 Haiwen Li
Haiwen Li Lingqiang Zhang
Lingqiang Zhang Lei Zhang
Lei Zhang Renzhi Han
Renzhi Han- 1Department of Surgery, Davis Heart and Lung Research Institute, Biomedical Sciences Graduate Program, Biophysics Graduate Program, The Ohio State University Wexner Medical Center, Columbus, OH, United States
- 2State Key Laboratory of Proteomics, National Center of Protein Sciences (Beijing), Beijing Institute of Lifeomics, Beijing, China
- 3Department of Anatomy and Neurobiology, Shanghai Yangzhi Rehabilitation Hospital, Shanghai Sunshine Rehabilitation Center, School of Medicine, Tongji University, Shanghai, China
Impaired biomolecules and cellular organelles are gradually built up during the development and aging of organisms, and this deteriorating process is expedited under stress conditions. As a major lysosome-mediated catabolic process, autophagy has evolved to eradicate these damaged cellular components and recycle nutrients to restore cellular homeostasis and fitness. The autophagic activities are altered under various disease conditions such as ischemia-reperfusion cardiac injury, sarcopenia, and genetic myopathies, which impact multiple cellular processes related to cellular growth and survival in cardiac and skeletal muscles. Thus, autophagy has been the focus for therapeutic development to treat these muscle diseases. To develop the specific and effective interventions targeting autophagy, it is essential to understand the molecular mechanisms by which autophagy is altered in heart and skeletal muscle disorders. Herein, we summarize how autophagy alterations are linked to cardiac and skeletal muscle defects and how these alterations occur. We further discuss potential pharmacological and genetic interventions to regulate autophagy activities and their applications in cardiac and skeletal muscle diseases.
Introduction
Autophagy is an evolutionarily conserved, catabolic process that digests undesirable cytoplasmic components and organelles in the lysosomes, allowing the cell to reuse the materials and maintain cellular homeostasis. Numerous studies have demonstrated the crucial roles of autophagy in many biological processes, such as development, aging, and immune responses (1–5). Emerging evidence has linked aberrant autophagy execution to many human diseases, such as cardiomyopathies and muscular dystrophies (1–5).
Based on the cargo sequestration methods, autophagy can be classified into three primary types: microautophagy, macroautophagy, and chaperone-mediated autophagy. Macroautophagy (henceforth termed autophagy) is well characterized among these types. Cells can sequester cytosolic materials into double-membrane vesicles (known as autophagosomes), and degrade these cargos by fusing with lysosomes during this process (6) (Figure 1). Based on the cargos, autophagy can be separated into bulk autophagy and selective autophagy such as ER-phagy, aggrephagy (7), and PINK1 (PTEN-induced kinase 1)/PRAK2 (parkin RBR E3 ubiquitin protein ligase)-mediated mitophagy (8) (Figure 2). This review mainly focuses on bulk autophagy and mitophagy in striated muscle diseases.
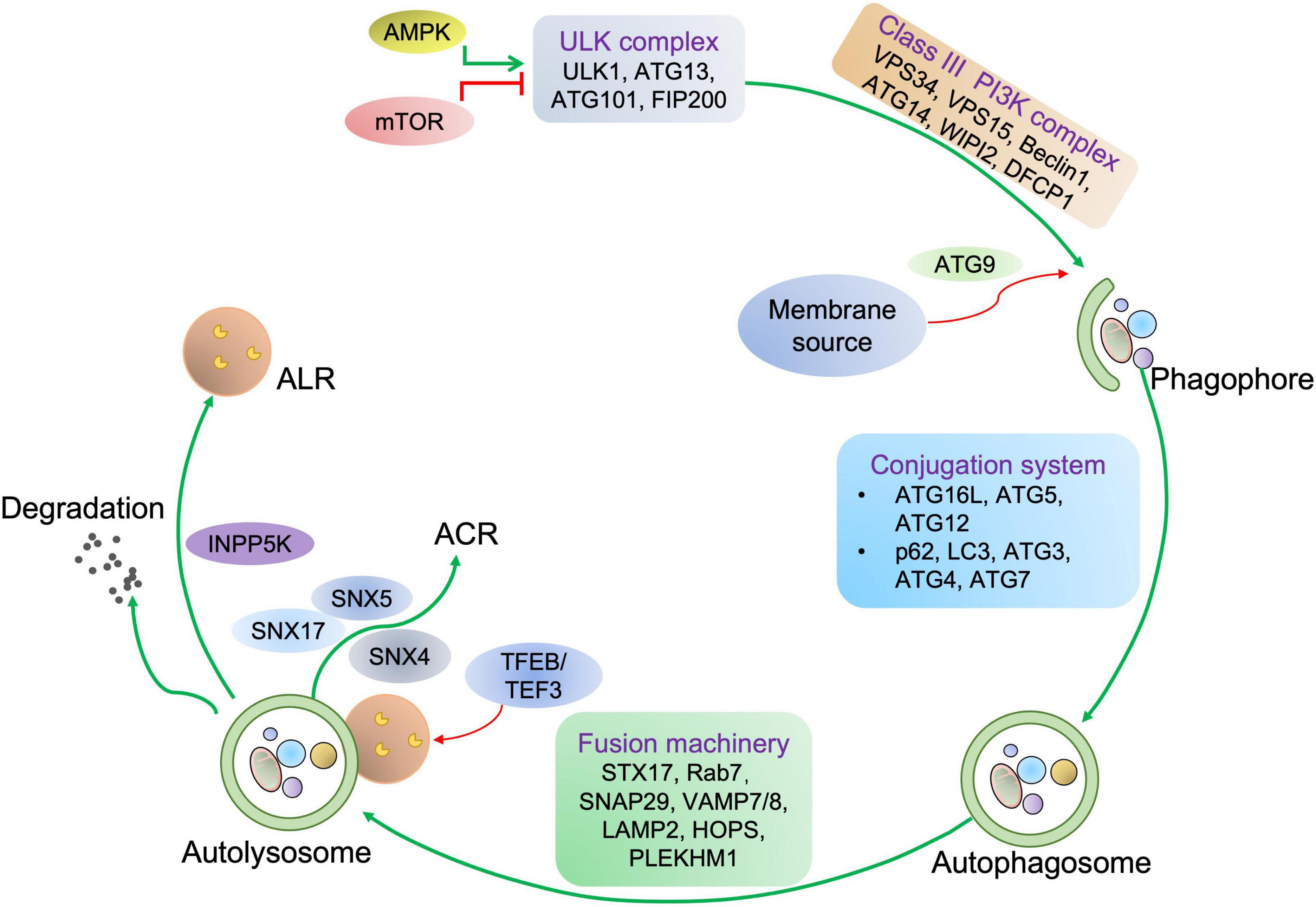
Figure 1. Autophagy process in mammalian cells. For autophagy initiation, environmental factors and metabolic stimuli are sensed by AMPK or mTORC1. The autophagic cargo undergoes a serial of processes, including the phagophore and autophagosome formation, autolysosome development before the cargo is degraded, concomitant with lysosome and autophagosome recycling. Once autophagy is induced, ULK1 is activated and then associated with the class III PI3K complex, eliciting the production of PI3P on the phagophore membrane, which further recruits WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) and double FYVE domain-containing protein 1 (DFCP1). Next, the ATG12-ATG5-ATG16L1 complex binding to WIPI2 is responsible for providing membranes from other organelles, including the plasma membrane, mitochondria, recycling endosomes, and Golgi complex. Then, ATG9 drives membrane expansion by delivering phospholipids. Moreover, the ATG12-ATG5-ATG16L1 complex promotes the conjugation of LC3, in which ATG4 cleaves LC3 to generate LC3-I, which further covalently bonds with phosphatidylethanolamine for LC3-II formation. LC3-II is a specific marker of autophagy where autophagosome-residing LC3-II can specifically associate with autophagy receptors with LC3-interacting motifs like p62. Finally, autophagosome-lysosome fusion is commonly mediated by UV radiation resistance-associated gene protein (UVRAG), homotypic fusion and protein sorting (HOPS), syntaxin-17 (STX17), vesicle-associated membrane protein 7/8 (VAMP7/8), synaptosome associated protein 29 (SNAP29), PLEKHM1, and the trafficking protein Rab-7. Concomitant with cargo removal, autophagosome effector proteins are either degraded or recycled by the Sorting Nexin 4/5/17 (SNX4/5/17) complex, and lysosomes maybe undergo the INPP5K-mediated ALR process.
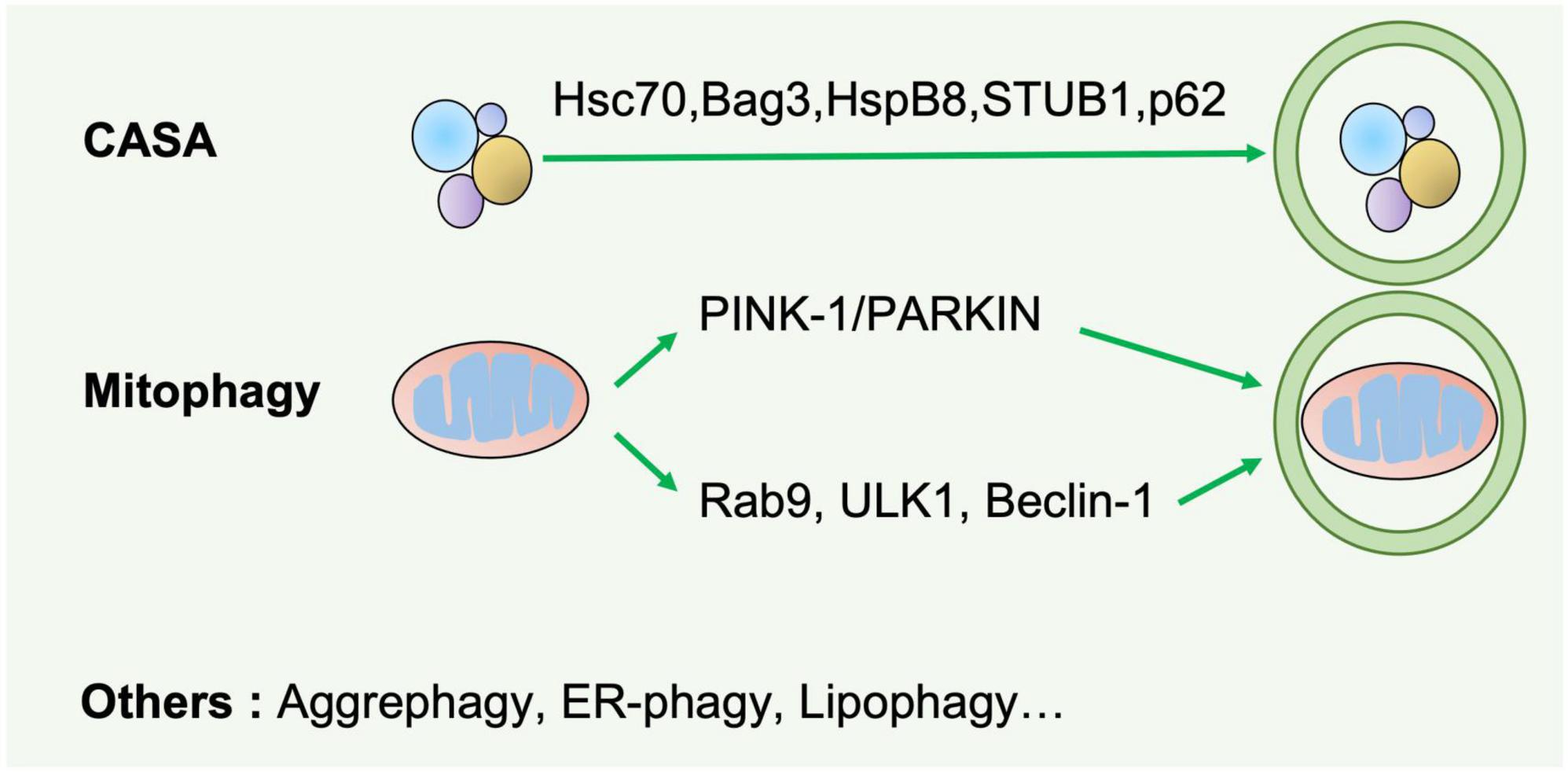
Figure 2. Selective autophagy. Selective autophagy delivers specific cargos for degradation and recycling, such as protein aggregates, mitochondria, and endoplasmic reticulum. The recognition of these cargoes requires selective autophagy receptors specifically binding LC3-II of autophagosomes and are thereby removed by lysosomes. CASA is a selective tension-induced autophagy pathway mediated by BAG3. Moreover, mitophagy is divided into two branches: the classical PINK1/PRAK2-mediated process and alternative mitophagy mediated by Rab9. Other types of selective autophagy include aggrephagy, ER-phagy, lipophagy et al.
As indicated in Figure 1, autophagy is a multiphasic process that involves the sequential and selective recruitment of autophagy-related (ATG) proteins. The complex process includes initiation/nucleation, phagophore formation, autophagosome formation, autophagosome-lysosome fusion, cargo degradation, and autophagic lysosome reformation (ALR) or emerging autophagosomal components recycling (ACR) (9). Different ATG proteins or complexes are involved in these steps. As shown in Figure 3, key upstream regulators of this process include the major inhibitor mammalian target of rapamycin (mTOR) and the primary activator AMP-activated kinase (AMPK). The main downstream phosphorylation substrates of AMPK are Unc-51-like kinase (ULK1) (10) and Forkhead box protein O (FoxO) (11, 12), in which the former is a crucial initiator of autophagy and the latter regulates the transcription of genes related to autophagy. Moreover, mTOR, particularly mTOCR1, suppresses autophagy through phosphorylating ULK1 at different sites (10) and transcription factor EB (TFEB)/transcription factor E3 (TFE3), two key proteins of lysosome biosynthesis (13, 14). The details of the autophagy process have been well reviewed in other studies (4, 15).
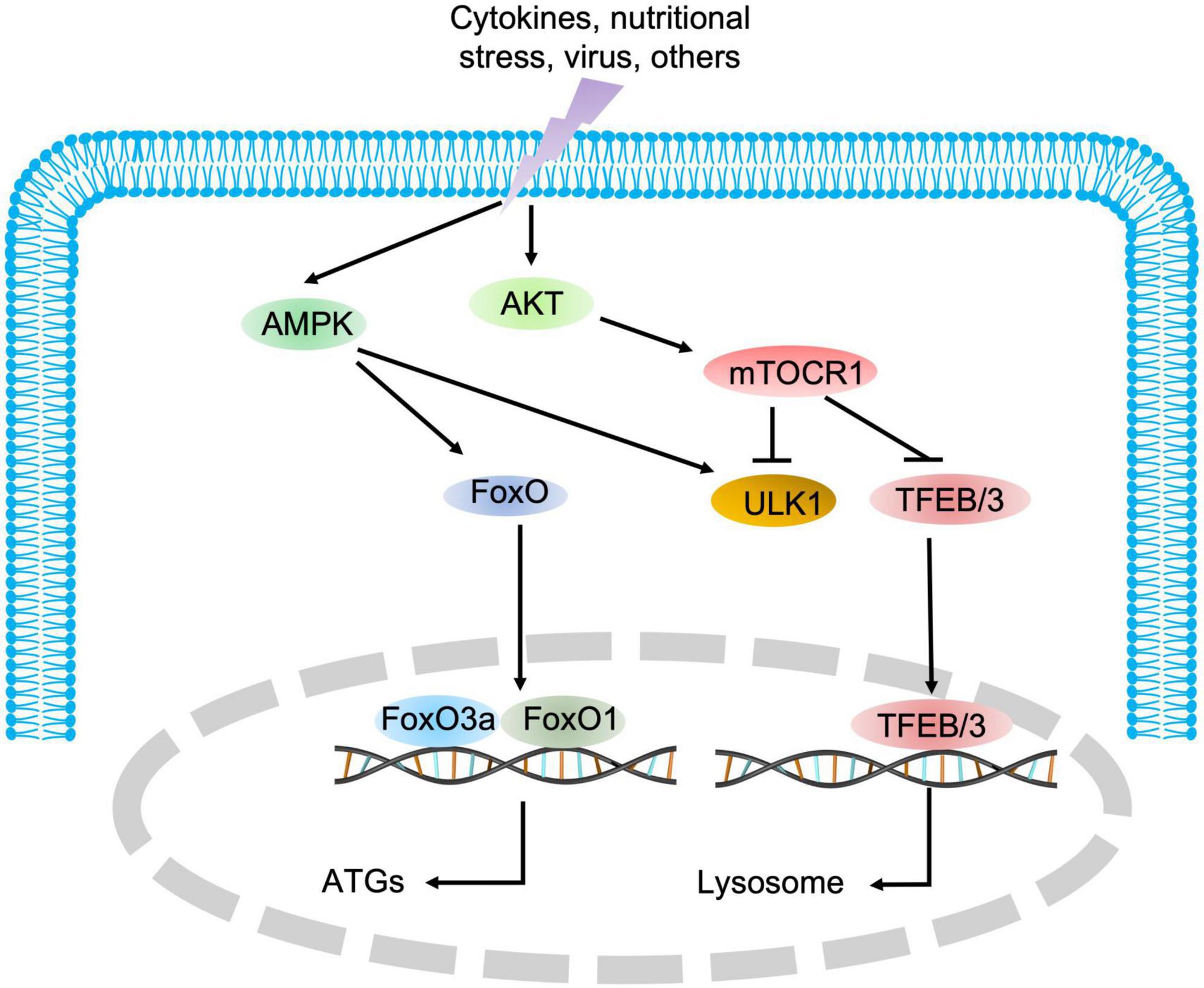
Figure 3. The regulatory signaling pathways of autophagy. AMPK and mTOR are the major positive and negative regulatory signaling pathways of autophagy, respectively. Once AMPK is activated, the downstream substrate ULK1 is phosphorylated, and FoxO transcription factors are translocated into the nucleus to promote the expression of autophagic genes. Conversely, mTOR blocks autophagic initiation by negatively phosphorylating ULK1 and inhibits lysosome biosynthesis by blocking TFEB/TFE3 translocation into the nucleus.
The role of autophagy in various pathophysiological processes has spurred great efforts toward identifying clinically druggable autophagic targets to prevent or cure human diseases, including cardiac and skeletal myopathies. Here, we systematically summarize the current insights into the role of autophagy in human diseases related to striated muscle and therapeutic strategies in preclinical development.
Aberrant autophagy in heart diseases
Heart disease is the leading cause of morbidity and death worldwide (16). Adult cardiomyocytes, the essential cellular component of cardiovascular system, are mostly long-lived and rarely renewed, implying that these cells heavily rely on intact autophagy to remove impaired proteins and organelles during their long life (5). Aberrant autophagy can lead to various heart defects.
Bulk autophagy in heart diseases
As illustrated by the genetic models of several essential or ancillary genes related to autophagy (Table 1), autophagy aberration predisposes the organisms to develop heart disorders under either basal or stress conditions (1, 17). For instance, three different cardiomyocyte-specific ATG5 conditional knockout (KO) mouse models display left ventricular dilatation and cardiac dysfunction without or with pressure overload (18, 19). Vacuolar protein sorting 34 (Vps34) negatively correlates with human hypertrophic cardiomyopathy (HCM) characterized by thickening of the heart muscle, in consistence with the observation that disruption of Vps34 causes cardiac hypertrophy in mice by accumulating ubiquitinated Crystallin Alpha B (CryAB) (20). Muscle-specific conditional KO of ATG14 causes early death and HCM with abnormal accumulation of autophagic cargoes in heart (21). Moreover, other core autophagy factors such as Beclin-1 (22), mTORC1 (23–25), and PLEKHM2 (Pleckstrin Homology and RUN Domain Containing M2) (26) are also essential for cardiac homeostasis, and their ectopic activity can cause heart defects.
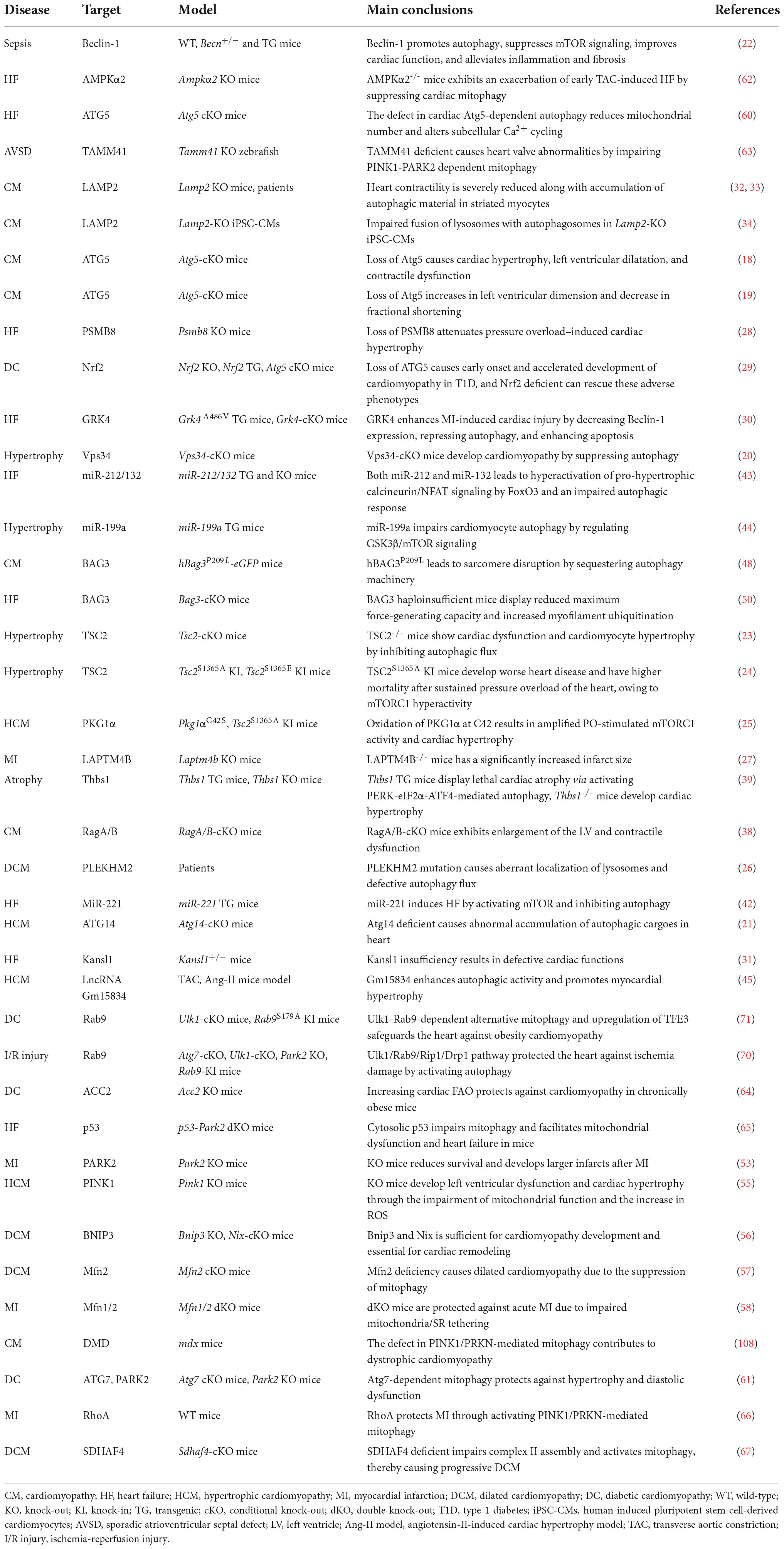
Table 1. Autophagy in heart diseases.
A large body of evidence has shown that alterations in regulatory proteins related to autophagy compromise cardiac function by modulating the core autophagy machinery. For example, mice with a disruption in lysosomal-associated transmembrane protein 4B (LAPTM4B) are susceptible to ischemia-reperfusion (I/R) injury by repressing mTORC1-mediated TFEB transcription (27). Upregulation of immunoproteasome catalytic subunit β5i leads to cardiac hypertrophy and heart failure (HF) by promoting ATG5 degradation (28), while Nrf2 ablation slows the progression of diabetic cardiomyopathy (DC) in cardiomyocyte-specific ATG5 KO mice (29). G protein-coupled receptor kinase 4 (GRK4) aggravates cardiomyocyte injury during myocardial infarction (MI) by inhibiting histone deacetylase 4 (HDAC4)-mediated Beclin-1 transcription, while MI-induced cardiac dysfunction and remodeling are improved by deleting cardiomyocyte-specific GRK4 (30). Moreover, other regulatory factors of autophagy, including KAT8 Regulatory NSL complex subunit 1 (KANSL1) (31), Lysosome-associated membrane protein 2 (LAMP2) (32–34), insulin-like growth factor 1 receptor (IGF1R) (35) and HDAC (36, 37), also play imperative roles in maintaining cardiac fitness, and their abnormality leads to heart diseases. These findings demonstrate that autophagy is important for cardiac function. However, in some cases, overactivation of autophagy can compromise cardiac fitness. For example, cardiac-specific knockout of the genes encoding the lysosomal proteins Rag family protein A/B (RagA/B) causes lysosomal storage disorder characterized by increased autophagosome accumulation due to the activation of yes-associated protein 1 (YAP1)-TFEB transcription (38). Furthermore, cardiomyocyte-specific transgenic thrombospondin-1 (Thbs1) mice develop lethal cardiac atrophy due to overactivation of PERK/ATF4-mediated autophagy (39).
MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) can modulate the expression of autophagy-related proteins and pathways (40) and are potential druggable targets for heart disease treatment (41). miR-221 induces HF by inhibiting mTOR-mediated autophagy, while rapamycin treatment abolishes the miR-221-induced suppression of autophagy and cardiac remodeling (42). The defective autophagic response and HF are caused when FoxO3 is inhibited by cardiomyocyte-specific overexpression of miR-212/132 (43) or mTORC1 is activated by miR-199a (44). Moreover, the suppression of lncRNA Gm15834 mitigates autophagy-mediated myocardial hypertrophy by downregulating ULK1 in mice (45).
Chaperone-assisted selective autophagy in heart diseases
The chaperone-assisted selective autophagy (CASA) machinery consists of the chaperones heat shock protein 70 (HSC70), heat shock protein beta-8 (HSPB8), co-chaperone Bcl2-associated athanogene 3 (BAG3), STIP1 homology and U-Box containing protein 1 (STUB1), and autophagic receptor sequestosome-1 (SQSTM1, also known as p62). CASA primarily mediates the autophagic degradation of filamin C, which is involved in actin–actin and actin–integrin interactions in muscle tissues (46, 47). Emerging evidence has demonstrated that BAG3 plays an essential role in maintaining cardiac function (46, 47). Human BAG3P209L-eGFP expression in mice causes the disintegration of Z-disc, accumulation of protein aggregates and development of early-onset restrictive cardiomyopathy with increased mortality, in line with the observation in BAG3P209L patients (48, 49). Histological and biochemical assays revealed the alterations in protein quality control system and autophagy in heart tissues from BAG3P209L-eGFP transgenic mice and patients (48, 49). Similarly, compromised CASA impairs cardiomyocyte contractility and leads to HF in BAG3 heterozygous KO mice (50). A recent study showed that loss-of-function of BAG5 (one of the BAG3 paralogs) also led to dilated cardiomyopathy (DCM), which is characterized by enlargement and dilation of the ventricles along with impaired contractility, in mice and humans partly by disrupting the interaction with HSC70 (51).
Mitophagy in heart diseases
Defects in mitophagy, a selective autophagy targeting mitochondria, have been closely linked to cardiac disorders (1). The classic PINK1/PRAK2-mediated mitophagy is essential for cardiac mitochondrial fitness and protects the heart from cardiomyopathy (52). Once mitochondria are damaged, PINK1 is increased and activated by autophosphorylation on the outer mitochondrial membrane (OMM). Activated PINK1 further phosphorylates ubiquitin, promoting the ubiquitin E3 ligase PRAK2 recruitment to mitochondria. Meanwhile, phospho-ubiquitin recruits and binds with autophagy receptors to initiate autophagosome formation. Parkin functions as an amplifier of mitophagy through further ubiquitination of mitochondrial proteins.
Park2 global knockout mice display a decrease in survival and develop larger infarcts than wild-type (WT) mice after MI (53), and cardiomyocyte-specific deletion of Park2 manifests cardiac hypertrophy at birth and early lethality (54). Systematic knockout of Pink1 leads to left ventricular defects and age-dependent cardiac hypertrophy by compromising mitochondrial fitness and increasing oxidative stress (55). Additionally, heart defects are also observed in the mouse models related to other key mitophagy factors, such as double KO of Bcl2 interacting protein 3 (BNIP3) and Bcl2 interacting protein 3 (Nix/BNIP3L) (56), cardiomyocyte-specific KO of mitophagy receptor Mitofusin 2 (Mfn2) (57), and inducible double KO of cardiac Mfn1/2 (58). As expected, the impairment in classic autophagy machinery including ATG5 (59, 60), ATG7 (61) as well as AMPKα2 (62) causes heart defects by altering mitophagy. In addition to the core components, the maintenance of heart fitness also requires the involvement of some other regulatory proteins of PINK1/PRAK2-mediated mitophagy such as TAM41 Mitochondrial Translocator Assembly and Maintenance Homolog (TAMM41) (63), acetyl-CoA carboxylase 2 (ACC2) (64), tumor protein p53 (p53) (65), Ras homolog family member A (RhoA) (66), and succinate dehydrogenase assembly factor 4 (SDHAF4) (67).
Mitophagy also plays a crucial role in preventing diabetes-induced cardiomyopathy (68), particularly for ULK1/Rab9 (Ras-related protein 9)-mediated mitophagy (69). As an alternative mitophagy, energy stress activates AMPK-mediated phosphorylation of Ulk1. Phosphorylated Ulk1 interacts with and further phosphorylates the Golgi-derived membrane-associated Rab9. Phosphorylated Rab9 forms a complex with receptor interacting protein kinase-1 (Rip1) and dynamin-related Protein 1 (Drp1), thereby catalyzing the phosphorylation of Drp1 by Rip1. Mitochondria with phosphorylated Drp1 are recognized and engulfed by Rab9-associated membranes, and finally degraded by lysosomes. Recent studies showed that Ulk1/Rab9-mediated mitophagy protected the heart against ischemic damage (70) and obesity-associated cardiomyopathy (71) in mice.
Targeting autophagy for the treatment of heart diseases
The abovementioned evidence indicates that autophagy is essential for cardiac homeostasis and function. Stimulation of autophagy can protect against cardiac defects, as supported by the fact that several autophagy activators manifest a potent therapeutic potential for cardiac disorders (72) (Figure 4 and Table 2).
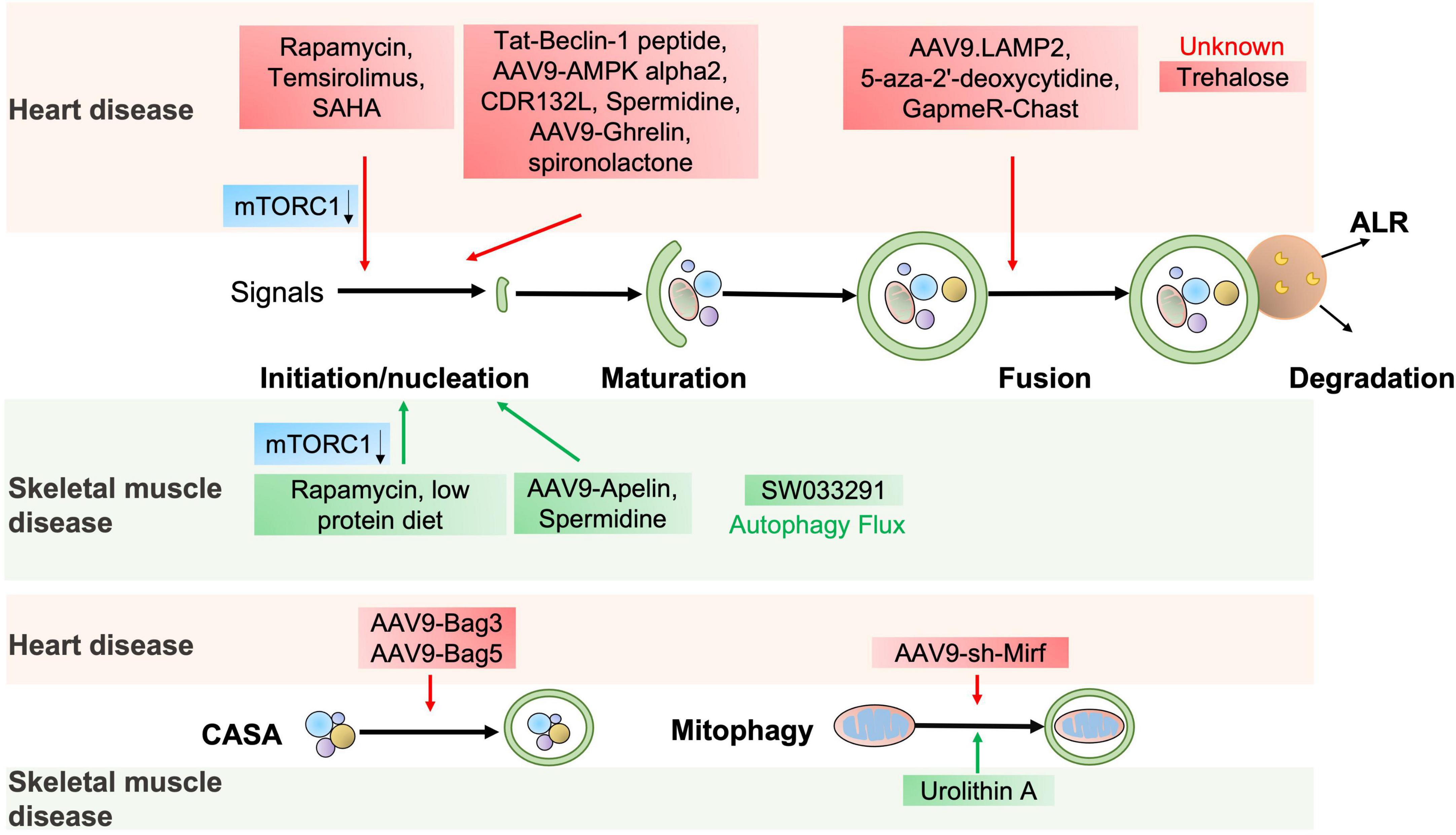
Figure 4. The potential autophagic targets for striated muscle disease treatment. The diagram includes current drug candidates (small molecules, peptides, ASO, and gene therapy) that act at different stages of the autophagy and might be beneficial in heart or skeletal muscle diseases.
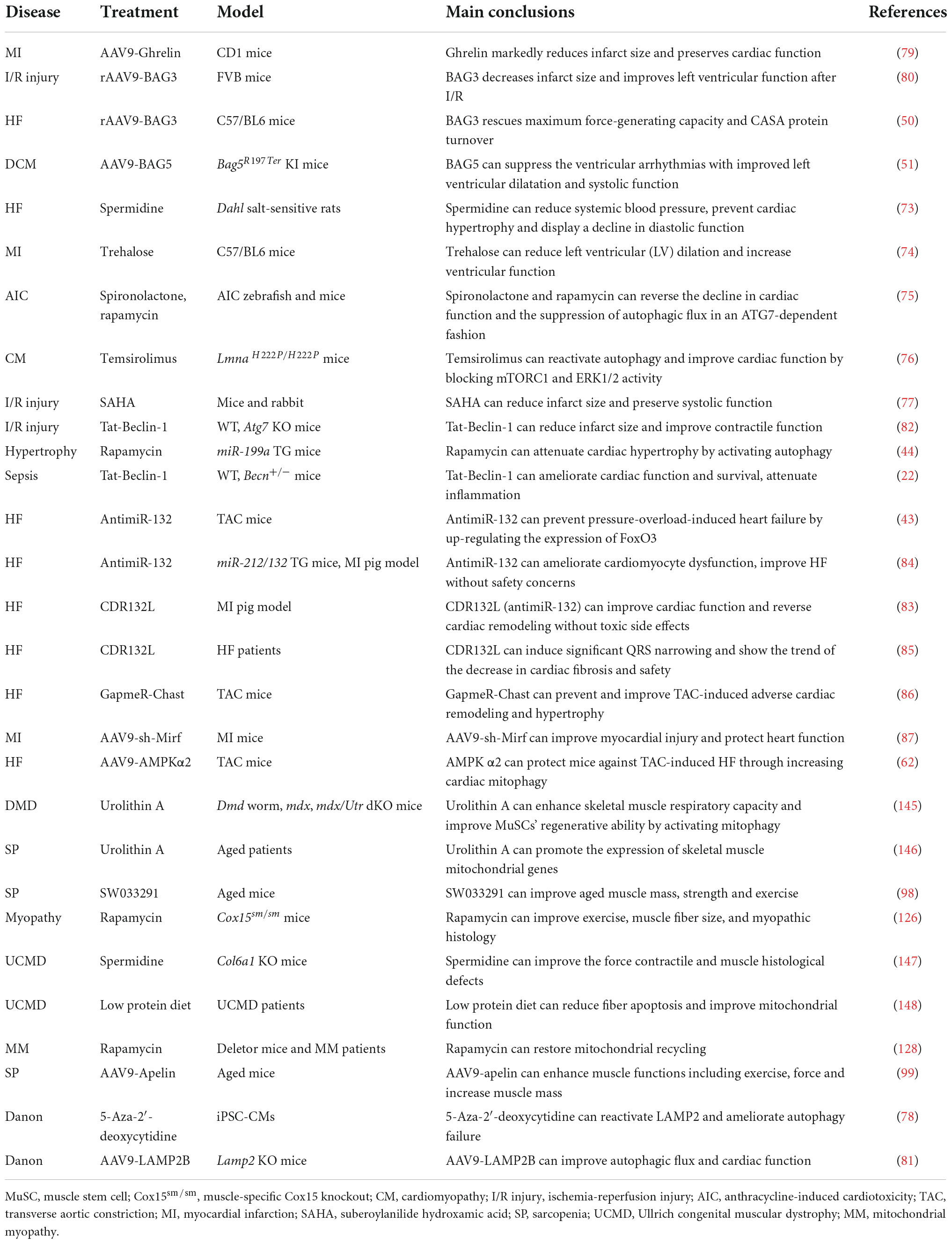
Table 2. Targeting autophagy for striated muscle disease treatment.
The autophagy agonist spermidine, a natural polyamine usually found in mammals, exerts cardioprotective effects including a decrease in cardiac hypertrophy and maintenance of diastolic function in mice and rats (73). Trehalose, a natural non-reducing disaccharide, significantly reduces ischemic remodeling, cardiac dysfunction, and HF in a chronic MI mouse model by activating TFEB-mediated autophagy (74). Anthracycline, including doxorubicin (DOX), is an effective antitumor drug, but the dose-dependent cardiotoxicity limits its application. Recent findings have revealed that anthracycline-induced cardiotoxicity (AIC) was associated with autophagy suppression (75). The Food and Drug Administration (FDA)-approved autophagy activators such as spironolactone, pravastatin, and minoxidil can mitigate AIC by activating ATG7-dependent autophagy (75). Moreover, the beneficial effects of treating autophagy-related heart diseases are also observed with other reagents, like rapamycin for cardiac hypertrophy (44), a rapamycin analog temsirolimus for LMNA-related heart defects (76), an FDA-approved HDAC inhibitor SAHA for MI (77), and a DNA demethylating agent 5-aza-2′-deoxycytidine for the heart defects related with Danon disease (78).
Emerging studies have shown that gene therapy may offer a promising approach for heart disease treatment. AAV9-Ghrelin preserves cardiac function and reduces infarct size after MI, via activating autophagy and eradicating damaged mitochondria after MI (79). The overexpression of rAAV9-BAG3 decreases infarct size and improves left ventricular function after I/R injury by activating autophagy and apoptosis (80). Moreover, similar improvements are also observed in AAV9-BAG3 for HF (48), AAV9-BAG5 for DCM (51), AAV9-LAMP2 for Danon disease (81), AAV9-AMPK α2 for transverse aortic constriction (TAC)-induced chronic HF (62), a cell-permeable Tat-Beclin-1 peptide for LPS-induced heart defects (22) and I/R injury (82).
Almost all aspects of cardiac cell function are regulated by a massive series of non-coding RNAs, including miRNAs and lncRNAs (41). Targeting non-coding RNAs of interest provides innovative therapeutic approaches for heart disease treatment by delivering short, antisense oligonucleotides (ASOs). Specific antagomirs against miR-132 safeguard against pressure-overload-induced HF by modulating FoxO3-mediated autophagy (43). As antimiR-132 (also known as CDR132L) shows high therapeutic efficacy in the mouse and pig models of HF (83, 84), this compound has recently entered the clinical trial stage in HF patients (85). LncRNA Chast induces cardiomyocyte hypertrophy and pathological heart remodeling in mice, as Chast impedes cardiomyocyte autophagy by negatively regulating the expression of the autophagy regulator PLEKHM1. Silencing of LncRNA Chast with ASO prevents and improves TAC-induced adverse cardiac remodeling without early signs of toxicity (86). Moreover, silencing of LncRNA 2810403D21Rik/Mirf mitigates cardiac injury and improves heart function in MI mice by promoting miR26a/USP15-mediated autophagy (87).
Aberrant autophagy in skeletal muscle diseases
Appropriate autophagy is not only essential for cardiac muscle homeostasis and function, but also for maintaining skeletal muscle structure and fitness under basal and stress conditions (88, 89). Autophagy defects lead to various skeletal muscle diseases, as shown in Table 3. Mutations in the core genes related to the autophagy process lead to muscle diseases, as evidenced by the fact that muscle-specific ATG7 deletion results in severe muscle atrophy and an age-dependent decline of force in mice (90) and muscle weakness in human patients (91). Similarly, mice with conditional knockout of ATG5 in skeletal muscle exhibit pronounced muscle wasting, kyphosis, and growth retardation (92). Interestingly, muscle-specific knockout of Vps15 causes the symptoms of autophagic vacuolar myopathy (AVM) with remarkable glycogen accumulation (93). Moreover, skeletal muscle defects are also caused by the mutations of other key autophagic genes, like Pik3c3 (also known as Vps34) (94), Atg14 (21), Ulk1, and Ulk2 (95). This notion that autophagy is required for muscle fitness is further substantiated by human skeletal muscle diseases with aberrant autophagy and/or accumulation of damaged organelles, such as sarcopenia, muscular dystrophies, and other myopathies (2, 4, 89, 96).
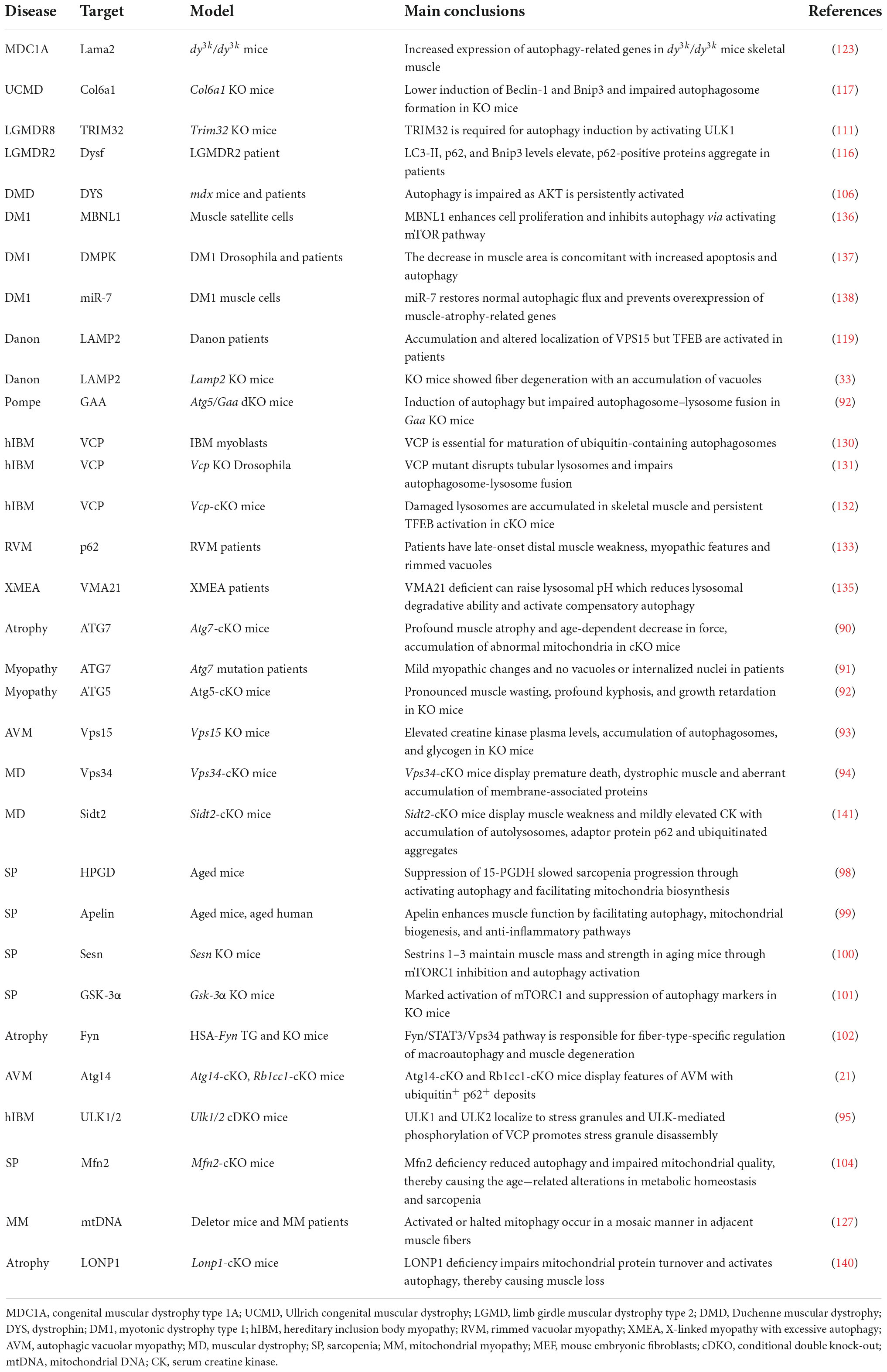
Table 3. Autophagy in skeletal muscle diseases.
Autophagy in sarcopenia
Sarcopenia, which commonly occurs in elders, is a progressive skeletal muscle disorder characterized by the accelerated loss of muscle mass and function closely linked to increased health concerns, including falls, functional decline, frailty, and even mortality (96). The etiology of sarcopenia is associated with multiple factors, including defective autophagy, where a time-dependent decline in autophagy activity causes stemness impairment in muscle satellite stem cells (96, 97). This tenet is further supported by recent findings demonstrating that suppression of the prostaglandin-degrading enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH or HPGD) slowed sarcopenia progression partly through activating autophagy (98) and that exerkine apelin reversed sarcopenia partially by triggering autophagy in mice and humans (99). Autophagy contributes to the maintenance of muscle mass and strength mediated by Sestrins 1–3 in aging mice (100). Glycogen synthase kinase-3 alpha (GSK3α) and Tyrosine-protein kinase (Fyn) are also involved in age-related alterations in sarcopenia by modulating autophagy (101, 102). Furthermore, mitophagy impairment has been associated with sarcopenia, as supported by the observation that the impairment of genes related to mitochondrial fusion or fission contributed to age-dependent muscle degeneration (103). For example, age-dependent loss or genetic disruption of Mfn2 in mouse skeletal muscle causes sarcopenia via inhibition of mitophagy (104).
Autophagy in muscular dystrophy
Duchenne muscular dystrophy (DMD) caused by DMD gene mutations is the most common childhood form of muscular dystrophy, with approximately 1 in 5,000 male births worldwide (105). DMD codes for the dystrophin protein, a cytoskeletal protein that functions in the muscle force transmission and sarcolemmal stability of muscle fibers. Loss of dystrophin leads to progressive muscle weakness and wasting, loss of ambulation, respiratory impairment, cardiomyopathy, and eventual death. A previous study demonstrated that autophagy was defective at late stages of disease progression in Dmd mice and DMD patients (106) and that autophagy impairment correlated with the decline in muscle regeneration and the increase in fibrotic tissue deposition in dystrophic muscles by modulating satellite cell activity (107). Autophagy induction is impaired as mTOR is constitutively activated, leading to the downregulation of LC3, Atg12, Bnip3, and Gabarapl1 in mdx mice (106). Moreover, PINK1/PRAK2-mediated mitophagy deficits also contribute to dystrophic phenotypes in a mdx mouse model (108).
Limb-girdle muscular dystrophies (LGMDs), the fourth most prevalent genetic muscle disease, are a group of genetically heterogeneous disorders characterized by progressive muscle weakness (5). LGMDs have more than 30 subtypes with variable severity and time of onset, and the pathological mechanism of some types has been associated with aberrant autophagy (109). LGMDR8 (110), characterized by impaired muscle regrowth and atrophy, is caused by mutations in the ubiquitin ligase Tripartite motif-containing protein 32 (TRIM32). TRIM32 is required for autophagy induction in response to atrophic stimuli in vivo by catalyzing unanchored K63-linked polyubiquitin of ULK1 and promoting the interaction of ULK1 with autophagy/Beclin 1 regulator 1 (AMBRA1) (111). LGMDR9 is an autosomal recessive disorder defined by proximal muscle weakness, calf hypertrophy, hypotonia and elevated CK level. LGMDR9 is caused by mutations in the fukutin-related protein gene (FKRP) encoding a glycosyltransferase involved in α-dystroglycan modification. A recent finding showed that Atg7 and LC3B-II were markedly increased, but p62 and mTOCR1 were decreased in LGMDR9 patients, indicating that autophagy activation has been linked with disease development (112). Conversely, another study found that autophagy was downregulated in patient-specific LGMDR9 iPSC-derived myotubes (113). LGMDR2 caused by DYSF mutation is an autosomal recessive disease, characterized by muscle inflammation, fibrosis and progressive weakness in the hip and shoulder area (114, 115). LGMDR2 patients display elevated LC3-II, p62, and Bnip3 levels (116).
Mutations of COL6A1 encoding collagen type VI has been linked to Ullrich congenital muscular dystrophy (UCMD) characterized by early-onset and generalized muscle weakness, and Bethlem myopathy (BM) characterized by proximal muscle weakness and flexion contractures. Autophagy defects are observed in Col6a1 deficient mice, in which abnormal AKT-mTOR pathway signaling pathway lowers the induction of Beclin-1 and Bnip3 and impairs autophagosome formation in muscle fibers (117). The massive accumulation of autophagosomes can cause autophagic vacuolar myopathies (AVMs) such as Danon disease (DD) and Pompe disease (4, 118). The causative defect of LAMP2 leads to Danon disease characterized by weakening of myocardial and skeletal muscles (32). Disruption of LAMP2 expression blocks the normal maturation of autophagosomes in Lamp2-deficient mice and impairs the fusion of autophagosome with lysosome in LAMP2-deficient human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) (34, 119, 120). Furthermore, the mutations in GAA encoding an acid alpha−glucosidase cause Pompe disease characterized by abnormal buildup of glycogen and muscle weakness. The fusion of autophagosome-lysosome is suppressed while autophagy initiation is induced in GAA mutant mouse model (92).
Although the abovementioned muscular dystrophies are associated with defective autophagy, excessive autophagy could cause muscular dystrophies (89, 121). Congenital muscular dystrophy type 1A (MDC1A) is caused by mutations in LAMA2 encoding the laminin α2 chain. MDC1A is characterized by clinically profound muscle hypotonia and progressive muscle weakness accompanied by contractures (122). Excessive autophagy appears to exacerbate the dystrophic pathologies in the Lama2-deficient mouse model and MDC1A patient tissues (123), as evidenced by the observation that a autophagy inhibitor 3-methyladenine (3-MA) improves MDC1A (123). However, the detailed relationship between Laminin α2 and autophagy remains elusive due to a lack of autophagic dynamics.
Autophagy in other myopathies
Mitochondrial myopathies (MM) are clinically and biochemically heterogeneous disorders characterized by ragged red fibers and peripheral and intermyofibrillar accumulations of abnormal mitochondria (124). The skeletal muscle-specific deletion of Cox15 encoding a Cytochrome C Oxidase Assembly protein, leads to severe myopathy in mice (125). Meanwhile, rapamycin can improve this myopathy by activating TFEB-mediated lysosome biosynthesis and autophagic flux (126). A recent study found that human patients with MM and Deletor mice (127), a model of adult-onset MM with multiple mtDNA deletions, exhibited overtly abnormal mitophagy by activating mTORC1 (128).
Defects in CASA cause myofibrillar myopathies characterized by Z-band disorganization and rimmed vacuoles (2). Under physiological conditions, CASA targets unfold filamin C for timely autophagic degradation. If CASA is defective, misfolded filamin C and other Z-disc proteins accumulate and impair the integrity of the Z-disc, causing myofibrillar machinery dysfunction (2).
Muscle cells from patients with inclusion body myopathy (IBM) build up ubiquitin-positive rimmed vacuoles and non-digested autophagic vacuoles (129, 130). One of the causative genes for hereditary inclusion body myopathy (hIBM) is VCP encoding valosin-containing protein (VCP), whose mutation disrupts the maturation of ubiquitin-containing autophagosomes (130) and the dynamic tubular lysosomal network in fruit flies (131), thereby impairing autophagosome-lysosome fusion. Skeletal muscle-specific KO of Vcp in adult mice causes necrotic myopathy with accumulating macroautophagic/autophagic proteins, damaged lysosomes, and persistent activation of TEFB-mediated lysosome biosynthesis (132). The dominantly inherited mutations in SQSTM1 have been linked to rimmed vacuolar myopathy (RVM) by blocking the aggregated and ubiquitinated proteins to the autophagosome for degradation (133) or perturbing the stress granule dynamics (134). X-linked myopathy with excessive autophagy (XMEA), a childhood onset disease characterized by progressive vacuolation and weakness of skeletal muscle, is attributed to the decrease in Vacuolar ATPase Assembly Factor 21 (VMA21), essential for lysosomal degradative ability by assembling the vacuolar ATPase (135). Moreover, the muscle integrity is also fine-tuned by other autophagic modulators, such as muscle blind-like 1 (MBNL1) (136), myotonic dystrophy protein kinase (DMPK) (137), miR-7 (138), inositol polyphosphate 5-phosphatase (INPP5K) (139), ion protease homolog (LONP1) (140) and Sid1 transmembrane family member 2 (Sidt2) (141).
Targeting autophagy for skeletal muscle disease treatment
Given that defective autophagy contributes to many skeletal muscle diseases, reactivating autophagy may be beneficial in treating these diseases, as shown in Figure 4 and Table 2. Small molecules, gene therapies, and ASO therapies targeting autophagy have been under development for myopathies (142, 143). Rapamycin improves the pathological manifestations caused by LMNA mutations (144), ameliorates the pathology of mitochondrial myopathy (126, 128), and mitigates the myopathic phenotype of Cox15sm/sm mice (126). Urolithin A, a natural microflora-derived metabolite that activates mitophagy, improves muscle function in worm and mouse models of DMD (145), and in elderly persons (146). SW033291, specifically inhibiting 15-PGDH-mediated PGE2 signaling, rejuvenates aged muscle mass, strength and exercise capacity partly by increasing autophagy (98). Moreover, the beneficial effects are also observed in other intervention approaches targeting autophagy, like an autophagy agonist spermidine or low protein diet for MDC1A (147, 148), and AAV9-Apelin for sarcopenia (99).
Summary and perspective
In summary, autophagy plays an important role in the pathogenesis of heart and skeletal muscle diseases. The above-mentioned signaling pathways and molecules are far from being exhaustive, which reflects the rapid development of the field and the complexity of the molecular regulation of autophagy but provides a framework to address the potential analogies between cardiac and skeletal muscle diseases. Some regulatory pathways of autophagy are shared by both cardiac and skeletal myocytes. First, the core machinery of autophagy (such as mTORC1 and AMPK) and CASA commonly play crucial roles in both cardiac and skeletal muscles, suggesting that they may be common therapeutic targets for diseases affecting these two tissues (Figure 4). Second, many muscular dystrophies also exhibit cardiomyopathies (Tables 1, 3). Third, although Rab9-mediated alternative mitophagy has been only demonstrated in the involvement of heart diseases until now, it does not rule out the possibility that this signaling pathway may also be involved in skeletal muscle diseases. Understanding autophagy alterations underlying these diseases has accelerated the development of pharmacological and genetic interventions. The introduction of novel animal models, therapeutic strategies and state-of-the-art approaches for autophagy studies will provide further insights into the roles of autophagy in muscles and facilitate the drug development in the future.
Despite of many studies linking autophagy alterations to various striated muscle pathologies, most employed global or conditional KO animal models to examine autophagy in a snapshot way at certain timepoints. These strategies are limited in several aspects. First, certain autophagy alterations may be a compensatory effect in genetic animal models, as organisms have evolved into sophisticated regulatory mechanisms to safeguard against genetic or environmental insults. Second, autophagy-independent functions of some autophagy-related genes may contribute to the outcomes. Third, autophagy is a highly dynamic process whereas a snapshot of autophagy may not reflect the entire picture. Manipulating autophagy-related genes at the adult stage, pharmacological interventions with high specificity as well as autophagic dynamics analysis will address these limitations in the future.
Traditional and novel experimental approaches studying autophagy in other tissues and diseases can be used to study striated muscle disorders. For instance, single-cell RNA sequencing can determine which cell types contribute to diseases, and establish the link between autophagy and cell types. Specific targets against autophagy in certain cell types will be more beneficial to treatment. Multiomics techniques will provide a broader landscape of the impact of autophagy abnormalities in striated muscle disorders. DNA sequencing applied to human biopsies may determine the relationship between the mutations in autophagy-relevant genes and myopathies. Moreover, High-throughput screening strategies based on cutting-edge CRISPR or RNAi will identify the factors involved in autophagy under physiological or pathophysiological settings of striated muscles.
Some traditional interventions including caloric restriction and small chemicals are not specific and may provoke side effects. Encouragingly, gene therapy and ASO are increasingly being explored to treat autophagy defects in genetic heart and skeletal muscle disorders. Moreover, modulating autophagy via novel approaches such as mRNA delivery and gene editing may provide increased efficacy and specificity for treating striated muscle diseases. The drug exploration will be profoundly energized via the introduction of novel models such as humanized animal models and human iPSC-derived organoids. Moreover, artificial intelligence and protein structure prediction will boost rationally design of drugs targeting autophagy with higher specificity and efficacy.
Author contributions
HL primarily wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
RH was supported by U.S. National Institutes of Health grants (R01HL116546, R01HL159900, and R01AR070752), a Parent Project Muscular Dystrophy award, and Miao family gift. RH had received commercial research support from Stealth BioTherapeutics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. (2017) 120:1812–24. doi: 10.1161/CIRCRESAHA.117.311082
2. Margeta M. Autophagy defects in skeletal myopathies. Annu Rev Pathol. (2020) 15:261–85. doi: 10.1146/annurev-pathmechdis-012419-032618
3. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. (2020) 19:12. doi: 10.1186/s12943-020-1138-4
4. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. (2021) 40:e108863. doi: 10.15252/embj.2021108863
5. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. (2018) 80:1–26. doi: 10.1146/annurev-physiol-021317-121427
6. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. (2020) 21:439–58. doi: 10.1038/s41580-020-0241-0
7. Ma X, Lu C, Chen Y, Li S, Ma N, Tao X, et al. CCT2 is an aggrephagy receptor for clearance of solid protein aggregates. Cell. (2022) 185:1325–45.e22. doi: 10.1016/j.cell.2022.03.005
8. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. (2018) 20:233–42. doi: 10.1038/s41556-018-0037-z
9. Zhou C, Wu Z, Du W, Que H, Wang Y, Ouyang Q, et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. Nat Cell Biol. (2022) 24:497–512. doi: 10.1038/s41556-022-00861-8
10. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
11. Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. (2007) 282:30107–19. doi: 10.1074/jbc.M705325200
12. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. (2015) 6:6670. doi: 10.1038/ncomms7670
13. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. (2012) 31:1095–108. doi: 10.1038/emboj.2012.32
14. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. (2012) 8:903–14. doi: 10.4161/auto.19653
15. Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol. (2021) 17:647–61. doi: 10.1038/s41574-021-00551-9
16. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke Statistics-2018 update: a report from the American Heart Association. Circulation. (2018) 137:e67–492. doi: 10.1161/CIR.0000000000000558
17. Orogo AM, Gustafsson AB. Therapeutic targeting of autophagy: potential and concerns in treating cardiovascular disease. Circ Res. (2015) 116:489–503. doi: 10.1161/CIRCRESAHA.116.303791
18. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. (2007) 13:619–24. doi: 10.1038/nm1574
19. Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. (2010) 6:600–6. doi: 10.4161/auto.6.5.11947
20. Kimura H, Eguchi S, Sasaki J, Kuba K, Nakanishi H, Takasuga S, et al. Vps34 regulates myofibril proteostasis to prevent hypertrophic cardiomyopathy. JCI Insight. (2017) 2:e89462. doi: 10.1172/jci.insight.89462
21. Li D, Vogel P, Li-Harms X, Wang B, Kundu M. ATG14 and RB1CC1 play essential roles in maintaining muscle homeostasis. Autophagy. (2021) 17:2576–85. doi: 10.1080/15548627.2021.1911549
22. Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, et al. Beclin-1-Dependent autophagy protects the heart during sepsis. Circulation. (2018) 138:2247–62. doi: 10.1161/CIRCULATIONAHA.117.032821
23. Taneike M, Nishida K, Omiya S, Zarrinpashneh E, Misaka T, Kitazume-Taneike R, et al. mTOR hyperactivation by ablation of tuberous sclerosis complex 2 in the mouse heart induces cardiac dysfunction with the increased number of small mitochondria mediated through the down-regulation of autophagy. PLoS One. (2016) 11:e0152628. doi: 10.1371/journal.pone.0152628
24. Ranek MJ, Kokkonen-Simon KM, Chen A, Dunkerly-Eyring BL, Vera MP, Oeing CU, et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature. (2019) 566:264–9. doi: 10.1038/s41586-019-0895-y
25. Oeing CU, Nakamura T, Pan S, Mishra S, Dunkerly-Eyring BL, Kokkonen-Simon KM, et al. PKG1alpha Cysteine-42 redox state controls mTORC1 activation in pathological cardiac hypertrophy. Circ Res. (2020) 127:522–33. doi: 10.1161/CIRCRESAHA.119.315714
26. Muhammad E, Levitas A, Singh SR, Braiman A, Ofir R, Etzion S, et al. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum Mol Genet. (2015) 24:7227–40. doi: 10.1093/hmg/ddv423
27. Gu S, Tan J, Li Q, Liu S, Ma J, Zheng Y, et al. Downregulation of LAPTM4B contributes to the impairment of the autophagic flux via unopposed activation of mTORC1 signaling during myocardial ischemia/reperfusion injury. Circ Res. (2020) 127:e148–65. doi: 10.1161/CIRCRESAHA.119.316388
28. Xie X, Bi HL, Lai S, Zhang YL, Li N, Cao HJ, et al. The immunoproteasome catalytic beta5i subunit regulates cardiac hypertrophy by targeting the autophagy protein ATG5 for degradation. Sci Adv. (2019) 5:eaau0495. doi: 10.1126/sciadv.aau0495
29. Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, et al. Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice. Diabetes. (2020) 69:2720–34. doi: 10.2337/db19-1176
30. Li L, Fu W, Gong X, Chen Z, Tang L, Yang D, et al. The role of G protein-coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur Heart J. (2021) 42:1415–30. doi: 10.1093/eurheartj/ehaa878
31. Li T, Lu D, Yao C, Li T, Dong H, Li Z, et al. Kansl1 haploinsufficiency impairs autophagosome-lysosome fusion and links autophagic dysfunction with Koolen-de Vries syndrome in mice. Nat Commun. (2022) 13:931. doi: 10.1038/s41467-022-28613-0
32. Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. (2000) 406:906–10. doi: 10.1038/35022604
33. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. (2000) 406:902–6. doi: 10.1038/35022595
34. Chi C, Leonard A, Knight WE, Beussman KM, Zhao Y, Cao Y, et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc Natl Acad Sci U S A. (2019) 116:556–65. doi: 10.1073/pnas.1808618116
35. Abdellatif M, Trummer-Herbst V, Martin Heberle A, Humnig A, Pendl T, Durand S, et al. Fine-Tuning cardiac insulin/insulin-like growth factor 1 receptor signaling to promote health and longevity. Circulation. (2022) 145:1853–66. doi: 10.1161/CIRCULATIONAHA.122.059863
36. Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. (2006) 113:2579–88. doi: 10.1161/CIRCULATIONAHA.106.625467
37. Morales CR, Li DL, Pedrozo Z, May HI, Jiang N, Kyrychenko V, et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci Signal. (2016) 9:ra34. doi: 10.1126/scisignal.aad5736
38. Ikeda S, Nah J, Shirakabe A, Zhai P, Oka SI, Sciarretta S, et al. YAP plays a crucial role in the development of cardiomyopathy in lysosomal storage diseases. J Clin Invest. (2021) 131:e143173. doi: 10.1172/JCI143173
39. Vanhoutte D, Schips TG, Vo A, Grimes KM, Baldwin TA, Brody MJ, et al. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat Commun. (2021) 12:3928. doi: 10.1038/s41467-021-24215-4
40. Akkoc Y, Gozuacik D. MicroRNAs as major regulators of the autophagy pathway. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118662. doi: 10.1016/j.bbamcr.2020.118662
41. Gao J, Chen X, Shan C, Wang Y, Li P, Shao K. Autophagy in cardiovascular diseases: role of noncoding RNAs. Mol Ther Nucleic Acids. (2021) 23:101–18. doi: 10.1016/j.omtn.2020.10.039
42. Su M, Wang J, Wang C, Wang X, Dong W, Qiu W, et al. Correction: MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. (2021) 28:420–2. doi: 10.1038/s41418-020-0582-4
43. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. (2012) 3:1078. doi: 10.1038/ncomms2090
44. Li Z, Song Y, Liu L, Hou N, An X, Zhan D, et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. (2017) 24:1205–13. doi: 10.1038/cdd.2015.95
45. Song C, Qi H, Liu Y, Chen Y, Shi P, Zhang S, et al. Inhibition of lncRNA Gm15834 attenuates autophagy-mediated myocardial hypertrophy via the miR-30b-3p/ULK1 axis in mice. Mol Ther. (2021) 29:1120–37. doi: 10.1016/j.ymthe.2020.10.024
46. Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. (2017) 127:3189–200. doi: 10.1172/JCI94310
47. Kirk JA, Cheung JY, Feldman AM. Therapeutic targeting of BAG3: considering its complexity in cancer and heart disease. J Clin Invest. (2021) 131:e149415. doi: 10.1172/JCI149415
48. Kimura K, Ooms A, Graf-Riesen K, Kuppusamy M, Unger A, Schuld J, et al. Overexpression of human BAG3(P209L) in mice causes restrictive cardiomyopathy. Nat Commun. (2021) 12:3575. doi: 10.1038/s41467-021-23858-7
49. Schanzer A, Rupp S, Graf S, Zengeler D, Jux C, Akinturk H, et al. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol Genet Metab. (2018) 123:388–99. doi: 10.1016/j.ymgme.2018.01.001
50. Martin TG, Myers VD, Dubey P, Dubey S, Perez E, Moravec CS, et al. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomeric protein turnover. Nat Commun. (2021) 12:2942. doi: 10.1038/s41467-021-23272-z
51. Hakui H, Kioka H, Miyashita Y, Nishimura S, Matsuoka K, Kato H, et al. Loss-of-function mutations in the co-chaperone protein BAG5 cause dilated cardiomyopathy requiring heart transplantation. Sci Transl Med. (2022) 14:eabf3274. doi: 10.1126/scitranslmed.abf3274
52. Dorn GW II. Parkin-dependent mitophagy in the heart. J Mol Cell Cardiol. (2016) 95:42–9. doi: 10.1016/j.yjmcc.2015.11.023
53. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. (2013) 288:915–26. doi: 10.1074/jbc.M112.411363
54. Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW II. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. (2015) 350:aad2459. doi: 10.1126/science.aad2459
55. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. (2011) 108:9572–7. doi: 10.1073/pnas.1106291108
56. Dorn GW II. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. (2010) 3:374–83. doi: 10.1007/s12265-010-9174-x
57. Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. doi: 10.1126/science.1231031
58. Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. (2016) 7:e2238. doi: 10.1038/cddis.2016.139
59. Zhao W, Li Y, Jia L, Pan L, Li H, Du J. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II. Free Radic Biol Med. (2014) 69:108–15. doi: 10.1016/j.freeradbiomed.2014.01.002
60. Ljubojevic-Holzer S, Kraler S, Djalinac N, Abdellatif M, Voglhuber J, Schipke J, et al. Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling. Cardiovasc Res. (2021) 118:1492–505. doi: 10.1093/cvr/cvab112
61. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. (2019) 124:1360–71. doi: 10.1161/CIRCRESAHA.118.314607
62. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res. (2018) 122:712–29. doi: 10.1161/CIRCRESAHA.117.312317
63. Yang RM, Tao J, Zhan M, Yuan H, Wang HH, Chen SJ, et al. TAMM41 is required for heart valve differentiation via regulation of PINK-PARK2 dependent mitophagy. Cell Death Differ. (2019) 26:2430–46. doi: 10.1038/s41418-019-0311-z
64. Shao D, Kolwicz SC Jr., Wang P, Roe ND, Villet O, Nishi K, et al. Increasing fatty acid oxidation prevents high-fat diet-induced cardiomyopathy through regulating parkin-mediated mitophagy. Circulation. (2020) 142:983–97. doi: 10.1161/CIRCULATIONAHA.119.043319
65. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. (2013) 4:2308. doi: 10.1038/ncomms3308
66. Tu M, Tan VP, Yu JD, Tripathi R, Bigham Z, Barlow M, et al. RhoA signaling increases mitophagy and protects cardiomyocytes against ischemia by stabilizing PINK1 protein and recruiting Parkin to mitochondria. Cell Death Differ. (2022): [Epub ahead of print]. doi: 10.1038/s41418-022-01032-w
67. Wang X, Zhang X, Cao K, Zeng M, Fu X, Zheng A, et al. Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy. Nat Commun. (2022) 13:3947. doi: 10.1038/s41467-022-31548-1
68. Kenny HC, Abel ED. Heart failure in Type 2 diabetes mellitus. Circ Res. (2019) 124:121–41. doi: 10.1161/CIRCRESAHA.118.311371
69. Dhingra R, Rabinovich-Nikitin I, Kirshenbaum LA. Ulk1/Rab9-mediated alternative mitophagy confers cardioprotection during energy stress. J Clin Invest. (2019) 129:509–12. doi: 10.1172/JCI125980
70. Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. (2019) 129:802–19. doi: 10.1172/JCI122035
71. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Alternative mitophagy protects the heart against obesity-associated cardiomyopathy. Circ Res. (2021) 129:1105–21. doi: 10.1161/CIRCRESAHA.121.319377
72. Frati G, Vecchione C, Sciarretta S. Novel beneficial cardiovascular effects of natural activators of autophagy. Circ Res. (2018) 123:947–9. doi: 10.1161/CIRCRESAHA.118.313530
73. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22:1428–38. doi: 10.1038/nm.4222
74. Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. (2018) 71:1999–2010. doi: 10.1016/j.jacc.2018.02.066
75. Wang Y, Lu X, Wang X, Qiu Q, Zhu P, Ma L, et al. atg7-Based autophagy activation reverses doxorubicin-induced cardiotoxicity. Circ Res. (2021) 129:e166–82. doi: 10.1161/CIRCRESAHA.121.319104
76. Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. (2012) 4:144ra102. doi: 10.1126/scitranslmed.3003875
77. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. (2014) 129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416
78. Ng KM, Mok PY, Butler AW, Ho JC, Choi SW, Lee YK, et al. Amelioration of X-Linked related autophagy failure in danon disease with DNA methylation inhibitor. Circulation. (2016) 134:1373–89. doi: 10.1161/CIRCULATIONAHA.115.019847
79. Ruozi G, Bortolotti F, Falcione A, Dal Ferro M, Ukovich L, Macedo A, et al. AAV-mediated in vivo functional selection of tissue-protective factors against ischaemia. Nat Commun. (2015) 6:7388. doi: 10.1038/ncomms8388
80. Su F, Myers VD, Knezevic T, Wang J, Gao E, Madesh M, et al. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury. JCI Insight. (2016) 1:e90931. doi: 10.1172/jci.insight.90931
81. Manso AM, Hashem SI, Nelson BC, Gault E, Soto-Hermida A, Villarruel E, et al. Systemic AAV9.LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease. Sci Transl Med. (2020) 12:eaax1744. doi: 10.1126/scitranslmed.aax1744
82. Xie M, Cho GW, Kong Y, Li DL, Altamirano F, Luo X, et al. Activation of autophagic flux blunts cardiac ischemia/reperfusion injury. Circ Res. (2021) 129:435–50. doi: 10.1161/CIRCRESAHA.120.318601
83. Batkai S, Genschel C, Viereck J, Rump S, Bar C, Borchert T, et al. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur Heart J. (2021) 42:192–201. doi: 10.1093/eurheartj/ehaa791
84. Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyongyosi M, et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat Commun. (2020) 11:633. doi: 10.1038/s41467-020-14349-2
85. Taubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. (2021) 42:178–88. doi: 10.1093/eurheartj/ehaa898
86. Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. (2016) 8:326ra22. doi: 10.1126/scitranslmed.aaf1475
87. Liang H, Su X, Wu Q, Shan H, Lv L, Yu T, et al. LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy. (2020) 16:1077–91. doi: 10.1080/15548627.2019.1659610
88. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. (2021) 22:733–50. doi: 10.1038/s41580-021-00392-4
89. Xia Q, Huang X, Huang J, Zheng Y, March ME, Li J, et al. The role of autophagy in skeletal muscle diseases. Front Physiol. (2021) 12:638983. doi: 10.3389/fphys.2021.638983
90. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab. (2009) 10:507–15. doi: 10.1016/j.cmet.2009.10.008
91. Collier JJ, Guissart C, Olahova M, Sasorith S, Piron-Prunier F, Suomi F, et al. Developmental consequences of defective ATG7-mediated autophagy in humans. N Engl J Med. (2021) 384:2406–17. doi: 10.1056/NEJMoa1915722
92. Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. (2008) 17:3897–908. doi: 10.1093/hmg/ddn292
93. Nemazanyy I, Blaauw B, Paolini C, Caillaud C, Protasi F, Mueller A, et al. Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med. (2013) 5:870–90. doi: 10.1002/emmm.201202057
94. Reifler A, Li X, Archambeau AJ, McDade JR, Sabha N, Michele DE, et al. Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am J Pathol. (2014) 184:1819–30. doi: 10.1016/j.ajpath.2014.02.012
95. Wang B, Maxwell BA, Joo JH, Gwon Y, Messing J, Mishra A, et al. ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol Cell. (2019) 74:742–57.e8. doi: 10.1016/j.molcel.2019.03.027
96. Cruz-Jentoft AJ, Sayer AA. Sarcopenia. Lancet. (2019) 393:2636–46. doi: 10.1016/s0140-6736(19)31138-9
97. Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature. (2016) 529:37–42. doi: 10.1038/nature16187
98. Palla AR, Ravichandran M, Wang YX, Alexandrova L, Yang AV, Kraft P, et al. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science. (2021) 371:eabc8059. doi: 10.1126/science.abc8059
99. Vinel C, Lukjanenko L, Batut A, Deleruyelle S, Pradere JP, Le Gonidec S, et al. The exerkine apelin reverses age-associated sarcopenia. Nat Med. (2018) 24:1360–71. doi: 10.1038/s41591-018-0131-6
100. Segales J, Perdiguero E, Serrano AL, Sousa-Victor P, Ortet L, Jardi M, et al. Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat Commun. (2020) 11:189. doi: 10.1038/s41467-019-13832-9
101. Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J Clin Invest. (2013) 123:1821–32. doi: 10.1172/JCI64398
102. Yamada E, Bastie CC, Koga H, Wang Y, Cuervo AM, Pessin JE. Mouse skeletal muscle fiber-type-specific macroautophagy and muscle wasting are regulated by a Fyn/STAT3/Vps34 signaling pathway. Cell Rep. (2012) 1:557–69. doi: 10.1016/j.celrep.2012.03.014
103. Leduc-Gaudet JP, Hussain SNA, Barreiro E, Gouspillou G. Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int J Mol Sci. (2021) 22:8179. doi: 10.3390/ijms22158179
104. Sebastian D, Sorianello E, Segales J, Irazoki A, Ruiz-Bonilla V, Sala D, et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. (2016) 35:1677–93. doi: 10.15252/embj.201593084
105. Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) 71:304–13. doi: 10.1002/ana.23528
106. De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. (2012) 3:e418. doi: 10.1038/cddis.2012.159
107. Fiacco E, Castagnetti F, Bianconi V, Madaro L, De Bardi M, Nazio F, et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. (2016) 23:1839–49. doi: 10.1038/cdd.2016.70
108. Kang C, Badr MA, Kyrychenko V, Eskelinen EL, Shirokova N. Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc Res. (2018) 114:90–102. doi: 10.1093/cvr/cvx201
109. Taghizadeh E, Rezaee M, Barreto GE, Sahebkar A. Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: a review. J Cell Physiol. (2019) 234:7874–84. doi: 10.1002/jcp.27907
110. Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J Mol Biol. (2005) 354:413–24. doi: 10.1016/j.jmb.2005.09.068
111. Di Rienzo M, Antonioli M, Fusco C, Liu Y, Mari M, Orhon I, et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci Adv. (2019) 5:eaau8857. doi: 10.1126/sciadv.aau8857
112. Franekova V, Storjord HI, Leivseth G, Nilssen O. Protein homeostasis in LGMDR9 (LGMD2I) - The role of ubiquitin-proteasome and autophagy-lysosomal system. Neuropathol Appl Neurobiol. (2021) 47:519–31. doi: 10.1111/nan.12684
113. Ortiz-Cordero C, Bincoletto C, Dhoke NR, Selvaraj S, Magli A, Zhou H, et al. Defective autophagy and increased apoptosis contribute toward the pathogenesis of FKRP-associated muscular dystrophies. Stem Cell Rep. (2021) 16:2752–67. doi: 10.1016/j.stemcr.2021.09.009
114. Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. (1998) 20:37–42. doi: 10.1038/1689
115. Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. (2003) 423:168–72. doi: 10.1038/nature01573
116. Fanin M, Nascimbeni AC, Angelini C. Muscle atrophy, ubiquitin-proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve. (2014) 50:340–7. doi: 10.1002/mus.24167
117. Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. (2010) 16:1313–20. doi: 10.1038/nm.2247
118. Castets P, Frank S, Sinnreich M, Ruegg MA. “Get the Balance Right”: pathological significance of autophagy perturbation in neuromuscular disorders. J Neuromuscul Dis. (2016) 3:127–55. doi: 10.3233/JND-160153
119. Nascimbeni AC, Fanin M, Angelini C, Sandri M. Autophagy dysregulation in Danon disease. Cell Death Dis. (2017) 8:e2565. doi: 10.1038/cddis.2016.475
120. Cenacchi G, Papa V, Pegoraro V, Marozzo R, Fanin M, Angelini C. Review: Danon disease: review of natural history and recent advances. Neuropathol Appl Neurobiol. (2020) 46:303–22. doi: 10.1111/nan.12587
121. Grumati P, Bonaldo P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells. (2012) 1:325–45. doi: 10.3390/cells1030325
122. Gawlik KI, Durbeej M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle. (2011) 1:9. doi: 10.1186/2044-5040-1-9
123. Carmignac V, Svensson M, Korner Z, Elowsson L, Matsumura C, Gawlik KI, et al. Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. (2011) 20:4891–902. doi: 10.1093/hmg/ddr427
124. Petty RK, Harding AE, Morgan-Hughes JA. The clinical features of mitochondrial myopathy. Brain. (1986) 109(Pt 5):915–38. doi: 10.1093/brain/109.5.915
125. Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. (2011) 14:80–90. doi: 10.1016/j.cmet.2011.04.011
126. Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C, et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med. (2018) 10:e8799. doi: 10.15252/emmm.201708799
127. Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. (2005) 102:17687–92. doi: 10.1073/pnas.0505551102
128. Mito T, Vincent AE, Faitg J, Taylor RW, Khan NA, McWilliams TG, et al. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab. (2022) 34:197–208.e5. doi: 10.1016/j.cmet.2021.12.017
129. Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. (2008) 1782:744–8. doi: 10.1016/j.bbadis.2008.09.003
130. Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. (2010) 6:217–27. doi: 10.4161/auto.6.2.11014
131. Johnson AE, Shu H, Hauswirth AG, Tong A, Davis GW. VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. Elife. (2015) 4:e07366. doi: 10.7554/eLife.07366
132. Arhzaouy K, Papadopoulos C, Schulze N, Pittman SK, Meyer H, Weihl CC. VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy. (2019) 15:1082–99. doi: 10.1080/15548627.2019.1569933
133. Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. (2015) 85:665–74. doi: 10.1212/WNL.0000000000001864
134. Lee Y, Jonson PH, Sarparanta J, Palmio J, Sarkar M, Vihola A, et al. TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J Clin Invest. (2018) 128:1164–77. doi: 10.1172/JCI97103
135. Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N, et al. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. (2013) 125:439–57. doi: 10.1007/s00401-012-1073-6
136. Song KY, Guo XM, Wang HQ, Zhang L, Huang SY, Huo YC, et al. MBNL1 reverses the proliferation defect of skeletal muscle satellite cells in myotonic dystrophy type 1 by inhibiting autophagy via the mTOR pathway. Cell Death Dis. (2020) 11:545. doi: 10.1038/s41419-020-02756-8
137. Bargiela A, Cerro-Herreros E, Fernandez-Costa JM, Vilchez JJ, Llamusi B, Artero R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis Model Mech. (2015) 8:679–90. doi: 10.1242/dmm.018127
138. Sabater-Arcis M, Bargiela A, Furling D, Artero R. miR-7 restores phenotypes in myotonic dystrophy muscle cells by repressing hyperactivated autophagy. Mol Ther Nucleic Acids. (2020) 19:278–92. doi: 10.1016/j.omtn.2019.11.012
139. McGrath MJ, Eramo MJ, Gurung R, Sriratana A, Gehrig SM, Lynch GS, et al. Defective lysosome reformation during autophagy causes skeletal muscle disease. J Clin Invest. (2021) 131:e135124. doi: 10.1172/JCI135124
140. Xu Z, Fu T, Guo Q, Zhou D, Sun W, Zhou Z, et al. Disuse-associated loss of the protease LONP1 in muscle impairs mitochondrial function and causes reduced skeletal muscle mass and strength. Nat Commun. (2022) 13:894. doi: 10.1038/s41467-022-28557-5
141. Liu H, Jiang W, Chen X, Chang G, Zhao L, Li X, et al. Skeletal muscle-specific Sidt2 knockout in mice induced muscular dystrophy-like phenotype. Metabolism. (2018) 85:259–70. doi: 10.1016/j.metabol.2018.05.004
142. Crudele JM, Chamberlain JS. AAV-based gene therapies for the muscular dystrophies. Hum Mol Genet. (2019) 28:R102–7. doi: 10.1093/hmg/ddz128
143. Fan J, Kou X, Jia S, Yang X, Yang Y, Chen N. Autophagy as a potential target for sarcopenia. J Cell Physiol. (2016) 231:1450–9. doi: 10.1002/jcp.25260
144. DuBose AJ, Lichtenstein ST, Petrash NM, Erdos MR, Gordon LB, Collins FS. Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc Natl Acad Sci U S A. (2018) 115:4206–11. doi: 10.1073/pnas.1802811115
145. Luan P, D’Amico D, Andreux PA, Laurila PP, Wohlwend M, Li H, et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med. (2021) 13:eabb0319. doi: 10.1126/scitranslmed.abb0319
146. Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. (2019) 1:595–603. doi: 10.1038/s42255-019-0073-4
147. Chrisam M, Pirozzi M, Castagnaro S, Blaauw B, Polishchuck R, Cecconi F, et al. Reactivation of autophagy by spermidine ameliorates the myopathic defects of collagen VI-null mice. Autophagy. (2015) 11:2142–52. doi: 10.1080/15548627.2015.1108508
Keywords: autophagy, mitophagy, muscular dystrophy, cardiomyopathy, myopathy, gene therapy, heart disease, skeletal muscle disease
Citation: Li H, Zhang L, Zhang L and Han R (2022) Autophagy in striated muscle diseases. Front. Cardiovasc. Med. 9:1000067. doi: 10.3389/fcvm.2022.1000067
Received: 21 July 2022; Accepted: 27 September 2022;
Published: 13 October 2022.
Edited by:
Dongtak Jeong, Hanyang University, South KoreaReviewed by:
Adam P. Lightfoot, Liverpool John Moores University, United KingdomJiling Liang, Wuhan Sports University, China
Copyright © 2022 Li, Zhang, Zhang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiwen Li, aGFpd2VuLmxpQG9zdW1jLmVkdQ==; Renzhi Han, cmVuemhpLmhhbkBvc3VtYy5lZHU=