequent side effects documented were nausea and fatigue, gastrointestinal disturbances, skin rash and photosensitivity reactions; serious side effects were rare, with fatal outcome observed in <1%.
The precise mechanism of action of this drug remains still unclear (77). Pirfenidone attenuates fibroblast proliferation, production of fibrosis-associated proteins (TGF-β, platelet-derived growth factor and β fibroblast growth factor) and cytokines (interleukin-1β and tumour necrosis factor-α), and reduces the increased biosynthesis and accumulation of extracellular matrix in response to pro-fibrotic mediators (i.e., TGF-β); it also blocks the proliferation and differentiation of fibroblasts into myofibroblasts by inhibiting several targets of TGF-β (Smad3, p38, Akt42), improves mitochondrial function and modulates lymphocyte activation (79). Pirfenidone has proven clinical effectiveness in IPF. Given the molecular overlap between pro-fibrotic pathways in lung and heart disease and the pleiotropic effects of Pirfenidone, this drug is being considered with increasingly interest as a potential treatment for cardiac disorders (7, 79) (Figure 2).
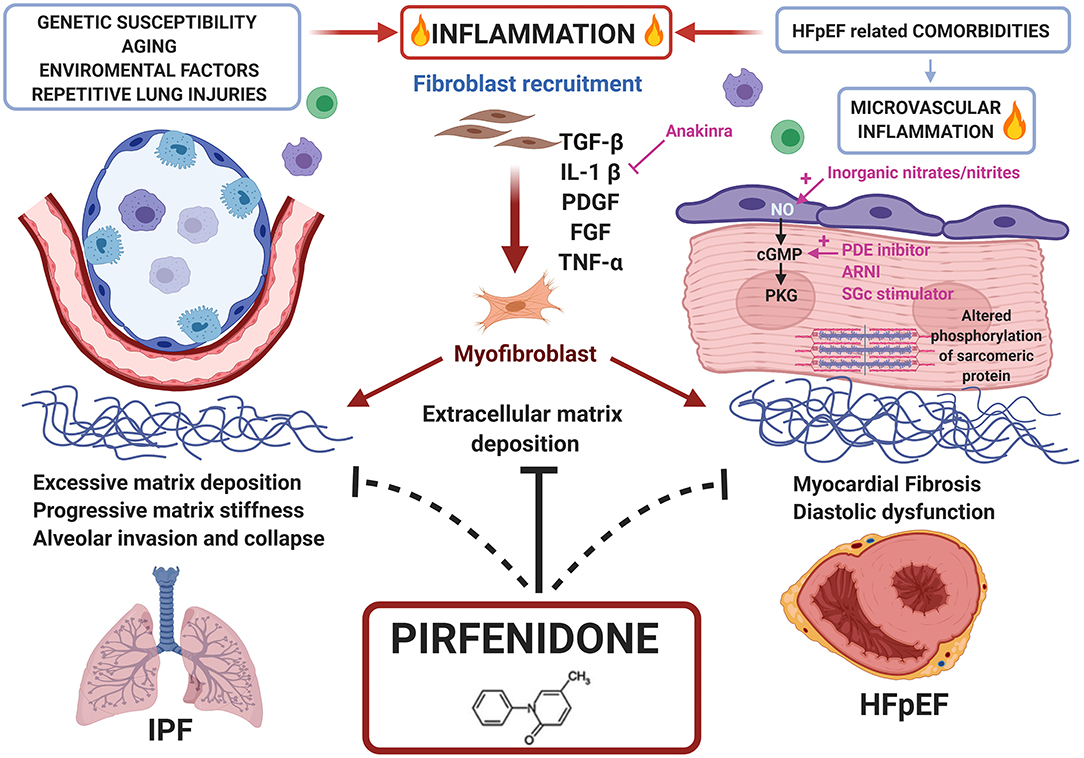
Figure 2. Common pathways between HFpEF and IPF and potential role of Pirfenidone as treatment for both diseases. Repetitive lung injuries over a genetically susceptible alveolar epithelium, activates inflammatory pathways and the overproduction of pro-fibrotic mediators like transforming growth factor (TGF)-β, enhancing fibroblast recruitment, and conversion to myofibroblast. Similarly, HFpEF comorbidities trigger microvascular inflammation converging to myocardial fibrosis. Pirfenidone, an antifibrotic and anti-inflammatory drug approved for clinical use in IPF, may be proposed for HFpEF treatment.
Pirfenidone and Myocardial Fibrosis: What Do We Know?
The available data on cardio-protective effects of Pirfenidone are still in a preclinical phase, but there are several evidences pointing to that direction (79).
Pirfenidone, in a dose- and time-dependent manner, reduces cardiac fibroblasts migratory ability, inhibits their proliferation and the process of myofibroblast differentiation (by inhibition of α-SMA expression) as well as the myocardial fibroblast synthesis and secretion of TGF-β1. Pirfenidone also regularizes ratios of myocardial MMPs and tissue inhibitors of metalloproteinases, enhancing myocardial renin-angiotensin system imbalance and cardiac fibroblast synthesis and secretion of IL-10, an anti-fibrotic cytokine (81–88).
In hypertensive mouse models (87), Pirfenidone showed to reverse and prevent cardiac remodelling and the increased cardiac stiffness. Similarly, Yamazaki et al. (88) found that Pirfenidone can prevent the progression of Ang II-induced cardiac hypertrophy and fibrosis, and Yamagami et al. (89) demonstrated that myocardial inflammation was alleviated in mice exposed to transverse aortic constriction. The Pirfenidone power to reduce cardiac fibrosis has been documented in streptozotocin-induced diabetes mice (86) and rats receiving intraperitoneal injections of doxorubicin too (83). In dog models (90) with HF induced by high-frequency left ventricular pacing, Pirfenidone showed a protective effect, preventing fibrosis of the atrial myocardial tissue. In a rat model (91) of myocardial infarction, Pirfenidone decreased scar size and myocardial fibrosis in the border zone, improving left ventricular systolic function, and reduced ventricular tachycardia susceptibility, suggesting a potential role of this drug also in this setting.
Pirfenidone and HFpEF: A Current Gap of Knowledge
To date, no data on Pifenidone effect on human myocardial fibrosis are available. Despite the prominent role of fibrosis in the pathophysiology of several cardiac disorders, and the evidence of the safety of therapy with Pifenidone, only a single study on the use of Pirfenidone for a cardiac condition has been started, the Efficacy and Safety of Pirfenidone in Patients With Heart Failure and Preserved Left Ventricular Ejection Fraction (PIROUETTE) trial (92). This is a randomised, double-blind, placebo-controlled phase II trial evaluating the efficacy and safety of 52 weeks of treatment with Pirfenidone in patients with HFpEF and myocardial fibrosis (defined as extracellular matrix volume ≥ 27% measured with cardiovascular magnetic resonance). The primary outcome of the study is the change in myocardial ECM volume. A sub-study will also investigate the relationship between myocardial fibrosis and myocardial energetics, and the related impact of Pifenidone. The trial is still ongoing and there is a growing interest about its results, it could lay the foundation for the improvement of outcome in HFpEF patients.
In humans, only two retrospective studies have investigated the effect of Pirfenidone on LV structure and function in patients with IPF treated with this drug.
In the first one, Alansari et al. (93) hypothesized that Pirfenidone could have had a more favourable effect on changes in echocardiographic parameters of LV structure and function in IPF patients responder compared to non-responders (defined as an absolute decline in forced vital capacity of more than 10% whilst being on the medication). After treatment, no significant differences in changes of echocardiographic parameters of LV structure, diastolic function, systolic function and GLS were observed between the two groups. In the second one (94), the same authors found that treatment with Pirfenidone was associated with decreases in indexed LV end diastolic and end systolic volumes. However, no improvements were noted in markers of LV diastolic, systolic function and strain. Both studies, however, were retrospective and meaningfully limited by their small sample size (27 and 24 subjects, respectively), while large and specifically designed prospective trials are needed to test properly the efficacy of Pirfenidone in HFpEF.
If cardiac fibrosis and microvascular inflammation are the common pathophysiological substrates in this conundrum, Pirfenidone could have a powerful role in the treatment of all the different HFpEF subtypes, targeting the “core mechanisms” they all share. However, differences in timing of fibrogenesis and in fibrotic burden in each HFpEF-related comorbidity are still poorly understood, being influenced by many co-factors. Nowadays circulating biomarkers are considered a powerful tool to depict the patients pro-inflammatory and pro-fibrotic profile. Moreover, CMR offers the unique possibility to non-invasively estimate the amount of myocardial fibrosis. We can speculate that a “risk stratification” strategy, using inflammation and fibrogenesis biomarkers and CMR, might identify patients who will benefit most from Pirfenidone: those showing an intense inflammatory and fibrogenetic activation before the evolution toward an advanced, irreversible, stage of diffuse myocardial fibrosis. Trials specifically testing Pirfenidone effects on patients with this profile might help to optimize treatment in HFpEF.
Conclusions
Up to now, HFpEF treatment has been borrowed from the HFrEF experience, with poor results. The complex and not completely understood HFpEF pathophysiology is probably the key to develop a tailored effective treatment. The new HFpEF paradigm states that the coronary microvascular endothelial inflammation is the main driving factor, activating complex molecular pathways that eventually converge to myocardial fibrosis. Coronary microvascular inflammation and myocardial fibrosis can be considered the fil rouge in the HFpEF conundrum, thus they can be considered reasonable targets treatment. Pirfenidone is a well-established drug for the treatment of IPF and in animal studies it showed its anti-inflammatory properties and its ability to reverse cardiac fibrosis. Taken together these data suggest that Pirfenidone could have a role in the treatment of HFpEF by targeting inflammation and myocardial fibrosis, however, at present, clinical trials are lacking. Large, specifically-designed studies with hard end-point in this setting are needed.
Author Contributions
FG and RL wrote sections of the manuscript. FC contributed to manuscript revision. All authors read and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis treatment of acute chronic heart failure: The Task Force for the diagnosis treatment of acute chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. (2016) 18:891–975. doi: 10.1093/eurheartj/ehw128
2. D'Amario D, Migliaro S, Borovac JA, Restivo A, Vergallo R, Galli M, et al. Microvascular dysfunction in heart failure with preserved ejection fraction. Front Physiol. (2019) 10:1347. doi: 10.3389/fphys.2019.01347
3. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. (2006) 355:251–9. doi: 10.1056/NEJMoa052256
4. Pfeffer MA, Shah AM, Borlaug BA. Heart failure with preserved ejection fraction in perspective. Circ Res. (2019) 124:1598–617. doi: 10.1161/CIRCRESAHA.119.313572
5. Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. (2002) 27:92–8. doi: 10.1007/s00059-002-2354-y
6. Zile MR, Baicu CF. Biomarkers of diastolic dysfunction and myocardial fibrosis: application to heart failure with a preserved ejection fraction. J Cardiovasc Transl Res. (2013) 6:501–15. doi: 10.1007/s12265-013-9472-1
7. Graziani F, Varone F, Crea F, Richeldi L. Treating heart failure with preserved ejection fraction: learning from pulmonary fibrosis. Eur J Heart Fail. (2018) 20:1385–91. doi: 10.1002/ejhf.1286
8. Jaroch J, Łoboz-Grudzień K. Novel paradigms for heart failure with preserved ejection fraction. Przegl Lek. (2016) 73:413–8.
9. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
10. Westermann D, Kasner M, Steendijk P, Spillmann F, Riad A, Weitmann K, et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation. (2008) 117:2051–60. doi: 10.1161/CIRCULATIONAHA.107.716886
11. Díez J. Profibrotic effects of angiotensin II in the heart: a matter of mediators. Hypertension. (2004) 43:1164–5. doi: 10.1161/01.HYP.0000128620.57061.67
12. Ammar KA, Redfield MM, Mahoney DW, Johnson M, Jacobsen SJ, Rodeheffer RJ. Central obesity: association with left ventricular dysfunction and mortality in the community. Am Heart J. (2008) 156:975–81. doi: 10.1016/j.ahj.2008.06.018
13. Redfield MM, Jacobsen SJ, Burnett JC Jr, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. (2003) 289:194–202. doi: 10.1001/jama.289.2.194
14. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. (2011) 4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451
15. Hage C, Michaëlsson E, Kull B, Miliotis T, Svedlund S, Linde C, et al. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. (2020) 7:1534–46. doi: 10.1002/ehf2.12700
16. Matsubara J, Sugiyama S, Nozaki T, Sugamura K, Konishi M, Ohba K, et al. Pentraxin 3 is a new inflammatory marker correlated with left ventricular diastolic dysfunction and heart failure with normal ejection fraction. J Am Coll Cardiol. (2011) 57:861–9. doi: 10.1016/j.jacc.2010.10.018
17. Edelmann F, Holzendorf V, Wachter R, Nolte K, Schmidt AG, Kraigher-Krainer E, et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail. (2015) 17:214–23. doi: 10.1002/ejhf.203
18. Shah KB, Kop WJ, Christenson RH, Diercks DB, Henderson S, Hanson K, et al. Prognostic utility of ST2 in patients with acute dyspnea and preserved left ventricular ejection fraction. Clin Chem. (2011) 57:874–82. doi: 10.1373/clinchem.2010.159277
19. Yamamoto E, Hirata Y, Tokitsu T, Kusaka H, Tabata N, Tsujita K, et al. The clinical significance of plasma neopterin in heart failure with preserved left ventricular ejection fraction. ESC Heart Fail. (2016) 3:53–9. doi: 10.1002/ehf2.12070
20. Tromp J, Khan MAF, Mentz RJ, O'connor CM, Metra M, Dittrich HC, et al. Biomarker profiles of acute heart failure patients with a mid-range ejection fraction. JACC Heart Fail. (2017) 5:507–17. doi: 10.1016/j.jchf.2017.04.007
21. Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, et al. Proteomic evaluation of the comorbidity-inflammation paradigm in heart failure with preserved ejection fraction: results from the PROMIS-HFpEF study. Circulation. (2020) 142:2029–44. doi: 10.1161/CIRCULATIONAHA.120.045810
22. Crea F, Bairey Merz CN, Beltrame JF, Kaski JC, Ogawa H, Ong P, et al. Coronary vasomotion disorders international study group (COVADIS). The parallel tales of microvascular angina and heart failure with preserved ejection fraction: a paradigm shift. Eur Heart J. (2017) 38:473–7. doi: 10.1093/eurheartj/ehw461
23. Pepine CJ, Petersen JW, Bairey Merz CN. A microvascular-myocardial diastolic dysfunctional state and risk for mental stress ischemia. A revised concept of ischemia during daily life. JACC Cardiovasc Imaging. (2014) 7:362–5. doi: 10.1016/j.jcmg.2013.11.009
24. Ikonomidis I, Tzortzis S, Triantafyllidi H, Parissis J, Papadopoulos C, Venetsanou K, et al. Association of impaired left ventricular twisting-untwisting with vascular dysfunction, neurohumoral activation and impaired exercise capacity in hypertensive heart disease. Eur J Heart Fail. (2015) 17:1240–51. doi: 10.1002/ejhf.403
25. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. (2015) 131:550–9. doi: 10.1161/CIRCULATIONAHA.114.009625
26. Shah SJ, Lam CSP, Svedlund S, Saraste A, Hage C, Tan RS, et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. (2018) 39:3439–50. doi: 10.1093/eurheartj/ehy531
27. Hage C, Svedlund S, Saraste A, Faxén UL, Benson L, Fermer ML, et al. Association of coronary microvascular dysfunction with heart failure hospitalizations and mortality in heart failure with preserved ejection fraction: a follow-up in the PROMIS-HFpEF study. J Card Fail. (2020) 26:1016–21. doi: 10.1016/j.cardfail.2020.08.010
28. Taqueti VR, Solomon SD, Shah AM, Desai AS, Groarke JD, Osborne MT, et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur Heart J. (2018) 39:840–9. doi: 10.1093/eurheartj/ehx721
29. Schelbert EB, Fridman Y, Wong TC, Abu Daya H, Piehler KM, Kadakkal A, et al. Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: association with baseline disease severity and subsequent outcome. JAMA Cardiol. (2017) 2:995–1006. doi: 10.1001/jamacardio.2017.2511
30. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. (2015) 131:1247–59. doi: 10.1161/CIRCULATIONAHA.114.013215
31. Rommel KP, von Roeder M, Latuscynski K, Oberueck C, Blazek S, Fengler K, et al. Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. (2016) 67:1815–25. doi: 10.1016/j.jacc.2016.02.018
32. Schelbert EB, Wong TC, Gheorghiade M. Think small and examine the constituents of left ventricular hypertrophy and heart failure: cardiomyocytes versus fibroblasts, collagen, and capillaries in the interstitium. J Am Heart Assoc. (2015) 4:e002491. doi: 10.1161/JAHA.115.002491
33. Roy C, Slimani A, de Meester C, Amzulescu M, Pasquet A, Vancraeynest D, et al. Associations and prognostic significance of diffuse myocardial fibrosis by cardiovascular magnetic resonance in heart failure with preserved ejection fraction. J Cardiovasc Magn Reson. (2018) 20:55. doi: 10.1186/s12968-018-0477-4
34. Borbély A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, et al. Cardiomyocyte stiffness in diastolic heart failure. Circulation. (2005) 111:774–81. doi: 10.1161/01.CIR.0000155257.33485.6D
35. Hara H, Takeda N, Komuro I. Pathophysiology and therapeutic potential of cardiac fibrosis. Inflamm Regen. (2017) 37:13. doi: 10.1186/s41232-017-0046-5
36. Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G, et al. NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol. (2013) 228:1433–42. doi: 10.1002/jcp.24296
37. Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen Y, et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther. (2014) 22:1122–33. doi: 10.1038/mt.2014.42
38. Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, et al. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. (2011) 45:287–94. doi: 10.1165/rcmb.2010-0323OC
39. Sassi Y, Avramopoulos P, Ramanujam D, Grüter L, Werfel S, Giosele S, et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat Commun. (2017) 8:1614. doi: 10.1038/s41467-017-01737-4
40. Wyman AE, Noor Z, Fishelevich R, Lockatell V, Shah NG, Todd NW, et al. Sirtuin 7 is decreased in pulmonary fibrosis and regulates the fibrotic phenotype of lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L945–58. doi: 10.1152/ajplung.00473.2016
41. Araki S, Izumiya Y, Rokutanda T, Ianni A, Hanatani S, Kimura Y, et al. Sirt7 contributes to myocardial tissue repair by maintaining transforming growth factor-β signaling pathway. Circulation. (2015) 132:1081–93. doi: 10.1161/CIRCULATIONAHA.114.014821
42. Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, et al. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol. (2016) 205:99–110. doi: 10.1016/j.ijcard.2015.12.008
43. Rong L, Wu J, Wang W, Zhao RP, Xu XW, Hu D. Sirt 1 activator attenuates thebleomycin-induced lung fibrosis in mice via inhibiting epithelial-to-mesenchymal transition (EMT). Eur Rev Med Pharmacol Sci. (2016) 20:2144–50.
44. Bindu S, Pillai VB, Kanwal A, Samant S, Mutlu GM, Verdin E, et al. SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L68–78. doi: 10.1152/ajplung.00188.2016
45. Guo X, Yan F, Shan X, Li J, Yang Y, Zhang J, et al. SIRT3 inhibits Ang II-induced transdifferentiation of cardiac fibroblasts through β-catenin/PPAR-γ signaling. Life Sci. (2017) 186:111–7. doi: 10.1016/j.lfs.2017.07.030
46. Zhang ZZ, Cheng YW, Jin HY, Chang Q, Shang QH, Xu YL, et al. The sirtuin 6 prevents angiotensin II-mediated myocardial fibrosis and injury by targeting AMPK-ACE2 signaling. Oncotarget. (2017) 8:72302–14. doi: 10.18632/oncotarget.20305
47. Tian K, Chen P, Liu Z, Si S, Zhang Q, Mou Y, et al. Sirtuin 6 inhibits epithelial to mesenchymal transition during idiopathic pulmonary fibrosis via inactivating TGF-β1/Smad3 signaling. Oncotarget. (2017) 8:61011–24. doi: 10.18632/oncotarget.17723
48. Cunningham JW, Claggett BL, O'Meara E, Prescott MF, Pfeffer MA, Shah SJ, et al. Effect of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFpEF. J Am Coll Cardiol. (2020) 76:503–14. doi: 10.1016/j.jacc.2020.05.072
49. Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J. PEP-CHF investigators. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. (2006) 27:2338–45. doi: 10.1093/eurheartj/ehl250
50. Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. I-PRESERVE investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. (2008) 359:2456–67. doi: 10.1056/NEJMoa0805450
51. Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, et al. CHARM investigators and committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. (2003) 362:777–81. doi: 10.1016/S0140-6736(03)14285-7
52. Kitzman DW, Hundley WG, Brubaker PH, Morgan TM, Moore JB, Stewart KP, et al. A randomized double-blind trial of enalapril in older patients with heart failure and preserved ejection fraction: effects on exercise tolerance and arterial distensibility. Circ Heart Fail. (2010) 3:477–85. doi: 10.1161/CIRCHEARTFAILURE.109.898916
53. Conraads VM, Metra M, Kamp O, De Keulenaer GW, Pieske B, Zamorano J, et al. Effects of the long-term administration of nebivolol on the clinical symptoms, exercise capacity, and left ventricular function of patients with diastolic dysfunction: results of the ELANDD study. Eur J Heart Fail. (2012) 14:219–25. doi: 10.1093/eurjhf/hfr161
54. Yamamoto K, Origasa H, Hori M, J-DHF Investigators. Effects of carvedilol on heart failure with preserved ejection fraction: the Japanese Diastolic Heart Failure Study (J-DHF). Eur J Heart Fail. (2013) 15:110–8. doi: 10.1093/eurjhf/hfs141
55. Edelmann F, Wachter R, Schmidt AG, Kraigher-Krainer E, Colantonio C, Kamke W, et al. Aldo-DHF Investigators. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA. (2013) 309:781–91. doi: 10.1001/jama.2013.905
56. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. (2014) 370:1383–92. doi: 10.1056/NEJMoa1313731
57. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. (2019) 381:1609–20. doi: 10.1056/NEJMoa1908655
58. Wachter R, Shah SJ, Cowie MR, Szecsödy P, Shi V, Ibram G, et al. Angiotensin receptor neprilysin inhibition versus individualized RAAS blockade: design and rationale of the PARALLAX trial. ESC Heart Fail. (2020) 7:856–64. doi: 10.1002/ehf2.12694
59. Ahmed A, Rich MW, Fleg JL, Zile MR, Young JB, Kitzman DW, et al. Effects of digoxin on morbidity and mortality in diastolic heart failure: the ancillary digitalis investigation group trial. Circulation. (2006) 114:397–403. doi: 10.1161/CIRCULATIONAHA.106.628347
60. Komajda M, Isnard R, Cohen-Solal A, Metra M, Pieske B, Ponikowski P, et al. prEserveD left ventricular ejectIon fraction chronic heart Failure with ivabradine studY (EDIFY) Investigators. Effect of ivabradine in patients with heart failure with preserved ejection fraction: the EDIFY randomized placebo-controlled trial. Eur J Heart Fail. (2017) 19:1495–503. doi: 10.1002/ejhf.876
61. Shah SJ, Voors AA, McMurray JJV, Kitzman DW, Viethen T, Bomfim Wirtz A, et al. Effect of neladenoson bialanate on exercise capacity among patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2019) 321:2101–12. doi: 10.1001/jama.2019.6717
62. Redfield MM, Anstrom KJ, Levine JA, Koepp GA, Borlaug BA, Chen HH, et al. Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med. (2015) 373:2314–24. doi: 10.1056/NEJMoa1510774
63. Borlaug BA, Anstrom KJ, Lewis GD, Shah SJ, Levine JA, Koepp GA, et al. Effect of inorganic nitrite vs placebo on exercise capacity among patients with heart failure with preserved ejection fraction: the INDIE-HFpEF randomized clinical trial. JAMA. (2018) 320:1764–73. doi: 10.1001/jama.2018.14852
64. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. RELAX trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2013) 309:1268–77. doi: 10.1001/jama.2013.2024
65. Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, et al. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. (2015) 36:2565–73. doi: 10.1093/eurheartj/ehv336
66. Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, Butler J, et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J. (2017) 38:1119–27. doi: 10.1093/eurheartj/ehw593
67. Armstrong PW, Lam CSP, Anstrom KJ, Ezekowitz J, Hernandez AF, O'Connor CM, et al. VITALITY-HFpEF study group. Effect of vericiguat vs placebo on quality of life in patients with heart failure and preserved ejection fraction: the VITALITY-HFpEF randomized clinical trial. JAMA. (2020) 324:1512–21. doi: 10.1001/jama.2020.15922
68. Udelson JE, Lewis GD, Shah SJ, Zile MR, Redfield MM, Burnett J Jr, et al. Effect of praliciguat on peak rate of oxygen consumption in patients with heart failure with preserved ejection fraction: the CAPACITY HFpEF randomized clinical trial. JAMA. (2020) 324:1522–31. doi: 10.1001/jama.2020.16641
69. Glezeva N, Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev. (2014) 19:681–94. doi: 10.1007/s10741-013-9405-8
70. Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. (2014) 113:321–7. doi: 10.1016/j.amjcard.2013.08.047
71. Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, et al. IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ Heart Fail. (2018) 11:e005036. doi: 10.1161/CIRCHEARTFAILURE.118.005036
72. Del Buono MG, Iannaccone G, Scacciavillani R, Carbone S, Camilli M, Niccoli G, et al. Heart failure with preserved ejection fraction diagnosis and treatment: an updated review of the evidence. Prog Cardiovasc Dis. (2020) 63:570–84. doi: 10.1016/j.pcad.2020.04.011
73. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
74. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
75. Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, et al. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl Sci. (2019) 4:575–91. doi: 10.1016/j.jacbts.2019.04.003
76. Cappetta D, De Angelis A, Ciuffreda LP, Coppini R, Cozzolino A, Miccichè A, et al. Amelioration of diastolic dysfunction by dapagliflozin in a non-diabetic model involves coronary endothelium. Pharmacol Res. (2020) 157:104781. doi: 10.1016/j.phrs.2020.104781
77. Richeldi L, Yasothan U, Kirkpatrick P. Pirfenidone. Nat Rev Drug Discov. (2011) 10:489–90. doi: 10.1038/nrd3495
78. Togami K, Kanehira Y, Tada H. Pharmacokinetic evaluation of tissue distribution of pirfenidone and its metabolites for idiopathic pulmonary fibrosis therapy. Biopharm Drug Dispos. (2015) 36:205–15. doi: 10.1002/bdd.1932
79. Aimo A, Cerbai E, Bartolucci G, Adamo L, Barison A, Lo Surdo G, et al. Pirfenidone is a cardioprotective drug: mechanisms of action and preclinical evidence. Pharmacol Res. (2020) 155:104694. doi: 10.1016/j.phrs.2020.104694
80. Cottin V, Koschel D, Günther A, Albera C, Azuma A, Sköld CM, et al. Long-term safety of pirfenidone: results of the prospective, observational PASSPORT study. ERJ Open Res. (2018) 4:00084–2018. doi: 10.1183/23120541.00084-2018
81. Shi Q, Liu X, Bai Y, Cui C, Li J, Li Y, et al. In vitro effects of pirfenidone on cardiac fibroblasts: proliferation, myofibroblast differentiation, migration and cytokine secretion. PLoS ONE. (2011) 6:e28134. doi: 10.1371/journal.pone.0028134
82. Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology. (2013) 126:1–11. doi: 10.1159/000351179
83. Giri SN, Al-Bayati MA, Du X, Schelegle E, Mohr FC, Margolin SB. Amelioration of doxorubicin-induced cardiac and renal toxicity by pirfenidone in rats. Cancer Chemother Pharmacol. (2004) 53:141–50. doi: 10.1007/s00280-003-0703-z
84. Van Erp C, Irwin NG, Hoey AJ. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve. (2006) 34:327–34. doi: 10.1002/mus.20590
85. Hisatomi K, Mukae H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S, et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm Med. (2012) 12:24. doi: 10.1186/1471-2466-12-24
86. Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol. (2001) 133:687–94. doi: 10.1038/sj.bjp.0704131
87. Mirkovic S, Seymour AM, Fenning A, Strachan A, Margolin SB, Taylor SM, et al. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol. (2002) 135:961–8. doi: 10.1038/sj.bjp.0704539
88. Yamazaki T, Yamashita N, Izumi Y, Nakamura Y, Shiota M, Hanatani A, et al. The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice. Hypertens Res. (2012) 35:34–40. doi: 10.1038/hr.2011.139
89. Yamagami K, Oka T, Wang Q, Ishizu T, Lee JK, Miwa K, et al. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am J Physiol Heart Circ Physiol. (2015) 309:H512–22. doi: 10.1152/ajpheart.00137.2015
90. Lee KW, Everett TH IV, Rahmutula D, Guerra JM, Wilson E, Ding C, et al. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. (2006) 114:1703–12. doi: 10.1161/CIRCULATIONAHA.106.624320
91. Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. (2010) 7:1438–45. doi: 10.1016/j.hrthm.2010.04.030
92. Lewis GA, Schelbert EB, Naish JH, Bedson E, Dodd S, Eccleson H, et al. Pirfenidone in heart failure with preserved ejection fraction-rationale and design of the PIROUETTE trial. Cardiovasc Drugs Ther. (2019) 33:461–70. doi: 10.1007/s10557-019-06876-y
93. Alansari S, Southern B, Riaz H, Sharma V, Borowski A, Tang W. Responses to pirfenidone treatment in patients with idiopathic pulmonary fibrosis is not associated with changes in echocardiographic parameters of left ventricular structure and function. J Card Fail. (2019) 25:S27. doi: 10.1016/j.cardfail.2019.07.074
Keywords: heart failure with preserved ejection fraction, pirfenidone, idiopatic pulmonary fibrosis, inflammation, heart failure
Citation: Graziani F, Lillo R and Crea F (2021) Rationale for the Use of Pirfenidone in Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 8:678530. doi: 10.3389/fcvm.2021.678530
Received: 09 March 2021; Accepted: 29 March 2021;
Published: 22 April 2021.
Edited by:
Alberto Aimo, Sant'Anna School of Advanced Studies, ItalyReviewed by:
Andrea Barison, Gabriele Monasterio Tuscany Foundation (CNR), ItalyValentina Barletta, Pisana University Hospital, Italy
Copyright © 2021 Graziani, Lillo and Crea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Graziani, ZnJhbmNlc2NhLmdyYXppYW5pQHBvbGljbGluaWNvZ2VtZWxsaS5pdA==