 Elysa Jui1†
Elysa Jui1† Kavya L. Singampalli1,2,3†
Kavya L. Singampalli1,2,3† Kevin Shani4
Kevin Shani4 Yao Ning3Jennifer P. Connell1
Yao Ning3Jennifer P. Connell1 Ravi K. Birla3
Ravi K. Birla3 Paul L. Bollyky5Christopher A. Caldarone6
Paul L. Bollyky5Christopher A. Caldarone6 Sundeep G. Keswani3
Sundeep G. Keswani3 K. Jane Grande-Allen1*
K. Jane Grande-Allen1*- 1Department of Bioengineering, Rice University, Houston, TX, United States
- 2Medical Scientist Training Program, Baylor College of Medicine, Houston, TX, United States
- 3Laboratory for Regenerative Tissue Repair, Division of Pediatric Surgery, Department of Surgery, Baylor College of Medicine and Texas Children's Hospital, Houston, TX, United States
- 4John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA, United States
- 5Division of Infectious Diseases, Department of Medicine, Stanford University School of Medicine, Stanford, CA, United States
- 6Division of Congenital Heart Surgery, Departments of Surgery and Pediatrics, Baylor College of Medicine and Texas Children's Hospital, Houston, TX, United States
Children with acquired heart disease face significant health challenges, including a lifetime of strict medical management, multiple cardiac surgeries, and a high mortality risk. Though the presentation of these conditions is diverse, a unifying factor is the role of immune and inflammatory responses in their development and/or progression. For example, infectious agents have been linked to pediatric cardiovascular disease, leading to a large health burden that disproportionately affects low-income areas. Other implicated mechanisms include antibody targeting of cardiac proteins, infection of cardiac cells, and inflammation-mediated damage to cardiac structures. These changes can alter blood flow patterns, change extracellular matrix composition, and induce cardiac remodeling. Therefore, understanding the relationship between the immune system and cardiovascular disease can inform targeted diagnostic and treatment approaches. In this review, we discuss the current understanding of pediatric immune-associated cardiac diseases, challenges in the field, and areas of research with potential for clinical benefit.
Introduction
Cardiovascular diseases (CVDs) comprise a group of disorders that affect the structure and/or function of the heart, including coronary artery disease, arrhythmia, peripheral arterial disease, and congenital heart disease (CHD), among others. Statistics indicate that a large portion of the health burden of CVD is concentrated in adults. However, there is also a significant burden of both acquired and congenital heart disease in the pediatric population, each accounting for hundreds of thousands of childhood deaths annually (1). CHD typically refers to structural disease that forms in utero, whereas acquired cardiovascular disease develops after birth. The list of acquired pediatric cardiovascular diseases is extensive, including rheumatic heart disease, myocarditis, vasculopathies, and cardiomyopathy (2); these conditions are often medically managed with serial imaging and medications (3).
Both congenital and acquired heart disease impose significant morbidity on children, including ICU stays, complex medical management, and a high risk of mortality (4). These diseases also impose long-term ramifications, including a risk of myocardial ischemia, valve damage, arrhythmia, and cardiomyopathy (2, 5). Furthermore, with the recent outbreak of COVID-19, it has been shown that pre-existing cardiovascular disease significantly increases the risk of mortality in COVID-19 patients (6), underscoring the importance of addressing these conditions.
Investigating the role of the immune response in the pathogenesis of CVD may be beneficial in children, as it has proven to be a fruitful avenue in adult cardiac disease. In adults, the role of the immune system has been extensively studied in valve disease and post-ischemic remodeling (7, 8). For example, after a myocardial infarction (MI), the inflammatory and cellular immune responses play a role in replacing damaged cardiomyocytes with scar tissue (9). Additionally, some cardiac arrhythmias following an MI have been associated with the systemic presence of inflammation and an increase in the presence of cardiac macrophages, supporting the involvement of macrophage-fibroblast crosstalk in the development of arrhythmias (10). Similar mechanisms may be relevant to pediatric cardiovascular disease as well, since changes such as remodeling, conduction disorders, and fibrosis are not unique to adults (9).
Building upon information from adult cardiac disease and the associated immune and inflammatory responses, we can further develop our understanding of pediatric disease. In this review, we will focus on the acquired pediatric cardiac diseases depicted in Figure 1 that have an established immune or inflammatory component in their development or progression. Due to the significant overlap between immune and inflammatory mechanisms in each condition, we have organized the diseases discussed by similarities in their pathogenesis. The following sections include an overview of subsets of acquired pediatric heart disease separated according to current knowledge of their underlying immune mechanisms, including pathogen-induced structural cardiac disease, myocardial inflammatory disease, inflammation-mediated cardiac fibrosis, and inflammation-mediated vascular disease, summarized in Table 1. Within each category, we highlight the role of both the immune and inflammatory responses in disease development. This understanding of disease pathophysiology can inform mechanisms underlying the development of pediatric heart disease, its progression, and best diagnostic and therapeutic practices.
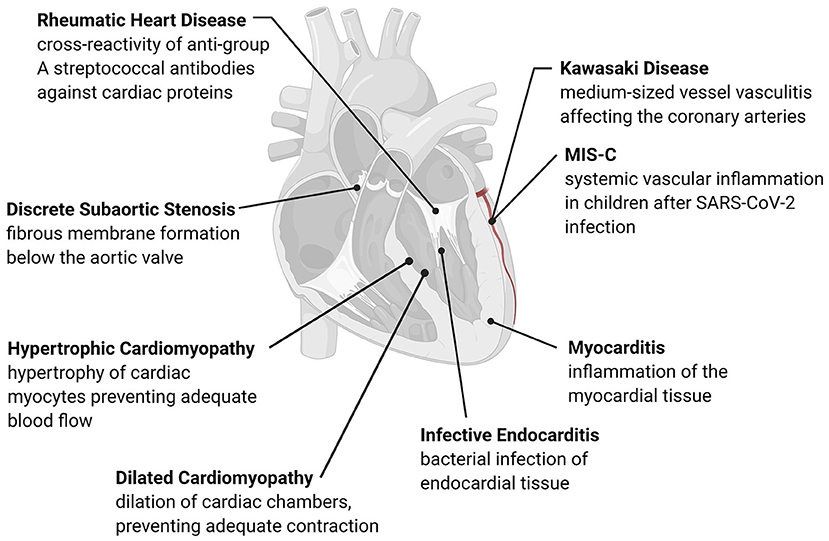
Figure 1. Overview of acquired pediatric heart disease. Acquired pediatric heart diseases can arise as a reaction to various stresses, including flow differences, structural changes, or pathogens. The development of many of these conditions has an immune or inflammatory trigger that causes remodeling of the cardiac structures or activation of native immune cells. Many of these conditions are progressive, leading to long-term complications such as valve damage, fibrosis, and adverse cardiac remodeling.
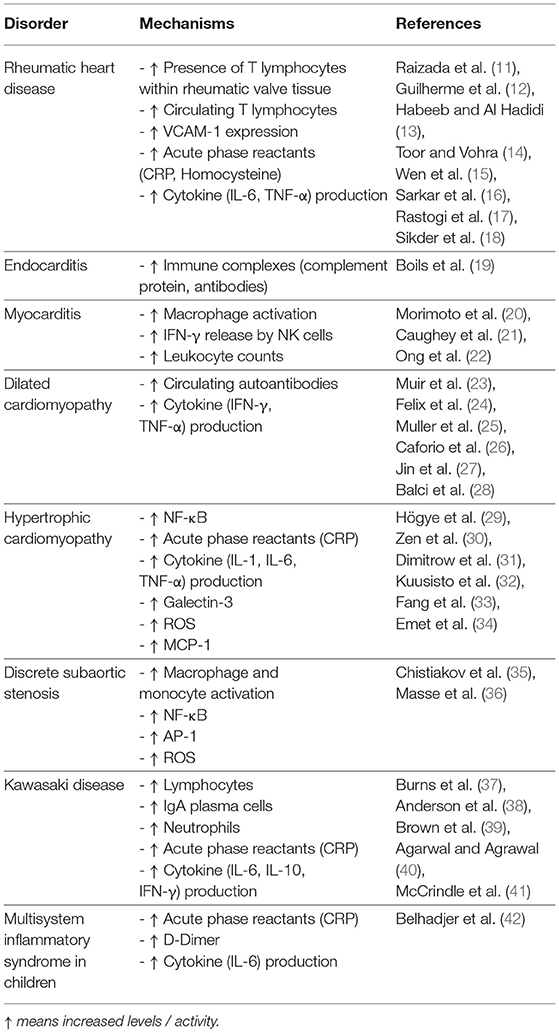
Table 1. Immune and Inflammatory markers associated with acquired heart disease.
Pathogen-Induced Acquired Heart Disease
Many acquired pediatric cardiac diseases are caused by known pathogens. In the case of rheumatic heart disease (RHD) and endocarditis, cardiac valve damage results from either direct cardiac infection, as seen in infective endocarditis, or complications caused by an immune response to bacterial pathogens, as seen in RHD (43–46). Defining the immune and inflammatory responses at play in these conditions can improve screening and diagnostic techniques and mitigate long-term valve damage to reduce the global health burden of pediatric heart disease.
Rheumatic Heart Disease
Antibody-mediated rheumatic heart disease (RHD) is one of the main causes of pediatric heart disease globally, accounting for over 200,000 new diagnoses and deaths each year (47). Unlike most pediatric cardiac diseases, the mechanisms underlying RHD and preventative treatments are relatively well-understood. RHD develops as a complication of inadequately treated Group A streptococcal (GAS) pharyngitis, where antibodies against streptococcal M protein cross-react with cardiac α-helical proteins in a process known as biomimicry (43), shown in Figure 2A. This reaction induces valvular fibrosis and calcification, leading to a characteristic “fish mouth” appearance of the mitral valve and clinical mitral valve stenosis (44).
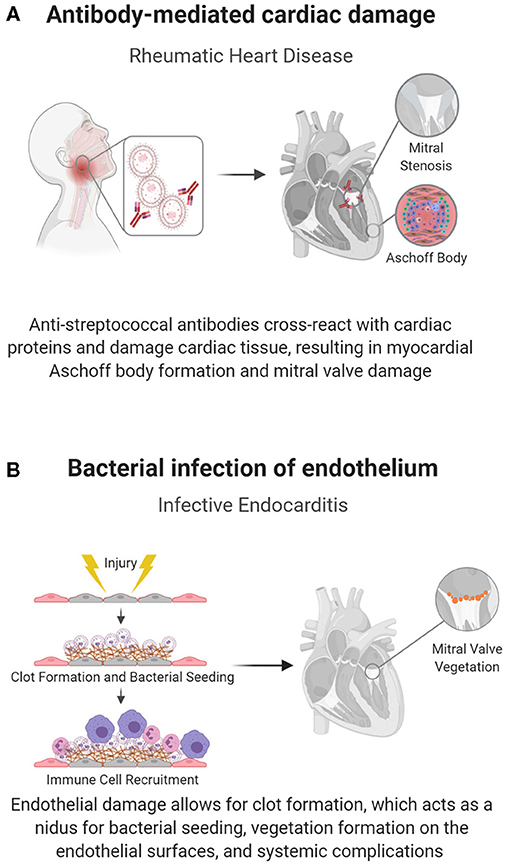
Figure 2. Pathogen-induced acquired pediatric heart disease. Some pediatric cardiac conditions have well-defined immune causes in response to an infectious organism. (A) In RHD, biomimicry, in which antibodies against Group A streptococcal bacteria cross react with cardiac proteins, causes cardiac damage, specifically mitral valve fibrosis and subsequent stenosis. (B) The endothelium can also become directly infected, as seen in endocarditis, in which flow induced damage creates a nidus for bacterial seeding.
Although the treatment of GAS pharyngitis with penicillin can prevent its progression into RHD, it is still a major cause of childhood death worldwide due to challenges in health care infrastructure and antibiotic availability (48). Understanding the role of the immune response in disease progression can therefore be beneficial as a target of future research in the diagnosis or treatment of RHD.
Recent studies have shown that in addition to biomimicry, cellular immune responses play a role in the development of RHD (49). These immune responses can cause carditis, but most significantly lead to mitral valve disease (50) as both antibodies and T-cells target cardiac myosin, valvular endothelial cells, the basement membrane, laminin, and vimentin (51). Damaged valve tissue in RHD is characterized by the abnormal presence of T-lymphocytes and the infiltration of helper CD4+ and cytotoxic CD8+ T-cells. The involvement of the immune system is further supported by the pathognomonic histologic finding of myocardial Aschoff bodies, granulomatous lesions that contain specific monocytes known as Anitschkow cells, and other immune cells (44). Patients with RHD also have circulating cross-reactive T-lymphocytes targeting both streptococcal antigens and cardiac tissue (12).
Beyond the initial antibody or lymphocyte insult to the valve, inflammation can promote disease progression. Damage to endothelial cells increases Vascular Cell Adhesion Molecule (VCAM) expression, allowing for improved T-cell adherence and localized, progressive inflammation (18). This inflammatory response involves the expression of proinflammatory molecules including Interleukin (IL)-6, Tumor Necrosis Factor (TNF)-α, IL-8, IL-2, and acute phase reactants (52); additionally, it is chronically supported by higher levels of the inflammatory markers C-reactive protein (CRP) and homocysteine (13). Peripheral levels of both IL-6 and TNF-α are positively correlated with severe valvular disease and calcification (53). Though larger scale studies of these and other markers are necessary to generalize the role of cytokines throughout the course of disease, they may be promising as predictors of the extent of valve damage, and thereby be beneficial to the early diagnosis and monitoring of RHD progression.
Infective Endocarditis
Infective endocarditis, or infection of the endocardial layer of the heart, is rare in children and mainly affects children with CHDs with prior surgical repairs, indwelling cardiac devices, or central venous catheters (45, 46). When it manifests, it can have devastating consequences due to bacterial biofilm formation, valvular damage, and potential infectious emboli and bacteremia, all of which necessitate prolonged antibiotic regimens (54).
The causative bacteria, typically gram-positive cocci (Staphylococcus aureus, viridans group streptococcus, enterococcus), have specialized adhesin proteins that facilitate adherence to the cardiac endothelium; however, adhesion is often dependent on endothelial damage, as seen in the abnormal flow patterns of CHD (45, 46). Damage to the endothelium exposes thromboplastin and tissue factor, inducing fibrin deposition, and the development of a non-bacterial thrombotic endocarditis (NBTE). Bacteria can then attach at the NBTE and replicate, initiating infective endocarditis (Figure 2B) (54). The infectious nidus can present either on the cardiac wall where abnormal flow jets cause endothelial damage or downstream of the damage, where slow flow patterns cause bacterial stasis, usually on valves in the left heart (45, 54). Structural heart disease also introduces a risk factor for endocarditis, as it is associated with areas of local flow disturbances. Accordingly, children with unrepaired ventricular septal defects (VSDs) have up to a 12% risk of contracting this otherwise rare condition (54). Furthermore, structural disease is often treated with implanted prosthetic devices, which can act as a nidus for bacterial seeding.
Accompanying the immune response to bacterial infection, a major immune complication of infective endocarditis is the development of glomerulonephritis. This outcome is caused by immune complex deposition (including complement proteins and/or antibodies) within the kidney, leading to vascular blockages and renal inflammation (19). Glomerulonephritis presents its own long-term challenges as children face chronic damage to their kidney function. Therefore, reducing the risk of these progressive complications is another area where understanding immune pathways can benefit children with heart disease.
Inflammation-Mediated Acquired Heart Disease
Myocarditis and dilated cardiomyopathy (DCM) are associated with cardiac inflammation, with DCM involving the remodeling and dilation of cardiac chambers which can result from prolonged myocarditis. Unlike the bacterial causes of RHD and endocarditis, myocarditis and DCM likely have viral triggers that cause myocardial inflammation, and introduce an autoimmune component targeting cardiac tissue (55–57). Understanding the specific immune and inflammatory mechanisms in myocarditis and DCM can lead to targeted treatments and the prevention of chronic complications from these conditions.
Myocarditis
One form of inflammatory heart disease that presents in a small percentage of children is myocarditis, a disease characterized by the inflammation of the myocardium (Figure 3A). Clinical manifestations vary and most patients experience mild flu-like symptoms or are completely asymptomatic. However, more severe cases may result in acute or chronic heart failure (58), or conduction disturbances such as atrioventricular (AV) blocks (20, 21). Complications from myocarditis are common in children, with up to a 25% mortality rate and only half recovering completely (57).
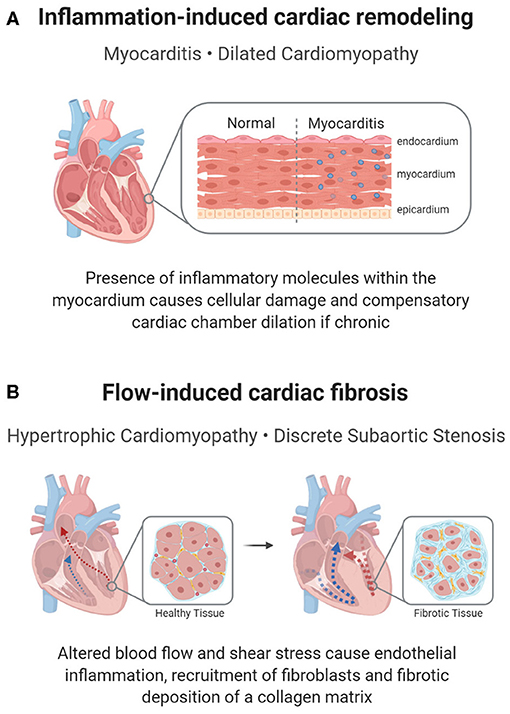
Figure 3. Inflammation-mediated pediatric cardiac disease. In addition to a cellular response, a molecular inflammatory response can lead to pediatric heart disease or its complications. (A) In myocarditis, inflammation of the cardiac muscle can cause reactionary remodeling, in some cases leading to DCM. (B) Remodeling can also be seen in shear stress induced inflammation, which propagates a pro-fibrotic environment in HCM or DSS.
The pathogenesis of viral and non-viral myocarditis involves infection of the myocardium, immune cell responses, activation of inflammatory pathways, tissue remodeling, and ultimately resolution. Myocarditis can have an infectious or non-infectious etiology. Non-infectious myocarditis, due in part to challenges in identifying infectious agents and data from animal models, is believed to have an autoimmune contribution to disease pathology and progression (56, 59). This is further supported by the association of autoimmune diseases, such as systemic lupus erythematosus (SLE), with myocarditis (56, 60). On the other hand, infectious myocarditis is caused by an identifiable viral agent, often the Coxsackie B virus in children (57). The pathogen triggers an immune response, resulting in myocardial edema and impairment of systolic and diastolic function (58). Infection by viral agents can also trigger macrophage activation and interferon (IFN)-γ release by natural killer (NK) cells, which, when unregulated, result in myocyte injury and cardiac dysfunction (22).
Despite the striking evidence of an immune response, immunosuppressive therapies, such as prednisone and cyclosporine, do not clearly improve the outcomes of myocarditis. In children, the administration of intravenous immunoglobulin (IVIG) has shown promising results in improving outcomes; however, this response has not yet been observed in adults (61, 62). Therefore, more specific anti-inflammatory therapies may be necessary, and the immune mechanisms involved in the propagation of myocarditis should be further investigated for novel treatment options.
Acute episodes of myocarditis can also progress to chronic disease or dilated cardiomyopathy (DCM), discussed below, due to the persistence of pro-inflammatory cytokines that are critical for removing the infectious agent (56). An area for further research is understanding the progression of acute myocarditis into DCM, the frequency of which may be much higher than currently predicted due to limits in viral detection and myocardial biopsy sampling. Furthermore, the histological evidence of myocarditis can deteriorate quickly, introducing challenges in accurately detecting myocarditis as a precursor to DCM. Consequently, only one-third of all cases of pediatric DCM have a known cause, making their subsequent treatment non-specific (58). Understanding this time course and defining key mechanistic targets and therapeutic windows are essential for improving outcomes.
Dilated Cardiomyopathy
As noted previously, an acute episode of myocarditis can result in DCM. Although cardiomyopathies are generally rare in children, their prognosis is extremely poor, due to limited treatment options and the limited ability to restore native cardiac function. Nearly 40% of children with either symptomatic DCM or hypertrophic cardiomyopathy (HCM) receive a transplant or die within 2 years due to cardiac complications, and survival has improved only slightly over the last two decades (63).
Of those DCM cases with a known etiology, half involve a viral pathogen. Overall, 20% of myocardial biopsies in DCM showed viral infection, with enterovirus and adenovirus being the causative agents (55). Additionally, viral pathogens are frequently credited for the pathogenesis of idiopathic dilated cardiomyopathy (IDCM), and studies have found elevated virus-specific antibody titers in many cases of IDCM (23). Regardless of a viral trigger, most cases of DCM present with cardiac dilation and defective left ventricular or biventricular contraction (64), and are commonly associated with inflammatory responses, including the presence of TNF-α and IFN-γ (28).
Similar to myocarditis, several studies have suggested that autoimmune mechanisms, in addition to a genetic pre-disposition, regulate the pathogenesis of pediatric IDCM. IDCM is hypothesized to be initiated by a pathogenic trigger in patients with pre-disposing human leukocyte antigen (HLA) alleles. Additionally, autoantibody presence in unaffected relatives of patients with IDCM and dysfunctional cardiac activity in 20% of first-degree relatives of DCM patients have been noted. These findings, in addition to associations between HLA-DR4 and both IDCM and anti-cardiac antibodies, support a genetic autoimmune cause (26, 27, 65). Recent studies have also shown that immunosuppression and immunoadsorption reduce the number of circulating autoantibodies and result in improved clinical outcomes, further supporting the relevance of immunological mechanisms in certain cases of DCM development (25). Therefore, investigation into the mechanisms behind the viral presence in DCM is warranted to address potential causative agents and offer prophylactic treatment approaches.
Inflammation-Mediated Fibrotic Disease
Hypertrophic cardiomyopathy (HCM) and discrete subaortic stenosis (DSS) are both caused by inflammation-mediated remodeling, similar to DCM. In HCM and DSS, localized inflammatory responses are caused by flow disturbances, which lead to the recruitment of inflammatory cells and ultimately, the formation of fibrotic tissue (35, 66, 67). Studying the causes of these conditions can allow for the development of better diagnostics and therapies to prevent chronic remodeling and functional cardiac changes in children.
Hypertrophic Cardiomyopathy
One of the more severe forms of inflammatory fibrotic remodeling is observed in hypertrophic cardiomyopathy (HCM), a pediatric condition in which septal hypertrophy causes tissue growth into the left ventricle. This growth leads to potential damage to the mitral valve and obstruction of the left ventricular outflow tract (LVOT) (68). HCM places children at risk of acute complications; in fact, HCM is the most common known cause of sudden cardiac death in young athletes, where it accounts for 36% of sudden cardiac deaths resulting from ventricular arrhythmias (5, 69). Many genetic mutations have been identified in children with HCM, typically in genes coding for sarcomere proteins important in cardiac contraction, including MYH7 and MYBPC3 (70); however, beyond a genetic pre-disposition, mechanisms of HCM progression have not been clearly defined.
In HCM, the tissue overgrowth includes both hypertrophic cardiomyocytes and fibrotic tissue, indicating that inflammation-induced fibrosis may contribute to hypertrophy (Figure 3B). Histologically, myectomy samples show interstitial and endocardial fibrosis and inflammation, with a disarray of myocytes (71). Clinically, children with HCM have a more pronounced cardiac presence of immune cells and inflammatory molecules (NF-κB, CRP, interleukins, TNF-α), with higher levels corresponding to increased fibrosis (32, 33). The inflammatory response is supported by genetic studies, which show an upregulation of pathways associated with immune cell activation and innate immune cell degranulation in children with HCM (72). Though many cellular processes could account for the inflammatory changes, a proposed mechanism for the inflammatory changes is an increase in neutrophil extracellular traps (NETs) in the left ventricle in children with HCM due to localized flow changes instigating a pro-thrombotic response, which attracts neutrophils (66). Within these NETs, neutrophils release their nuclear contents into the extracellular matrix (ECM) and subsequently trap cells, including inflammatory and fibrotic cells, leading to inflammation, hypoxic and reperfusion injury and fibrosis (73). While localized inflammation, fibrosis, and thrombotic responses caused by NETs have been studied in HCM, further research into the role of NETs in childhood disease pathogenesis could provide a novel approach to defining HCM-associated fibrosis (66).
Markers of endothelial dysfunction, remodeling, and immune cell infiltration are present at even early stages of HCM (74), potentially enabling earlier detection. Galectin-3, a systemic marker of cardiac fibrosis, is elevated in patients with HCM and is significantly higher in patients with a history of cardiac arrest, syncope, fatal arrhythmias, or sudden cardiac death (34). The extent of fibrosis can also be correlated with increases in Stromal Cell-Derived Factor (SDF) and Macrophage Chemoattractant Protein-1 (MCP-1), which increase immune cell recruitment (33), and chronic systemic increases in pro-fibrotic and inflammatory cytokines such as IL-1, TNF-α, and CRP (32).
Inflammatory changes are more prominent in the case of hypertrophic obstructive cardiomyopathy, where myocyte hypertrophy blocks the forward flow of blood. The obstruction-generated flow disruption alters the mechanical stresses experienced by cardiac cells and also leads to changes in the cytokine profile, which subsequently induces myofibroblast differentiation and collagen deposition (75). These cytokine changes include elevations in IL-6 in left ventricular dilation (30) increased MCP-1, and subsequent macrophage recruitment in systolic dysfunction (76), and higher levels of reactive oxygen species (ROS), which disrupt typical endothelial function (31). Since the increased immune and inflammatory presence can act as positive feedback to drive pathologic changes in HCM, the study of molecules along this pathway as therapeutic targets has the potential to prevent adverse cardiac remodeling.
Discrete Subaortic Stenosis
Similar inflammatory and fibrotic mechanisms have been suggested to cause discrete subaortic stenosis (DSS), a condition in which a fibrotic membrane forms below the aortic valve, obstructing blood flow into the aorta (36, 77). In DSS, an irregular LVOT architecture, such as increased mitral and aortic valve separation or a steep aortoseptal angle, results in abnormal flow patterns and changes in wall shear stress (77). Consequently, this disturbed flow is believed to induce a proliferative fibrotic response from the endothelial cells and resident fibroblasts, resulting in the characteristic membrane formation of DSS (67) (Figure 3B). Histological analysis of septal myectomy in these patients shows endocardial and interstitial fibrosis, evidence of inflammation, vacuolization, and glycosaminoglycan and proteoglycan deposition (78), which implies that the development of this membrane may partly be due to an immune etiology.
Although the mechanism of membrane formation in DSS has not been specifically elucidated, there are hypotheses that indicate that shear stress-induced endothelial damage may cause a fibrotic response, similar to its effects in other tissues. Specifically, it has been shown in vasculature that at areas of elevated flow with high wall shear stress (WSS), the interfacial force caused by blood flow and positive WSS spatial gradients encourage cellular damage and ECM degradation (36, 79). This flow condition then results in the increased endothelial release of microRNA (miR)-155 and miR-205/712 which activate pro-inflammatory phenotypes in macrophages and monocytes, respectively. Additionally, the pro-inflammatory factors NF-κB and AP-1 are upregulated in this state, which increases the expression of adhesion molecules that promote leukocyte attachment to endothelial cells (35).
These inflammatory changes can be mitigated by altering flow characteristics from disturbed to steady flow. Specifically, laminar flow characteristics upregulate the anti-inflammatory factors Krüppel-like factor 2 (KLF2) and nuclear factor erythroid 2-related factor 2 (Nrf2), which can reduce inflammation through the suppression of NF-κB and Activator Protein (AP)-1. Though the inflammatory pathways that respond to endothelial damage have not been studied extensively in pediatric heart disease, studies focused on endothelial responses to changes in wall shear stress suggest a contributory role in the pathogenesis of DSS. Therefore, it is important to better define the interaction between biomechanical forces and fibrosis to understand the fibrotic mechanisms involved, and ultimately prevent the development or progression of these fibrotic conditions.
Inflammation-Mediated Vascular Disease
Similar to the acquired pediatric cardiac diseases discussed above, pediatric vascular disease also has a likely pathogenic trigger. Both Kawasaki Disease (KD) and multisystem inflammatory syndrome in children (MIS-C) are believed to have a viral cause, leading to inflammatory responses and vascular damage (80, 81). In both cases, the inflammation can have profound implications on the heart, leading to acute conditions, such as heart failure, and long-term complications, including coronary artery aneurysms (81, 82).
Kawasaki Disease
Kawasaki disease (KD), an acute vasculitis that typically develops in children within the first few years of life as a complication of an infection (80), is the primary cause of acquired pediatric heart disease (83). KD is a clinical diagnosis in children who present with prolonged high fever, rash, extremity swelling, and inflammation of the lips, mouth, and throat. If untreated, it is a critical risk factor for subsequent coronary artery aneurysms and myocardial infarction in early adulthood (58, 84).
The acute inflammatory symptoms of KD suggest an infectious cause, which is further supported by their resolution within 14 days and the young age group in which it presents, namely children under age 5. KD is rarely seen in infants, who are protected by maternal antibodies, and in adults who have likely developed immunity to the causative agent (40). Though no specific pathogenic trigger for KD has been elucidated, there is compelling evidence of immune-mediated damage. For example, the clinical symptoms of a rash and red tongue have been compared to a toxic shock-like reaction, as seen with bacterial superantigen infection. The vascular invasion of cytotoxic T-cells in KD, on the other hand, support a viral etiology (40).
Animal models of KD facilitate research of the immune responses seen in this condition. In murine models, the phenotype of KD is created by exposing mice to extracts from bacteria, such as Lactobacillus casei (85, 86), or yeast, such as Candida albicans (87). These models provide a promising route to understand the cytokine profiles that lead to vascular damage, such as increased IL-1 and granulocyte macrophage colony stimulating factor (GM-CSF) (86, 88), and possible therapeutic options (85, 87).
Through clinical studies and studies in animal models, researchers have found that the complications of KD have substantial immune and inflammatory components. Along with the clinical presentation of fever, rash, and conjunctivitis, there is a markedly elevated expression of markers of systemic inflammation (i.e., CRP and IFN-γ) and the upregulation of inflammatory cytokine (IL-6, IL-10) production (40, 41). Vascular damage, which is a critical consequence of KD, may involve increased T-cell activity, increased effector T-cell specific cytokines, and reduced regulatory T-cell responses (40). In cases of coronary damage, additional immune cells, such as neutrophils, lymphocytes (specifically cytotoxic CD8+ T-cells), and IgA plasma cells are seen at the coronary lesion sites (39). These T-cells and macrophages within the coronary arteries secrete TNF-α, which activates matrix metalloprotease (MMP)-9 to degrade extracellular matrix components, reducing the integrity of vasculature and pre-disposing children to aneurysm formation (40) (Figure 4A). Therefore, understanding and treating the long-term vascular inflammatory profile could prevent the development of coronary complications and potential cardiac ischemia resulting from KD.
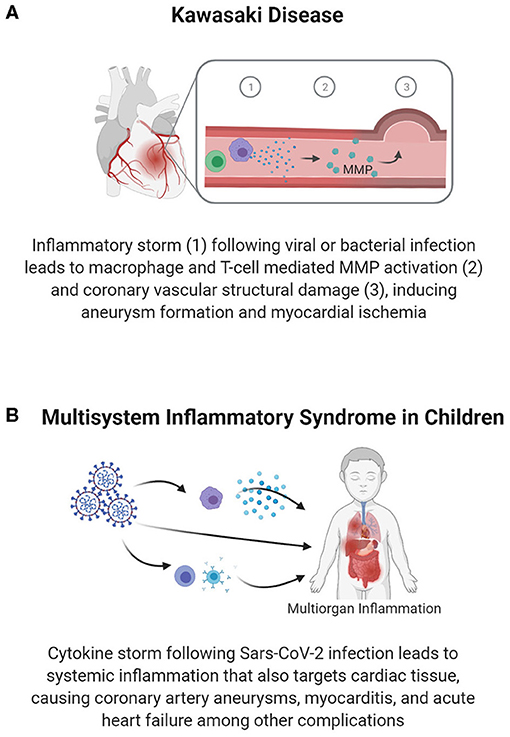
Figure 4. Vascular inflammation-mediated pediatric cardiovascular disease. Inflammation following infection can lead to vascular damage and cardiac pathology. Inflammation and an associated immune response within the vasculature can cause vascular structural damage, as seen in coronary aneurysms in Kawasaki disease (A), which reduces blood flow in the coronary arteries and predisposes individuals to premature MI. In Multisystem Inflammatory Syndrome in Children (B), systemic inflammation causes damage to various organ systems, including targeting vascular and cardiac tissues to cause myocarditis, coronary artery aneurysms, and heart failure.
The role of the immune system is further supported by the use of IVIG as an effective form of treatment (58). It is hypothesized that IVIG may target pathogenic antigens, regulate cytokine production, and/or suppress effector T-cell activity and antibody production (40). Better elucidating these mechanisms along with the cause of KD can allow for potential advancements in disease intervention, especially for KD that is refractory to IVIG treatment or KD that is not treated acutely.
Multisystem Inflammatory Syndrome in Children
In the midst of the COVID-19 pandemic, a sudden spike in cases of a Kawasaki-like disease known as multisystem inflammatory syndrome in children (MIS-C) has been reported (89, 90). The similarities of MIS-C to KD and the pathways causing MIS-C are still being studied. Nonetheless, the role of a robust inflammatory response has been identified. As shown in Figure 4B, current data indicates multi-organ inflammatory involvement in MIS-C, with children suffering from gastrointestinal, respiratory, hematologic, neurologic, and cardiac symptoms presenting approximately 4 weeks after infection with SARS-CoV-2 (81, 91). Relative to KD, patients with MIS-C are more likely to present with myocardial complications, including ventricular dysfunction, arrhythmia, and shock (81). The collective set of symptoms leads to 75% of children with MIS-C being admitted to the PICU, and 4–5% requiring EMCO (82).
The cardiac complications of MIS-C vary, but range from pericarditis to vascular damage, including coronary artery aneurysms, and heart failure (81). The more severe end of the spectrum includes children who present with febrile cardiogenic shock or left ventricular dysfunction in conjunction with systemic inflammation and endothelial dysfunction (42), as evidenced by the elevated inflammatory markers CRP and D-Dimer. The immune and inflammatory responses are characterized by an IL-6 and IFN-γ cytokine storm, macrophage activation, elevated neutrophil counts, lymphopenia, and an elevated neutrophil: lymphocyte ratio (92, 93). Additionally, >70% of children with MIS-C show increased biomarkers of cardiac damage or dysfunction, such as natriuretic peptides and/or cardiac troponin (82). Recent studies have also correlated the need for intensive care in children with MIS-C and the levels of systemic inflammatory markers (94); though these are correlations based on current clinical data, these inflammatory markers may have utility in predicting the severity of MIS-C associated cardiac complications in children as further studies are conducted.
Similar to KD, treatment for the majority of patients with MIS-C consists primarily of IVIG administration, whereas patients with persistent inflammatory states are treated with an IL-1 receptor antagonist (42). There is a greater resistance to IVIG in MIS-C relative to KD (82), necessitating improved therapeutics. Therefore, in the face of this novel immune complication, a better understanding of the underlying immune mechanisms can help predict long-term cardiac complications and appropriate treatment regimens.
Conclusion
Despite their prevalence and high clinical impact, the mechanisms causing many pediatric heart diseases have yet to be elucidated. This knowledge gap may be due to the heterogeneity of the causes of these diseases and the broad spectrum of phenotypes. Despite the limited understanding of these conditions, there is substantial support for the role of immune and inflammatory responses in causing these heart diseases or their complications. The major mechanism linking the two is an increase in pro-inflammatory mediators and immune cell recruitment, which can foster an autoimmune response against cardiac proteins, induce cardiac remodeling, cause fibrosis due to changes in flow patterns, and/or stimulate vascular inflammatory responses. However, complicating our basic understanding of these disease processes is the complexity of the immune system and the diverse outcomes that can result from an immune response, making the determination of causative immune pathways difficult.
Applying immune mechanisms from adult cardiac disease can provide the foundation to studying immune and inflammatory mechanisms in children. For example, cardiac remodeling after a myocardial infarction (MI) in adults relies on a delicate balance of pro- and anti-inflammatory markers and the temporal control of immune cell recruitment, such as macrophages, neutrophils, and T-lymphocytes (7, 9, 95). Similar mechanisms can be explored in pediatric conditions involving cardiac remodeling or fibrotic healing responses. Moreover, adult cardiac disease can inform us on research methods applicable to pediatric diseases. For example, the development of murine models of MI has allowed for in vivo studies of immune responses in cardiac tissue (96). Additionally, immune specific modeling, such as multi-scale modeling of leukocyte transendothelial migration (TEM) during atherogenesis combine spatiotemporal events that occur at the cellular level to identify the role of leukocytes in atherosclerotic plaque evolution (97).These techniques not only provide the potential to extrapolate findings from adults to better model pediatric disease, but also prevent the development of adult-onset complications, including MI or peripheral vascular disease.
Furthermore, as pediatric heart diseases become better understood, and the role of the immune system and inflammation clarified, there may be a potential to treat pediatric heart disease with immune-modulating therapies. For example, targeted anti-inflammatories that mitigate the immune response have shown benefits in reducing adverse cardiac remodeling in adults (98, 99). Some of these therapies may also be applicable in children and can help reduce the long-term risks of pediatric heart disease; however, the use of current and future immunomodulatory therapies in the children relies on a more holistic understanding of pediatric heart disease. Therefore, it is imperative that we clarify the precise contribution of the immune response in individual cardiac conditions to improve the quality-of-life and reduce the risk of fatal cardiac complications in children with heart disease.
Author Contributions
KG-A conceptualized the article. EJ and KLS conducted the research and drafted the manuscript. All authors critically edited the article for intellectual merit.
Funding
This work was supported by the National Institutes of Health (R01 HL140305 to KG-A and SK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EA declared a shared affiliation, with no collaboration, with the authors KLS and YN to the handling editor at the time of the review.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The illustrations were created using Biorender.com.
Abbreviations
CRP, C-reactive protein; ECM, extracellular matrix; IFN, interferon; IL, interleukin; IVIG, intravenous immunoglobulin; MCP, macrophage chemoattractant protein; MMP, matrix metalloprotease; NF-κB, Nuclear Factor–κB; ROS, reactive oxygen species; TNF, tumor necrosis factor.
References
1. Curry C, Zuhlke L, Mocumbi A, Kennedy N. Acquired heart disease in low-income and middle-income countries. Arch Dis Child. (2018) 103:73–7. doi: 10.1136/archdischild-2016-312521
2. Agarwal N, Taneja S. Acquired heart disease in children is not necessarily rheumatic. Trop Doct. (2019) 49:181–4. doi: 10.1177/0049475519833513
3. Moller JH, Taubert KA, Allen HD, Clark EB, Lauer MR. Cardiovascular health and disease in children: current status. A Special Writing Group from the Task Force on Children and Youth, American Heart Association. Circulation. (1994) 89:923–30. doi: 10.1161/01.CIR.89.2.923
4. Leong K, Kane JM, Joy FB. Acquired cardiac disease in the pediatric intensive care unit. Pediatr Ann. (2018) 47:e280–5. doi: 10.3928/19382359-20180620-01
5. Maurizi N, Passantino S, Spaziani G, Girolami F, Arretini A, Targetti M, et al. Long-term outcomes of pediatric-onset hypertrophic cardiomyopathy and age-specific risk factors for lethal arrhythmic events. JAMA Cardiol. (2018) 3:520–5. doi: 10.1001/jamacardio.2018.0789
6. Alsaied T, Aboulhosn JA, Cotts TB, Daniels CJ, Etheridge SP, Feltes TF, et al. Coronavirus disease 2019 (COVID-19) pandemic implications in pediatric and adult congenital heart disease. J Am Heart Assoc. (2020) 9:e017224. doi: 10.1161/JAHA.120.017224
7. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11:255–65. doi: 10.1038/nrcardio.2014.28
8. Mathieu P, Bouchareb R, Boulanger CM. Innate and adaptive immunity in calcific aortic valve disease. J Immunol Res. (2015) 2015:851945. doi: 10.1155/2015/851945
9. Lai SL, Marin-Juez R, Stainier DYR. Immune responses in cardiac repair and regeneration: a comparative point of view. Cell Mol Life Sci. (2019) 76:1365–80. doi: 10.1007/s00018-018-2995-5
10. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. (2018) 18:733–44. doi: 10.1038/s41577-018-0065-8
11. Raizada V, Williams RC Jr., Chopra P, Gopinath N, Prakash K, Sharma KB, et al. Tissue distribution of lymphocytes in rheumatic heart valves as defined by monoclonal anti-T cell antibodies. Am J Med. (1983) 74:90–6. doi: 10.1016/0002-9343(83)91124-5
12. Guilherme L, Cunha-Neto E, Coelho V, Snitcowsky R, Pomerantzeff PM, Assis RV, et al. Human heart-infiltrating T-cell clones from rheumatic heart disease patients recognize both streptococcal and cardiac proteins. Circulation. (1995) 92:415–20. doi: 10.1161/01.CIR.92.3.415
13. Habeeb NM, Al Hadidi SI. Ongoing inflammation in children with rheumatic heart disease. Cardiol Young. (2011) 21:334–9. doi: 10.1017/S1047951111000047
14. Toor D, Vohra H. Immune responsiveness during disease progression from acute rheumatic fever to chronic rheumatic heart disease. Microbes Infect. (2012) 14:1111–7. doi: 10.1016/j.micinf.2012.07.003
15. Wen Y, Zeng Z, Gui C, Li L, Li W. Changes in the expression of Th17 cell-associated cytokines in the development of rheumatic heart disease. Cardiovasc Pathol. (2015) 24:382–7. doi: 10.1016/j.carpath.2015.07.006
16. Sarkar S, Rastogi M, Chaudhary P, Kumar R, Arora P, Sagar V, et al. Association of rheumatic fever & rheumatic heart disease with plausible early & late-stage disease markers. Indian J Med Res. (2017) 145:758–66. doi: 10.4103/ijmr.IJMR_1554_14
17. Rastogi M, Sarkar S, Makol A, Sandip Singh R, Saikia UN, Banerjee D, et al. Anti-endothelial cell antibody rich sera from rheumatic heart disease patients induces proinflammatory phenotype and methylation alteration in endothelial cells. Genes Dis. (2018) 5:275–89. doi: 10.1016/j.gendis.2018.02.002
18. Sikder S, Rush CM, Govan BL, Alim MA, Ketheesan N. Anti-streptococcal antibody and T-cell interactions with vascular endothelial cells initiate the development of rheumatic carditis. J Leukoc Biol. (2020) 107:263–71. doi: 10.1002/JLB.4MA0919-096RR
19. Boils CL, Nasr SH, Walker PD, Couser WG, Larsen PC. Update on endocarditis-associated glomerulonephritis. Kidney Int. (2015) 87:1241–9. doi: 10.1038/ki.2014.424
20. Morimoto S, Kato S, Hiramitsu S, Uemura A, Ohtsuki M, Kato Y, et al. Role of myocardial interstitial edema in conduction disturbances in acute myocarditis. Heart Vessels. (2006) 21:356–60. doi: 10.1007/s00380-006-0922-4
21. Caughey RW, Humphrey JM, Thomas EP. High-degree atrioventricular block in a child with acute myocarditis. Ochsner J. (2014) 14:244–7.
22. Ong S, Rose NR, Cihakova D. Natural killer cells in inflammatory heart disease. Clin Immunol. (2017) 175:26–33. doi: 10.1016/j.clim.2016.11.010
23. Muir P, Nicholson F, Tilzey AJ, Signy M, English TA, Banatvala EJ. Chronic relapsing pericarditis and dilated cardiomyopathy: serological evidence of persistent enterovirus infection. Lancet. (1989) 1:804–7. doi: 10.1016/S0140-6736(89)92270-8
24. Felix SB, Staudt A, Dorffel WV, Stangl V, Merkel K, Pohl M, et al. Hemodynamic effects of immunoadsorption and subsequent immunoglobulin substitution in dilated cardiomyopathy: three-month results from a randomized study. J Am Coll Cardiol. (2000) 35:1590–8. doi: 10.1016/S0735-1097(00)00568-4
25. Muller J, Wallukat G, Dandel M, Bieda H, Brandes K, Spiegelsberger S, et al. Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy. Circulation. (2000) 101:385–91. doi: 10.1161/01.CIR.101.4.385
26. Caforio AL, Mahon NG, Baig MK, Tona F, Murphy RT, Elliott PM, McKenna JW. Prospective familial assessment in dilated cardiomyopathy: cardiac autoantibodies predict disease development in asymptomatic relatives. Circulation. (2007) 115:76–83. doi: 10.1161/CIRCULATIONAHA.106.641472
27. Jin B, Luo XP, Ni HC, Shen W, Shi HM, Li Y. A meta-analysis of HLA-DR polymorphism and genetic susceptibility to idiopathic dilated cardiomyopathy. Mol Biol Rep. (2012) 39:221–6. doi: 10.1007/s11033-011-0729-y
28. Balci SO, Col-Araz N, Baspinar O, Sever T, Balat A, Pehlivan S. Cytokine gene polymorphisms in childhood dilated cardiomyopathy: interferon- gamma, tumor necrosis factor-alpha and transforming growth factor - beta 1 genes are associated with the disease in Turkish Patients. Iran J Pediatr. (2013) 23:603–4.
29. Högye M, Mándi Y, Csanády M, Sepp R, Buzás K. Comparison of circulating levels of interleukin-6 and tumor necrosis factor-alpha in hypertrophic cardiomyopathy and in idiopathic dilated cardiomyopathy. Am J Cardiol. (2004) 94:249–51. doi: 10.1016/j.amjcard.2004.03.078
30. Zen K, Irie H, Doue T, Takamiya M, Yamano T, Sawada T, et al. Analysis of circulating apoptosis mediators and proinflammatory cytokines in patients with idiopathic hypertrophic cardiomyopathy: comparison between nonobstructive and dilated-phase hypertrophic cardiomyopathy. Int Heart J. (2005) 46:231–44. doi: 10.1536/ihj.46.231
31. Dimitrow PP, Undas A, Wołkow P, Tracz W, Dubiel SJ. Enhanced oxidative stress in hypertrophic cardiomyopathy. Pharmacol Rep. (2009) 61:491–5. doi: 10.1016/S1734-1140(09)70091-X
32. Kuusisto J, Karja V, Sipola P, Kholova I, Peuhkurinen K, Jaaskelainen P, et al. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart. (2012) 98:1007–13. doi: 10.1136/heartjnl-2011-300960
33. Fang L, Ellims AH, Beale AL, Taylor AJ, Murphy A, Dart MA. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am J Transl Res. (2017) 9:5063–73.
34. Emet S, Dadashov M, Sonsoz MR, Cakir MO, Yilmaz M, Elitok A, et al. Galectin-3: a novel biomarker predicts sudden cardiac death in hypertrophic cardiomyopathy. Am J Med Sci. (2018) 356:537–43. doi: 10.1016/j.amjms.2018.08.013
35. Chistiakov DA, Orekhov AN, Bobryshev VY. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol (Oxf). (2017) 219:382–408. doi: 10.1111/apha.12725
36. Masse DD, Shar JA, Brown KN, Keswani SG, Grande-Allen KJ, Sucosky P. Discrete subaortic stenosis: perspective roadmap to a complex disease. Front Cardiovasc Med. (2018) 5:122. doi: 10.3389/fcvm.2018.00122
37. Burns JC, Kushner HI, Bastian JF, Shike H, Shimizu C, Matsubara T, et al. Kawasaki disease: a brief history. Pediatrics. (2000) 106:E27. doi: 10.1542/peds.106.2.e27
38. Anderson MS, Burns J, Treadwell TA, Pietra BA, Glode PM. Erythrocyte sedimentation rate and C-reactive protein discrepancy and high prevalence of coronary artery abnormalities in Kawasaki disease. Pediatr Infect Dis J. (2001) 20:698–702. doi: 10.1097/00006454-200107000-00011
39. Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley HA. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. (2001) 184:940–3. doi: 10.1086/323155
40. Agarwal S, Agrawal KD. Kawasaki disease: etiopathogenesis and novel treatment strategies. Expert Rev Clin Immunol. (2017) 13:247–58. doi: 10.1080/1744666X.2017.1232165
41. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of kawasaki disease: a scientific statement for health professionals from the american heart association. Circulation. (2017) 135:e927–99. doi: 10.1161/CIR.0000000000000484
42. Belhadjer Z, Meot M, Bajolle F, Khraiche D, Legendre A, Abakka S, et al. Acute heart failure in multisystem inflammatory syndrome in children (MIS-C) in the context of global SARS-CoV-2 pandemic. Circulation. (2020) 142:429–36. doi: 10.1161/CIRCULATIONAHA.120.048360
43. Guilherme L, Kalil J, Cunningham M. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity. (2006) 39:31–9. doi: 10.1080/08916930500484674
44. Yanagawa B, Butany J, Verma S. Update on rheumatic heart disease. Curr Opin Cardiol. (2016) 31:162–8. doi: 10.1097/HCO.0000000000000269
45. Baltimore RS, Gewitz M, Baddour LM, Beerman LB, Jackson MA, Lockhart PB, et al. Infective endocarditis in childhood: 2015 update: a scientific statement from the American heart association. Circulation. (2015) 132:1487–515. doi: 10.1161/CIR.0000000000000298
46. Kelchtermans J, Grossar L, Eyskens B, Cools B, Roggen M, Boshoff D, et al. Clinical characteristics of infective endocarditis in children. Pediatr Infect Dis J. (2019) 38:453–8. doi: 10.1097/INF.0000000000002212
47. Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. (2005) 5:685–94. doi: 10.1016/S1473-3099(05)70267-X
48. Marijon E, Mirabel M, Celermajer DS, Jouven X. Rheumatic heart disease. Lancet. (2012) 379:953–64. doi: 10.1016/S0140-6736(11)61171-9
49. Toor D, Sharma N. T cell subsets: an integral component in pathogenesis of rheumatic heart disease. Immunol Res. (2018) 66:18–30. doi: 10.1007/s12026-017-8978-z
50. Kanagasingam A, Francis GR, Komagarajah B, Ladchumanan D, Sivapramyan A, Packiyarajah P, et al. Pattern of rheumatic valvular involvement and its contribution towards valvular malfunction in young adults. Ceylon Med J. (2019) 64:91–7. doi: 10.4038/cmj.v64i3.8951
51. Delunardo F, Scalzi V, Capozzi A, Camerini S, Misasi R, Pierdominici M, et al. Streptococcal-vimentin cross-reactive antibodies induce microvascular cardiac endothelial proinflammatory phenotype in rheumatic heart disease. Clin Exp Immunol. (2013) 173:419–29. doi: 10.1111/cei.12135
52. Davutoglu V, Celik A, Aksoy M. Contribution of selected serum inflammatory mediators to the progression of chronic rheumatic valve disease, subsequent valve calcification and NYHA functional class. J Heart Valve Dis. (2005) 14:251–6.
53. Diamantino Soares C, Araujo Passos LS, Sable C, Beaton A, Ribeiro VT, Gollob KJ, et al. Circulating cytokines predict severity of rheumatic heart disease. Int J Cardiol. (2019) 289:107–9. doi: 10.1016/j.ijcard.2019.04.063
54. Knirsch W, Nadal D. Infective endocarditis in congenital heart disease. Eur J Pediatr. (2011) 170:1111–27. doi: 10.1007/s00431-011-1520-8
55. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, et al. Detection of viruses in myocardial tissues by polymerase chain reaction. Evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. (2003) 42:466–72. doi: 10.1016/S0735-1097(03)00648-X
56. Bracamonte-Baran W, Cihakova D. Cardiac autoimmunity: myocarditis. Adv Exp Med Biol. (2017) 1003:187–221. doi: 10.1007/978-3-319-57613-8_10
57. Dancea B. Myocarditis in infants and children: a review for the paediatrician. Paediatr Child Health. (2001) 6:543–5. doi: 10.1093/pch/6.8.543
58. Luce WA. Immune mechanisms in pediatric cardiovascular disease. In: Immune Dysfunction and Immunotherapy in Heart Disease.
59. Maisch B. Cardio-immunology of myocarditis: focus on immune mechanisms and treatment options. Front Cardiovasc Med. (2019) 6:48. doi: 10.3389/fcvm.2019.00048
60. Cooper LT, Jr., Berry GJ, Shabetai R. Idiopathic giant-cell myocarditis—natural history and treatment. N Engl J Med. (1997) 336:1860–6. doi: 10.1056/NEJM199706263362603
61. Huang X, Sun Y, Su G, Li Y, Shuai X. Intravenous immunoglobulin therapy for acute myocarditis in children and adults. Int Heart J. (2019) 60:359–65. doi: 10.1536/ihj.18-299
62. Yen CY, Hung MC, Wong YC, Chang CY, Lai CC, Wu GK. Role of intravenous immunoglobulin therapy in the survival rate of pediatric patients with acute myocarditis: a systematic review and meta-analysis. Sci Rep. (2019) 9:10459. doi: 10.1038/s41598-019-46888-0
63. Lipshultz SE, Cochran TR, Briston DA, Brown SR, Sambatakos PJ, Miller TL, et al. Pediatric cardiomyopathies: causes, epidemiology, clinical course, preventive strategies and therapies. Fut Cardiol. (2013) 9:817–48. doi: 10.2217/fca.13.66
64. Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. (2019) 5:32. doi: 10.1038/s41572-019-0084-1
65. Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American heart association. Circulation. (2019) 140:e9–68. doi: 10.1161/CIR.0000000000000682
66. Becker RC, Phillip Owens A, III, Sadayappan S. The potential roles of Von Willebrand factor and neutrophil extracellular traps in the natural history of hypertrophic and hypertensive cardiomyopathy. Thromb Res. (2020) 192:78–87. doi: 10.1016/j.thromres.2020.05.003
67. Shar JA, Brown KN, Keswani SG, Grande-Allen J, Sucosky P. Impact of aortoseptal angle abnormalities and discrete subaortic stenosis on left-ventricular outflow tract hemodynamics: preliminary computational assessment. Front Bioeng Biotechnol. (2020) 8:114. doi: 10.3389/fbioe.2020.00114
68. Moak JP, Kaski PJ. Hypertrophic cardiomyopathy in children. Heart. (2012) 98:1044–54. doi: 10.1136/heartjnl-2011-300531
69. Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller OF. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation. (2009) 119:1085–92. doi: 10.1161/CIRCULATIONAHA.108.804617
70. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121:749–70. doi: 10.1161/CIRCRESAHA.117.311059
71. Lamke GT, Allen RD, Edwards WD, Tazelaar HD, Danielson KG. Surgical pathology of subaortic septal myectomy associated with hypertrophic cardiomyopathy. A study of 204 cases (1996-2000). Cardiovasc Pathol. (2003) 12:149–58. doi: 10.1016/S1054-8807(03)00036-X
72. Chen R, Ge T, Jiang W, Huo J, Chang Q, Geng J, Shan Q. Identification of biomarkers correlated with hypertrophic cardiomyopathy with co-expression analysis. J Cell Physiol. (2019) 234:21999–2008. doi: 10.1002/jcp.28762
73. Becker RC, Owens AP III, Sadayappan S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J Thromb Thrombolysis. (2020) 49:177–83. doi: 10.1007/s11239-019-02026-1
74. Fernlund E, Gyllenhammar T, Jablonowski R, Carlsson M, Larsson A, Arnlov J, et al. Serum biomarkers of myocardial remodeling and coronary dysfunction in early stages of hypertrophic cardiomyopathy in the young. Pediatr Cardiol. (2017) 38:853–63. doi: 10.1007/s00246-017-1593-x
75. Westermann D. Does inflammation trigger fibrosis in hypertrophic cardiomyopathy: a burning question? Heart. (2012) 98:965–6. doi: 10.1136/heartjnl-2012-301730
76. Iwasaki J, Nakamura K, Matsubara H, Nakamura Y, Nishii N, Banba K, et al. Relationship between circulating levels of monocyte chemoattractant protein-1 and systolic dysfunction in patients with hypertrophic cardiomyopathy. Cardiovasc Pathol. (2009) 18:317–22. doi: 10.1016/j.carpath.2008.12.004
77. Yap SC, Roos-Hesselink JW, Bogers AJ, Meijboom JF. Steepened aortoseptal angle may be a risk factor for discrete subaortic stenosis in adults. Int J Cardiol. (2008) 126:138–9. doi: 10.1016/j.ijcard.2007.01.078
78. Butany J, Vaideeswar P, David ET. Discrete subaortic membranes in adults–a clinicopathological analysis. Cardiovasc Pathol. (2009) 18:236–42. doi: 10.1016/j.carpath.2008.06.013
79. Dolan JM, Meng H, Singh S, Paluch R, Kolega J. High fluid shear stress and spatial shear stress gradients affect endothelial proliferation, survival, and alignment. Ann Biomed Eng. (2011) 39:1620–31. doi: 10.1007/s10439-011-0267-8
80. Ramphul K, Mejias GS. Kawasaki disease: a comprehensive review. Arch Med Sci Atheroscler Dis. (2018) 3:e41–5. doi: 10.5114/amsad.2018.74522
81. Alsaied T, Tremoulet AH, Burns JC, Saidi A, Dionne A, Lang SM, et al. Review of cardiac involvement in multisystem inflammatory syndrome in children. Circulation. (2020) 143:78–88. doi: 10.1161/CIRCULATIONAHA.120.049836
82. Rodriguez-Gonzalez M, Castellano-Martinez A, Cascales-Poyatos HM, Perez-Reviriego AA. Cardiovascular impact of COVID-19 with a focus on children: a systematic review. World J Clin Cases. (2020) 8:5250–83. doi: 10.12998/wjcc.v8.i21.5250
83. Noval Rivas M, Arditi M. Kawasaki disease: pathophysiology and insights from mouse models. Nat Rev Rheumatol. (2020) 16:391–405. doi: 10.1038/s41584-020-0426-0
84. Pilania RK, Singh S. Kawasaki disease. In Cimaz R, editor. Periodic Non-Periodic Fevers. Cham: Springer (2019). p. 45–63. doi: 10.1007/978-3-030-19055-2_4
85. Gorelik M, Lee Y, Abe M, Andrews T, Davis L, Patterson J, et al. IL-1 receptor antagonist, anakinra, prevents myocardial dysfunction in a mouse model of Kawasaki disease vasculitis and myocarditis. Clin Exp Immunol. (2019) 198:101–10. doi: 10.1111/cei.13314
86. Matundan HH, Sin J, Rivas MN, Fishbein MC, Lehman TJ, Chen S, et al. Myocardial fibrosis after adrenergic stimulation as a long-term sequela in a mouse model of Kawasaki disease vasculitis. JCI Insight. (2019) 4:e126279. doi: 10.1172/jci.insight.126279
87. Hashimoto Y, Fukazawa R, Nagi-Miura N, Ohno N, Suzuki N, Katsube Y, et al. Interleukin-1 beta inhibition attenuates vasculitis in a mouse model of Kawasaki disease. J Nippon Med Sch. (2019) 86:108–16. doi: 10.1272/jnms.JNMS.2019_86-206
88. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks PI. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. (2016) 213:1983–98. doi: 10.1084/jem.20151853
89. Peck JL. COVID-19: impacts and implications for pediatric practice. J Pediatr Health Care. (2020) 34:619–29. doi: 10.1016/j.pedhc.2020.07.004
90. Viner RM, Whittaker E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet. (2020) 395:1741–3. doi: 10.1016/S0140-6736(20)31129-6
91. Henderson LA, Yeung MRS. MIS-C: early lessons from immune profiling. Nat Rev Rheumatol. (2020) 17:75–6. doi: 10.1038/s41584-020-00566-y
92. Garcia-Salido A, de Carlos Vicente JC, Belda Hofheinz S, Balcells Ramirez J, Slocker Barrio M, Leoz Gordillo I, et al. Spanish Pediatric Intensive Care Society working group on: Severe manifestations of SARS-CoV-2 in children and adolescents: from COVID-19 pneumonia to multisystem inflammatory syndrome: a multicentre study in pediatric intensive care units in Spain. Crit Care. (2020) 24:666. doi: 10.1186/s13054-020-03332-4
93. Hobbs CV, Khaitan A, Kirmse BM, Borkowsky W. COVID-19 in children: a review and parallels to other hyperinflammatory syndromes. Front Pediatr. (2020) 8:593455. doi: 10.3389/fped.2020.593455
94. Valverde I, Singh Y, Sanchez-de-Toledo J, Theocharis P, Chikermane A, Di Filippo S, et al. Acute cardiovascular manifestations in 286 children with multisystem inflammatory syndrome associated with COVID-19 Infection in Europe. Circulation. (2020) 143:21–32. doi: 10.2139/ssrn.3634853
95. Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, Hausenloy JD. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
96. Oppi S, Luscher TF, Stein S. Mouse models for atherosclerosis research-which is my line? Front Cardiovasc Med. (2019) 6:46. doi: 10.3389/fcvm.2019.00046
97. Bhui R, Hayenga NH. An agent-based model of leukocyte transendothelial migration during atherogenesis. PLoS Comput Biol. (2017) 13:e1005523. doi: 10.1371/journal.pcbi.1005523
98. Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol. (2005) 204:428–36. doi: 10.1002/jcp.20307
99. Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. (2005) 111:2430–7. doi: 10.1161/01.CIR.0000165066.71481.8E
Keywords: pediatric heart disease, pediatric vascular disease, immune response, inflammation, cardiac remodeling
Citation: Jui E, Singampalli KL, Shani K, Ning Y, Connell JP, Birla RK, Bollyky PL, Caldarone CA, Keswani SG and Grande-Allen KJ (2021) The Immune and Inflammatory Basis of Acquired Pediatric Cardiac Disease. Front. Cardiovasc. Med. 8:701224. doi: 10.3389/fcvm.2021.701224
Received: 27 April 2021; Accepted: 30 June 2021;
Published: 27 July 2021.
Edited by:
Juan Pablo Kaski, University College London, United KingdomReviewed by:
Alexandros Protonotarios, University College London, United KingdomElumalai Appachi, Baylor College of Medicine, United States
Copyright © 2021 Jui, Singampalli, Shani, Ning, Connell, Birla, Bollyky, Caldarone, Keswani and Grande-Allen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: K. Jane Grande-Allen, Z3JhbmRlQHJpY2UuZWR1
†These authors have contributed equally to this work