 Mengfei Cheng
Mengfei Cheng Fang Yang2†
Fang Yang2† Jiahui Liu
Jiahui Liu Shuai Jiang
Shuai Jiang Mei Dong
Mei Dong- 1Department of Pharmacy, Harbin Medical University Cancer Hospital, Harbin, China
- 2The First Department of Respiratory Medical Oncology, Harbin Medical University Cancer Hospital, Harbin, China
- 3Department of Clinical Laboratory, Harbin Medical University Cancer Hospital, Harbin, China
With the development of anti-tumor drugs, tyrosine kinase inhibitors (TKIs) are an indispensable part of targeted therapy. They can be superior to traditional chemotherapeutic drugs in selectivity, safety, and efficacy. However, they have been found to be associated with serious adverse effects in use, such as myocardial infarction, fluid retention, hypertension, and rash. Although TKIs induced arrhythmia with a lower incidence than other cardiovascular diseases, much clinical evidence indicated that adequate attention and management should be provided to patients. This review focuses on QT interval prolongation and atrial fibrillation (AF) which are conveniently monitored in clinical practice. We collected data about TKIs, and analyzed the molecule mechanism, discussed the actual clinical evidence and drug-drug interaction, and provided countermeasures to QT interval prolongation and AF. We also pooled data to show that both QT prolongation and AF are related to their multi-target effects. Furthermore, more than 30 TKIs were approved by the FDA, but most of the novel drugs had a small sample size in the preclinical trial and risk/benefit assessments were not perfect, which led to a suspension after listing, like nilotinib. Similarly, vandetanib exhibits the most significant QT prolongation and ibrutinib exhibits the highest incidence in AF, but does not receive enough attention during treatment.
Introduction
Protein kinases are enzymes that catalyze adenosine triphosphate (ATP) γ-phosphate transfer to tyrosine residues of the substrate protein and regulate many essential cellular biochemical functions including differentiation, proliferation, and death (1). More than 500 kinases have been discovered, and depending on their substrate specificity, they can be divided into two categories: catalytic tyrosine/tyrosine-like phosphorylation and serine/threonine phosphorylation (2). More than 30% of proteins may be modified by the kinases in the human body (3) and over 50% of proto-oncogene and oncogene products have protein tyrosine kinase activity (4). In this way, tyrosine kinase inhibitors (TKIs) by competitive inhibition of the ATP binding pocket, inhibit tumor proliferation, which has been widely used in cancer target therapy (1, 5).
TKIs can be divided into monoclonal antibody drugs and small molecule TKIs. Monoclonal antibody drugs are mainly bound to unique epitopes on the extracellular matrix to regulate downstream signal transduction, thereby inhibiting the proliferation, invasion, and angiogenesis of tumor cells. Small molecule TKIs act by intracellular inhibition of phosphorylation (6). So, inherently, small molecule TKIs are less selective than monoclonal antibodies and may lead to more adverse effects (AEs).
After the first TKI drug imatinib was approved in the US in 2001, a total of more than 30 TKIs were approved by the FDA up to 2020 (7). Although all the approved TKIs can inhibit BCR-ABL1, they still have different targeted sites and distinctive potency and activity. First-generation TKIs, like imatinib, dramatically improved the 5-year survival from 11 to 90% in Philadelphia chromosome-positive chronic myeloid leukemia (CML) patients (8). Second-generation TKIs, such as nilotinib (9) and dasatinib (10), exhibited the ability to overcome imatinib resistance and a more rapid molecule response. Third-generation TKI ponatinib is the only drug that works against BCR-ABL1T315I mutation (11).
As successful as TKIs are, they still face some challenges, such as AEs caused by drug poor selectivity, drug resistance, and other reasons. According to the FDA's Adverse Effects Reporting System (FARES) database's cardiovascular (CV) toxicity section, TKIs were considered to be the suspected drug in 83.2% of CV events. And torsade de pointes/QT prolongation was considered the only acute event, which had a 6.8% incidence, higher than other anticancer drugs (1.4%) (12). Similarly, correlations were found between QT prolongation with increased risk of polymorphic ventricular tachycardia, which leads to lethal arrhythmia and subsequent sudden cardiac death (SCD) (3, 7, 12). The mechanisms and countermeasures of TKI-induced arrhythmia are still unknown. To this purpose, we wrote this paper to provide a broad overview for the potential of approved TKIs in prolonging QT interval and atrial toxicity and systemic and comprehensive treatment for patients.
As shown in Figure 1, the data for this review were identified by searches of PubMed and references from relevant articles using the search terms “TKIs,” “QT prolongation,” “atrial fibrillation,” and “on and off-target.” We identified 6,151 records through the PubMed database. We removed 2,759 records as they were review papers (n = 1,267), meta-analyses (n = 1,181), or case reports (n = 311). A total of 2,931 studies on TKIs combined with other anticancer drugs were also removed. Then, 407 records were excluded by reading the abstract. Finally, 54 records were enrolled. Only articles published in English between 2005 and 2021 were included.
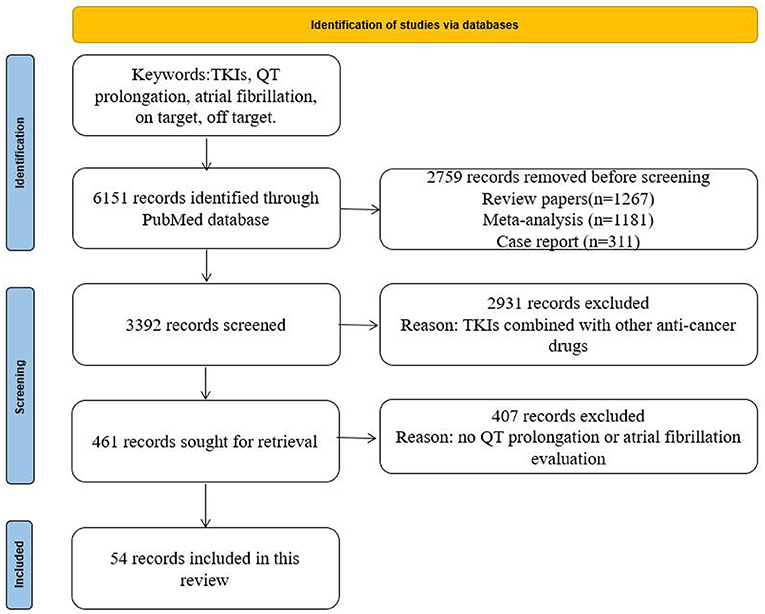
Figure 1. Search strategy and selection criteria.
Potential Molecule Mechanism of QT Prolongation and Atrial Toxicity
Most TKIs are multi-target drugs, and the target receptors include vascular endothelial growth factor receptor (VEGFR), BCR-ABL, platelet-derived growth factor receptor (PDGFR), epidermal growth factor receptor (EGFR), and c-KIT, etc. (13, 14). Then they regulate the downstream signaling pathways, for example, PI3K, MEK, and AKT, etc. (14, 15). Only a few TKIs have only one or two targets, such as axitinib, bosutinib, and gefitinib (3). Due to the numerous targets of TKIs, the potential mechanisms of TKI-related side effects previously proposed can be divided into: “on-target” and “off-target” (14, 16) effects.
On-Target
On-target means that the targets of TKIs exist in the tumor cell, but also play an important role in other normal organ cells, which may damage the biochemical function of normal cells (17, 18). A typical example of an on-target effect was observed in the Lu et al. trial. They designed an experiment to expose canine ventricular myocytes to drugs that have been demonstrated to prolong QT interval. As the result showed, the inhibition of the PI3K signaling pathway was the actual reason for QT prolongation. After blocking PI3K signaling, with the increase of persistent sodium current (INaP) and the decrease of L-type calcium current (ICaL) and potassium current (IKr and IKs), the total sodium and potassium current change accounted for over 70% of the whole prolongation, not just the potassium channels. And then, PI3K and its second message manager phosphatidylinositol 3,4,5-trisphosphate (PIP3) could affect multiple ion channels, similarly, resulting in action potential duration (APD). They also confirmed this result by breeding mice with reduced PI3K signaling showed QT prolongation (19).
Then, in McMullen's trial, they found that mice with decreased activity of the PI3K-Akt pathway was associated with higher susceptibility to AF and the same observation was relevant in humans. This pathway is an important regulator of cardiac protection under stress conditions. Other experiments also made a hypothesis that AF was related to ROS signaling, which would occur in abnormal Ca2+ release and atrial remodeling (20).
Off-Target
Off-target refers to a non-selective TKI acting on normal organ cells. However, the target does not exist in tumor cells (21, 22). Generally, whether a TKI drug has the effect of QT prolongation, we can first observe whether there are fluorophenyl or fluoromethyl-phenyl rings in the molecular structure of the drugs (23). Drugs inhibit the hERG (human ether-a-go-go) subunit with which the channel conducts the main ventricular repolarization potassium current (IKr) potential during phases 2–3 of the action potential (3, 24, 25). And hERG is regulated by cAMP and cAMP-dependent PKA. Vandetanib has been demonstrated to have values of hERG IC50 of 0.4 μM, and its metabolites are also active. In in vivo studies, a dose-dependent increase in QT prolongation has been demonstrated in dogs (3).
For instance, ibrutinib influences tumor cells by inhibiting Bruton tyrosine kinase (BTK); it also has off-target effects on Tec protein tyrosine kinase (TEC) (26). Both BTK and TEC transcripts have been demonstrated to express in cardiac tissue and at a higher expression when AF occurs rather than sinus rhythm. The PI3K-Akt pathway is regulated by BTK and TEC, and plays an important role in cardiac protection under conditions of stress (27).
Even if some drugs act on the same target or class of targets, it cannot be proved that these drugs have the effect of a prolonged QT interval. For example, sorafenib, vandetanib, and axitinib act on VEGFR, but only sorafenib and vandetanib will induce QT prolongation (23).
Selected Representative Drugs
Vandetanib
Vandetanib has the most significant prolongation of corrected QT interval according to Fridericia's formula (QTcF). It is an oral multitarget TKI drug, which inhibits VEGFR-2, EGFR, and the activity of tyrosine kinase. It was approved for the treatment of metastatic medullary thyroid cancer (MTC) by the FDA in 2011 (28).
In an early report, Ghatalia et al. initiated a meta-analysis about QTcF interval prolongation with VEGFR TKIs (29). They studied 13 clinical trials that included 4,204 patients with multi-tumor types who received 100 or 300 mg of vandetanib daily. The incidence of QT prolongation ranged from 0.3 to 23.9%, AF incidence ranged from 0.43 to 1.79%, and all-grade arrhythmia ranged from 0 to 1.69%. A high dose of vandetanib was associated with a high risk of QT prolongation by the authors. Then, under an approved dosage, phase II trial, multicenter, open-label study, 17 patients with metastatic or recurrent NSCLC with a RET rearrangement and against platinum-based doublet chemotherapy were treated with 300 mg of vandetanib once daily. A total of 6 out of 17 patients had grade 3 AEs, 2 were QT prolongation (11%) (30). In another phase III, double-blind, placebo-controlled clinical trial, 331 patients with advanced or metastatic MTC were randomized 2:1 to receive 300 mg of vandetanib daily orally (n = 231) or placebo (n = 100). During the treatment, most AEs could be well-managed by dose interruption or reduction, QT prolongation occurred in 14 patients in all grades (31).
Vandetanib is metabolized by cytochrome P450 enzyme (CYP) 3A4, which inhibits or promotes activity by many common drugs (32). In a phase I trial, healthy individuals received 200 mg of itraconazole daily and were given a single dose of 300 mg of vandetanib on day 1 and day 4. A slight increase (9%) was observed in the serum concentration of vandetanib (33). Other substrates of CYP3A4, such as ketoconazole and rifampicin should be considered in the drug combination. In addition, other drugs which may induce QT prolongation need to be considered in the drug combination.
Ibrutinib
Ibrutinib has the highest incidence of AF. It is an oral irreversible small molecule inhibitor of Bruton's tyrosine kinase (BTK), which is inhibited by a covalent bond with the specific cysteine Cys-481 of BTK, thereby inhibiting the proliferation and survival of malignant B cells, as well as reducing their migration and substrate adhesion (34, 35). Through the inhibition of BTK, downstream signaling pathways (MAPK, PI3K, and NF-?B) and phosphorylation functions (PLCγ, ERK, and AKT) are influenced (35–37).
In a preclinical in vitro study, it was demonstrated that there were effects on hERG, but no specific risks for human cardiac issues, by the authors. In the in vivo safety study of dogs, ibrutinib may increase PR interval, decrease heart rate, and shorten heart rate-corrected QT interval (38). Besides, its use is associated with atrial toxicity. The possible mechanisms are still not entirely clear, but it has been demonstrated that AF is an off-target effect (39). Xiao et al., who used a mouse model and conducted chemo-proteomic analysis of cardiac lysates, found that C-terminal Src kinase (CSK) was the most likely target for ibrutinib-induced AF (40). Jiang et al. created a C57BI/6 mice model where an ibrutinib group received 25 mg/kg/d of ibrutinib and a control group was treated with hydroxypropy1-β-cyclodextrin for 4 weeks. Compared with the control group, the ibrutinib group displayed Ca2+ dysregulation in atrial myocytes, it increased spontaneous Ca2+ release, CaMKII level, phosphorylated CaMKII, and other related sites, and reduced sarcoplasmic Ca2+ capacity (41); both may lead to AF. Ibrutinib is an independent risk factor for the development of atrial arrhythmias, with an incidence of AF of more than 10–15% (42–44). Based on Alexandre's paper, ibrutinib is the most frequent anticancer drug to cause AF (45). In addition to straightforward arrhythmias, other potential effects of ibrutinib on ECG are little known. In early clinical trials, ~6–16% of participants had an increased risk of AF (46). A review of 16 studies showed that the incidence of ibrutinib-associated AF was 5.77 per 100 person-years (27). Fradley et al. enrolled 137 patients who were treated with ibrutinib, 21 pre- and post-ibrutinib ECG readings were obtained. Compared with the pre ibrutinib ECG, after administration, the ECG showed QT interval shortening from 446 to 437 ms, based on Bazett's formula (47). In another phase II clinical trial, a mean 7.5 ms shortening of the corrected QT interval was found after ibrutinib treatment (34). However, no significant QT effects were found in healthy subjects. Ibrutinib showed concentration-dependent mild shortening of the QT interval and PR prolongation, but seemed to have no significant clinical meaning (38).
Ibrutinib is metabolized by CYP3A4, therefore the coadministration of calcium channel blockers and other enzyme inhibitors should be fully considered. Besides, ibrutinib may increase the P-glycoprotein (P-gp) substrate level, such as digoxin and omeprazole, etc. (27, 48, 49).
Ponatinib
Ponatinib is a third-generation TKI, which was designed by a computational and structure-based approach (50). It was created with a unique carbon-carbon triple bond linkage that overcomes the steric hindrance caused by the T315I mutant in CML or Ph+ acute lymphoblastic leukemia (ALL) (50, 51). The toxicity is mostly explained as a lack of selectivity, and on and off-target effects. Ponatinib inhibits over 60 kinases, including PDGFR, c-KIT, VEGFR, and EGFR. It also acts on perturbation of pro-survival signaling pathways, the AKT and ERK pathways impact cardiac function (52, 53). And in the Sharma et al. trial, they used human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) to evaluate the 21 approved TKIs, and demonstrated that ponatinib is the most toxic (54).
In a pre-clinical trial, ponatinib was not associated with cardiotoxicity. However, it exhibited a high occurrence of cardiac events in follow-up trials (11, 55). One year after being approved by the FDA, ponatinib was suspended because of safety concerns (56). At the dose of 50 mg/kg, the mean tumor volume decreased by 96%, so 45 mg once daily has been suggested, but the label cautions that an optimal dosage has not been identified (52, 57). In Sonnichsen's trial, 39 patients received ponatinib treatment at 30-, 45- and 60-mg dose levels, and the QTcF changes from baseline showed −10.9, −3.9, and −5.0 ms. Seventy-five patients in different dose levels were enrolled to evaluate the PK-PD effect, no significant correlation was found between drug exposure and QT changes (50). For the FDA CV events report for TKIs in 2020, 14.4% were found to be related to ponatinib (12). And in another clinical trial, 78 patients with CML were treated with ponatinib, the most common CAEs were arrhythmia (9%), higher than hypertension (7.7%) and myocardial infarction (3.8%) (58). In a ponatinib vs. imatinib phase III trial, 307 newly diagnosed CML patients were assigned to receive ponatinib (n = 155) or imatinib (n = 152). The results showed that no significant differences were observed in major molecule response at 12 months, but three serious AFs were observed in the ponatinib group, while no AFs occurred in the imatinib group (59).
Ponatinib is mostly metabolized by CYP3A4/5, but also by the substrates of CYP2C6 and CYP2C8. When co-administered with ketoconazole, the AUC0−∞, AUC0−t, and Cmax indicated increased exposures to ponatinib of 78, 70, and 47%, respectively. So, a dose decreased to 30 from 45 mg daily could be considered when combined with strong CYP3A4 inhibitors (60, 61).
Nilotinib
Nilotinib also has strong cardiotoxicity. Its ability to prolong QT interval and induce AF is just after that of vandetanib and ibrutinib. It is an orally administered small molecule TKI that was designed with a lipophilic structure which competitively binds to the inactive conformation of the ABL kinase domain and leads to a higher and faster molecule response than imatinib in patients (62, 63). Nilotinib is more selective than imatinib, and does not have an effect on Src, EGFR, and VEGF kinase at a concentration <3,000 nM (64–66). However, it targets PDGFRa, PDGFRb, c-kit, DDR, and colony-stimulating factor receptor 1, which is similar to imatinib (65).
Under the multi-target effect, nilotinib induce cardiotoxicity. In a preclinical safety study, nilotinib inhibited the hERG channel at an IC50 value of 0.13 μM (3), and exhibited the signs of QT prolongation in an isolated rabbit heart, but no toxicity in neonatal rat ventricular myocytes (67). There was no evidence that nilotinib had an effect on QTc in dogs at the dose up to 300 mg (3). In a response and safety phase I study, 33 patients with CML-BP were enrolled and received second-line nilotinib treatment. Thirteen patients (39%) achieved a hematologic response. As for arrhythmia, the QTcF increased by 5–15 ms in the study group, and one patient developed AF (grade = 2) (9). Then, in a phase II study, 280 patients with CML were enrolled, 6-month major cytogenetic responses were achieved in 48% of patients, and only 1% (3 of 280) had QTcF > 500 ms (62). In another phase II study, 44 patients with CML-AP or CML-CP were enrolled, and 6.1% displayed QT prolongation at all grades (68). A positive correlation was found between QT prolongation and nilotinib exposure in many trials (69–71). In patients, as Cmax increased by 1,000 ng/ml, the QTcF also increased by 4.2 ms; an increase of 1,000 ng/ml in Ctrough brought on an increase of 6.9 ms in QTcF (70, 71). Contrary to the earlier study, the 2020 FDA CV events report showed that QTc prolongation (any grade) induced by TKI was almost 28.8%, among these events 38.7% was due to nilotinib (12). It was significantly higher than early clinical trials. Alexandre et al. used the World Health Organization (WHO) individual case safety report database, vigibase, to identify the correlation between anticancer drugs and AF. As the results showed, 11,757 of 2,124,646 AF cases were associated with 176 anticancer drugs, and nilotinib accounted for 241 cases (2%) (45).
Nilotinib is also metabolized in the liver and is the competitive inhibitor of CYP3A4/5, CYP2C8, CYP2C9, CYP2D6, and uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) (72, 73). When co-administered with ketoconazole, the area under the curve (AUC) increased by 3-fold and Cmax by 1.8-fold (74). Other drugs like rifampicin and esomeprazole reduced the plasma concentration to different degrees (72, 75).
Dasatinib
Dasatinib seems to rarely cause QT interval prolongation events, and pooled safety data suggest that the overall risk for cardiotoxicity is minimal in dasatinib. But it still occurs in clinical use (76). Dasatinib is an effective BCR-ABL inhibitor in the treatment of CML and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) after relapse or resistance to imatinib (77, 78). It acts on the targets BCR-ABL, c-Kit, Src family kinases (SFKS), and PDGFR-α/β. By acting on BCR-ABL and the Src family, dasatinib inhibits the downstream PI3K signaling pathway (3, 79), which may induce cardiotoxicity.
The IC50 of the effect on hERG is 14.3 μM, which is safer than vandetanib in QT effects. In an in vivo study, no QT prolongation was found in monkeys using a body-weight dose strategy (10 or 70 mg/kg). In a clinical trial, 2,182 patients were treated with dasatinib, 21 displayed QT ≥ 500 ms, all patients had a mean increase from baseline of 3–5 ms (3). In another retrospective study, 115 cancer patients received dasatinib treatment, 41.7% of patients showed QT prolongation and the mean (SD) of pre- and post- therapeutic QT interval change was 30 ms (12).
Dasatinib influences CYP3A4 and CYP2C8 enzymes and the drug transporter P-gp. In drug-drug interaction, omeprazole, esomeprazole, and pantoprazole will inhibit the P-gp to decrease absorption and lead to higher exposure. Metoclopramide has an additive effect on dasatinib which will increase the incidence of QT interval (80, 81). In a phase I PK and drug interaction study, 17 patients were enrolled to determine whether the coadministration of ketoconazole affected the PK of dasatinib (82). Rhe result showed that ketoconazole led to an increase in dasatinib exposure and may correlate to an ~6 ms prolongation in QT interval.
The current study showed that QT prolongation occurred in nearly 30% of patients who received TKIs treatment, and 20% were high grade (7). Onco-cardiology is essential for cancer treatment because the incidence of cancer and cardiovascular diseases (CVD) is concentrated in middle-aged and senior patients. During the cancer treatment, it may aggravate CVD, and its side effects may lead to failure of cancer treatment (6). Other unexpected side effects include: Human epidermal growth factor receptor 2 (HER2) and trastuzumab are associated with left ventricular ejection fraction (LVEF) decrease and congestive heart failure (CHF) (83); anti-VEGF drugs were found to significantly increase the incidence of hypertension (84). However, arrhythmia could be induced by lots of targets.
These five TKIs all have obvious characteristics, mechanisms, aim targets, and AEs. Vandetanib and ibrutinib were found to have high incidences of arrhythmia but dasatinib was recommended not to be much concerns about cardiotoxicity. All of them exhibit an effect on QT interval and atria toxicity in clinical application. And, according to the FDA and WHO databases, the impact of some TKIs in QT and AF is much higher than in previous clinical trials (12, 45). Although QT interval prolongation has a lower mortality than hypertension or coronary heart disease, it is convenient as a monitoring strategy of CVD, and evaluation of QT interval changes can maximize the optimization of drug cessation or reduction. The main information of the selected representative drugs is summarized in Table 1.
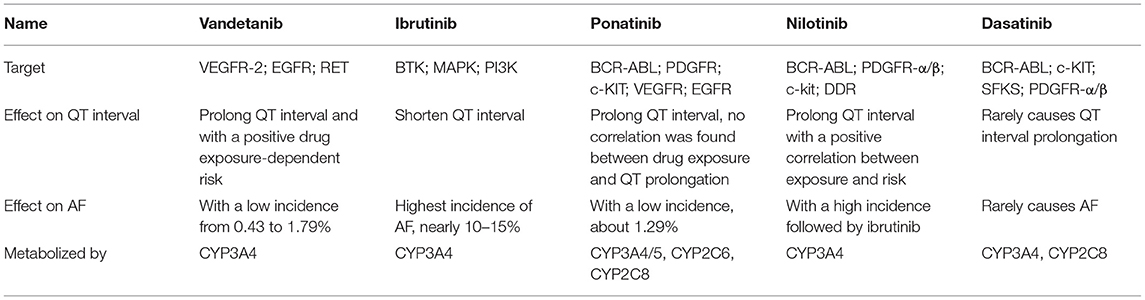
Table 1. Target, effect on QT interval/AF, and metabolism of selected representative TKIs.
Management
Arrhythmia induced by TKIs is easily monitored and can be an early warning of other serious CVD. For the agents with risk factors, routine monitoring methods such as ECG, blood pressure, electrolytes, and cardiac biomarkers are recommended during the course of treatment. In addition, collecting past medical history and physical function evaluation of patients are recommended to identify those at heightened risk for cardiovascular events. Once the arrhythmia occurs, beta-blockers and type I and III antiarrhythmic drugs are helpful for patients.
Primarily, CVD risk factors should be carefully evaluated before deciding to use TKIs. When patients are diagnosed with underlying diseases, like hypertension, coronary heart disease, diabetes, and other CVDs, they need to be carefully monitored based on their cardiac function. Baseline electrocardiograms (ECGs) should be obtained which will evaluate the risk of arrhythmia. Myoglobin (MYO), B-type natriuretic peptide (BNP), and other biochemical indexes should be obtained and corrected (85–87).
Second, electrolytes should be monitored, especially Na+, K+, and Ca2+ which have an influence on heart rhythm. Abnormal electrolytes will be a potential risk of arrhythmia and should be corrected immediately (87, 88).
Third, drug-drug interaction should be fully considered. Most TKIs are metabolized by CYP enzymes and transported by P-gp; other drugs influencing CYP enzymes and the competitive bond to P-gp should be fully considered during coadministration. And drugs that have been proved to prolong the QT interval or induce AF should be avoided (89, 90).
Fourth, reducing doses or stopping TKI treatment in time are of vital importance, especially when the QT interval is ≥500 ms, or the change of QT is > 60 ms compared with baseline. TKIs could be restarted when the QT is < 450 ms. If ventricular tachycardia, syncope, or other serious cardiovascular adverse reactions occurred again, the drugs should be stopped permanently (76, 91).
Fifth, after taking medicine, if a faint, headache, or irregular heartbeat occurs, healthcare should be provided immediately. The decision of heart rate or rhythm control should be patient-centered and symptom-oriented (87), beta-blockers may be the first choice for heart rate control, and type Ic and type III antiarrhythmic drugs are helpful for heart rhythm (87, 91).
Conclusion
To conclude, we focused on the molecule mechanism, clinical outcome, coadministration, and countermeasures of TKI drugs in arrhythmia. There is commonality and variability coexistence in TKI class, studies showed that QT prolongation is the most significant in vandetanib, AF most occurs in ibrutinib, and nilotinib has a high incidence of QT prolongation and AF. Their actual incidence and life-threatening status are higher than preclinical trials, lots of them do not get a black box warning from the FDA. But CVD caused by antitumor drugs needs to be avoided during treatment. Therefore, early diagnosis, convenient monitoring measures, and appropriate treatment methods should be provided to patients. So, ECGs monitoring should be more widely used in cancer patients, existing guidelines should be more specific, more real-world clinical trials need to be done, and on-target and off-target toxicity should be completely understood in the future.
Author Contributions
MC and FY collected data and wrote the paper. FY, JL, DY, SZ, and YY collected the literature and information. SJ, MD, and JL reviewed the paper. All authors read and approved the final manuscript.
Funding
This work was supported by the Key Program of Harbin Medical University Cancer Hospital Haiyan Fund (no. JJZD2019-03) and the General Program of Harbin Medical University Cancer Hospital Haiyan Fund (no. JJMS2021-25).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. (2005) 353:172–87. doi: 10.1056/NEJMra044389
2. Grimminger F, Schermuly RT, Ghofrani HA. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat Rev. (2010) 9:956–70. doi: 10.1038/nrd3297
3. Shah RR, Morganroth J, Shah DR. Cardiovascular safety of tyrosine kinase inhibitors: with a special focus on cardiac repolarisation (QT interval). Drug Saf. (2013) 36:295–316. doi: 10.1007/s40264-013-0047-5
4. Drake JM, Lee JK, Witte ON. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol Cell Biol. (2014) 34:1722–32. doi: 10.1128/MCB.01592-13
5. Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today. (2016) 21:5–10. doi: 10.1016/j.drudis.2015.07.008
6. Chen YC, Chung CC, Lin YK, Chen YJ. Genetic and ethnic modulation of cardiovascular toxicity of vascular endothelial growth factor inhibitors. Ann Med. (2018) 50:46–56. doi: 10.1080/07853890.2017.1383629
7. Abu Rmilah AA, Lin G, Begna KH, Friedman PA, Herrmann J. Risk of QTc prolongation among cancer patients treated with tyrosine kinase inhibitors. Int J Cancer. (2020) 147:3160–7. doi: 10.1002/ijc.33119
8. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. (2013) 122:872–84. doi: 10.1182/blood-2013-05-501569
9. Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. (2006) 354:2542–51. doi: 10.1056/NEJMoa055104
10. Shah NP, Rousselot P, Schiffer C, Rea D, Cortes JE, Milone J, et al. Dasatinib in imatinib-resistant or -intolerant chronic-phase, chronic myeloid leukemia patients: 7-year follow-up of study CA180-034. Am J Hematol. (2016) 91:869–74. doi: 10.1002/ajh.24423
11. Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. (2018) 132:393–404. doi: 10.1182/blood-2016-09-739086
12. Cirmi S, El Abd A, Letinier L, Navarra M, Salvo F. Cardiovascular toxicity of tyrosine kinase inhibitors used in chronic myeloid leukemia: an analysis of the FDA Adverse Event Reporting System Database (FAERS). Cancers. (2020) 12:826. doi: 10.3390/cancers12040826
13. Kim SY, Kim SM, Chang H, Kim BW, Lee YS, Chang HS, Park CS. Safety of tyrosine kinase inhibitors in patients with differentiated thyroid cancer: real-world use of lenvatinib and sorafenib in Korea. Front Endocrinol. (2019) 10:384. doi: 10.3389/fendo.2019.00384
14. Hu W, Hirakawa B, Jessen B, Lee M, Aguirre S. A tyrosine kinase inhibitor-induced myocardial degeneration in rats through off-target phosphodiesterase inhibition. J Appl Toxicol. (2012) 32:1008–20. doi: 10.1002/jat.2801
15. Wu MD, Moslehi JJ, Lindner JR. Arterial thrombotic complications of tyrosine kinase inhibitors. Arterioscler Thromb Vasc Biol. (2021) 41:3–10. doi: 10.1161/ATVBAHA.120.314694
16. Shah RR, Morganroth J. Update on cardiovascular safety of tyrosine kinase inhibitors: with a special focus on QT interval, left ventricular dysfunction and overall risk/benefit. Drug Saf. (2015) 38:693–710. doi: 10.1007/s40264-015-0300-1
17. Herman SEM, Montraveta A, Niemann CU, Mora-Jensen H, Gulrajani M, Krantz F, et al. The Bruton Tyrosine Kinase (BTK) inhibitor acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin Cancer Res. (2017) 23:2831–41. doi: 10.1158/1078-0432.CCR-16-0463
18. Guo Y, Liu Y, Hu N, Yu D, Zhou C, Shi G, et al. Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton's tyrosine kinase. J Med Chem. (2019) 62:7923–40. doi: 10.1021/acs.jmedchem.9b00687
19. Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS, Lin RZ. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Trans Med. (2012) 4:131ra150. doi: 10.1126/scitranslmed.3003623
20. McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. (2014) 124:3829–30. doi: 10.1182/blood-2014-10-604272
21. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. (2020) 34:966–84. doi: 10.1038/s41375-020-0776-2
22. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. (2009) 9:28–39. doi: 10.1038/nrc2559
23. Himmel HM, Hoffmann M. QTc shortening with a new investigational cancer drug: a brief case study. J Pharmacol Toxicol Methods. (2010) 62:72–81. doi: 10.1016/j.vascn.2010.05.012
24. Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, clinical significance. Physiol Rev. (2012) 92:1393–478. doi: 10.1152/physrev.00036.2011
25. Cohen IS, Lin RZ, Ballou LM. Acquired long QT syndrome and phosphoinositide 3-kinase. Trends Cardiovasc Med. (2017) 27:451–9. doi: 10.1016/j.tcm.2017.05.005
26. Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. (2016) 9:80. doi: 10.1186/s13045-016-0313-y
27. Ganatra S, Sharma A, Shah S, Chaudhry GM, Martin DT, Neilan TG, et al. Ibrutinib-associated atrial fibrillation. JACC Clin Electrophysiol. (2018) 4:1491–500. doi: 10.1016/j.jacep.2018.06.004
28. Commander H, Whiteside G, Perry C. Vandetanib: first global approval. Drugs. (2011) 71:1355–65. doi: 10.2165/11595310-000000000-00000
29. Ghatalia P, Je Y, Kaymakcalan MD, Sonpavde G, Choueiri TK. QTc interval prolongation with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Br J Cancer. (2015) 112:296–305. doi: 10.1038/bjc.2014.564
30. Lee SH, Lee JK, Ahn MJ, Kim DW, Sun JM, Keam B, et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: a phase II clinical trial. Ann Oncol. (2017) 28:292–7. doi: 10.1093/annonc/mdw559
31. Thornton K, Kim G, Maher VE, Chattopadhyay S, Tang S, Moon YJ, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: Food US, and Drug Administration drug approval summary. Clin Cancer Res. (2012) 18:3722–30. doi: 10.1158/1078-0432.CCR-12-0411
32. Weil A, Martin P, Smith R, Oliver S, Langmuir P, Read J, Molz KH. Pharmacokinetics of vandetanib in subjects with renal or hepatic impairment. Clin Pharmacokin. (2010) 49:607–18. doi: 10.2165/11534330-000000000-00000
33. Martin P, Oliver S, Robertson J, Kennedy SJ, Read J, Duvauchelle T. Pharmacokinetic drug interactions with vandetanib during coadministration with rifampicin or itraconazole. Drugs R&D. (2011) 11:37–51. doi: 10.2165/11586980-000000000-00000
34. Cameron F, Sanford M. Ibrutinib: first global approval. Drugs. (2014) 74:263–71. doi: 10.1007/s40265-014-0178-8
35. Deeks ED. Ibrutinib: a review in chronic lymphocytic leukaemia. Drugs. (2017) 77:225–36. doi: 10.1007/s40265-017-0695-3
36. Cheng S, Ma J, Guo A, Lu P, Leonard JP, Coleman M, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. (2014) 28:649–57. doi: 10.1038/leu.2013.358
37. Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, et al. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. (2014) 123:3286–95. doi: 10.1182/blood-2014-02-548610
38. de Jong J, Hellemans P, Jiao JJ, Huang Y, Mesens S, Sukbuntherng J, et al. Ibrutinib does not prolong the corrected QT interval in healthy subjects: results from a thorough QT study. Cancer Chemother Pharmacol. (2017) 80:1227–37. doi: 10.1007/s00280-017-3471-x
39. Mulder TA, Peña-Pérez L, Berglöf A, Meinke S, Estupiñán HY, Heimersson K, et al. Ibrutinib has time-dependent on- and off-target effects on plasma biomarkers and immune cells in chronic lymphocytic leukemia. HemaSphere. (2021) 5:e564. doi: 10.1097/HS9.0000000000000564
40. Xiao L, Salem JE, Clauss S, Hanley A, Bapat A, Hulsmans M, et al. Ibrutinib-mediated atrial fibrillation attributable to inhibition of C-terminal Src kinase. Circulation. (2020) 142:2443–55. doi: 10.1161/CIRCULATIONAHA.120.049210
41. Jiang L, Li L, Ruan Y, Zuo S, Wu X, Zhao Q, et al. Ibrutinib promotes atrial fibrillation by inducing structural remodeling and calcium dysregulation in the atrium. Heart Rhythm. (2019) 16:1374–82. doi: 10.1016/j.hrthm.2019.04.008
42. Fradley MG, Gliksman M, Emole J, Viganego F, Rhea I, Welter-Frost A, et al. Rates and risk of atrial arrhythmias in patients treated with ibrutinib compared with cytotoxic chemotherapy. Am J Cardiol. (2019) 124:539–44. doi: 10.1016/j.amjcard.2019.05.029
43. Leong DP, Caron F, Hillis C, Duan A, Healey JS, Fraser G, et al. The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood. (2016) 128:138–40. doi: 10.1182/blood-2016-05-712828
44. Wiczer TE, Levine LB, Brumbaugh J, Coggins J, Zhao Q, Ruppert AS, et al. Cumulative incidence, risk factors, and management of atrial fibrillation in patients receiving ibrutinib. Blood Adv. (2017) 1:1739–48. doi: 10.1182/bloodadvances.2017009720
45. Alexandre J, Salem JE, Moslehi J, Sassier M, Ropert C, Cautela J, et al. Identification of anticancer drugs associated with atrial fibrillation - analysis of the WHO pharmacovigilance database. Eur Heart J Cardiovasc Pharmacother. (2020) 7:312–20. doi: 10.1093/ehjcvp/pvaa037
46. Brown JR, Moslehi J, O'Brien S, Ghia P, Hillmen P, Cymbalista F, et al. Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica. (2017) 102:1796–805. doi: 10.3324/haematol.2017.171041
47. Fradley MG, Welter-Frost A, Gliksman M, Emole J, Viganego F, Lee DH, et al. Electrocardiographic changes associated with ibrutinib exposure. Cancer Control. (2020) 27:1073274820931808. doi: 10.1177/1073274820931808
48. de Zwart L, Snoeys J, De Jong J, Sukbuntherng J, Mannaert E, Monshouwer M. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin Pharmacol Therap. (2016) 100:548–57. doi: 10.1002/cpt.419
49. de Jong J, Hellemans P, De Wilde S, Patricia D, Masterson T, Manikhas G, et al. A drug-drug interaction study of ibrutinib with moderate/strong CYP3A inhibitors in patients with B-cell malignancies. Leuk Lymphoma. (2018) 59:2888–95. doi: 10.1080/10428194.2018.1460474
50. Sonnichsen D, Dorer DJ, Cortes J, Talpaz M, Deininger MW, Shah NP, et al. Analysis of the potential effect of ponatinib on the QTc interval in patients with refractory hematological malignancies. Cancer Chemother Pharmacol. (2013) 71:1599–607. doi: 10.1007/s00280-013-2160-7
51. Massaro F, Molica M, Breccia M. Ponatinib: a review of efficacy and safety. Curr Cancer Drug Targets. (2018) 18:847–56. doi: 10.2174/1568009617666171002142659
52. Singh AP, Umbarkar P, Tousif S, Lal H. Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: emphasis on ponatinib. Int J Cardiol. (2020) 316:214–21. doi: 10.1016/j.ijcard.2020.05.077
53. Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, et al. Ponatinib (AP24534). a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Therap. (2012) 11:690–9. doi: 10.1158/1535-7163.MCT-11-0450
54. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9:aaf2584. doi: 10.1126/scitranslmed.aaf2584
55. Breccia M, Pregno P, Spallarossa P, Arboscello E, Ciceri F, Giorgi M, et al. Identification, prevention and management of cardiovascular risk in chronic myeloid leukaemia patients candidate to ponatinib: an expert opinion. Ann Hematol. (2017) 96:549–58. doi: 10.1007/s00277-016-2820-x
56. Ariad suspends ponatinib sales. Cancer Discov. (2014) 4:6–7. doi: 10.1158/2159-8290.CD-NB2013-163
57. O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534. a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. (2009) 16:401–12. doi: 10.1016/j.ccr.2009.09.028
58. Chan O, Talati C, Isenalumhe L, Shams S, Nodzon L, Fradley M, Pinilla-Ibarz J. Side-effects profile and outcomes of ponatinib in the treatment of chronic myeloid leukemia. Blood Adv. (2020) 4:530–8. doi: 10.1182/bloodadvances.2019000268
59. Lipton JH, Chuah C, Guerci-Bresler A, Rosti G, Simpson D, Assouline S, et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. Lancet Oncol. (2016) 17:612–21. doi: 10.1016/S1470-2045(16)00080-2
60. Chien JY, Lucksiri A, Ernest CS 2nd, Gorski JC, Wrighton SA, Hall SD. Stochastic prediction of CYP3A-mediated inhibition of midazolam clearance by ketoconazole. Drug Metab Disposition. (2006) 34:1208–19. doi: 10.1124/dmd.105.008730
61. Narasimhan NI, Dorer DJ, Niland K, Haluska F, Sonnichsen D. Effects of ketoconazole on the pharmacokinetics of ponatinib in healthy subjects, J Clin Pharmacol. (2013) 53:974–81. doi: 10.1002/jcph.109
62. Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. (2007) 110:3540–6. doi: 10.1182/blood-2007-03-080689
63. Garnock-Jones KP. Nilotinib: in the first-line treatment of newly diagnosed Philadelphia chromosome-positive chronic myeloid leukaemia in chronic phase. Drugs. (2011) 71:1579–90. doi: 10.2165/11207770-000000000-00000
64. Manley PW, Stiefl N, Cowan-Jacob SW, Kaufman S, Mestan J, Wartmann M, et al. Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg Med Chem. (2010) 18:6977–86. doi: 10.1016/j.bmc.2010.08.026
65. Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. (2005) 7:129–41. doi: 10.1016/j.ccr.2005.01.007
66. Sacha T, Saglio G. Nilotinib in the treatment of chronic myeloid leukemia. Future Oncol. (2019) 15:953–65. doi: 10.2217/fon-2018-0468
67. Shopp GM, Helson L, Bouchard A, Salvail D, Majeed M. Liposomes ameliorate Crizotinib- and Nilotinib-induced inhibition of the cardiac IKr channel and QTc prolongation. Anticancer Res. (2014) 34:4733–40.
68. Takahashi N, Miura M, Kuroki J, Mitani K, Kitabayashi A, Sasaki O, et al. Multicenter phase II clinical trial of nilotinib for patients with imatinib-resistant or -intolerant chronic myeloid leukemia from the East Japan CML study group evaluation of molecular response and the efficacy and safety of nilotinib. Biomarker Res. (2014) 2:6. doi: 10.1186/2050-7771-2-6
69. Kim TD, le Coutre P, Schwarz M, Grille P, Levitin M, Fateh-Moghadam S, et al. Clinical cardiac safety profile of nilotinib. Haematologica. (2012) 97:883–9. doi: 10.3324/haematol.2011.058776
70. Larson RA, Yin OQ, Hochhaus A, Saglio G, Clark RE, Nakamae H, et al. Population pharmacokinetic and exposure-response analysis of nilotinib in patients with newly diagnosed Ph+ chronic myeloid leukemia in chronic phase. Euro J Clin Pharmacol. (2012) 68:723–33. doi: 10.1007/s00228-011-1200-7
71. Giles FJ, Yin OQ, Sallas WM, le Coutre PD, Woodman RC, Ottmann OG, et al. Nilotinib population pharmacokinetics and exposure-response analysis in patients with imatinib-resistant or -intolerant chronic myeloid leukemia. Euro J Clin Pharmacol. (2013) 69:813–23. doi: 10.1007/s00228-012-1385-4
72. Tian X, Zhang H, Heimbach T, He H, Buchbinder A, Aghoghovbia M, et al. Clinical pharmacokinetic and pharmacodynamic overview of Nilotinib, a selective tyrosine kinase inhibitor. J Clin Pharmacol. (2018) 58:1533–40. doi: 10.1002/jcph.1312
73. Shibata T, Minami Y, Mitsuma A, Morita S, Inada-Inoue M, Oguri T, et al. Association between severe toxicity of nilotinib and UGT1A1 polymorphisms in Japanese patients with chronic myelogenous leukemia. Int J Clin Oncol. (2014) 19:391–6. doi: 10.1007/s10147-013-0562-5
74. Tanaka C, Yin OQ, Smith T, Sethuraman V, Grouss K, Galitz L, et al. Effects of rifampin and ketoconazole on the pharmacokinetics of nilotinib in healthy participants. J Clin Pharmacol. (2011) 51:75–83. doi: 10.1177/0091270010367428
75. Zhang H, Sheng J, Ko JH, Zheng C, Zhou W, Priess P, et al. Inhibitory effect of single and repeated doses of nilotinib on the pharmacokinetics of CYP3A substrate midazolam. J Clin Pharmacol. (2015) 55:401–8. doi: 10.1002/jcph.434
76. Chaar M, Kamta J, Ait-Oudhia S. Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities. Onco Targets Ther. (2018) 11:6227–37. doi: 10.2147/OTT.S170138
77. Foa R, Vitale A, Vignetti M, Meloni G, Guarini A, De Propris MS, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. (2011) 118:6521–8. doi: 10.1182/blood-2011-05-351403
78. Foà R, Bassan R, Vitale A, Elia L, Piciocchi A, Puzzolo MC, et al. Dasatinib-Blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. (2020) 383:1613–23. doi: 10.1056/NEJMoa2016272
79. Barbarin A, Abdallah M, Lefevre L, Piccirilli N, Cayssials E, Roy L, et al. Innate T-alphabeta lymphocytes as new immunological components of anti-tumoral “off-target” effects of the tyrosine kinase inhibitor dasatinib. Sci Rep. (2020) 10:3245. doi: 10.1038/s41598-020-60195-z
80. Haouala A, Widmer N, Duchosal MA, Montemurro M, Buclin T, Decosterd LA. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. (2011) 117:e75–87. doi: 10.1182/blood-2010-07-294330
81. Kamath AV, Wang J, Lee FY, Marathe PH. Preclinical pharmacokinetics and in vitro metabolism of dasatinib (BMS-354825): a potent oral multi-targeted kinase inhibitor against SRC and BCR-ABL. Cancer Chemother Pharmacol. (2008) 61:365–76. doi: 10.1007/s00280-007-0478-8
82. Johnson FM, Agrawal S, Burris H, Rosen L, Dhillon N, Hong D, et al. Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer. (2010) 116:1582–91. doi: 10.1002/cncr.24927
83. Chen T, Xu T, Li Y, Liang C, Chen J, Lu Y, et al. Risk of cardiac dysfunction with trastuzumab in breast cancer patients: a meta-analysis. Cancer Treatm Rev. (2011) 37:312–20. doi: 10.1016/j.ctrv.2010.09.001
84. Abdel-Qadir H, Ethier JL, Lee DS, Thavendiranathan P, Amir E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: a systematic review and meta-analysis. Cancer Treatm Rev. (2017) 53:120–7. doi: 10.1016/j.ctrv.2016.12.002
85. Chang HM, Moudgil R, Scarabelli T, Okwuosa TM, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J Am Coll Cardiol. (2017) 70:2536–51. doi: 10.1016/j.jacc.2017.09.1096
86. Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. (2016) 37:2768–801. doi: 10.1093/eurheartj/ehw211
87. Wu Q, Bai B, Tian C, Li D, Yu H, Song B, Li B, Chu X. The molecular mechanisms of cardiotoxicity induced by HER2, VEGF, and tyrosine kinase inhibitors: an updated review. Cardiovasc Drugs Ther. (2021). doi: 10.1007/s10557-021-07181-3
88. Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 2. J Am Coll Cardiol. (2017) 70:2552–65. doi: 10.1016/j.jacc.2017.09.1095
89. Suttorp M, Bornhäuser M, Metzler M, Millot F, Schleyer E. Pharmacology and pharmacokinetics of imatinib in pediatric patients. Expert Rev Clin Pharmacol. (2018) 11:219–31. doi: 10.1080/17512433.2018.1398644
90. Rimassa L, Danesi R, Pressiani T, Merle P. Management of adverse events associated with tyrosine kinase inhibitors: improving outcomes for patients with hepatocellular carcinoma. Cancer Treat Rev. (2019) 77:20–8. doi: 10.1016/j.ctrv.2019.05.004
Keywords: TKIs, QT prolongation, atrial fibrillation, molecule mechanism, therapeutic strategies, pharmacokinetics
Citation: Cheng M, Yang F, Liu J, Yang D, Zhang S, Yu Y, Jiang S and Dong M (2021) Tyrosine Kinase Inhibitors-Induced Arrhythmias: From Molecular Mechanisms, Pharmacokinetics to Therapeutic Strategies. Front. Cardiovasc. Med. 8:758010. doi: 10.3389/fcvm.2021.758010
Received: 13 August 2021; Accepted: 25 October 2021;
Published: 19 November 2021.
Edited by:
Feng Cao, People's Liberation Army General Hospital, ChinaReviewed by:
Concetta Zito, University of Messina, ItalyHari Deshpande, Yale University, United States
Copyright © 2021 Cheng, Yang, Liu, Yang, Zhang, Yu, Jiang and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuai Jiang, amR5aG11M0AxNjMuY29t; Mei Dong, MTM4MDQ1NjczNzBAMTYzLmNvbQ==
†These authors have contributed equally to this work