 Po-Yen Hsu
Po-Yen Hsu Laurent Désaubry
Laurent Désaubry Canan G. Nebigil
Canan G. Nebigil- INSERM UMR 1260, Regenerative Nanomedicine, University of Strasbourg, FMTS (Fédération de Médecine Translationnelle de l'Université de Strasbourg), Strasbourg, France
Vascular toxicity is a frequent adverse effect of current anticancer chemotherapies and often results from endothelial dysfunction. Vascular endothelial growth factor inhibitors (VEGFi), anthracyclines, plant alkaloids, alkylating agents, antimetabolites, and radiation therapy evoke vascular toxicity. These anticancer treatments not only affect tumor vascularization in a beneficial manner, they also damage ECs in the heart. Cardiac ECs have a vital role in cardiovascular functions including hemostasis, inflammatory and coagulation responses, vasculogenesis, and angiogenesis. EC damage can be resulted from capturing angiogenic factors, inhibiting EC proliferation, survival and signal transduction, or altering vascular tone. EC dysfunction accounts for the pathogenesis of myocardial infarction, atherothrombosis, microangiopathies, and hypertension. In this review, we provide a comprehensive overview of the effects of chemotherapeutic agents on vascular toxicity leading to hypertension, microvascular rarefaction thrombosis and atherosclerosis, and affecting drug delivery. We also describe the potential therapeutic approaches such as vascular endothelial growth factor (VEGF)-B and prokineticin receptor-1 agonists to maintain endothelial function during or following treatments with chemotherapeutic agents, without affecting anti-tumor effectiveness.
Introduction
Anticancer chemotherapies target the vasculature of both tumor and unfortunately other organs. Additionally, mechanism-independent (“off-target”) effects of chemotherapies also account for the development of the vascular toxicity. Vascular toxicity occurs during acute chemotherapeutic regimen, and after once treatments have ceased, persists into survival. The susceptibility to develop vascular complications following chemotherapeutics also relates to many factors such as cardiovascular risk and pre-existing vascular diseases, as well as genetic predispositions.
Chemotherapeutics-mediated vascular toxicity often results from loss of endothelial cell (EC) functions (1). ECs sense hemodynamic changes, and accordingly respond to stimuli by the release of vasoactive substances like vasorelaxants such as nitric oxide, (NO), prostacyclin, (PGI2), vasoconstrictors such as endothelin-1, (ET-1), anti-thrombotic (plasminogen activators), and angiogenic factors such as vascular endothelial growth factor (VEGF) (2) and prokineticins (3). Disturbance of NO/ET-1 balance is a characteristic of endothelial dysfunction and play an important role in the progression of vascular diseases.
Chemotherapeutics-mediated EC dysfunction in the heart is initially asymptomatic. The long-term consequences of cancer treatments can lead to the onset of cardiovascular disorders such as hypertension, coronary artery disease, and heart failure. Indeed, progressive EC damages make ECs more vulnerable to chronic inflammatory stressors and hyperlipidemia insults (4). EC dysfunction further promotes thrombus formation, and inflammation by releasing plasminogen activator inhibitor 1 (PAI1), platelet-activated factor 4 (PF-4), and interleukins (IL-1 and IL-6) to accelerate atherosclerosis formation. Chemotherapeutics can also have direct pro-coagulant, anti-angiogenesis, and vasoconstriction effects (Figure 1).
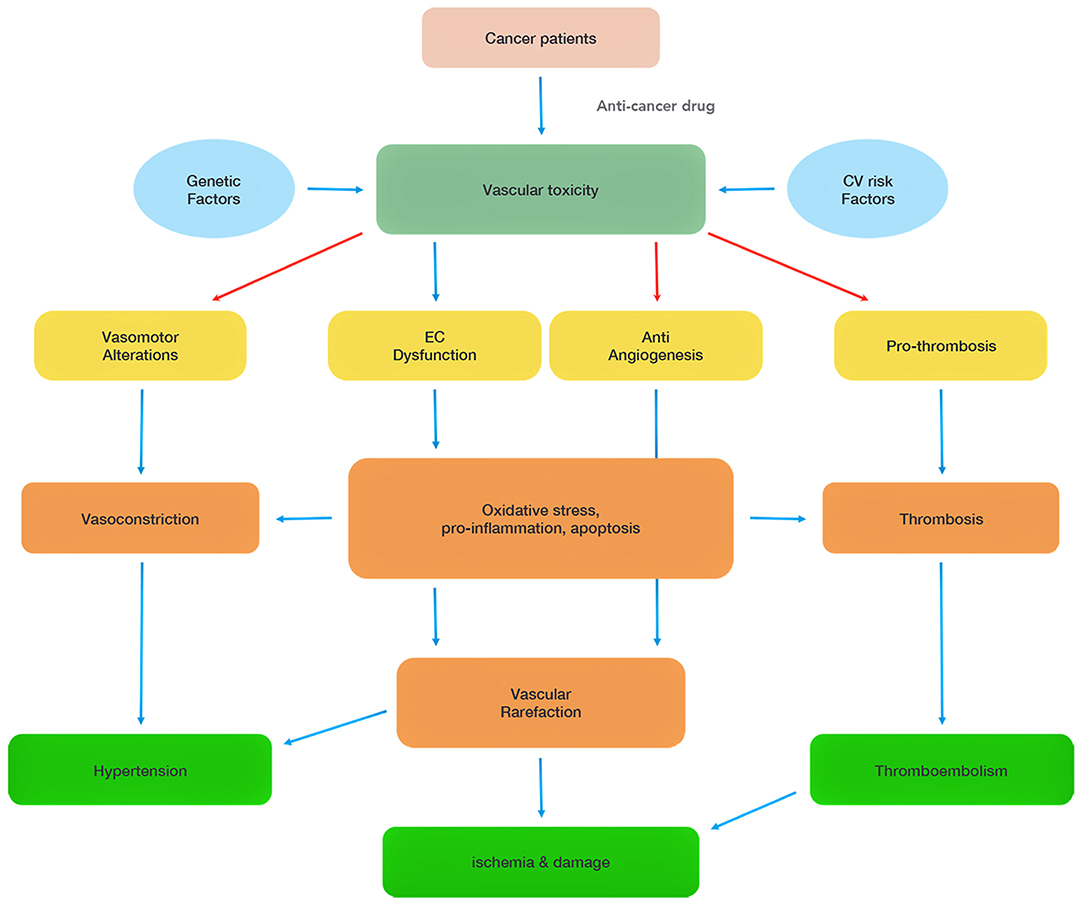
Figure 1. Summary of chemotherapy-associated vascular toxicity. Vascular toxicity can be attributed to four main mechanisms (vasomotor alterations, endothelial cell (EC) dysfunction, anti-angiogenesis, pro-thrombosis), which induce hypertension, ischemia and thromboembolism, and damage heart functions. The red arrows highlight direct effects of some of the chemotherapeutics.
Vascular damage in the cardiovascular system can be caused not only by anti-angiogenic chemotherapy (inhibitors of vascular endothelial growth factor (VEGFi), but also by anti-tumor antibiotics (bleomycin and anthracyclines) (5, 6). The first line of treatments includes monoclonal antibodies (e.g., bevacizumab), and multiple kinase inhibitors such as sunitinib, a multi-targeted inhibitor, or sorafenib (7). In addition, plant alkaloids (taxanes, vinca alkaloids), alkylating agents (cisplatin, cyclophosphamide), antimetabolites (5-fluorouracil), and radiation therapy also foster vascular damages (8) (Figure 2).
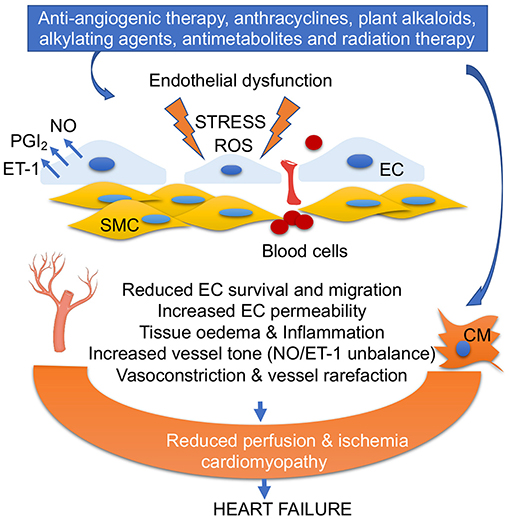
Figure 2. Anticancer drug-mediated endothelial dysfunction. Oxidative stress, ROS accumulation, alteration of PGI2/ET-1 ratio, and low NO levels in ECs impair EC survival and migration, increase fenestration and blood cell infiltration into vascular smooth muscles (SMC), trigger inflammation, and promote vasoconstriction. EC dysfunction and vascular rarefaction reduce myocardial perfusion, leading ischemia, and cardiomyopathy. Anticancer drug-mediated cardiomyocyte (CM) damage together with EC dysfunction leads to heart failure.
Hereafter, we concentrate on these anti-cancer drugs-mediated vascular damages that evoke cardiovascular diseases and impair drug delivery.
Anticancer Therapy-Mediated Oxidative Stress and Vascular Injury
Many chemotherapeutics induce accumulation of the reactive oxygen species (ROS) products (9) that disrupt intracellular homeostasis and damage proteins, lipids, and DNA in the vascular cells. ROS such as superoxide radical anions (O2.−), lipid radicals (ROO.−), hydroxyl radicals (HO.−), and nitric oxide (NO) are formed by all vascular layers, including endothelium, smooth muscle, and adventitia (10). ROS induces VEGF expression in vascular endothelial and smooth muscle cells by upregulating hypoxia-inducible transcription factors (HIF-1). VEGF further stimulates the accumulation of ROS through activation of NADPH oxidase (11). The NO itself has a cardiovascular protective properties (12). However, when NO combines with ROS, it generates peroxynitrite radicals (ONOO.−) that promote inflammation, apoptosis, necrosis, and ultimately toxicity (13).
A high production of ROS is also a major promoter for the lipid peroxidation of unsaturated fatty acids, leading to apoptosis, autophagy, and ferroptosis (14). Lipid peroxidation followed by the activation of phospholipase A2 initiates the activation of arachidonic acid (AA) pathway. Thus, lipid peroxidation is not only responsible for the generation of prostaglandins, but also for the induction of inflammation and apoptosis in vascular ECs (14). ROS also promotes peroxidation of a mitochondrion-specific inner membrane phospholipid, cardiolipin to activate intrinsic apoptosis (15). Lipid peroxidation products can bind to specific mitochondrial and autophagy-related proteins driving autophagic cell death (16). Elevated intracellular iron concentration elevates ROS levels that cause lipid peroxidation and consequently to ferroptosis-mediated cell death (17).
Reactive nitrogen species (RNS) are formed by the reaction between ROS and NO that damage mitochondrial DNA. Excessive ROS also induces senescence in endothelial, vascular smooth muscle cell (VSMC), and endothelial progenitor cells (18). Indeed, accumulation of ROS and oxidative stress reduces NO bioavailability and consequently results in development of hypertension (19) (Figure 2).
Anticancer Therapy-Mediated Endothelial Dysfunction and Hypertension
Approximately 25% of cancer patients develop hypertension due to adverse effects of VEGFi, TKI, anthracyclines, alkylating agents, and antimetabolites (20). The pathophysiology of hypertension induced by these agents is not fully elucidated. Several mechanisms have been proposed based on the preclinical and clinical studies, including; (1) increased total peripheral resistance induced by endothelial dysfunction due to predominantly the reduced production of vasodilators (NO and PGI2), the increased production of vasoconstrictors (ET-1) and the reduced nitric oxide bioavailability, (2) increase in vascular tone, (3) vascular rarefaction, (4) and renal thrombotic microangiopathy, leading to proteinuria and hypertension, (5) natriuresis and impaired lymphatic function could also contribute to development of hypertension (21).
Inhibitors of VEGF (VEGFi) or Tyrosine Kinase (TKI)
Approximately 80% of patients treated with VEGFi or TKI manifest hypertension (22). VEGF signaling promotes production of NO and the vasodilatory prostanoid prostacyclin (PGI2) through activation of phospholipase A2 via PLCγ/PKC pathways (23). After VEGF binding, VEGF receptor (VEGFR) activates phosphoinositol-3 kinase (PI3K)/serine-threonine protein kinase B (Akt) survival pathway in ECs. Thus, interruption of the VEGF signaling pathway by anticancer drugs leads to development of hypertension. Similarly, the VEGF trap aflibercept promotes hypertension (24), interrupting VEGF-mediated vasodilatory, and survival signaling (25). VEGFi-induced vascular toxicities can also be due to accumulation of ROS and down-regulation of nuclear factor erythroid 2-related factor 2 (Nrf2) that regulates antioxidant genes (26). Prohypertensive effects of VEGFi can also be promoted by microparticles of injured ECs (27).
TKIs stimulate ROS accumulation and reduce NO levels (28). For example, vatalanib or sunitinib increases ROS accumulation in both VSMCs and ECs by inhibiting NO synthase (NOS) thereby reducing NO levels and decreasing endothelium-dependent vasorelaxation (29). Sunitinib-induced hypertension may not depend on endothelium, but may be due to decreased arterioles diameters. Indeed, it inhibits platelet-derived growth factor receptor (PDGFR) that causes coronary microvascular dysfunction due to loss of pericytes, leading to the mechanical instability of the capillary wall in cardiac and other tissues (30).
Anthracyclines
They cause ≈20% increase in carotid artery stiffness in patients. Anthracyclines also led to a 3-fold increase in vascular stiffness with a 10-year follow-up period in adolescent childhood cancer survivors, indicating that alterations in vascular integrity persist years to decades following anthracycline chemotherapy (31). Anthracycline-induced endothelial toxicity and hypertension can be caused by several mechanisms. The first one is an oxidative stress-mediated process (32). Indeed, doxorubicin binds to endothelial (eNOS) and decreases NO levels, leading to the production of superoxide. Reduced concentration of NO shifts endothelium to a pro-coagulant status and impairs vasodilatation (33). Recently, doxorubicin has been shown to stabilize NRF2 in the cytoplasm thereby reducing detoxification pathway in mice heart (6). Doxorubicin also induces mitochondrial DNA damage in an RNS/ROS-independent manner, along with a possible decrease in B-cell lymphoma (Bcl)-2, that leads to apoptosis of the ECs. The EC death further reduces the availability of NO, ET-1, PGI2, and neuregulin (NRG)-1 to cardiomyocytes. Indeed, accumulation of ROS and oxidative stress reduces NO bioavailability and consequently results in development of hypertension (34). The second mechanism is apoptosis due to DNA interference (35). Doxorubicin -mediated topoisomerase II-β inhibition and DNA-binding directly induce DNA damage and apoptosis in ECs (36). Doxorubicin also reduces the tight junction protein zona occludens (ZO)-1 in ECs, thereby, increasing microvascular permeability (37). Anthracyclines at the accumulative dosage dysregulate renin-angiotensin-aldosterone (RAA) system (38), that play significant role in the development of hypertension (39).
Alkylating Agents
Cyclophosphamide or its metabolites reduce vasoactive substance NO, increase ET-1 and inducible (i) NOS (40). They activate the toll-like receptor 4 (TLR-4) and causes subsequent activation of mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinases (JNK) (41). Once activated, these signaling pathways increase the expression of tissue necrosis factor alpha (TNFα), cyclooxygenase-2 (cox-2), prostaglandins (PGs), and interleukins (ILs). Cyclophosphamide also decreases fatty acid binding protein (H-FABP) and carnitine palmitoyl transferase-I (CPT-1) levels, resulting in the accumulation of free fatty acids and reduction of ATP production (42). Reduced ATP levels lead to the accumulation of intracellular calcium, which activates transforming growth factor beta (TGF-β) and the production of pro-inflammatory cytokines. Cyclophosphamide is associated with development of interstitial pneumonia and pulmonary fibrosis, leading to vascular sclerosis, and pulmonary hypertension (40).
Patients treated with cisplatin-based chemotherapy also develop persistent hypertension due to endothelial cell activation, damage, and subsequent endothelial dysfunction (43). Cisplatin induces release of inflammatory substances such as IL-1 and IL-6 from ECs to produce hydrogen peroxide that provoke oxidative stress, and mitochondrial DNA lesions, orchestrating cell death (44).
Antimetabolites
5-Fluorouracil (5-FU) induces ultrastructural changes in the endothelium of the heart, as well as in various organs by promoting both accumulation of ROS and autophagy process in ECs (45). Its vascular adverse effects include angina with coronary artery spasm and rarely hypertension (46).
In general, anticancer therapies increases blood pressure, therefore, an antihypertensive therapy can be required in case of diastolic blood pressure (DBP) increase >20 mmHg after initiation of anticancer therapy, yet DBP remains within normal limits (47).
Anticancer Therapy-Mediated Microvascular Rarefaction
Anticancer drugs induce capillary rarefaction that is described as a reduction of the density of arterioles and capillaries. One of the causes of microvascular rarefaction is a decrease of survival rate of microvascular EC. The second mechanism involve endothelial dysfunction that participates to thrombosis, leading to a further reduction in vascular perfusion, and micro vessel destruction (48). The molecular mechanisms of capillary rarefaction associated with the loss of pericytes due to inhibition of platelet derived growth factor (PDGF) receptor (PDGFR), and inhibition of angiogenesis by blocking VEGF signaling pathway.
VEGFi and TKIs
Prolonged TKI treatments lead to capillary rarefaction, due to endothelial dysfunction (25). In addition, disruption of both endothelium-dependent and -independent vasodilatation can also promote intense vasoconstriction and microvascular rarefaction. The vascular rarefaction may also be a consequence of VEGFi-associated hypertension (49). Bevacizumab promotes retinal microvascular dysfunction in humans (50). On the other hand, microvascular rarefaction increases peripheral resistance in the microcirculation, thereby, reducing blood flow and further elevating blood pressure.
Anthracyclines
A recent preclinical study has shown that chronic treatment with doxorubicin promotes vessel rarefaction in the heart (6). Moreover, a low dose of doxorubicin inhibits EC motility in vitro without causing apoptosis. However, whether doxorubicin provoke hypertension in these mice has not been studied.
Alkylating Agents
Cyclophosphamide causes extravasation of proteins, toxic metabolites, and erythrocytes, which breaks-down ECs, promotes hemorrhage, blocks the small arteries, and induces displacement of vascular ECs that directly damages the blood vessels and cardiac cells (51). Cyclophosphamide may reduce VEGF levels that is associated with microvascular rarefaction.
Cisplatin inhibits EC proliferation and motility in vitro and causes apoptosis (52). Cisplatin also inhibits angiogenesis (53). Thus, both EC dysfunction and anti-angiogenic effects of platinum derivatives promote vascular rarefaction.
Anticancer Therapy-Mediated Hypercoagulation, Thrombosis, and Atherosclerosis
Cancer patients exhibit an increased risk of arterial and venous thrombotic events. Approximately cancer patients develop the risk of arterial (2–5%) and venous (4–20%) thromboembolism during the anti-angiogenic therapies (54). The mechanisms that underline the chemotherapeutic–associated thrombosis is not fully understood. It appears that targeted therapies-mediated thromboembolism is associated with on-target effects. However, conventional chemotherapies-mediated thromboembolism attributed to off-target effects. Based on the preclinical and clinical studies, the proposed mechanisms include; (1) the activation or disruption of the endothelium, (2) decrease in anticoagulants and increase in procoagulants, such as TF (tissue factor), cytokine-controlled defective anticoagulant pathways, and changes in the fibrinolytic pathways, and (3) the activation of platelets (55).
VEGFis and TKIs
They impair the VEGF-mediated tissue-type plasminogen activator (t-PA) release (56), and elevate platelets and coagulation factors to induce thrombosis (57). Additionally, TKIs increase hematocrit and reduce NO- and PGI2-mediated anti-platelet activity (58). Accordingly, a meta-analysis in patients receiving TKIs demonstrated that the risk of myocardial infarction increased by 3.5-fold, and the development of arterial thrombosis by 1.8-fold, in the treated group (59). VEGFR inhibitors accelerate atherosclerosis and increase the risk of cholesterol embolization syndrome, leading to acute cardiovascular complications (60).
Anthracyclines
Doxorubicin has been shown to a significantly increase a risk of venous thrombosis by 16.0% (47). Several preclinical and clinical studies have showed that doxorubicin-mediated thrombogenic effects are resulted from an elevated prothrombotic state induced by (1) endothelial injury, (2) the down-regulation of the endothelium-based protein C anticoagulant pathway due to the reduced levels of endothelial protein C receptor in ECs, (3) an increased TF procoagulant activity, and (4) activated platelets (61). In patients with breast cancer, doxorubicin increases levels of thrombin-antithrombin complexes, protein C, and activated protein C (62). Its prothrombotic effects are also due to phosphatidylserine-bearing microparticle (MP) generation, promoting intracellular Ca2+ increase and ATP depletion in platelets (63). A dysfunction of the NADP-dependent mitochondrial enzyme aldehyde dehydrogenase-2 (ALDH2) in ECs is also involved in the development of doxorubicin-mediated vascular damage and thrombosis (64). Altered levels of endothelium-derived NRG-1, PGI2, and ET-1 from ECs can also contribute to anti-platelet activity of doxorubicin (65).
Alkylating Agents
Cyclophosphamide and its toxic metabolites stimulate activation and release of platelet factor 4 (PF-4) that initiates the cascade of thrombosis and the binding of oxidized low-density lipoprotein (LDL) to ECs, and aggravates monocyte adhesion to endothelium (41). Cyclophosphamide-induced intrapapillary micro emboli is prominent cause of the ischemic myocardial damage (41). It also fosters acute pericarditis, myocardial hemorrhage, and atrophic and focal necrosis with interstitial edema (66).
Cisplatin facilities endothelial damage, hypercoagulation measured by increased levels of thrombin-antithrombin complexes and D-dimer, and platelet aggregation via activation of the arachidonic acid pathway that forms several inflammatory and thrombogenic molecules (67). However, the absolute risk of venous thrombosis associated with this class of agent remains low.
Antimetabolites
5-FU damages ECs and provokes severe vessel leakage and subsequent thrombus formation (68, 69). Patients receiving a cisplatin-based regimen with epirubicin and 5-FU or capecitabine exhibited an incidence of venous thrombosis of 15.1% (70).
In general, anticancer agents have more pronounced effect of the incidence of venous thrombosis than arterial thrombosis. An anticoagulation therapy may be required.
Anticancer Therapy-Mediated Impaired Vasculature and Drug Delivery
Anti-angiogenic agents alone or in combination with other chemotherapeutics are widely used to inhibit tumor growth by targeting vascular network (71). Some types of cancers are sensitive to anti-angiogenic therapy, while other types of cancers are completely insensitive. Adaptation to microenvironment, such as metabolic changes (72) or autophagy (73), can determine whether a tumor is sensitive to anti-cancer treatments. Some tumors can initially respond, but then develop acquired resistance during the anti-angiogenic treatment due to activation of alternative pathways, such as vessel co-option and vessel mimicry (74). Development of hypoxia in tumors reduces the activity of the prolyl hydroxylase domain proteins (PHD1–3), and prevents the degradation of HIF-1α and HIF-2α (75). High levels of HIFs in turn increases the transcription of HIF-driven hypoxia-related genes, including the potent angiogenic factors, VEGF to form a neovascular network to further increase tumor growth. Indeed, long-term anti-angiogenic therapy promotes genetic instability in tumor ECs, and causes vascular permeability and metastasis (76). Additionally, tumor-associated macrophages can trans-differentiate into ECs (77). In this case, tumors become highly vascularized and also resistant to chemotherapies. Tumor cells including infiltrated immature myeloid cells (78), fibroblasts (79), and endothelial progenitor cells (80) integrate into vessels or release pro-angiogenic growth factors, such as prokineticin-2 (3) or PDGF-C (79), leading to worse outcomes of drug delivery, invasion, and metastasis (Figure 3).
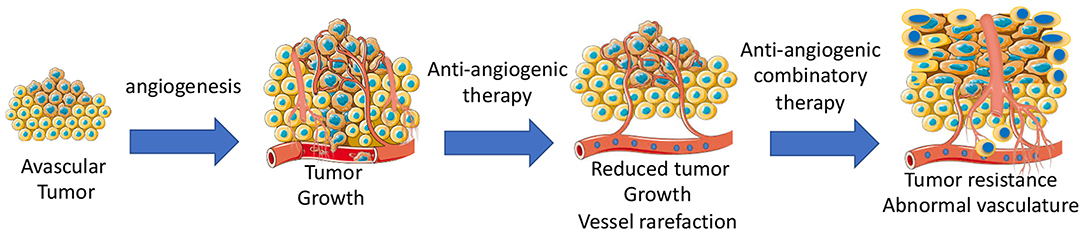
Figure 3. Development of angiogenesis and anti-angiogenic therapy-mediated development of tumor resistance due to abnormal tumor vasculatures.
Detection of Endothelial Damage and Thrombosis
Endothelial damages alter the expression of adhesion molecules and increase levels of pro-inflammatory cytokines (81). Thus, expression of adhesion molecules such as E-selectin, endothelin-1, and vascular cell adhesion molecule-1 (VCAM-1) are biomarkers of endothelial damage (82). The elevated levels of pro-inflammatory cytokines such as C-reactive protein (CRP) and IL-6 are also indicators of EC damage (83). Because asymmetric dimethylarginine (ADMA) synthesized via arginine methylation inhibits eNOS and promotes superoxide generation (84), ADMA is a marker of ROS generation. Indeed, activated ECs initiates procoagulant activity by releasing endothelium-derived glycoproteins such as von Willebrand factor (vWF), NRG-1, soluble thrombomodulin (sTM), and tissue plasminogen factor (t-PA) (85). Thus, increase levels of vWF, NRG-1 sTM, and t-PA are also the indicators of procoagulant activity and thrombosis.
The detection of micro vessel architectural parameters by Magnetic Resonance Imaging (MRI), Vessel Architectural Imaging (VAI), Microvascular Density (MVD), Positron Emission Tomography (PET), 3D ultrasonography, and CT is necessary in the clinic to asses vascular damage and select a proper timing window for tumor vascular normalization by anti-angiogenic therapies (86).
Drugs Protecting Endothelial Cell Damage Induced By Chemotherapeutic Agents
Angiotensin converting enzyme inhibitors (ACEi), NO donors, antioxidants, and statins have direct effects on ECs, while angiotensin receptor blockers (ARBs), renin inhibitors, beta blockers, and estrogens indirectly affect EC function. Beta blockers, thiazide diuretics, mineralocorticoid receptor antagonists are used as additional antihypertensive agents. Here we focus on the first group of the EC protective drugs.
ACEis
ACEis ameliorates the left ventricular ejection fraction (LVEF) decline, when they are administered together or after anthracyclines. However, the vascular protective effects of ACEi, zofenoprilat, but not other ACEi (i.e., captopril or enalaprilat) are related with activation of survival pathways in cardiac cells, and its antioxidant and ROS scavenger properties. More specifically, zofenoprilat up-regulates the expression of eNOS, FGF-2, and telomerase (TERT) transcripts, thereby, promoting cell survival, rescuing damaged ECs, and inducing physiological angiogenesis without altering vascularization at tumors (87). Thus, zofenoprilat exerts its EC protective effects through off-target mechanisms, and may even maximize cytotoxic drug delivery to tumor cells (8).
Nitric Oxide Donors, Antioxidants, and Statin
Novel NO donor drugs metal-nonoates (88) and the mitochondrial aldehyde dehydrogenase (ALDH2) activator, called Alda-1 may restore eNOS functioning, and FGF-2 production and release, thereby, protecting ECs against anticancer drug-mediated damages (89). ALDH2 plays a central role in the vasodilator actions of nitroglycerin, restores mitochondrial functions, and promotes vascular recovery of ischemic myocardium (90). However, high ALDH2 metabolic activities have been observed in tumor ECs as well. Thus, whether ALDH2 mitigates anti-cancer drug efficacy in tumor should be tested.
Many animal models showed that vitamin E, vitamin C, vitamin A, coenzyme Q, and flavonoids can reduce the anthracycline-mediated cardiovascular toxicity. However, clinical use of antioxidants to protect the heart during anthracycline chemotherapy is paved away due to reduce cytotoxic efficacy toward cancer cells (91).
All FDA-approved statins are effective in lowering serum cholesterol by inhibiting activity of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), a rate-limiting enzyme of the mevalonate pathway, in the liver. Therefore, they are integrated into cancer patient care to protect against atherosclerosis development during anticancer therapies. However, epidemiologic studies demonstrated that statin type, dose, and treatment duration, statin sensitivity, and toxicity are all important variables to evaluate statins beneficial effects in adverse effects of anticancer drugs (92).
New Horizons in Therapeutic Strategies: Pro-Angiogenic Therapy to Prevent Vascular Toxicity Without Altering Anti-Neoplastic Properties of Chemotherapeutics
VEGF-B-Mediated Endothelial Protection Against Doxorubicin-Mediated Cardiotoxicity
Vascular endothelial growth factor-B (VEGF-B) promotes coronary arteriogenesis, physiological cardiac hypertrophy, and ischemia resistance. It also prevents doxorubicin-induced cardiotoxicity and congestive heart failure. A recent preclinical study has shown that pretreatment of tumor-bearing mice with an adeno-associated viral vector expressing VEGF-B completely inhibits the doxorubicin-induced cardiac atrophy and whole-body wasting (93). VEGF-B also alleviates capillary rarefaction in the heart and improves cardiac function in doxorubicin-treated mice. Indeed, VEGF-B protects EC from apoptosis and restores tube-formation capacity of ECs without altering anti-tumor role of doxorubicin. Importantly, VEGF-B does not affect serum or tissue concentrations of doxorubicin. By inhibiting doxorubicin-induced endothelial damage, VEGF-B could provide a novel therapeutic possibility for the prevention of chemotherapy-associated cardiotoxicity in cancer patients.
Prokineticin Receptor-1 Signaling Inhibits Dose- and Time-Dependent Anthracycline-Induced Cardiovascular Toxicity via Myocardial and Vascular Protection
Prokineticins (PROK1 and PROK2) are neuropeptides/hormones that are mainly released by macrophages and reproduction organs in the peripheral system (94). They utilize two G-protein–coupled receptors (GPCRs) namely prokineticin receptors (PKR1 and PKR2). Expression of PROK2 and PKR1 levels are altered in patients with abdominal aortic rupture (8), during end-stage cardiac failure (95) after acute myocardial infarction (96), and in adipose tissues from obese patients (97). Interestingly, PKR1 gene transfer improves survival and heart function in a mouse model of myocardial infarction (95) and promotes coronary arteriogenesis (98). However, PKR2 overexpression in cardiomyocytes promotes pathological cardiac hypertrophy and causes vascular leakage (99, 100). These receptors have also divergent effects on ECs (101). Thus, a non-peptide agonist specific for PKR1, called IS20, was developed to mimic the cardioprotective effects of PROK2 against heart failure developed by myocardial infarction (102) and anthracyclines (6) in mice.
A recent preclinical study has demonstrated that prolonged exposure to low-dose doxorubicin does not induce apoptosis in ECs, but impairs angiogenesis (6). Importantly, IS20 restores doxorubicin-mediated cardiovascular toxicity by activating Akt or MAPK pathways. Genetic or pharmacological inactivation of PKR1 abolishes these effects of IS20. Mice exposed to chronic doxorubicin treatment exhibit apoptosis in cardiac cells, vascular rarefaction and fibrosis, consequently impaired systolic and diastolic cardiac function, and reduced survival rate. IS20 reverses these detrimental effects of doxorubicin. IS20 also does not alter the cytotoxicity or antitumor effects of doxorubicin in breast cancer lines or in a mouse model of breast cancer. Altogether, this study provides evidence that PKR-1 is a promising target to combat cardiovascular toxicity of cancer treatments (6).
Conclusion and Perspectives
Anticancer treatments induce vascular damage, hypertension, and thrombosis, which affect survival and quality of life of the patient (Table 1). Therefore, pre-existing hypertension and a thrombosis risk assessment should be conducted before starting any type of chemotherapies (103). A continued characterization of changes of microvessel network patterns and blood pressure by anticancer drugs is necessary to prevent development of hypertension and organ damages, especially during the 1st cycle of therapy when the patients experience a secondary elevation in blood pressure.
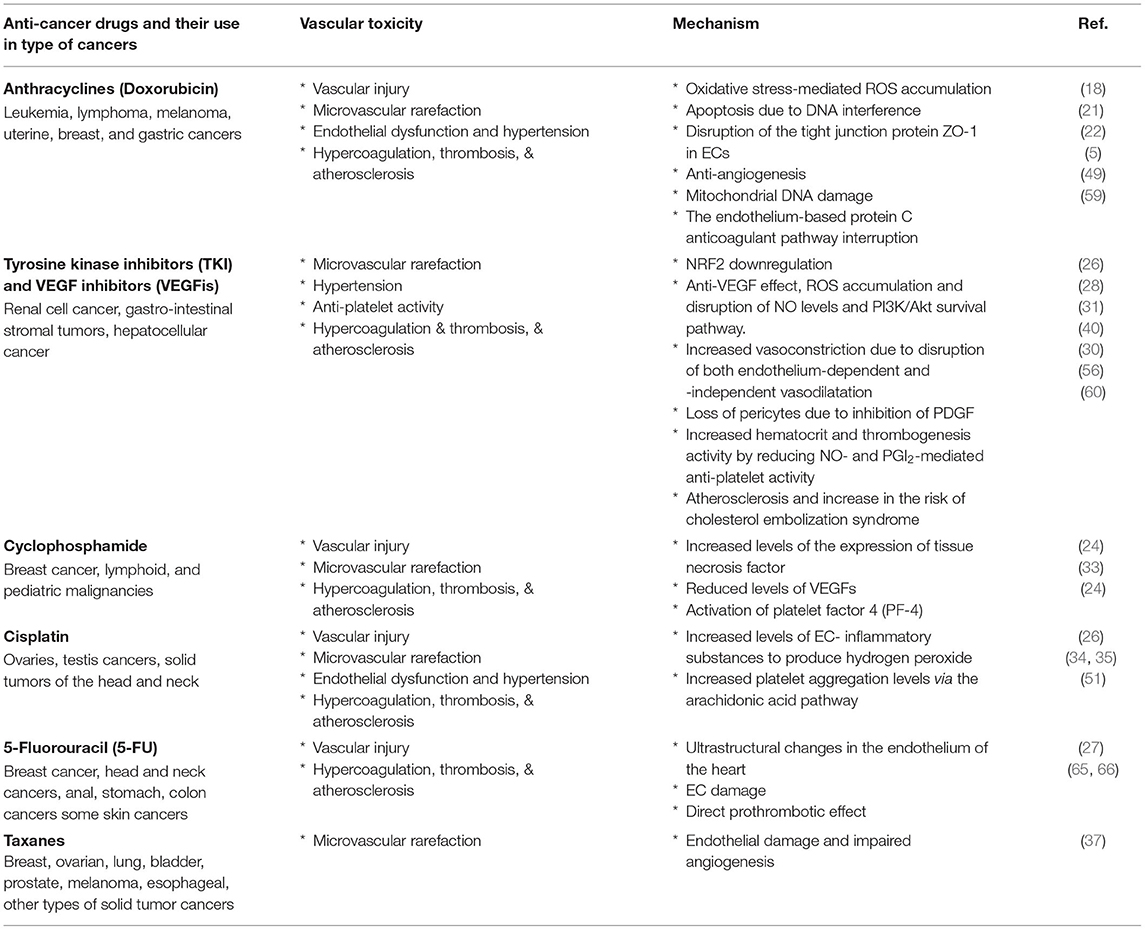
Table 1. Vascular damages and diseases induced by the widely prescribed anticancer drugs.
Several mechanisms for anticancer drug-mediated vascular toxicity have been identified (104), however, there are still many unknown molecular processes that need to be unraveled to better understand exactly how anticancer treatments provoke vascular damages. Endothelial metabolism and new signaling pathways could be novel targets of the vascular protectant.
Identification of underlying pathological mechanisms of development of vascular toxicity is a key element to optimize benefits in tumor development and drug delivery of chemotherapies.
The improvement in cancer therapy of the past two decades is due to the development of numerous novel targeted therapies. These drugs are also used in combination with other new anti-cancer drugs including inhibitors of immune check points, poly (ADP-ribose) polymerase (PARP), and histone deacetylase (HDAC). However, most of these treatments also induce vascular toxicity, leading to hypertension, thromboembolism, vasculitis, development of atherosclerotic plaques, and fibrotic heart disease. More clinical trials of cancer therapies are needed to be better document the vascular complications of the chemotherapeutics.
Some of the new cardiovascular protectants including GPCR-targeted compounds are potential drug candidates to improve management and prevention of the cardio vascular toxicity of anti-cancer therapy (105). Whether, these potential vascular protective agents minimize thrombotic risk associated with chemotherapies should also be examined. Further, studies are also necessary to examine their effects on the efficacy of anti-tumor drugs.
Author Contributions
AM, P-YH, and CGN: create the figure and contribute to the writing. LD, NB-J, and CGN: conribute writing. CGN: conribution to editing, designing, and orginization of the idea. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by grants from the ANR-16-ECVD-000, ERA-CVD (JTC 2016) Transnational Research Projects on Cardiovascular Diseases and Fondation de France (R20085MM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Prof. Pascal Dollé (IGBMC, Illkirch) for his kind editing and discussions. The drawings in Graphical (Figures 1–5) were produced and adapted, using Servier Medical Art illustration resources (www.servier.com) and Biorender (www.Biorender.com) (Figure 4).
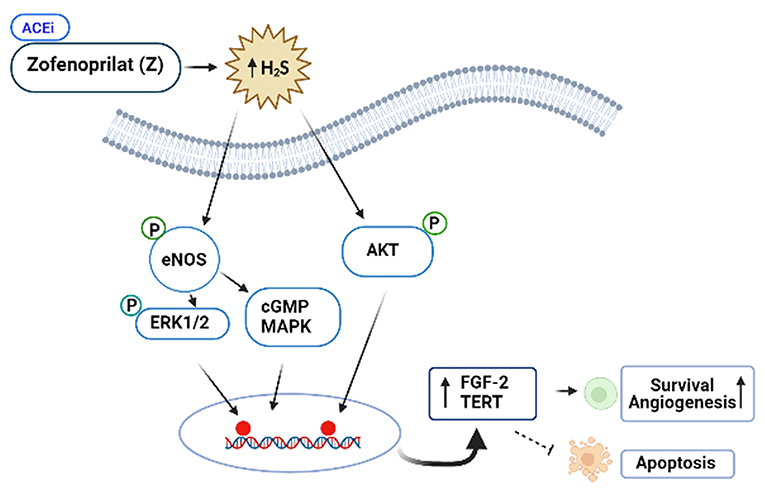
Figure 4. Protective role of the SH-containing ACi, Zofenoprilat, against anticancer-mediated vascular toxicity. Zofenoprilat is the most effective clinically used vascular protectant. It increases survival of ECs and promotes angiogenesis, inhibits apoptosis. The mechanism involves activation of NOS, ERK1/2, cGMP and Akt kinases and increases in the expression levels of fibroblast growth factor (FGF-2) and telomerase reverse transcriptase (TERT). These effects appear to be the off-target effects of zofenoprilat via increasing production of H2S, independent of ACEi properties.
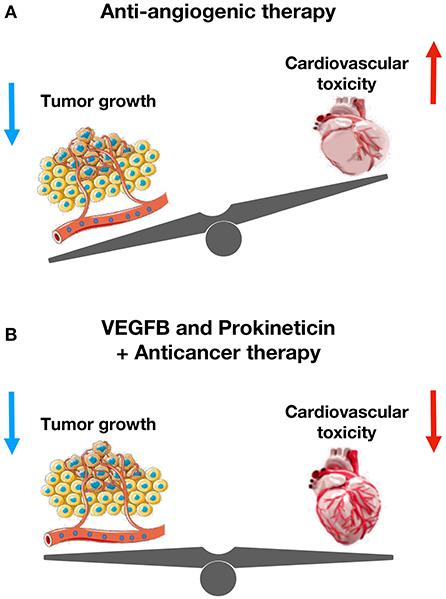
Figure 5. The cardioprotective role of VEGFB and prokineticin against anti-cancer drugs-mediated cardiovascular toxicity. (A) Anti-angiogenic therapy reduces tumor size and increases cardiovascular toxicity. (B) VEGFB and Prokineticin receptor agonist protects cardiovascular system without altering anti-tumor effect of chemotherapeutics.
References
1. Gao Y, Galis ZS. Exploring the role of endothelial cell resilience in cardiovascular health and disease. Arterioscler Thromb Vasc Biol. (2021) 41:179–85. doi: 10.1161/ATVBAHA.120.314346
2. Kruger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. (2019) 20:4411. doi: 10.3390/ijms20184411
3. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. (2007) 450:825–31. doi: 10.1038/nature06348
4. Cameron AC, Touyz RM, Lang NN. Vascular complications of cancer chemotherapy. Can J Cardiol. (2016) 32:852–62. doi: 10.1016/j.cjca.2015.12.023
5. Lv H, Tan R, Liao J, Hao Z, Yang X, Liu Y, et al. Doxorubicin contributes to thrombus formation and vascular injury by interfering with platelet function. Am J Physiol Heart Circ Physiol. (2020) 319:H133–43. doi: 10.1152/ajpheart.00456.2019
6. Gasser A, Chen Y-W, Audebrand A, Daglayan A, Charavin M, Escoubet B, et al. Prokineticin receptor-1 signaling inhibits dose- and time-dependent anthracycline-induced cardiovascular toxicity via myocardial and vascular protection. JACC Cardiooncol. (2019) 1:84–102. doi: 10.1016/j.jaccao.2019.06.003
7. Versmissen J, Mirabito Colafella KM, Koolen SLW, Danser AHJ. Vascular cardio-oncology: vascular endothelial growth factor inhibitors and hypertension. Cardiovasc Res. (2019) 115:904–14. doi: 10.1093/cvr/cvz022
8. Morbidelli L, Donnini S, Ziche M. Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents. Cardiooncology. (2016) 2:3. doi: 10.1186/s40959-016-0010-6
9. Varricchi G, Ameri P, Cadeddu C, Ghigo A, Madonna R, Marone G, et al. Antineoplastic drug-induced cardiotoxicity: a redox perspective. Front Physiol. (2018) 9:167. doi: 10.3389/fphys.2018.00167
10. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
11. Moris D, Spartalis M, Spartalis E, Karachaliou GS, Karaolanis GI, Tsourouflis G, et al. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann Transl Med. (2017) 5:326. doi: 10.21037/atm.2017.06.27
12. Forstermann U, Li H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol. (2011) 164:213–23. doi: 10.1111/j.1476-5381.2010.01196.x
13. Lim W, Kim JH, Gook E, Kim J, Ko Y, Kim I, et al. Inhibition of mitochondria-dependent apoptosis by 635-nm irradiation in sodium nitroprusside-treated SH-SY5Y cells. Free Radic Biol Med. (2009) 47:850–7. doi: 10.1016/j.freeradbiomed.2009.06.023
14. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. (2019) 2019:5080843. doi: 10.1155/2019/5080843
15. Zhong H, Xiao M, Zarkovic K, Zhu M, Sa R, Lu J, et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: a novel link between oxidative stress and cancer. Free Radic Biol Med. (2017) 102:67–76. doi: 10.1016/j.freeradbiomed.2016.10.494
16. Xie Y, Li J, Kang R, Tang D. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. (2020) 8:431. doi: 10.3389/fcell.2020.00431
17. Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
18. Liang Y, Li J, Lin Q, Huang P, Zhang L, Wu W, et al. Research progress on signaling pathway-associated oxidative stress in endothelial cells. Oxid Med Cell Longev. (2017) 2017:7156941. doi: 10.1155/2017/7156941
19. Schulz E, Jansen T, Wenzel P, Daiber A, Munzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal. (2008) 10:1115–26. doi: 10.1089/ars.2007.1989
20. Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am J Kidney Dis. (2007) 49:186–93. doi: 10.1053/j.ajkd.2006.11.039
21. Narayan V, Ky B. Common cardiovascular complications of cancer therapy: epidemiology, risk prediction, and prevention. Annu Rev Med. (2018) 69:97–111. doi: 10.1146/annurev-med-041316-090622
22. Small HY, Montezano AC, Rios FJ, Savoia C, Touyz RM. Hypertension due to antiangiogenic cancer therapy with vascular endothelial growth factor inhibitors: understanding and managing a new syndrome. Can J Cardiol. (2014) 30:534–43. doi: 10.1016/j.cjca.2014.02.011
23. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. (2006) 7:359–71. doi: 10.1038/nrm1911
24. Sinicrope FA, Williamson EE, Borgeson DD. Aflibercept and its role in the treatment of colorectal cancer–letter. Clin Cancer Res. (2013) 19:6057. doi: 10.1158/1078-0432.CCR-13-2056
25. Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. (2008) 19:927–34. doi: 10.1093/annonc/mdm550
26. Neves KB, Rios FJ, van der Mey L, Alves-Lopes R, Cameron AC, Volpe M, et al. VEGFR (Vascular Endothelial Growth Factor Receptor) inhibition induces cardiovascular damage via redox-sensitive processes. Hypertension. (2018) 71:638–47. doi: 10.1161/HYPERTENSIONAHA.117.10490
27. Neves KB, Rios FJ, Jones R, Evans TRJ, Montezano AC, Touyz RM. Microparticles from vascular endothelial growth factor pathway inhibitor-treated cancer patients mediate endothelial cell injury. Cardiovasc Res. (2019) 115:978–88. doi: 10.1093/cvr/cvz021
28. Teppo HR, Soini Y, Karihtala P. Reactive oxygen species-mediated mechanisms of action of targeted cancer therapy. Oxid Med Cell Longev. (2017) 2017:1485283. doi: 10.1155/2017/1485283
29. Thijs AM, van Herpen CM, Verweij V, Pertijs J, van den Broek PH, van der Graaf WT, et al. Impaired endothelium-dependent vasodilation does not initiate the development of sunitinib-associated hypertension. J Hypertens. (2015) 33:2075–82. doi: 10.1097/HJH.0000000000000662
30. Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med. (2013) 5:187ra69. doi: 10.1126/scitranslmed.3005066
31. Parr SK, Liang J, Schadler KL, Gilchrist SC, Steele CC, Ade CJ. Anticancer therapy-related increases in arterial stiffness: a systematic review and meta-analysis. J Am Heart Assoc. (2020) 9:e015598. doi: 10.1161/JAHA.119.015598
32. Wolf M, Baynes J. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochim Biophys Acta. (2006) 1760:267–71. doi: 10.1016/j.bbagen.2005.10.012
33. He H, Wang L, Qiao Y, Zhou Q, Li H, Chen S, et al. Doxorubicin induces endotheliotoxicity and mitochondrial dysfunction via ROS/eNOS/NO pathway. Front Pharmacol. (2019) 10:1531. doi: 10.3389/fphar.2019.01531
34. Vaitiekus D, Muckiene G, Vaitiekiene A, Maciuliene D, Vaiciuliene D, Ambrazeviciute G, et al. Impact of arterial hypertension on doxorubicin-based chemotherapy-induced subclinical cardiac damage in breast cancer patients. Cardiovasc Toxicol. (2020) 20:321–7. doi: 10.1007/s12012-019-09556-3
35. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
36. Wojcik T, Buczek E, Majzner K, Kolodziejczyk A, Miszczyk J, Kaczara P, et al. Comparative endothelial profiling of doxorubicin and daunorubicin in cultured endothelial cells. Toxicol In Vitro. (2015) 29:512–21. doi: 10.1016/j.tiv.2014.12.009
37. Wilkinson EL, Sidaway JE, Cross MJ. Cardiotoxic drugs herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biol Open. (2016) 5:1362–70. doi: 10.1242/bio.020362
38. Sobczuk P, Czerwinska M, Kleibert M, Cudnoch-Jedrzejewska A. Anthracycline-induced cardiotoxicity and renin-angiotensin-aldosterone system-from molecular mechanisms to therapeutic applications. Heart Fail Rev. (2020) 1–25. doi: 10.1007/s10741-020-09977-1
39. Kuriakose RK, Kukreja RC, Xi L. Potential therapeutic strategies for hypertension-exacerbated cardiotoxicity of anticancer drugs. Oxid Med Cell Longev. (2016) 2016:8139861. doi: 10.1155/2016/8139861
40. Soultati A, Mountzios G, Avgerinou C, Papaxoinis G, Pectasides D, Dimopoulos MA, et al. Endothelial vascular toxicity from chemotherapeutic agents: preclinical evidence and clinical implications. Cancer Treat Rev. (2012) 38:473–83. doi: 10.1016/j.ctrv.2011.09.002
41. Iqubal A, Iqubal MK, Sharma S, Ansari MA, Najmi AK, Ali SM, et al. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: old drug with a new vision. Life Sci. (2019) 218:112–31. doi: 10.1016/j.lfs.2018.12.018
42. Kurauchi K, Nishikawa T, Miyahara E, Okamoto Y, Kawano Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res Notes. (2017) 10:406. doi: 10.1186/s13104-017-2726-2
43. de Vos FY, Nuver J, Willemse PH, van der Zee AG, Messerschmidt J, Burgerhof JG, et al. Long-term survivors of ovarian malignancies after cisplatin-based chemotherapy cardiovascular risk factors and signs of vascular damage. Eur J Cancer. (2004) 40:696–700. doi: 10.1016/j.ejca.2003.11.026
44. Herradon E, Gonzalez C, Uranga JA, Abalo R, Martin MI, Lopez-Miranda V. Characterization of cardiovascular alterations induced by different chronic cisplatin treatments. Front Pharmacol. (2017) 8:196. doi: 10.3389/fphar.2017.00196
45. Sudhoff T, Enderle MD, Pahlke M, Petz C, Teschendorf C, Graeven U, et al. 5-Fluorouracil induces arterial vasocontractions. Ann Oncol. (2004) 15:661–4. doi: 10.1093/annonc/mdh150
46. Shiga T, Hiraide M. Cardiotoxicities of 5-fluorouracil and other fluoropyrimidines. Curr Treat Options Oncol. (2020) 21:27. doi: 10.1007/s11864-020-0719-1
47. Zangari M, Siegel E, Barlogie B, Anaissie E, Saghafifar F, Fassas A, et al. Thrombogenic activity of doxorubicin in myeloma patients receiving thalidomide: implications for therapy. Blood. (2002) 100:1168–71. doi: 10.1182/blood-2002-01-0335
48. Chen N, Ren M, Li R, Deng X, Li Y, Yan K, et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol Cancer. (2015) 14:140. doi: 10.1186/s12943-015-0418-x
49. Steeghs N, Gelderblom H, Roodt JO, Christensen O, Rajagopalan P, Hovens M, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clin Cancer Res. (2008) 14:3470–6. doi: 10.1158/1078-0432.CCR-07-5050
50. Reimann M, Folprecht G, Haase R, Trautmann K, Ehninger G, Reichmann H, et al. Anti-Vascular endothelial growth factor therapy impairs endothelial function of retinal microcirculation in colon cancer patients - an observational study. Exp Transl Stroke Med. (2013) 5:7. doi: 10.1186/2040-7378-5-7
51. Dhesi S, Chu MP, Blevins G, Paterson I, Larratt L, Oudit GY, et al. Cyclophosphamide-induced cardiomyopathy: a case report, review, and recommendations for management. J Investig Med High Impact Case Rep. (2013) 1:80346. doi: 10.1177/2324709613480346
52. Ramer R, Schmied T, Wagner C, Haustein M, Hinz B. The antiangiogenic action of cisplatin on endothelial cells is mediated through the release of tissue inhibitor of matrix metalloproteinases-1 from lung cancer cells. Oncotarget. (2018) 9:34038–55. doi: 10.18632/oncotarget.25954
53. Muscella A, Vetrugno C, Biagioni F, Calabriso N, Calierno MT, Fornai F, et al. Antitumour and antiangiogenic activities of [Pt(O,O'-acac)(gamma-acac)(DMS)] in a xenograft model of human renal cell carcinoma. Br J Pharmacol. (2016) 173:2633–44. doi: 10.1111/bph.13543
54. Grover SP, Hisada YM, Kasthuri RS, Reeves BN, Mackman N. Cancer therapy-associated thrombosis. Arterioscler Thromb Vasc Biol. (2021) 41:1291–305. doi: 10.1161/ATVBAHA.120.314378
55. Levi M, Sivapalaratnam S. An overview of thrombotic complications of old and new anticancer drugs. Thromb Res. (2020) 191(Suppl. 1):S17–21. doi: 10.1016/S0049-3848(20)30391-1
56. Oliver JJ, Webb DJ, Newby DE. Stimulated tissue plasminogen activator release as a marker of endothelial function in humans. Arterioscler Thromb Vasc Biol. (2005) 25:2470–9. doi: 10.1161/01.ATV.0000189309.05924.88
57. Elice F, Rodeghiero F, Falanga A, Rickles FR. Thrombosis associated with angiogenesis inhibitors. Best Pract Res Clin Haematol. (2009) 22:115–28. doi: 10.1016/j.beha.2009.01.001
58. Eremina V, Quaggin SE. Biology of anti-angiogenic therapy-induced thrombotic microangiopathy. Semin Nephrol. (2010) 30:582–90. doi: 10.1016/j.semnephrol.2010.09.006
59. Faruque LI, Lin M, Battistella M, Wiebe N, Reiman T, Hemmelgarn B, et al. Systematic review of the risk of adverse outcomes associated with vascular endothelial growth factor inhibitors for the treatment of cancer. PLoS ONE. (2014) 9:e101145. doi: 10.1371/journal.pone.0101145
60. Mir O, Mouthon L, Alexandre J, Mallion JM, Deray G, Guillevin L, et al. Bevacizumab-induced cardiovascular events: a consequence of cholesterol emboli syndrome? J Natl Cancer Inst. (2007) 99:85–6. doi: 10.1093/jnci/djk011
61. Woodley-Cook J, Shin LY, Swystun L, Caruso S, Beaudin S, Liaw PC. Effects of the chemotherapeutic agent doxorubicin on the protein C anticoagulant pathway. Mol Cancer Ther. (2006) 5:3303–11. doi: 10.1158/1535-7163.MCT-06-0154
62. Mukherjee SD, Swystun LL, Mackman N, Wang JG, Pond G, Levine MN, et al. Impact of chemotherapy on thrombin generation and on the protein C pathway in breast cancer patients. Pathophysiol Haemost Thromb. (2010) 37:88–97. doi: 10.1159/000324166
63. Kim SH, Lim KM, Noh JY, Kim K, Kang S, Chang YK, et al. Doxorubicin-induced platelet procoagulant activities: an important clue for chemotherapy-associated thrombosis. Toxicol Sci. (2011) 124:215–24. doi: 10.1093/toxsci/kfr222
64. Ge W, Yuan M, Ceylan AF, Wang X, Ren J. Mitochondrial aldehyde dehydrogenase protects against doxorubicin cardiotoxicity through a transient receptor potential channel vanilloid 1-mediated mechanism. Biochim Biophys Acta. (2016) 1862:622–34. doi: 10.1016/j.bbadis.2015.12.014
65. Luu AZ, Chowdhury B, Al-Omran M, Teoh H, Hess DA, Verma S. Role of endothelium in doxorubicin-induced cardiomyopathy. JACC Basic Transl Sci. (2018) 3:861–70. doi: 10.1016/j.jacbts.2018.06.005
66. Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, et al. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients' blood cells: implications for cancer chemoimmunotherapy. Clin Cancer Res. (2013) 19:4249–61. doi: 10.1158/1078-0432.CCR-12-3666
67. Oppelt P, Betbadal A, Nayak L. Approach to chemotherapy-associated thrombosis. Vasc Med. (2015) 20:153–61. doi: 10.1177/1358863X14568705
68. Sara JD, Kaur J, Khodadadi R, Rehman M, Lobo R, Chakrabarti S, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. (2018) 10:80140. doi: 10.1177/1758835918780140
69. Sorrentino MF, Kim J, Foderaro AE, Truesdell AG. 5-fluorouracil induced cardiotoxicity: review of the literature. Cardiol J. (2012) 19:453–8. doi: 10.5603/CJ.2012.0084
70. Cunningham D, Sirohi B, Pluzanska A, Utracka-Hutka B, Zaluski J, Glynne-Jones R, et al. Two different first-line 5-fluorouracil regimens with or without oxaliplatin in patients with metastatic colorectal cancer. Ann Oncol. (2009) 20:244–50. doi: 10.1093/annonc/mdn638
71. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
72. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017) 17:457–74. doi: 10.1038/nrc.2017.51
73. Chandra A, Rick J, Yagnik G, Aghi MK. Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin Cancer Biol. (2020) 66:75–88. doi: 10.1016/j.semcancer.2019.08.031
74. Lupo G, Caporarello N, Olivieri M, Cristaldi M, Motta C, Bramanti V, et al. Anti-angiogenic therapy in cancer: downsides and new pivots for precision medicine. Front Pharmacol. (2016) 7:519. doi: 10.3389/fphar.2016.00519
75. Macklin PS, McAuliffe J, Pugh CW, Yamamoto A. Hypoxia and HIF pathway in cancer and the placenta. Placenta. (2017) 56:8–13. doi: 10.1016/j.placenta.2017.03.010
76. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int J Mol Sci. (2018) 19:1232. doi: 10.3390/ijms19041232
77. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. (2014) 5:75. doi: 10.3389/fphys.2014.00075
78. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. (2013) 19:1114–23. doi: 10.1038/nm.3291
79. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. (2009) 15:21–34. doi: 10.1016/j.ccr.2008.12.004
80. Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. (2006) 313:1785–7. doi: 10.1126/science.1127592
81. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. (2007) 115:1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859
82. Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. (2017) 174:1591–619. doi: 10.1111/bph.13517
83. Meroni PL, Borghi MO, Raschi E, Ventura D, Sarzi Puttini PC, Atzeni F, et al. Inflammatory response and the endothelium. Thromb Res. (2004) 114:329–34. doi: 10.1016/j.thromres.2004.06.045
84. Landim MB, Casella Filho A, Chagas AC. Asymmetric dimethylarginine (ADMA) and endothelial dysfunction: implications for atherogenesis. Clinics. (2009) 64:471–8. doi: 10.1590/S1807-59322009000500015
85. Goncharov NV, Nadeev AD, Jenkins RO, Avdonin PV. Markers and biomarkers of endothelium: when something is rotten in the state. Oxid Med Cell Longev. (2017) 2017:9759735. doi: 10.1155/2017/9759735
86. Sandoo A, Kitas GD. A methodological approach to non-invasive assessments of vascular function and morphology. J Vis Exp. (2015) 45:267–73. doi: 10.3791/52339
87. Donnini S, Terzuoli E, Ziche M, Morbidelli L. Sulfhydryl angiotensin-converting enzyme inhibitor promotes endothelial cell survival through nitric-oxide synthase, fibroblast growth factor-2, and telomerase cross-talk. J Pharmacol Exp Ther. (2010) 332:776–84. doi: 10.1124/jpet.109.159178
88. Monti M, Solito R, Puccetti L, Pasotti L, Roggeri R, Monzani E, et al. Protective effects of novel metal-nonoates on the cellular components of the vascular system. J Pharmacol Exp Ther. (2014) 351:500–9. doi: 10.1124/jpet.114.218404
89. Solito R, Corti F, Chen CH, Mochly-Rosen D, Giachetti A, Ziche M, et al. Mitochondrial aldehyde dehydrogenase-2 activation prevents beta-amyloid-induced endothelial cell dysfunction and restores angiogenesis. J Cell Sci. (2013) 126:1952–61. doi: 10.1242/jcs.117184
90. Putman DM, Liu KY, Broughton HC, Bell GI, Hess DA. Umbilical cord blood-derived aldehyde dehydrogenase-expressing progenitor cells promote recovery from acute ischemic injury. Stem Cells. (2012) 30:2248–60. doi: 10.1002/stem.1206
91. Vincent DT, Ibrahim YF, Espey MG, Suzuki YJ. The role of antioxidants in the era of cardiooncology. Cancer Chemother Pharmacol. (2013) 72:1157–68. doi: 10.1007/s00280-013-2260-4
92. Longo J, van Leeuwen JE, Elbaz M, Branchard E, Penn LZ. Statins as anticancer agents in the era of precision medicine. Clin Cancer Res. (2020) 26:5791–800. doi: 10.1158/1078-0432.CCR-20-1967
93. Rasanen M, Degerman J, Nissinen TA, Miinalainen I, Kerkela R, Siltanen A, et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc Natl Acad Sci U S A. (2016) 113:13144–49. doi: 10.1073/pnas.1616168113
94. Nebigil CG. Prokineticin receptors in cardiovascular function: foe or friend? Trends Cardiovasc Med. (2009) 19:55–60. doi: 10.1016/j.tcm.2009.04.007
95. Urayama K, Guilini C, Messaddeq N, Hu K, Steenman M, Kurose H, et al. The prokineticin receptor-1 (GPR73) promotes cardiomyocyte survival and angiogenesis. FASEB J. (2007) 21:2980–93. doi: 10.1096/fj.07-8116com
96. Nguyen TL, Gasser A, Nebigil CG. Role of Prokineticin Receptor-1 in Epicardial Progenitor Cells. J Dev Biol. (2020) 8:32. doi: 10.3390/jdb8040032
97. Szatkowski C, Vallet J, Dormishian M, Messaddeq N, Valet P, Boulberdaa M, et al. Prokineticin receptor 1 as a novel suppressor of preadipocyte proliferation and differentiation to control obesity. PLoS ONE. (2013) 8:e81175. doi: 10.1371/journal.pone.0081175
98. Arora H, Boulberdaa M, Qureshi R, Bitirim V, Gasser A, Messaddeq N, et al. Prokineticin receptor-1 signaling promotes epicardial to mesenchymal transition during heart development. Sci Rep. (2016) 6:25541. doi: 10.1038/srep25541
99. Urayama K, Dedeoglu DB, Guilini C, Frantz S, Ertl G, Messaddeq N, et al. Transgenic myocardial overexpression of prokineticin receptor-2 (GPR73b) induces hypertrophy and capillary vessel leakage. Cardiovasc Res. (2009) 81:28–37. doi: 10.1093/cvr/cvn251
100. Demir F, Urayama K, Audebrand A, Toprak-Semiz A, Steenman M, Kurose H, et al. Pressure overload-mediated sustained PKR2 (Prokineticin-2 Receptor) signaling in cardiomyocytes contributes to cardiac hypertrophy and endotheliopathies. Hypertension. (2021) 77:1559–70. doi: 10.1161/HYPERTENSIONAHA.120.16808
101. Guilini C, Urayama K, Turkeri G, Dedeoglu DB, Kurose H, Messaddeq N, et al. Divergent roles of prokineticin receptors in the endothelial cells: angiogenesis and fenestration. Am J Physiol Heart Circ Physiol. (2010) 298:H844–52. doi: 10.1152/ajpheart.00898.2009
102. Gasser A, Brogi S, Urayama K, Nishi T, Kurose H, Tafi A, et al. Discovery and cardioprotective effects of the first non-Peptide agonists of the G protein-coupled prokineticin receptor-1. PLoS ONE. (2015) 10:e0121027. doi: 10.1371/journal.pone.0121027
103. Abdol Razak NB, Jones G, Bhandari M, Berndt MC, Metharom P. Cancer-associated thrombosis: an overview of mechanisms, risk factors, and treatment. Cancers. (2018) 10:380. doi: 10.3390/cancers10100380
104. Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. (2014) 3:e000665. doi: 10.1161/JAHA.113.000665
Keywords: vascular toxicity, anti-cancer drugs, cardiotoxicity, hypertension, thrombosis
Citation: Hsu P-Y, Mammadova A, Benkirane-Jessel N, Désaubry L and Nebigil CG (2021) Updates on Anticancer Therapy-Mediated Vascular Toxicity and New Horizons in Therapeutic Strategies. Front. Cardiovasc. Med. 8:694711. doi: 10.3389/fcvm.2021.694711
Received: 13 April 2021; Accepted: 18 June 2021;
Published: 27 July 2021.
Edited by:
Margaret Rose Cunningham, University of Strathclyde, United KingdomReviewed by:
Paola Rizzo, University of Ferrara, ItalyKerstin Timm, University of Oxford, United Kingdom
Copyright © 2021 Hsu, Mammadova, Benkirane-Jessel, Désaubry and Nebigil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Canan G. Nebigil, bmViaWdpbEB1bmlzdHJhLmZy