Abstract
Cardiac amyloidosis is a rare, debilitating, and usually fatal disease increasingly recognized in clinical practice despite patients presenting with non-specific symptoms of cardiomyopathy. The current standard of care (SoC) focuses on preventing further amyloid formation and deposition, either with anti-plasma cell dyscrasia (anti-PCD) therapies in light-chain (AL) amyloidosis or stabilizers of transthyretin (TTR) in transthyretin amyloidosis (ATTR). The SoC is supplemented by therapies to treat the complications arising from organ dysfunction; for example, heart failure, arrhythmia, and proteinuria. Advancements in treatments have improved patient survival, especially for those whose disease is detected and for whom treatment is initiated at an early stage. However, there still are many unmet medical needs, particularly for patients with severe disease for whom morbidity and mortality remain high. There currently are no approved treatments to reverse amyloid infiltration and deplete the amyloid fibrils already deposited in organs, which can continue to cause progressive dysfunction. Anti-fibril therapies aimed at removing the deposited fibrils are being investigated for safety and efficacy in improving outcomes for patients with severe disease. However, there is no clinical evidence yet that removing deposited amyloid fibrils will improve organ function, thereby improving quality of life or extending life. Nevertheless, anti-fibril therapies are actively being investigated in clinical trials to evaluate their ability to complement and synergize with current SoC.
Introduction
Amyloidosis is a rare and debilitating disease caused by misfolded proteins that self-aggregate into amyloid fibrils and deposit into various organs (1, 2). Cardiac amyloidosis (CA) results when amyloid fibrils deposit in the interstitial spaces of the myocardium (1, 2). CA is associated with long delays in diagnosis and may be followed by short period between diagnosis and death, especially among patients with advanced disease (3). Progressive deposition of amyloid fibrils in the myocardium results in a loss of cardiac architecture and function, leading to poorer quality of life, increased hospitalizations, and death (1, 2, 4, 5). In light chain (AL) amyloidosis, the circulating amyloid precursor also contributes to cardiac dysfunction through direct toxicity (6). Although the epidemiology of CA is not fully established, it is believed that CA is underrepresented as a cause of heart failure (7, 8).
The 2 main forms of CA are AL and transthyretin (TTR) amyloidosis (ATTR; Figure 1A) (1–4, 6–8). The fibrils in AL amyloidosis consist of misfolded immunoglobulin light chains resulting from clonal B-cell proliferation or plasma cell dyscrasia (PCD) originating in the bone marrow (1–4, 6–8). The fibrils in ATTR amyloidosis consist of misfolded TTR forming due to dissociation of either the wild-type protein (ATTRwt) or facilitated by mutations in the TTR gene (ATTRv) (1–4, 6–8). Both forms of CA may be difficult to diagnose due to non-specific symptoms and overlap with other cardiomyopathies causing delayed initiation of treatment and, consequently, poorer prognosis (9, 10). Survival was significantly better among patients with AL amyloidosis diagnosed in < 6 months from symptom onset with >52% of patients surviving over the 5-year period of the study than for those whose diagnosis took longer who also had significantly increased risk of death as shown by >63% of patients dying during the study period (9). Undiagnosed and delayed diagnosis of CA results in high morbidity and high mortality whereas early diagnosis has both clinical and quality of life (QoL) benefits for patients with AL, ATTRwt, or ATTRv CA (10–12). Using a disease simulation model, early diagnosis and timely treatment have been shown to extend the calculated life expectancy from the onset of symptoms by more than 5 and more than 7 years among patients with ATTRwt and ATTRv amyloidosis, respectively (12).
Figure 1
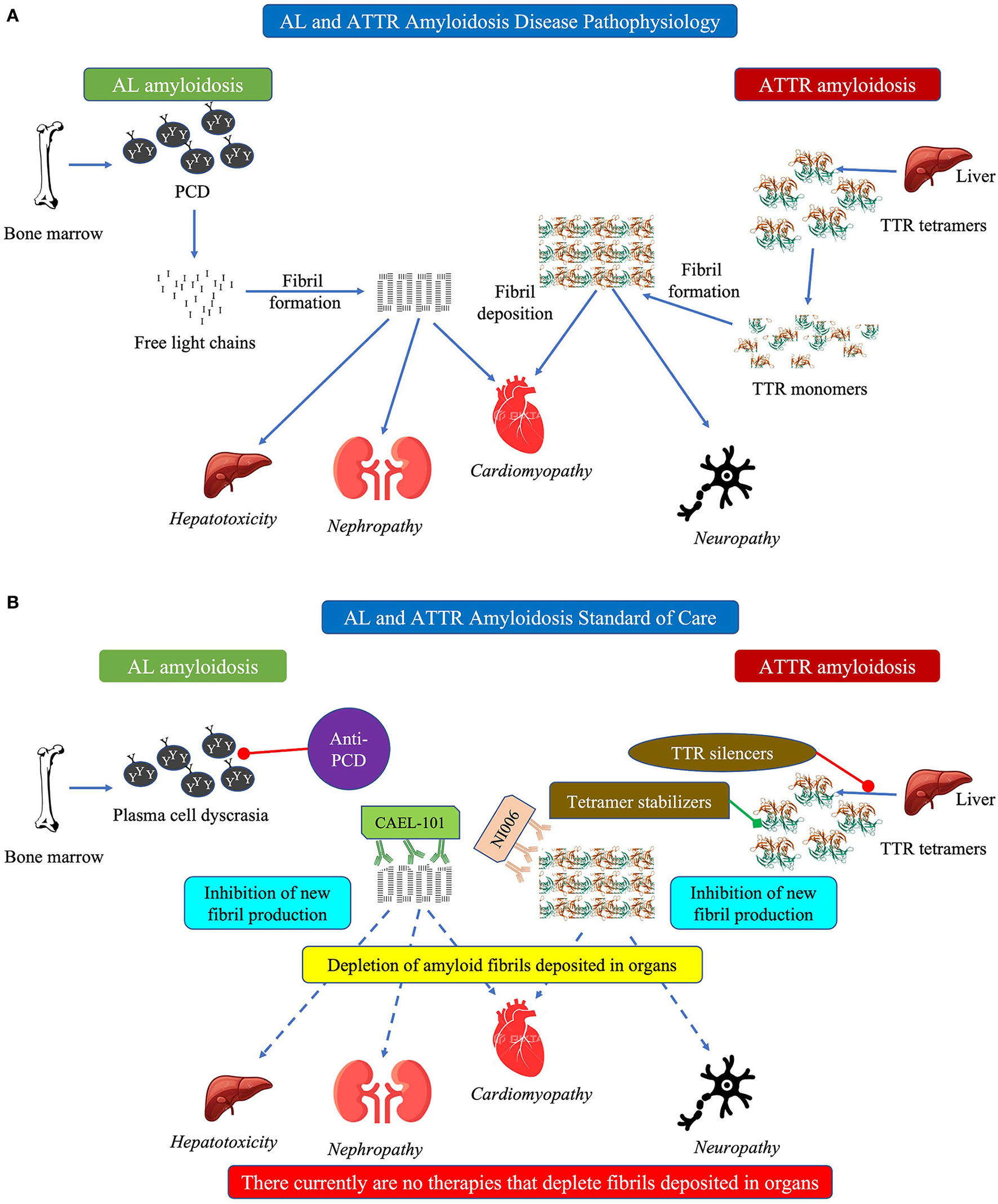
(A) Schematic diagram showing pathophysiology of cardiac amyloidosis and (B) schematic diagram showing current standard of care in cardiac amyloidosis. In (A) this schematic figure shows the pathophysiology of how both AL (left side) and ATTR (right side) amyloidosis can cause cardiomyopathy. In (B) this schematic figure shows the current standard of care for AL amyloidosis (anti-PCD) and ATTR amyloidosis (TTR silencers, tetramer stabilizers). Text in italics indicate manifestations of the disease. AL, light chain amyloidosis; ATTR, transthyretin amyloidosis; PCD, plasma cell dyscrasia; TTR, transthyretin.
Standard of care and limitations
Treatments for AL, ATTRwt, and ATTRv amyloidosis are very different. Thus, it is critical to identify the amyloidosis type and characterize the fibrils prior to treatment initiation to ensure patients receive the correct treatment (7, 8, 13). Treatment of CA is largely risk-adapted based on the disease burden and stratification of patients. For both AL and ATTR amyloidosis, the standard of care (SoC) focuses on preventing further generation and deposition of amyloid fibrils combined with supportive care (Figure 1B) (1, 7, 8, 13, 14). In addition, both AL and ATTR amyloidosis need a multidisciplinary team of specialists, specific to each patient, to manage the primary disease and the myriad comorbidities that occur (15–17).
AL amyloidosis
The basis of SoC is increased overall survival and improved organ function when amyloidogenic light chain synthesis is suppressed or stopped (18). Therefore, the focus of current, risk-adapted therapy is to prevent more amyloid fibril generation (1, 7, 8, 13, 14). The source of amyloidogenic light chains is a clonal expansion of a plasma cell, similar to multiple myeloma (1, 3, 8, 13, 14). Therefore, most therapeutic agents currently used to treat AL amyloidosis are those with proven efficacy in treating multiple myeloma (19). Validated criteria for hematologic response, classified as complete response (CR), very good partial response (VGPR), and partial response (PR), and organ response have been published previously (20). The main options are autologous stem cell transplant (ASCT) or anti-PCD chemotherapy/immunotherapy to eliminate the underlying PCD (21, 22). However, very few patients are candidates for ASCT as strict eligibility criteria, including age < 70 years, low troponin and natriuretic peptides values, preserved cardiac, hepatic and renal function, are essential to reduce transplant-related mortality (22–27). Many patients eligible for ASCT benefit from induction therapy with anti-PCD regimens to prepare them for transplant and to improve outcomes (23, 25, 28). Patients obtaining good hematologic response with induction therapy may not need ASCT (26, 29).
For patients who are ineligible for ASCT, and those who decline the procedure, anti-PCD chemotherapy is the only option (23, 28). Current guidelines recommend a combination of cyclophosphamide, bortezomib, dexamethasone (CyBorD), and daratumumab as first-line therapy for newly diagnosed patients with AL amyloidosis (27, 28). Cyclophosphamide is an alkylating agent that causes damage to DNA strands resulting in apoptosis of the cell (30). Bortezomib is a proteasome inhibitor (31). Proteasomes are a multi-subunit enzyme complexes found in large numbers in the cell and are involved in reducing proteotoxicity and regulating proteins that control cell-cycle progression and apoptosis (32, 33). Inhibition of the catalytic core of proteasomes results in accumulation of ubiquitinated proteins and cellular apoptosis (31). Amyloid-generating plasma cells are particularly sensitive to proteasome inhibition because they rely on the proteasome to reduce the toxic effects of the amyloidogenic light chains and prevent apoptosis (34). Dexamethasone induces cellular apoptosis via the nuclear glucocorticoid receptor (35). However, dexamethasone use is associated with an increased risk of a cardiac event and death among patients with severe CA (stage IIIb per the European modification of the Mayo 2004 staging system) (36, 37). There is the potential need for monitoring in an intensive care unit for all high-risk patients, e.g., those in Stage IIIb and IV, receiving chemotherapy (38, 39). Daratumumab is a monoclonal antibody (mAb) that binds to CD38, a transmembrane glycoprotein expressed on the surface of plasma cells, causing apoptosis (40). It is the only agent specifically approved for treating AL amyloidosis when administered with CyBorD. Efficacy of CyBorD-daratumumab is very high, with 78% of patients achieving significant hematologic response, defined CR or VGPR (41–43). In a small group of patients with AL amyloidosis, median survival was 655 days for those treated with CyBorD (n = 15) compared with 178 days for those treated with melphalan-dexamethasone (n = 10) (44).
However, a survey of patients with AL amyloidosis reported more than 30% reducing treatments and more than 20% discontinuing at least 1 treatment due to adverse events (AE), requiring patients to receive other drug combinations (25, 45). In regions where daratumumab or bortezomib are not available, other combinations of alkylating agents, steroids, and immunomodulatory agents often are used as first-line therapy (46). Patients who are refractory to first-line anti-PCD therapy or who relapse are treated with immunomodulatory agents (e.g., thalidomide, lenalidomide, and pomalidomide), usually in combination with dexamethasone, to overcome resistance to alkylating agents and proteasome inhibitors (23, 47). Regardless of the anti-PCD treatment regimen, all patients require comprehensive supportive care from diagnosis onward to maintain organ function as best possible (48).
Of note, all anti-PCD therapeutic agents are chemotherapeutic agents that cause cell death and consequently can have relevant toxicity. Alkylating agents like cyclophosphamide and melphalan can have severe side effects, including hematopoietic, gastrointestinal, hepatic, gonadal, pulmonary, renal, cardiac, and neural toxicity (30). Treatment with bortezomib can result in peripheral neuropathy (49). The combination of bortezomib and dexamethasone can increase plasma levels of N-terminal pro-brain natriuretic peptide (NT-proBNP), a biomarker for cardiomyopathy, and risk of death especially among patients with advanced disease (36, 37). Immunomodulators are associated with increased cardiomyopathy, thromboembolic complications, myelosuppression, immunosuppression, renal failure, and may aggravate heart failure (18, 23, 25, 47, 50). Despite recent advances, only about 50% of patients achieve complete hematologic response with the currently available therapies. Unless effective rescue treatment is employed, the disease can continue to progress in many patients, particularly those who do not attain at least hematologic VGPR, indicating a huge therapeutic gap. Isatuximab, an anti-CD38 antibody similar to daratumumab, is under investigation to treat the PCD underlying AL amyloidosis (51). However, like all other anti-PCD therapies, it does not address removal of the fibrils already deposited in organs (51).
ATTR amyloidosis
The current SoC for ATTR amyloidosis involves disease-modifying therapy to address the underlying disease, symptomatic therapy to manage cardiovascular and neurologic complications, supportive care, and genetic counseling (52–54). The goal of specific treatment in ATTR amyloidosis is to stabilize the TTR tetramer or stop amyloid fibril production (55).
The liver produces about 95% of TTR measured in the serum (52, 55). Hence, liver transplantation historically has been the first-line therapy to eliminate the main source of amyloidogenic TTR (52, 53, 55, 56). However, progression of CA after liver transplantation limits its utility, especially among patients with advanced disease (53, 55–57). This may be due to the continued presence of small amyloid fibril fragments that stimulate the aggregation of larger, pathogenic fibrils—a process termed “amyloid seeding” (58).
Silencers
TTR expression can also be decreased pharmacologically using agents that “silence” or block the synthesis of the TTR protein (Figure 1B) (59). Antisense oligonucleotides (ASO), such as inotersen, are single-stranded deoxyribonucleotide strands that are complementary to the mRNA target and block protein production of the target, TTR in this case (59, 60). Inotersen, administered subcutaneously, stabilized cardiac symptoms in patients with ATTR cardiomyopathy (ATTR-CM) (61). Whereas treatment with inotersen significantly improves neurological symptoms, in rare cases it can cause severe thrombocytopenia and glomerulonephritis resulting in a boxed warning (53, 56, 61, 62). To prescribe inotersen in the USA, physicians must be trained and certified in Risk Evaluation and Mitigation Strategy (REMS) of the drug and their patients must be enrolled in the REMS program and undergo regular monitoring.
Small interfering RNA (siRNA), such as patisiran, are a class of short double-stranded non-coding RNA molecules that recognize and degrade target mRNA, TTR mRNA in this case (59, 63, 64). Treatment with patisiran, currently only approved for treating ATTR polyneuropathy (ATTR-PN), also might be associated with cardiac amyloid regression in a proportion of patients, as evidenced by reduced extracellular volume and disease stabilization with significant differences in NT-proBNP, left ventricular wall thickness, global longitudinal strain, and cardiac output. In these patients, changes also were associated with improved overall survival and lower cardiovascular-related hospitalizations compared with placebo (65, 66). In the APOLLO-B study (NCT03997383), compared with placebo patisiran significantly improved the functional capacity, measured with 6-min walk test (6MWT), and quality of life (QoL) of patients with ATTR-CM at 12 months with no additional safety signals (reported at XVIII International Symposium on Amyloidosis, 4–8 September 2022, Heidelberg, Germany). However, there were no statistically significant benefits observed in composite secondary endpoints, including all-cause mortality. Although patisiran has fewer concerns about AE than inotersen, it needs to be administered by a trained healthcare professional and patients are exposed for long periods to corticosteroids and antihistamines to limit infusion reactions (67).
Since a normal physiological function of TTR is transporting vitamin A, reduction of TTR, due to either inotersen or patisiran, results in severe vitamin A depletion and requires daily supplementation to maintain normal levels (53, 56, 61, 62). Both drugs show improvement among patients with low to moderate disease burden. Recent data suggest patisiran also may have benefits among patients with ATTR-CM.
Vutrisiran is a second-generation siRNA formulation of patisiran and can block the expression of both ATTRwt and ATTRv genes (68). It has been approved for treatment of polyneuropathy. Compared with placebo, vutrisiran was shown to reduce serum levels of NT-proBNP, improve some echocardiographic parameters, and improve scintigraphy tracer uptake in ATTRv patients with polyneuropathy. It is currently under investigation for treatment of patients with ATTR-CM (NCT04153149).
Eplontersen is a ligand conjugated ASO with the same primary sequence as inotersen (69). Conjugation with ligand facilitates targeted uptake of the drug by hepatocytes, which has the potential for greater efficacy and lower toxicity than the unconjugated drug, inotersen (69). Eplontersen is reported to significantly lower TTR levels from baseline and be well tolerated. It is under investigation for treatment of both ATTR-CM and ATTR-PN. In the NEURO-TTRansform trial (NCT04136184), compared with placebo eplontersen slowed the progression of neuropathic disease and improved QoL among patients with hereditary ATTR-PN (reported at XVIII International Symposium on Amyloidosis). No specific safety concerns were reported.
Stabilizers
Another approach to treating ATTR amyloidosis is to stabilize the TTR tetramer protein complex, thereby preventing its dissociation into amyloidogenic TTR monomers and oligomers (Figure 1B) (70). Native TTR tetramer stabilization requires both thyroxine-binding pockets of the TTR tetramer to be occupied, thus requiring high concentrations of TTR stabilizers to prevent its dissociation (66).
Diflunisal, an oral non-steroidal anti-inflammatory drug (NSAID), stabilizes TTR tetramers (52). However, long-term use may be associated with increased fluid retention leading to heart failure, gastritis, peptic ulcer disease, and worsening of renal dysfunction (71). Therefore, diflunisal should be used with caution in older adults and in patients with severe congestive heart disease. Moreover, renal insufficiency limits its use in patients with ATTR-CM or ATTR-PN with cardiac or renal impairment requiring careful patient selection and management of drug-associated AEs (52).
Tafamidis is an orally bioavailable agent, and the only one approved to treat ATTR-CM (72). It occupies the thyroxine-binding sites in wild-type and several variants of TTR with high affinity and selectivity, thereby preventing their dissociation (73). Compared with placebo, treatment with tafamidis was associated with TTR stabilization in almost all patients, significantly lower all-cause mortality, lower rate of cardiovascular-related hospitalizations, and less decrease in 6MWT functional capacity indicating that tafamidis stabilized disease, delayed disease progression, and slowed decline in patient QoL (72, 74). However, a pre-specified subgroup of patients with New York Heart Association (NYHA) class III heart failure, representative of patients with advanced disease, had a higher rate of cardiovascular-related hospitalizations compared with placebo, i.e., an inverse relationship between effectiveness and NYHA class (72). However, improved survival was observed at 5-year follow-up among patients in NYHA class III compared with those who received placebo initially (reported at European Society of Cardiology Heart Failure Congress, 26–29 May 2022, Barcelona, Spain). Patients in NYHA class IV, representative of very advanced disease, were excluded from this trial. In all clinical studies, tafamidis demonstrated an appropriate benefit-to-risk ratio.
Acoramidis (AG10/ALXN2060) is a new TTR stabilizer under investigation for treatment of ATTR amyloidosis (75). Acoramidis binds TTR with greater selectivity than either tafamidis or diflunisal and increases serum levels of TTR tetramers and is well tolerated (75–77). In the ongoing open-label extension of the phase 2 trial (NCT03536767), serum TTR levels were below the lower limit of normal in 40.4% of patients and there was a median decrease from baseline of 479 pg/mL in serum NT-proBNP levels (78). Two randomized, double-blind, placebo-controlled, phase 3 studies (NCT03860935 and NCT04622046) are in progress to determine the efficacy and safety of acoramidis in patients with ATTR-CM.
Another stabilizer under investigation for treatment of ATTR amyloidosis is tolcapone (79). Like acoramidis, tolcapone binds TTR with greater selectivity than either tafamidis or diflunisal and increases serum levels of TTR tetramers (79, 80). However, tolcapone has a boxed warning for potentially fatal acute fulminant liver failure and is not suggested for patients with ATTR-CM (77).
Future anti-fibril therapies
Despite advances in treatment options, there are as yet no approved treatments for removal of amyloid fibrils already deposited in the organs, especially the heart (61, 81). These fibrils can continue to cause progressive damage to the organs resulting in death. The current hypothesis is that removal or depletion of these amyloid fibrils will decrease organ damage and restore function, particularly that of the heart resulting in improved survival (Figures 2A,B).
Figure 2
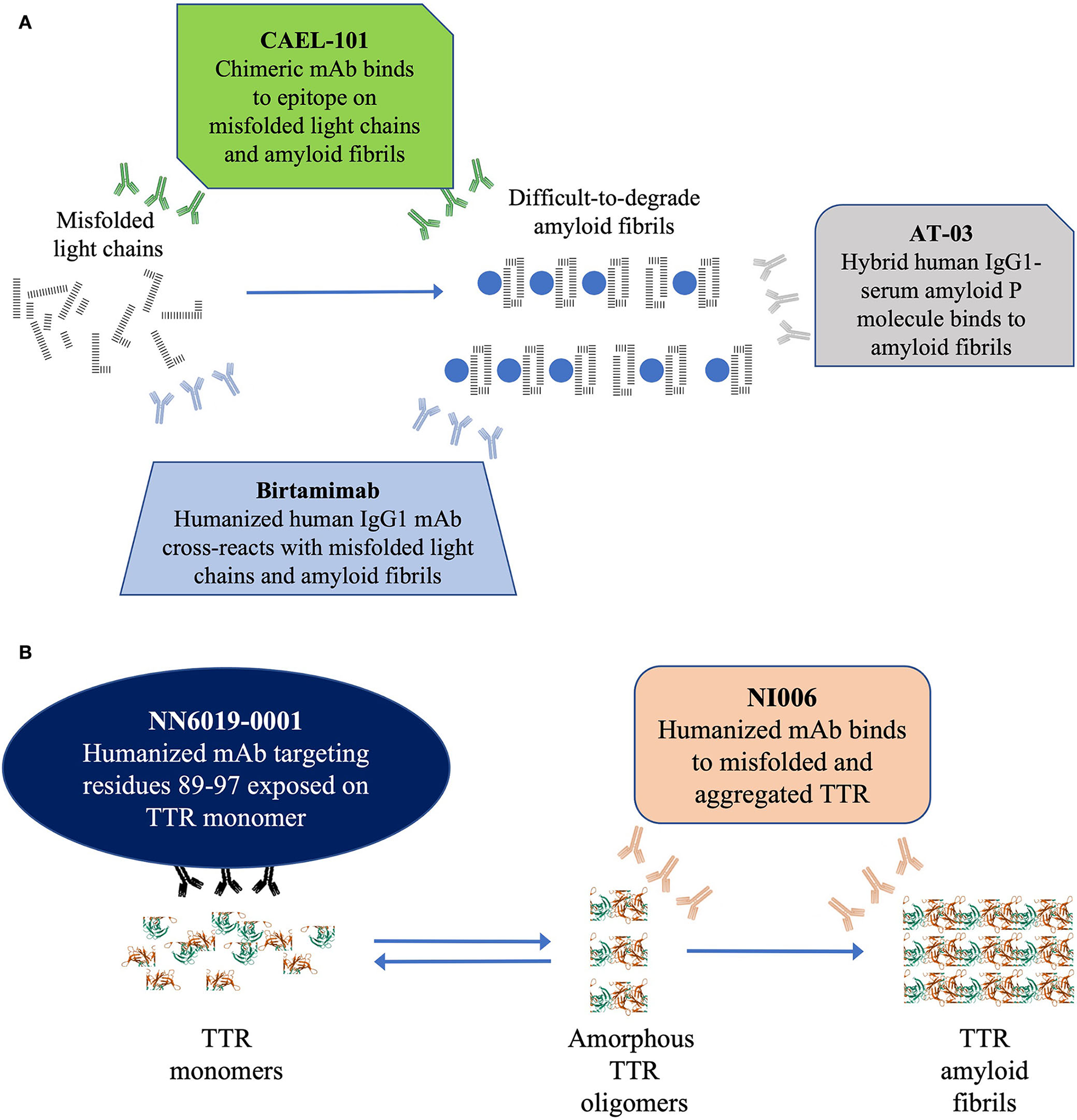
(A) Schematic diagram depicting the 3 anti-fibril mAb therapies currently under investigation for AL amyloidosis and (B) Schematic diagram depicting the 2 anti-fibril mAb therapies currently under investigation for ATTR amyloidosis. In (A) this schematic figure shows the 3 mAbs currently under investigation for depletion of light-chain fibrils in AL amyloidosis. In (B) this schematic figure shows the 2 mAbs currently under investigation for depletion of TTR fibrils in ATTR amyloidosis. AL, light chain amyloidosis; IgG1, immunoglobulin G1; mAb, monoclonal antibody.
AL amyloidosis
There currently are 3 mAbs, birtamimab, CAEL-101, and AT-03 (Figure 2A), under investigation as anti-fibril agents (82–84). It is hoped that these antibodies provide direct proof of concept by depleting the deposits of light chain amyloid fibrils from organs improving their function.
Birtamimab, a fully humanized mAb developed to recognize a cryptic epitope on serum amyloid A protein, cross reacts with light chain amyloid fibrils and activates macrophage-mediated degradation and clearance of the fibrils (83). In a phase 1/2 trial, birtamimab was well tolerated at all doses administered up to 24 mg/kg (85). In the phase 2b PRONTO trial (NCT02632786), birtamimab failed to improve cardiac response, 6MWT, and NT-proBNP levels in previously treated patients with AL amyloidosis (86). Furthermore, a futility analysis of the phase 3 VITAL (NCT02312206) trial showed that birtamimab did not reduce all-cause mortality in newly diagnosed patients resulting in termination of the trial (87). However, a post-hoc analyses showed promising results among patients in Mayo 2012 Stage IV (85, 87, 88). A double-blind, placebo-controlled, phase 3 trial (AFFIRM-AL; NCT04973137) is currently recruiting patients to confirm these results (89).
CAEL-101, a chimeric mAb developed to recognize a cryptic epitope on immunoglobulin light chains, binds to misfolded free light chains and amyloid fibrils deposited in organs (90–92). In phase 1 trials, CAEL-101 demonstrated reductions in biomarkers of cardiomyopathy and nephropathy (93–95). The ongoing phase 2 trial demonstrated that CAEL-101 was well tolerated when administered with anti-PCD therapy that included daratumumab or as monotherapy after cessation of anti-PCD therapy. There are 2 concurrent randomized, double-blind, placebo-controlled, phase 3 trials actively recruiting patients with advanced cardiac disease 2015 European Modification of Mayo 2004 Stages IIIA (NCT04512235) and IIIB (NCT04504825) (96).
AT-03, a hybrid human mAb against serum amyloid P protein, binds all types of amyloid fibrils (84). AT-03 has recently completed a phase 1 biodistribution study (NCT05201911), the results of which have not yet been reported.
ATTR amyloidosis
There currently are 2 mAbs under investigation to deplete or remove TTR amyloid fibrils, NI006 and NN6019-0001 (formerly known as PRX004; Figure 2B) (73, 97). It is hoped that these antibodies will provide direct proof of concept by depleting TTR amyloid fibril deposits from organs and improving their function.
NI006 is a humanized mAb that selectively binds to a cryptic epitope that is exposed in misfolded TTR oligomers and aggregated TTR fibrils (97). In preclinical studies, NI006 bound with high affinity to both ATTRwt and ATTRv amyloid fibrils and facilitated their elimination via activation of phagocytic cells. A phase 1 study (NCT04360434) is in progress to determine the dosage and safety of NI006 in patients with ATTR-CM.
NN6019-0001 binds to a cryptic epitope that is exposed when TTR tetramers dissociate into monomers (73). NN6019-0001 binds and neutralizes the various prefibrillar species of TTR and prevents the formation of new amyloid fibrils (73). In phase 1 trials, NN6019-0001 was well tolerated at all doses and showed improvement in both global longitudinal strain and neurologic symptoms (reported at XVIII International Symposium on Amyloidosis, 4–8 September 2022, Heidelberg, Germany). A placebo-controlled, phase 2 study (NCT05442047) is currently recruiting patients to determine the efficacy and safety of NN6019-0001 at 10 and 60 mg/kg.
Conclusions
Although there have been considerable advances in the treatment of CA, there are still opportunities for improvement. Overall CR among patients with AL amyloidosis still hovers around 50% and does not always translate into organ response. Despite advances, disease continues to progress for many patients. Current therapies to treat both AL and ATTR amyloidosis are focused on eliminating or stabilizing the source of the amyloidogenic protein. However, there are no approved therapies that deplete or eliminate already deposited amyloid fibrils, which can continue to cause progressive organ damage, especially the heart, causing early death. There are promising studies focusing on amyloid fibril depletion in both forms of CA that are expected to add to the armamentarium available and to elucidate whether removal of preexisting fibrils will improve survival and QoL of patients.
Statements
Author contributions
CCQ was invited to contribute. All authors provided critical input into the concept and content of this review article. All authors approved the submission of the final article.
Funding
This manuscript was funded by Alexion AstraZeneca Rare Disease. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
Medical writing support was provided by Mukund Nori, PhD, MBA, CMPP, of rareLife solutions, Westport, CT, USA, and funded by Alexion, AstraZeneca Rare Disease.
Conflict of interest
CCQ, JC, DS, and MM are employed by Alexion, AstraZeneca Rare Disease. MF: consultant (Akcea, Alexion, Alnylam, Eidos, Ionis, Intellia, Pfizer, Prothena, and Sanofi); advisory board (Akcea, Alnylam, Eidos, Intellia, Pfizer, and Prothena). TD: consultant (Alnylam, GlaxoSmithKline, Pfizer, and Prothena); honoraria (Alnylam, Pfizer, and Prothena; research grants (GlaxoSmithKline and Pfizer); clinical trial support (Alnylam, Ionis, and Pfizer). PG-P: consultant (Alexion, Alnylam, AstraZeneca, Attralus, Bridgebio, Intellia, Ionis, Neurimmune, NovoNordisk, and Pfizer); speakers bureau (Alnylam, Bridgebio, Ionis, and Pfizer); grant support to his institution (Alnylam and Pfizer). MM: advisory board or DSMB (Alnylam, Eidos, Intellia, Ionis, NovoNordisk); research grants (Eidos, Janssen, Pfizer); clinical trial support (Alnylam, Attralus, Eidos, Ionis, Pfizer). GP: advisory board (Alexion, Argobio, Janssen, Protego); honoraria (Alexion, Argobio, The Binding Site Group, Janssen, Pfizer, Protego, Prothena, Sebia, and Siemens); research funding (Gate Bioscience, The Binding Site Group). The remaining author declare that the article was developed in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
GriffinJMRosenblumHMaurerMS. Pathophysiology and therapeutic approaches to cardiac amyloidosis. Circ Res. (2021) 128:1554–75. 10.1161/CIRCRESAHA.121.318187
2.
Nativi-NicolauJMaurerMS. Amyloidosis cardiomyopathy: Update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. (2018) 33:571–9. 10.1097/HCO.0000000000000547
3.
MacedoAVSSchwartzmannPVde GusmaoBMMeloMDTCoelho-FilhoOR. Advances in the treatment of cardiac amyloidosis. Curr Treat Options Oncol. (2020) 21:36. 10.1007/s11864-020-00738-8
4.
KnightDSZumboGBarcellaWSteedenJAMuthuranguVMartinez-NaharroAet al. Cardiac structural and functional consequences of amyloid deposition by cardiac magnetic resonance and echocardiography and their prognostic roles. JACC Cardiovasc Imag. (2019) 12:823–33. 10.1016/j.jcmg.2018.02.016
5.
LaneTFontanaMMartinez-NaharroAQuartaCCWhelanCJPetrieAet al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. (2019) 140:16–26. 10.1161/CIRCULATIONAHA.118.038169
6.
MerliniGDispenzieriASanchorawalaVSchonlandSOPalladiniGHawkinsPNet al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. (2018) 4:38. 10.1038/s41572-018-0034-3
7.
Garcia-PaviaPRapezziCAdlerYAradMBassoCBrucatoAet al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. (2021) 42:1554–68. 10.1093/eurheartj/ehab072
8.
ManolisASManolisAAManolisTAMelitaH. Cardiac amyloidosis: An underdiagnosed/underappreciated disease. Eur J Intern Med. (2019) 67:1–13. 10.1016/j.ejim.2019.07.022
9.
SchulmanAConnorsLHWeinbergJMendelsonLMJoshiTSheltonACet al. Patient outcomes in light chain (AL) amyloidosis: the clock is ticking from symptoms to diagnosis. Eur J Haematol. (2020) 105:495–501. 10.1111/ejh.13472
10.
Pour-GhazIBathAKayaliSAlkhatibDYedlapatiNRheaIet al. A review of cardiac amyloidosis: presentation, diagnosis, and treatment. Curr Probl Cardiol. (2022) 2022:101366. 10.1016/j.cpcardiol.2022.101366
11.
CohenOCWechalekarAD. Systemic amyloidosis: Moving into the spotlight. Leukemia. (2020) 34:1215–28. 10.1038/s41375-020-0802-4
12.
RozenbaumMHLargeSBhambriRStewartMYoungRDoornewaardAVet al. Estimating the health benefits of timely diagnosis and treatment of transthyretin amyloid cardiomyopathy. J Comp Eff Res. (2021) 10:927–38. 10.2217/cer-2021-0071
13.
IhneSMorbachCObiciLPalladiniGStörkS. Amyloidosis in heart failure. Curr Heart Fail Rep. (2019) 16:285–303. 10.1007/s11897-019-00446-x
14.
SiddiqiOKRubergFL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. (2018) 28:10–21. 10.1016/j.tcm.2017.07.004
15.
KoikeHOkumuraTMuroharaTKatsunoM. Multidisciplinary approaches for transthyretin amyloidosis. Cardiol Ther. (2021) 10:289–311. 10.1007/s40119-021-00222-w
16.
SarosiekSSanchorawalaV. Treatment options for relapsed/refractory systemic light-chain (AL) amyloidosis: current perspectives. J Blood Med. (2019) 10:373–80. 10.2147/JBM.S183857
17.
WechalekarADHawkinsPNGillmoreJD. Perspectives in treatment of AL amyloidosis. Br J Haematol. (2008) 140:365–77. 10.1111/j.1365-2141.2007.06936.x
18.
SanchorawalaV. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. (2006) 1:1331–41. 10.2215/CJN.02740806
19.
RubinJMaurerMS. Cardiac amyloidosis: overlooked, underappreciated, and treatable. Annu Rev Med. (2020) 71:203–19. 10.1146/annurev-med-052918-020140
20.
PalladiniGMilaniPMerliniG. Management of AL amyloidosis in 2020. Blood. (2020) 136:2620–7. 10.1182/blood.2020006913
21.
ManwaniRCohenOSharpleyFMahmoodSSachchithananthamSFoardDet al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood. (2019) 134:2271–80. 10.1182/blood.2019000834
22.
GertzMA. Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am J Hematol. (2013) 88:416–25. 10.1002/ajh.23400
23.
DispenzieriABuadiFKumarSKReederCBSherTLacyMQet al. Treatment of immunoglobulin light chain amyloidosis: Mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus statement. Mayo Clin Proc. (2015) 90:1054–81. 10.1016/j.mayocp.2015.06.009
24.
ManwaniRFoardDMahmoodSSachchithananthamSLaneTQuartaCet al. Rapid hematologic responses improve outcomes in patients with very advanced (stage iiib) cardiac immunoglobulin light chain amyloidosis. Haematologica. (2018) 103:e165–e8. 10.3324/haematol.2017.178095
25.
MuchtarEDispenzieriAGertzMAKumarSKBuadiFKLeungNet al. Treatment of AL amyloidosis: Mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus statement 2020 update. Mayo Clin Proc. (2021) 96:1546–77. 10.1016/j.mayocp.2021.03.012
26.
SanchorawalaVBoccadoroMGertzMHegenbartUKastritisELandauHet al. Guidelines for high dose chemotherapy and stem cell transplantation for systemic AL amyloidosis: EHA-ISA working group guidelines. Amyloid. (2022) 29:1–7. 10.1080/13506129.2021.2002841
27.
WechalekarADCibeiraMTGibbsSDJaccardAKumarSMerliniGet al. Guidelines for non-transplant chemotherapy for treatment of systemic AL amyloidosis: EHA-ISA working group. Amyloid. (2022) 2022:1–15. 10.1080/13506129.2022.2093635
28.
National Comprehensive Cancer Network.Systemic Light Chain Amyloidosis: NCCN Evidence Blocks™ v2: NCCN.org.Pennsylvania: National Comprehensive Cancer Network (2021). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/amyloidosis_blocks.pdf (accessed March 12, 2021).
29.
BassetMMilaniPNuvoloneMBenignaFRodigariLFoliAet al. Sequential response-driven bortezomib-based therapy followed by autologous stem cell transplant in AL amyloidosis. Blood Adv. (2020) 4:4175–9. 10.1182/bloodadvances.2020002219
30.
ColvinM. Alkylating agents. In: Holland-Frei Cancer Medicine Hamilton.ON: BC Decker (2003). 10.1016/B0-12-227555-1/00001-0
31.
DriscollJJGirniusS. Proteasome inhibitors to treat AL amyloidosis. In: Exploring New Findings on Amyloidosis.London: Intechopen (2016). 10.5772/63467
32.
TanakaK. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. (2009) 85:12–36. 10.2183/pjab.85.12
33.
ThibaudeauTASmithDM. A practical review of proteasome pharmacology. Pharmacol Rev. (2019) 71:170–97. 10.1124/pr.117.015370
34.
OlivaLOrfanelliUResnatiMRaimondiAOrsiAMilanEet al. The amyloidogenic light chain is a stressor that sensitizes plasma cells to proteasome inhibitor toxicity. Blood. (2017) 129:2132–42. 10.1182/blood-2016-08-730978
35.
KervoëlenCMénoretEGomez-BougiePBatailleRGodonCMarionneau-LambotSet al. Dexamethasone-induced cell death is restricted to specific molecular subgroups of multiple myeloma. Oncotarget. (2015) 6:26922–34. 10.18632/oncotarget.4616
36.
BézardMOghinaSVitielloDKharoubiMKordeliEGalatAet al. Dexamethasone is associated with early deaths in light chain amyloidosis patients with severe cardiac involvement. PLoS ONE. (2021) 16:e0257189. 10.1371/journal.pone.0257189
37.
Le BrasFMolinier-FrenkelVGuellichADupuisJBelhadjKGuendouzSet al. Sequential cyclophosphamide-bortezomib-dexamethasone unmasks the harmful cardiac effect of dexamethasone in primary light-chain cardiac amyloidosis. Eur J Cancer. (2017) 76:183–7. 10.1016/j.ejca.2017.02.004
38.
KastritisEWechalekarASchönlandSSanchorawalaVMerliniGPalladiniGet al. Challenges in the management of patients with systemic light chain (AL) amyloidosis during the covid-19 pandemic. Br J Haematol. (2020) 190:346–57. 10.1111/bjh.16898
39.
BazziTKropmanKBenjaminMAl-RammahiA. Light chain amyloidosis presenting as a septic shock: a case report and review of literature. Cureus. (2022) 14:e30263. 10.7759/cureus.30263
40.
van de DonkNWCJRichardsonPGMalavasiF. Cd38 antibodies in multiple myeloma: back to the future. Blood. (2018) 131:13–29. 10.1182/blood-2017-06-740944
41.
KastritisEPalladiniGMinnemaMCWechalekarADJaccardALeeHCet al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. (2021) 385:46–58. 10.1056/NEJMoa2028631
42.
PalladiniGKastritisEMaurerMSZonderJAMinnemaMCWechalekarADet al. Daratumumab plus cybord for patients with newly diagnosed AL amyloidosis: safety run-in results of andromeda. Blood. (2020) 136:71–80. 10.1182/blood.2019004460
43.
SidiqiMHGertzMA. Daratumumab for the treatment of AL amyloidosis. Leuk Lymphoma. (2019) 60:295–301. 10.1080/10428194.2018.1485914
44.
BrennanXWithersBJabbourAMillikenSKotlyarEFayKet al. Efficacy of bortezomib, cyclophosphamide and dexamethasone in cardiac AL amyloidosis. Intern Med J. (2022) 52:1826–30. 10.1111/imj.15926
45.
RizioAAWhiteMKMcCauslandKLQuockTPGuthrieSDYokotaMet al. Treatment tolerability in patients with immunoglobulin light-chain amyloidosis. Am Health Drug Benefits. (2018) 11:430–7.
46.
LiuBWangYBaiMWangDZhaoJZhangMet al. Cyclophosphamide + thalidomide + dexamethasone vs. melphalan + dexamethasone for the treatment of amyloid light-chain amyloidosis with kidney involvement: a retrospective study in Chinese patients. Clin Ther. (2019) 41:1186–98. 10.1016/j.clinthera.2018.12.003
47.
MerliniGAL. amyloidosis: from molecular mechanisms to targeted therapies. Hematol Am Soc Hematol Educ Prog. (2017) 2017:1–12. 10.1182/asheducation-2017.1.1
48.
CibeiraMTOrtiz-PérezJTQuintanaLFFernádez de LarreaCTovarNBladéJ. Supportive care in AL amyloidosis. Acta Haematol. (2020) 143:335–42. 10.1159/000506760
49.
ReeceDESanchorawalaVHegenbartUMerliniGPalladiniGFermandJ-Pet al. Weekly and twice-weekly bortezomib in patients with systemic AL amyloidosis: results of a phase 1 dose-escalation study. Blood. (2009) 114:1489–97. 10.1182/blood-2009-02-203398
50.
MilaniPSharpleyFSchonlandSOBassetMMahmoodSNuvoloneMet al. Pomalidomide and dexamethasone grant rapid haematologic responses in patients with relapsed and refractory AL amyloidosis: a European retrospective series of 153 patients. Amyloid. (2020) 27:231–6. 10.1080/13506129.2020.1767566
51.
PopkovaTHajekRJelinekT. Monoclonal antibodies in the treatment of AL amyloidosis: co-targetting the plasma cell clone and amyloid deposits. Br J Haematol. (2020) 189:228–38. 10.1111/bjh.16436
52.
SekijimaYUedaMKoikeHMisawaSIshiiTAndoY. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. (2018) 13:6. 10.1186/s13023-017-0726-x
53.
YamamotoHYokochiT. Transthyretin cardiac amyloidosis: An update on diagnosis and treatment. ESC Heart Fail. (2019) 6:1128–39. 10.1002/ehf2.12518
54.
AndoYAdamsDBensonMDBerkJLPlante-BordeneuveVCoelhoTet al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. (2022) 29:143–55. 10.1080/13506129.2022.2052838
55.
CuddySAMFalkRH. Amyloidosis as a systemic disease in context. Can J Cardiol. (2020) 36:396–407. 10.1016/j.cjca.2019.12.033
56.
KitaokaHIzumiCIzumiyaYInomataTUedaMKuboTet al. JCS 2020 guideline on diagnosis and treatment of cardiac amyloidosis. Circ J. (2020) 84:1610–71. 10.1253/circj.CJ-20-0110
57.
AndoYCoelhoTBerkJLCruzMWEriczonBGIkedaSet al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. (2013) 8:31. 10.1186/1750-1172-8-31
58.
MorfinoPAimoAPanichellaGRapezziCEmdinM. Amyloid seeding as a disease mechanism and treatment target in transthyretin cardiac amyloidosis. Heart Fail Rev. (2022) 27:2187–200. 10.1007/s10741-022-10237-7
59.
KittlesonMMMaurerMSAmbardekarAVBullock-PalmerRPChangPPEisenHJet al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. (2020) 142:e7–e22. 10.1161/CIR.0000000000000792
60.
DhuriKBechtoldCQuijanoEPhamHGuptaAVikramAet al. Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. (2020) 9:4. 10.3390/jcm9062004
61.
RubergFLGroganMHannaMKellyJWMaurerMS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. (2019) 73:2872–91. 10.1016/j.jacc.2019.04.003
62.
BensonMDWaddington-CruzMBerkJLPolydefkisMDyckPJWangAKet al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. (2018) 379:22–31. 10.1056/NEJMoa1716793
63.
DanaHChalbataniGMMahmoodzadehHKarimlooRRezaieanOMoradzadehAet al. Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci. (2017) 13:48–57.
64.
HabtemariamBAKarstenVAttarwalaHGoelVMelchMClausenVAet al. Single-dose pharmacokinetics and pharmacodynamics of transthyretin targeting n-acetylgalactosamine-small interfering ribonucleic acid conjugate, vutrisiran, in healthy subjects. Clin Pharmacol Ther. (2021) 109:372–82. 10.1002/cpt.1974
65.
SolomonSDAdamsDKristenAGroganMGonzalez-DuarteAMaurerMSet al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. (2019) 139:431–43. 10.1161/CIRCULATIONAHA.118.035831
66.
FontanaMMartinez-NaharroAChackoLRowczenioDGilbertsonJAWhelanCJet al. Reduction in CMR derived extracellular volume with patisiran indicates cardiac amyloid regression. JACC Cardiovasc Imag. (2021) 14:189–99. 10.1016/j.jcmg.2020.07.043
67.
GillmoreJDGaneETaubelJKaoJFontanaMMaitlandMLet al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. (2021) 385:493–502. 10.1056/NEJMoa2107454
68.
AdamsDTournevILTaylorMSCoelhoTPlanté-BordeneuveVBerkJLet al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. (2022) 2022:1–9. 10.1080/13506129.2022.2091985
69.
CoelhoTAndoYBensonMDBerkJLWaddington-CruzMDyckPJet al. Design and rationale of the global phase 3 neuro-ttransform study of antisense oligonucleotide AKCEA-TTR-LRx (ion-682884-cs3) in hereditary transthyretin-mediated amyloid polyneuropathy. Neurol Ther. (2021) 10:375–89. 10.1007/s40120-021-00235-6
70.
BenbrahimMNormanKSanchorawalaVSiddiqiOKHughesD. A review of novel agents and clinical considerations in patients with ATTR cardiac amyloidosis. J Cardiovasc Pharmacol. (2021) 77:544–8. 10.1097/FJC.0000000000001004
71.
DharmarajanKMaurerMS. Transthyretin cardiac amyloidoses in older north americans. J Am Geriatr Soc. (2012) 60:765–74. 10.1111/j.1532-5415.2011.03868.x
72.
MaurerMSSchwartzJHGundapaneniBElliottPMMerliniGWaddington-CruzMet al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. (2018) 379:1007–16. 10.1056/NEJMoa1805689
73.
GalantNJBugyei-TwumARakhitRWalshPSharpeSArslanPEet al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: a potential diagnostic and therapeutic for TTR amyloidoses. Sci Rep. (2016) 6:25080. 10.1038/srep27679
74.
CoelhoTMaiaLFMartins da SilvaAWaddington CruzMPlante-BordeneuveVLozeronPet al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. (2012) 79:785–92. 10.1212/WNL.0b013e3182661eb1
75.
PenchalaSCConnellySWangYParkMSZhaoLBaranczakAet al. Ag10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated v122i transthyretin. Proc Natl Acad Sci U S A. (2013) 110:9992–7. 10.1073/pnas.1300761110
76.
FoxJCHellawellJLRaoSO'ReillyTLumpkinRJerneliusJet al. First-in-human study of AG10, a novel, oral, specific, selective, and potent transthyretin stabilizer for the treatment of transthyretin amyloidosis: a phase 1 safety, tolerability, pharmacokinetic, and pharmacodynamic study in healthy adult volunteers. Clin Pharmacol Drug Dev. (2020) 9:115–29. 10.1002/cpdd.700
77.
YadavJDOtheeHChanKAManDCBelliveauPPTowleJ. Transthyretin amyloid cardiomyopathy-current and future therapies. Ann Pharmacother. (2021) 55:1502–14. 10.1177/10600280211000351
78.
MasriAArasMFalkRHGroganMJacobyDJudgeDPet al. Long-term safety and tolerability of acoramidis (AG10) in symptomatic transthyretin amyloid cardiomyopathy: updated analysis from an ongoing phase 2 open-label extension study. J Am Coll Cardiol. (2022) 79(9_Supplement):227. 10.1016/S0735-1097(22)01218-9
79.
Sant'AnnaRGallegoPRobinsonLZPereira-HenriquesAFerreiraNPinheiroFet al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat Commun. (2016) 7:10787. 10.1038/ncomms10787
80.
GamezJSalvadoMReigNSunePCasasnovasCRojas-GarciaRet al. Transthyretin stabilization activity of the catechol-o-methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: proof-of-concept study. Amyloid. (2019) 26:74–84. 10.1080/13506129.2019.1597702
81.
GertzMA. Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care. (2017) 23:S107–S12.
82.
O'NuallainBAllenAKennelSJWeissDTSolomonAWallJS. Localization of a conformational epitope common to non-native and fibrillar immunoglobulin light chains. Biochemistry. (2007) 46:1240–7. 10.1021/bi0616605
83.
WallJSKennelSJWilliamsARicheyTStuckeyAHuangYet al. AL amyloid imaging and therapy with a monoclonal antibody to a cryptic epitope on amyloid fibrils. PLoS ONE. (2012) 7:e52686. 10.1371/journal.pone.0052686
84.
SiracCJaccardACodoRBenderSMartinez-RivasGBridouxFet al. Pre-clinical characterization of a novel fusion protein (AT-03), with pan-amyloid binding and removal. Blood. (2021) 138:1207. 10.1182/blood-2021-151908
85.
GertzMALandauHComenzoRLSeldinDWeissBZonderJet al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol. (2016) 34:1097–103. 10.1200/JCO.2015.63.6530
86.
BalSLandauH. AL amyloidosis: untangling new therapies. Hematology. (2021) 2021:682–8. 10.1182/hematology.2021000305
87.
GertzMACohenADComenzoRLDu MondCKastritisELandauHJet al. Results of the phase 3 vital study of neod001 (birtamimab) plus standard of care in patients with light chain (AL) amyloidosis suggest survival benefit for Mayo stage IV patients. Blood. (2019) 134(Supplement_1):3166. 10.1182/blood-2019-124482
88.
GertzMALandauHJWeissBM. Organ response in patients with AL amyloidosis treated with neod001, an amyloid-directed monoclonal antibody. Am J Hematol. (2016) 91:E506–E8. 10.1002/ajh.24563
89.
GertzMASanchorawalaVWechalekarADAndoYKohYNieCet al. Birtamimab in patients with Mayo stage IV AL amyloidosis: rationale for confirmatory affirm-AL phase 3 study. J Clin Oncol. (2022) 40(16_suppl):TPS8076-TPS. 10.1200/JCO.2022.40.16_suppl.TPS8076
90.
SolomonAWeissDTWallJS. Therapeutic potential of chimeric amyloid-reactive monoclonal antibody 11-1f4. Clin Cancer Res. (2003) 9:3831s−8s.
91.
SolomonAWeissDTWallJS. Immunotherapy in systemic primary (AL) amyloidosis using amyloid-reactive monoclonal antibodies. Cancer Biother Radiopharm. (2003) 18:853–60. 10.1089/108497803322702824
92.
WallJSKennelSJStuckeyACLongMJTownsendDWSmithGTet al. Radioimmunodetection of amyloid deposits in patients with AL amyloidosis. Blood. (2010) 116:2241–4. 10.1182/blood-2010-03-273797
93.
EdwardsCVGouldJLangerALMaparaMRadhakrishnanJMaurerMSet al. Interim analysis of the phase 1a/b study of chimeric fibril-reactive monoclonal antibody 11-1f4 in patients with AL amyloidosis. Amyloid. (2017) 24:58–9. 10.1080/13506129.2017.1292900
94.
EdwardsCVGouldJLangerALMaparaMRadhakrishnanJMaurerMSet al. Analysis of the phase 1a/b study of chimeric fibril-reactive monoclonal antibody 11-1f4 in patients with AL amyloidosis. Blood. (2016) 128:643. 10.1182/blood.V128.22.643.643
95.
EdwardsCVRaoNBhutaniDMaparaMRadhakrishnanJShamesSet al. Phase 1a/b study of monoclonal antibody CAEL-101 (11-1f4) in patients with AL amyloidosis. Blood. (2021) 138:2632–41. 10.1182/blood.2020009039
96.
WechalekarADSilowskyJDanielEHarnettMSpectorMSobolovSBet al. Cardiac Amyloid Reaching for Extended Survival (CARES): study design of two placebo-controlled, double-blind, randomized, international phase 3 trials assessing CAEL-101 in patients with Mayo stage IIIa or stage IIIb AL amyloidosis. Blood. (2021) 138(Supplement 1):1673. 10.1182/blood-2021-152488
97.
MichalonAHagenbuchAHuyCVarelaECombaluzierBDamyTet al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat Commun. (2021) 12:3142. 10.1038/s41467-021-23274-x
Summary
Keywords
cardiac light chain amyloidosis, cardiac amyloidosis (CA), cardiac amyloidosis—ATTR, standard of care (SoC), treatment gaps, future treatments
Citation
Quarta CC, Fontana M, Damy T, Catini J, Simoneau D, Mercuri M, Garcia-Pavia P, Maurer MS and Palladini G (2022) Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front. Cardiovasc. Med. 9:1073503. doi: 10.3389/fcvm.2022.1073503
Received
20 October 2022
Accepted
24 November 2022
Published
20 December 2022
Volume
9 - 2022
Edited by
Gianfranco Sinagra, University of Trieste, Italy
Reviewed by
Paolo Morfino, Sant'Anna School of Advanced Studies, Italy; Alberto Cipriani, University Hospital of Padua, Italy
Updates
Copyright
© 2022 Quarta, Fontana, Damy, Catini, Simoneau, Mercuri, Garcia-Pavia, Maurer and Palladini.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Cristina Quarta candidacristina.quarta@alexion.com
This article was submitted to General Cardiovascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.