Abstract
Mitral valve prolapse (MVP) due to myxomatous degeneration is one of the most important chronic degenerative cardiovascular diseases in people and dogs. It is a common cause of heart failure leading to significant morbidity and mortality in both species. Human MVP is usually classified into primary or non-syndromic, including Barlow’s Disease (BD), fibro-elastic deficiency (FED) and Filamin-A mutation, and secondary or syndromic forms (typically familial), such as Marfan syndrome (MFS), Ehlers-Danlos syndrome, and Loeys–Dietz syndrome. Despite different etiologies the diseased valves share pathological features consistent with myxomatous degeneration. To reflect this common pathology the condition is often called myxomatous mitral valve degeneration (disease) (MMVD) and this term is universally used to describe the analogous condition in the dog. MMVD in both species is characterized by leaflet thickening and deformity, disorganized extracellular matrix, increased transformation of the quiescent valve interstitial cell (qVICs) to an activated state (aVICs), also known as activated myofibroblasts. Significant alterations in these cellular activities contribute to the initiation and progression of MMVD due to the increased expression of transforming growth factor-β (TGF-β) superfamily cytokines and the dysregulation of the TGF-β signaling pathways. Further understanding the molecular mechanisms of MMVD is needed to identify pharmacological manipulation strategies of the signaling pathway that might regulate VIC differentiation and so control the disease onset and development. This review briefly summarizes current understanding of the histopathology, cellular activities, molecular mechanisms and pathogenesis of MMVD in dogs and humans, and in more detail reviews the evidence for the role of TGF-β.
Introduction
Mitral valve prolapse (MVP) is one of the most common cardiac valvular abnormality in dogs and humans, and is a major source of morbidity and mortality and a common cause of heart failure, ventricular dysfunction, arrhythmias and sudden cardiac death (1–6). In the dog the condition is more commonly called myxomatous mitral valve disease (MMVD). Since this mitral valvulopathy in both species has various synonyms and myxomatous changes is the predominant pathological finding, in this review the terms MVP and MMVD will be used interchangeably and when needed for human and dog, respectively. MVP affects to 2–3% of the human population, and more than 10% of individuals over the age of 65 years have mitral valve insufficiency (7, 8). It accounts for 7% of deaths in dogs before 10 years of age and its prevalence is very high, estimated between 30 and 70% of all elderly dogs, with the greater prevalence in small breed dogs and in certain predisposed breeds such as the cavalier King Charles spaniel (CKCS) (9–11). Some forms of MVP will have a congenital or genetic basis, meaning MVP can be further characterized into primary non-syndromic, and secondary syndromic forms (4). Syndromic MVP is commonly associated with global genetic connective tissue disorders, such as Marfan syndrome (MFS), Loeys-Dietz syndrome and Ehlers-Danlos syndrome (12–14). It can also appear as isolated non-syndromic MVP, typically in a familial setting. The first confirmed non-syndromic genetic mutation was for the X-linked FLNA (Filamin-A) gene mutation, which causes valvular defects and progressive myxomatous degeneration and MVP in mice and humans (15–17). For the non-syndromic variants two disease types are reported including Barlow’s Disease (BD), where there is myxomatous degeneration and end-stage fibrosis, and fibroelastic deficiency (FED) where there is only myxomatous degeneration with valve thinning rather than thickening (18). FED is typically only seen in the very elderly, while Barlow’s Disease (BD) has more commonality with canine MMVD in terms of its slowly acquired development and progression, despite the lack of fibrosis in the dog. Lastly, MMVD has also been described in mice and pigs, induced by gene interference or surgical intervention, but the extent of MMVD in other species as a consequence of aging is unknown (19–21). Details of MMVD in both species are summarized in Table 1.
TABLE 1
| Classification | Histopathological features | Abnormal signaling | Cellular changes | ||
| Human MVP | Primary (Non-syndromic) | Barlow’s Disease (BD) | 1. Typical lesions of myxomatous degeneration 2. End-stage fibrosis 3. Leaflet thickening | 1. TGF-β signaling 2. 5-HT (serotonin) signaling 3. BMP signaling 4.Wnt/β-catenin | 1. Increased aVICs from qVICs 2. Increased macrophage infiltration 3. EndoMT |
| Fibroelastic deficiency (FED) | 1. Typical lesions of myxomatous degeneration 2. Deficiency in collagen 3. Valve thinning | TGF-β signaling | Increased aVICs from qVICs | ||
| Filamin-A mutation | Typical lesions of myxomatous degeneration | 1. TGF-β signaling, 2. Ras/Mek/Erk signaling 3. 5-HT (serotonin) signaling | 1. Increased aVICs from qVICs | ||
| Secondary (Syndromic forms) | Marfan syndrome (MFS) | Typical lesions of myxomatous degeneration | 1. TGF-β signaling 2. BMP signaling 3. Wnt/β-catenin | 1. Increased aVICs from qVICs 2. Increased macrophage infiltration 3. EndoMT | |
| Ehlers-Danlos syndrome | Typical lesions of myxomatous degeneration | ||||
| Loeys–Dietz syndrome | Typical lesions of myxomatous degeneration | TGF-β signaling | |||
| Canine MMVD | 1. Typical lesions of myxomatous degeneration 2. Lack of any fibrotic changes | 1. TGF-β signaling 2. 5-HT (serotonin) signaling 3. BMP signaling | 1. Increased aVICs from qVICs 2. EndoMT 3. Lack of inflammatory infiltration | ||
Characteristics of various forms of MMVD in the human and dogs.
For both syndromic and non-syndromic forms in both species the diseased valves appear histologically similar and exhibit features of myxomatous (myxoid) degeneration. Progressive deterioration of the mitral valves is typically characterized by increased valvular nodularity, leaflet thickening and deformity, excessive accumulation of proteoglycans (GAGs), collagen and elastin fragmentation, increased expression of proteolytic enzymes, disorganized extracellular matrix (ECM) and increased numbers of activated valve interstitial cells (aVICs; activated myofibroblasts) and with more obvious macrophage infiltration in human valves (3, 5, 22–25). Furthermore, in human valves there can be additional fibrosis in end-stage disease (BD) characterized by fibrotic layers on the valve surface, a change not seen in the dog (26–29).
Although, the histopathological changes of myxomatous degeneration are well characterized, some aspects of the underlying molecular changes and its contribution to pathogenesis are still to be identified (22, 30, 31). Most MMVD seen in human and veterinary clinical practice are sporadic and of unknown etiology, although a genetic or inherited basis suspected. However, there is increasing evidence for a primary role for members of the transforming growth factor (TGF)-β superfamily in the pathogenesis and progression of various MMVD forms (5, 32–37). This is not surprising as the TGF-β superfamily are important in the regulation of most cellular events, such as proliferation, differentiation, migration, autophagy, apoptosis and senescence in a variety of cardiovascular diseases (38, 39). While much is known about the cellular and molecular events of the disease in both species, the exact molecular and regulatory mechanisms are not yet elaborated, especially with regard to early disease onset and progression (3–5, 37). In human MVP most studies of non-syndromic forms have been restricted to examining end-stage valves obtained at surgery (29, 34, 35, 40). Information on earlier onset is restricted mainly to inherited connective tissue disorders, such as MFS, osteogenesis imperfecta, and Ehlers-Danlos syndrome, although myxomatous degeneration in those patients can appear with advanced age (17).
CD45+ hematopoietic cells have been detected in human, sheep, and murine MMVD valves related to non-infective causes, although MMVD in the absence of infective endocarditis has traditionally been regarded as a non-inflammatory disease (34, 41–44). The majority of these cells are characterized as macrophages implicating macrophage infiltration as a potential secondary driver of MMVD progression. CD45 expression of mitral VECs can be induced by TGF-β signaling in sheep MMVD (25, 43, 45). TGF-β signaling is enhanced in MMVD in mouse, humans and dogs and has been associated with ECM dysregulation and increased macrophage numbers in diseased valves (4, 32, 34, 46, 47). These data suggest that TGF-β signaling might be associated with the emergence of an inflammatory micro-environment comprised of increased recruitment of pro-inflammatory macrophages from the circulation and immunogenic ECM remodeling, somewhat analogous to features of calcific aortic valve disease (CAVD) (6, 25). In CAVD TGF-β signaling is involved in the pathogenesis, as for MMVD a promoter role in the early VIC activation as shown by up-regulation of α-smooth muscle actin (α-SMA). In dogs with advanced disease there is evidence of a marginal increase in the number of mast cells in affected valves (24), but no involvement of the resident macrophage population or recruitment of inflammatory cells are found in severe MMVD (elderly dogs) (48, 49). Whether there is a macrophage population present in dogs or only during early stage of the disease has yet to be elucidated.
Valve Structure, Pathology, and Cell and Molecular Changes
The cross-sectional structure of the normal mitral leaflets is similar to the aortic valve, with at least three layers, atrialis, spongiosa, and fibrosa identified, a fourth ventricularis proposed by some, and each with different thickness and cell and ECM composition, and both sides lined with valve endothelial cells (VECs). The thin atrialis contains a large amount of elastin with a mixed amount of scattered collagen fibers and valve interstitial cells (VICs) (50, 51). The spongiosa contains loosely arranged collagen fibers and is rich in glycosaminoglycans (GAGs), such as hyaluronan, and various proteoglycans (52). This layer, consisting mainly of collagen I and III and small numbers of thin elastin fibers. The majority of cells in the spongiosa, and throughout the whole valve, are quiescent VICs (qVICs) along with few mast cells (24, 53–55). The dense fibrosa layer is composed of tightly packed collagen bundles arranged parallel to the leaflet free edge and within which are scattered VICs (Figure 1).
FIGURE 1
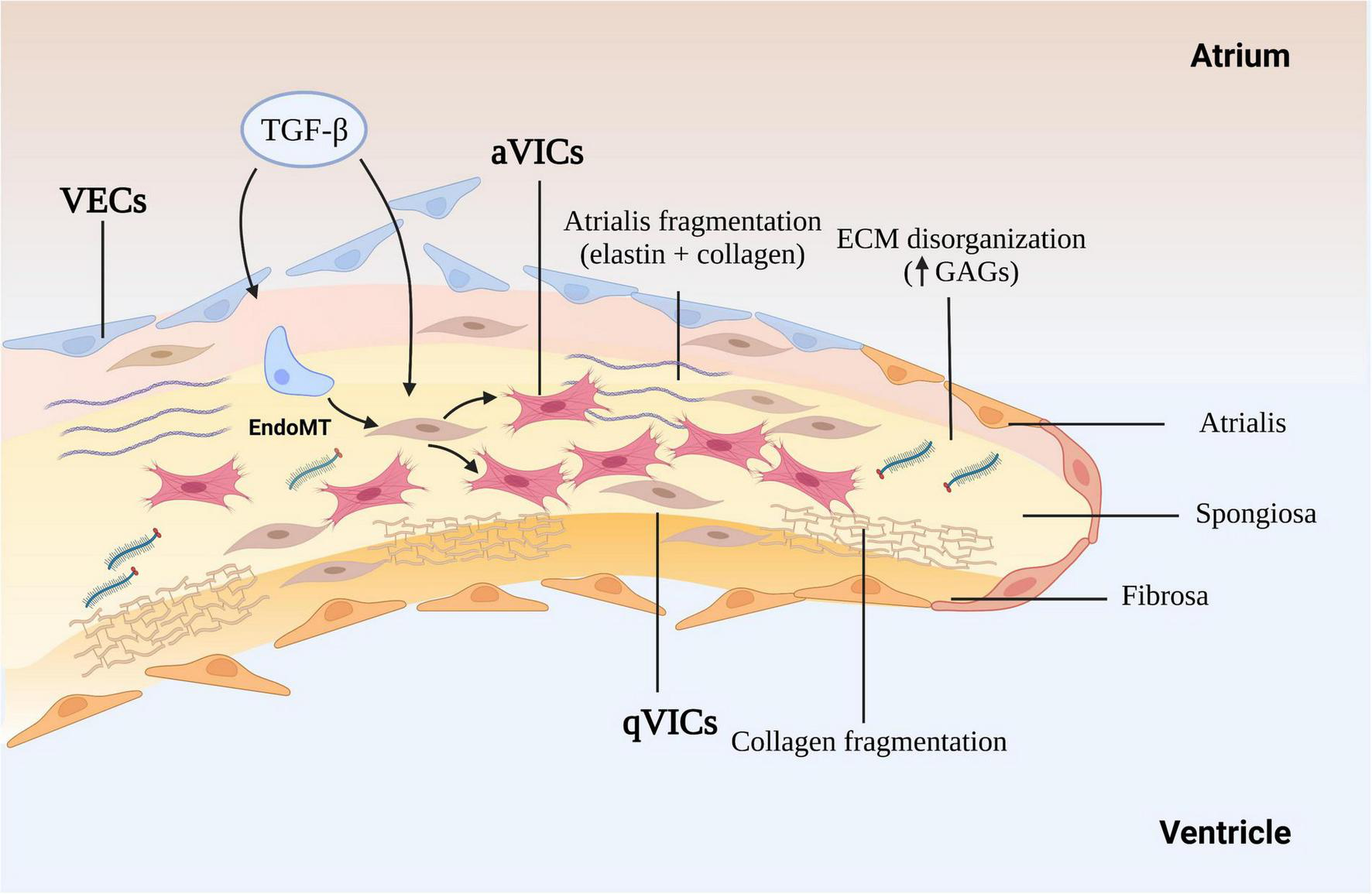
The mechanism of myxomatous degeneration. The schematic shows the endothelial to mesenchymal transition (EndoMT) process and the activation of quiescent valve interstitial cells to myofibroblasts that affect matrix production and remodeling, driving collagen and elastin fragmentation and loss. ECM, extracellular matrix; GAGs, glycosaminoglycans; MMP, matrix metalloprotease; TGF-β, transforming growth factor-β; qVICs, quiescent valvular interstitial cells; aVICs, activated valvular interstitial cells.
The gross and histopathological changes in both species are reasonably well characterized, and will be only briefly describe here, but to give contextual background to the role of TGF-β in the disease process (5, 22, 37, 56–58). Changes the endothelium include endothelial loss, cellular pleomorphism, endothelial-to-mesenchymal transition (EndoMT), disruption of the basement membrane and accumulation of aVICs in the sub-endothelium (23, 46, 55). The myxomatous degeneration itself is characterized by expansion of the spongiosa and separation as well as fragmentation of the dense collagen bundles in the fibrosa (5, 59). There is a reduction in connective tissue density, accumulation of a myxoid extracellular matrix rich in GAGs, loss of mature collagen and replacement with immature fibrillar collagen lacking cross-links structural, in all the layers of leaflet as well as the chordae tendineae (60). The main cellular event is the phenotypic differentiation of the VICs from a quiescent phenotype (qVICs) to an activated myofibroblast (aVICs) in the spongiosa and fibrosa, with accumulation of aVICs in the sub-endothelium (46, 61). Furthermore, there is evidence of aVIC persistence with dysregulation of apoptosis, and transcriptomic evidence of altered gene expression associated with cellular senescence (37, 62). Rather than aVICs being cleared for the tissue as would normally happen, their persistence might contribute to the aberrant matric remodeling typical of the disease. The aVICs are presumed responsible for valve matrix degrading, and at a rate exceeding that of production of new collagen and elastin (63). This matrix degeneration is presumed to be due to increased production of various proteolytic enzymes including matrix metalloproteinases (MMP-1, MMP-2, MMP-9, and MMP-13). It should be noted that human valvulopathies heighten the risk of developing endocarditis which is not the case in the dog. The reason for this species difference is unknown. Overall, these pathological changes account for the reduced tensile strength, distorted valve shape and mechanics typical of the disease resulting in the mitral regurgitation and heart failure.
In addition to the naturally occurring forms of MMVD there are various rodent models available, and comment will be made on how they inform thinking on the role of TGF-β (Table 2). Many of these models are transgenic and questions can be raised to their validity as models of what is a chronic degenerative disease, but that discussion is beyond the scope of this review.
TABLE 2
| Classification | In vivo animal models | In vitro culture models |
| Myxomatous mitral valve degeneration (MMVD) | 1.Mouse treated with AngII (84) 2.Mouse treated with nordexfenfluramine (NDF) (133, 134) 3. FVB mouse (135) | 1.Porcine 3D mitral VIC culture system (136) 2.Porcine mitral VIC culture system (100) 3.Human mitral valve tissue culture system (34) 4.Human mitral VIC culture system (34, 114) |
| Barlow’s Disease (BD) | Dogs with spontaneous MMVD (5, 37) | 1.Canine 2D mitral VIC and VEC co-culture system (36) 2.Canine 3D tissue-engineered fibrin-based cell culture system (137) |
| Filamin-A mutation | Filamin-A KO mouse (89) | |
| Marfan syndrome (MFS) (32, 107) | Fbn1C1039G/+ mouse Fbn1C1039G/C1039G mouse | |
| Ehlers-Danlos syndrome (138) | Col3a1± mouse Col5a1± mouse | |
| Loeys–Dietz syndrome (117) | TGF-βR1± mouse TGF-βR2± mouse TGF-βR1M318R/+ mouse TGF-βR2G357W/+ mouse |
In vivo and in vitro models for MMVD.
The Role of Transforming Growth Factor-β in Myxomatous Mitral Valve Degeneration
Effects of Transforming Growth Factor-β on Valvular Interstitial Cells
As mentioned previously, VICs exist in all layers as two distinct phenotypes, qVICs and aVICs, and predominantly in healthy and diseased valves, respectively (64). During development endothelial cells invade the endocardial cushion and are transformed into embryonic progenitor mesenchymal cells to induce ECM remodeling under the regulation of the TGF-β family and bone morphogenetic proteins (BMPs) (65). Once the valve is formed qVICs predominate and maintain valve structure and function (24, 48, 54). VICs in normal valves have a quiescent vimentin+/alpha-smooth muscle actin (αSMA)- phenotype and are presumed to operate in a homeostatic role controlling ECM remodeling and repair (66). TGF-β up-regulation appears to have an important role among various biological pathways in the pathogenesis of the multiple forms of MMVD. Specifically, TGF-β is known to activate qVICs toward a pathologic synthetic phenotype, as shown both in animal models and in human and canine in vitro studies (32, 36, 48). Similarly, antagonism of the TGF-βR II receptor by SB431542 transitions aVICs to the qVIC phenotype in a canine low serum culture system, where VICs were isolated from diseased canine mitral valves and maintained in 2% (v/v) FBS media (36). Members of the TGF-β superfamily are overexpressed in surgically excised human diseased valves where aVICs predominate and this is associated with increased expression of MMPs, presumably driving degeneration of collagen and elastin structures (22, 34, 35, 67, 68). Finally, examining transcriptomic data from human valves, upregulation of BMP 4 has been shown to mediate the activation of VICs from healthy quiescent cells to a pathologic synthetic phenotype (67).
Transforming Growth Factor-β Control of EndoMT in Myxomatous Mitral Valve Degeneration
One of the important, but less documented, changes to the endothelium in MMVD is induction of EndoMT, accompanied by activation of transcriptional regulatory mechanisms important in heart valve development, and is seen in both human and canine MMVD (46, 69–71). This is associated with co-expression of hyaluronic acid synthase (HAS)-2 and α-SMA in endothelial cells and increased expression of Sox9, and increased expression of HAS-2 in stromal interstitial cells (46, 52, 72). There are other lines of evidence suggesting a role for EndoMT in MMVD, with a role for TGF-β. Cultured endocardial cells derived from mature ovine valves have been shown to transdifferentiate via TGF-β signaling into mesenchymal cells that express α-SMA, and human valve endothelial cells adopt a mesenchymal phenotype after exposure to TGF-β (73, 74). Clonal expansion has shown endothelial-like cells have a strong response to TGF-β, which can then be inhibited by vascular endothelial growth factor (VEGF) (74). In vitro mitral valve endothelial cells have been shown to be important for maintaining the quiescence of valve interstitial cells and thereby reciprocally preventing TGF-β-driven EndoMT of endothelial cells (75). Lastly, inhibition of TGF-β by the angiotensin two receptor antagonist losartan can reduce EndoMT of mitral valve endothelial cells in vitro in a sheep model and can decrease leaflet thickness and block TGF-β signaling and downstream targets (43). Nevertheless, evidence for EndoMT is restricted to valve transcriptomic data in the dog and human, and found in valve tissue in the dog using IHC, but to what extent this might contribute to the valve VIC population and to disease pathogenesis in vivo is unknown (Figure 2).
FIGURE 2
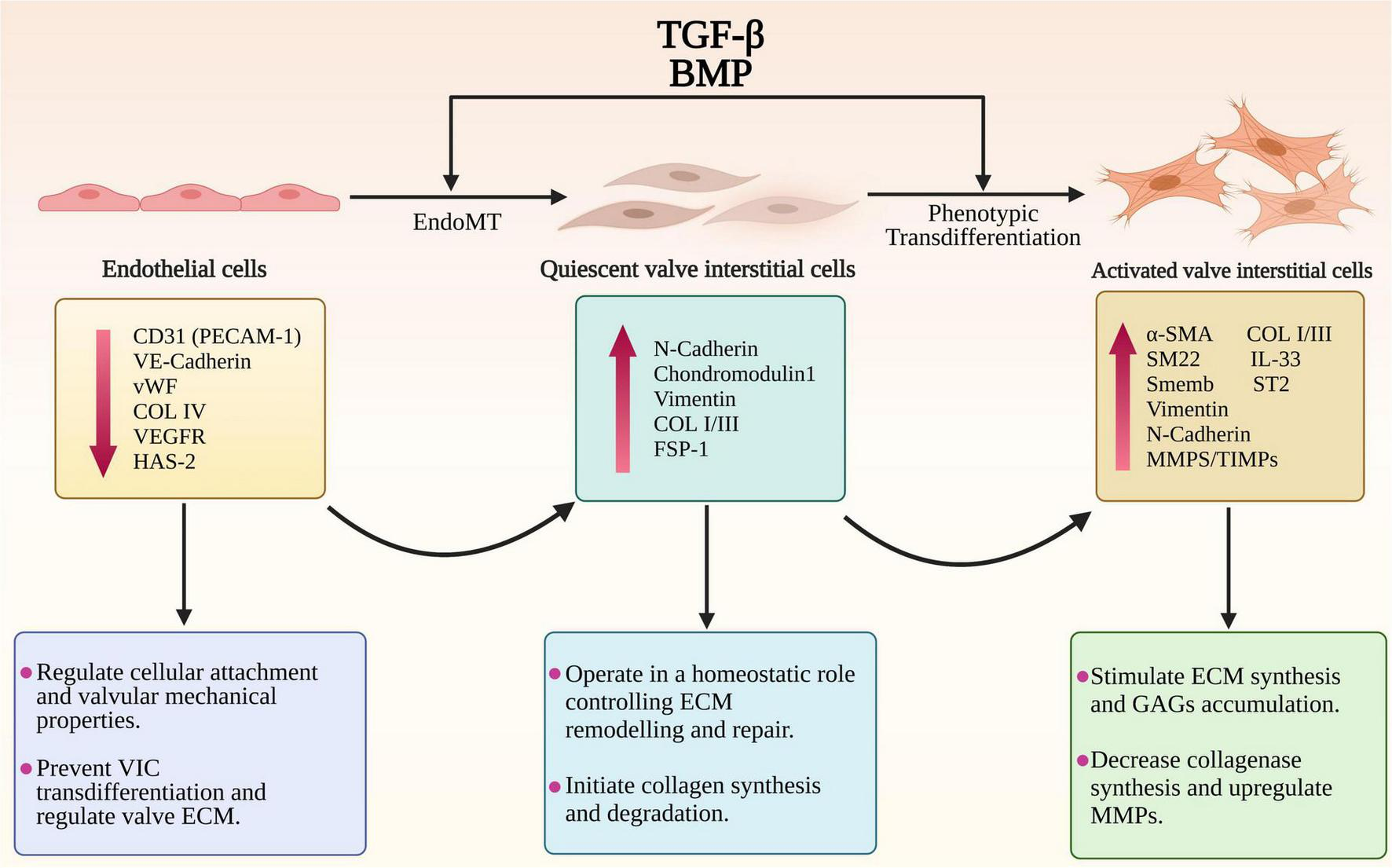
The endothelial to mesenchymal transition (EndoMT) and phenotypic trans-differentiation from quiescent valve interstitial cells (qVICs) to activated valve interstitial cells (aVICs) in the progression of MMVD. The diagram illustrates the phenotypic, gene expression changes and cellular functional changes occurring during MMVD. The phenotypic conversion of endothelial cells into qVICs includes increased production of N-cadherin, vimentin and fibroblast-specific protein-1 (FSP-1). These events are accompanied by downregulation of the markers of endothelial cells such as CD31/platelet-endothelial cell adhesion molecule-1 (CD31/PECAM-1), vascular-endothelial cadherin (VE-cadherin), COL4, vascular epidermal growth factor receptor (VEGFR), and von Willebrand factor (vWF). Furthermore, the TGF-β-activated VICs show a significant increase in unique markers including α-smooth muscle actin (α-SMA), SM22 and Smemb (embryonic smooth muscle myosin) as well as the secretory proteins regulating the ECM disorganization including MMPs/TIMPs, IL-1β, IL-11, and IL-33.
Transforming Growth Factor-β Initiated Signaling Pathways in Myxomatous Mitral Valve Degeneration
The TGF-β family consists of a large variety of pleiotropic multifunctional proteins that play significant roles in embryonic development, autoimmunity, cancer, fibrotic disorders, and cardiovascular diseases (34, 76–79). TGF-βs and BMPs are considered as the most important initiators of the signaling pathway (80–83). Although the effect of TGF-βs and BMPs on the initiation and progression of MMVD has been shown, the molecular and regulatory events involved are highly complex resulting from a large number of signaling interactions promoted by diverse molecular mechanisms. Many of these interaction still need to be elucidated.
TGF-β-mediated valvular fibrosis is only observed in end-stage human MMVD and seen as fibrotic over-lays developing on the atrial and ventricular sides of leaflets. However the myxomatous degeneration found in both species is believed to be highly TGF-β-dependent (5, 37). Various studies have suggested the important roles for the members of TGF-β family in the initiation and development of MMVD in humans, mouse and dog (32–37, 47, 48, 67). Pathway analysis by transcriptomic profiling in human and canine valve tissue has identified TGF-β signaling as the dominant pathway in both the development and progression of MMVD (37, 84). Increased expression of multiple TGF-β isoforms in parallel with the accumulation of ECM components and transformation of VICs into myofibroblasts is observed in the surgically excised samples of myxomatous valves from human and dog (34, 37, 47, 85, 86). In a transgenic fibrillin (Fbn)-1-deficient mouse Marfan Disease model, where TGF-β signaling was potentiated, VICs are phenotypical altered with associated myxomatous ECM remodeling (32, 87). Transformation of qVICs to the diseased activated myofibroblast phenotype can be blocked with TGF-β-neutralizing antibodies (NeuAb), antagonism of the TGF-β RII receptor, antagonizing TGF-β signaling and blocking Smad phosphorylation (32, 34–36). In an in vitro Marfan Disease model an exon encoded Fbn-1 sequence triggers release of endogenous TGF-β1 and stimulates TGF-β receptor-mediated Smad2 signaling in the presence of cell layer ECM (88). However, it has been shown that non-Smad (non-canonical) signaling pathways are also implicated in MMVD progression, including regulation by several molecular mediators such as filamin A (FLN-A) and scleraxis (Scx) (86, 89).
Canonical Transforming Growth Factor-β-Mediated Signaling Pathways in Myxomatous Mitral Valve Degeneration
Substitution of an epidermal growth factor-like domain in the fibrillin-1 (Fbn-1) gene with a cysteine (C1039G) in transgenic mice will result in increased release of activated TGF-βs to initiate the aberrant signaling that contributes to myxomatous degeneration (32). Significant increased expression of TGF-βs, latency-associated peptides (LAPs), latent TGF-β activator integrins and phosphorylated SMAD2/3 has been reported in MMVD valve tissues using transcriptomic analysis and on histopathology, indicating the key contribution of canonical TGF-β-mediated signaling to MMVD (34, 37, 90, 91). Understanding how TGF-β signaling can control MMVD pathogenesis requires some explanation of the complex pathways involved. Briefly, the TGF-β signaling pathway is initiated by binding of activated TGF-β ligands with TGF-β receptor complexes in the cell membrane. Subsequent to intracellular biosynthesis TGF-β homodimers are secreted extracellularly as inactive protein complexes, which maintain latency through the non-covalent binding with the pro-peptide latency-associated peptide (LAP). To exert its diverse biological functions TGF-β requires to be liberated from the latent complex and activated extracellularly before binding to its receptors. For example, Fbn-1, as a structural component of the ECM microfibrils, can release (activate) latent TGF-βs from the microfibrils by means of the substitution of Fbn-1 fragments for latent TGF-β-binding proteins (LTBPs) and by its complex interactions with various activators such as integrins (37, 88, 92–95). TGF-β release can also be controlled by torqueing mechanics where the bound protein complex is linked to cells (in this case VICs) by various integrins. Following the activation/release of TGF-βs these homodimers bind to their corresponding transmembrane receptor complexes (TGF-βR I and TGF-βR II) and initiate a complicated intracellular signaling cascade of molecular and regulatory events (96–98). The active receptor complex recruits and activates the downstream signaling effector proteins Smad2 and Smad3 (the canonical pathway) and they form an intracellular complex with co-Smad (Smad4, the mediator Smad) enabling its translocation from the cytoplasm into the nucleus. In the nucleus the Smad complex interacts with the Smad binding elements (SBE) to invoke transcription of various TGF-β-responsive genes, including those involved in ECM remodeling, VIC differentiation and EndoMT (99) (Figure 3).
FIGURE 3
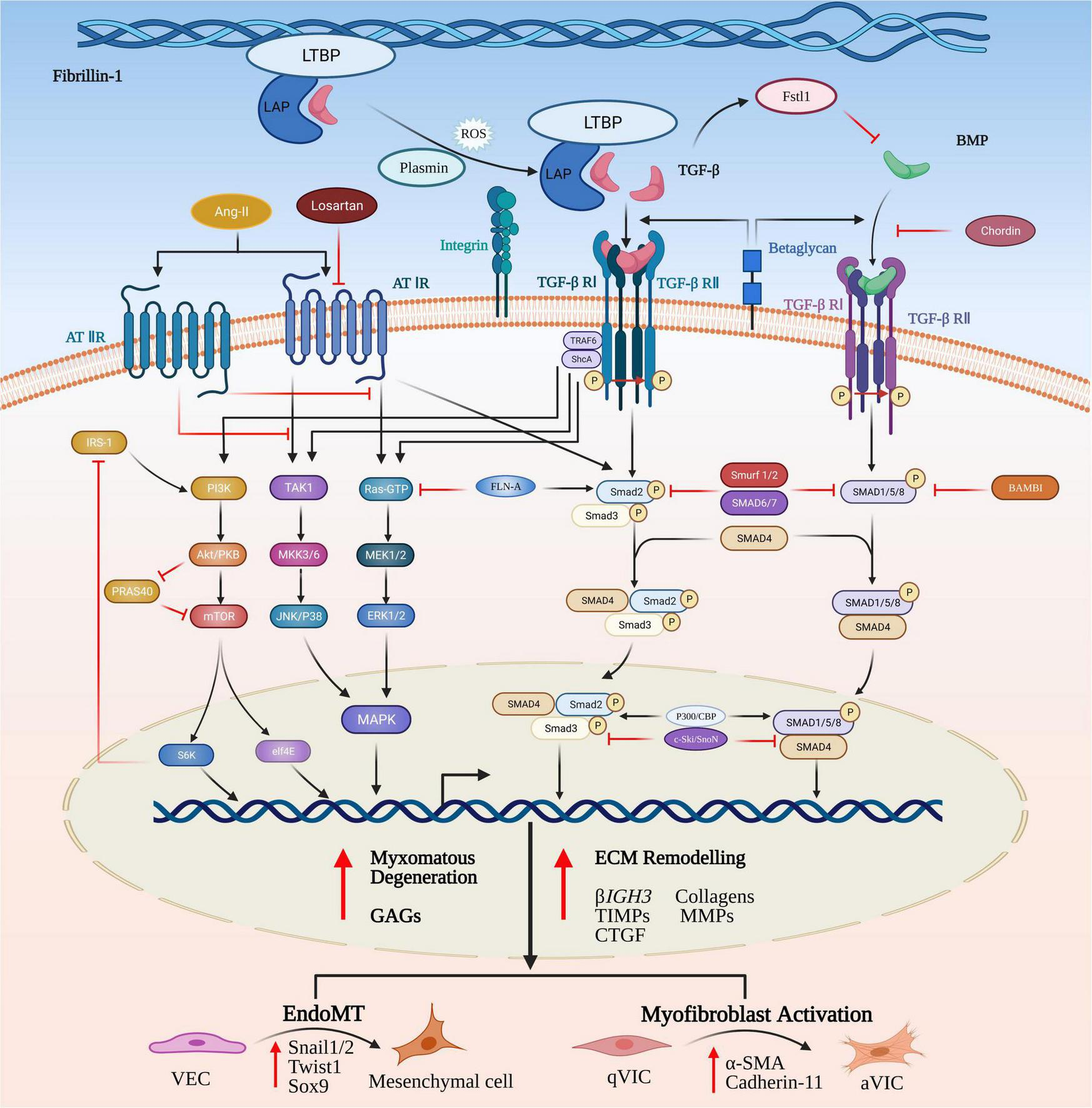
Canonical and non-canonical TGF-β and BMP signaling pathways in MMVD. TGF-β homodimers are secreted extracellularly as inactive protein complexes, maintaining latency through binding with the latency-associated peptide (LAP). Latent TGF-β-binding proteins (LTBPs) form a bridge between fibrillin-1 (Fbn-1) and LAP and serve to sequester the complex into the extracellular matrix (ECM). The complex can be released from the microfibrils by its complex interactions with activators such as reactive oxygen species (ROS), plasmin and integrins. Activation of the latent complex mechanically triggers the complex deformation (torqueing) and the release of active TGF-β. The Smad-mediated TGF-β cascade is initiated by the binding of activated TGF-β homodimers to their corresponding transmembrane heterodimeric TGF-β I/II receptor complex. The active receptor complex recruits and phosphorylates the downstream signaling effector proteins, Smad2 and Smad3. Upon phosphorylation, Smad2/3 is released and forms an intracellular complex with Smad4 that translocates from the cytoplasm into the nucleus, where it interacts with Smad binding elements of TGF-β target genes involved in induction of ECM remodeling, VIC differentiation, EndoMT and myxomatous alterations. The non-canonical pathways are comprised of phosphatidylinositol 3-kinase (PI3K) and the kinases members from mitogen-activated protein kinase (MAPK) family. TGF-β can activate all three known MAPK pathways, referred to as TGF-β activated kinase 1 (TAK1)-mediated p38 activation, c-Jun amino-terminal kinase (JNK) MAPK, and extracellular signal-regulated kinase (ERK). Signal transduction through these pathways modulates non-Smad-mediated TGF-β-responsive cellular activities dependent on a specific cellular type or context. In a similar manner, BMPs bind to their transmembrane complexes to initiate its phosphorylation, but with higher affinity with type I receptor, assisted by type III co-receptors, endoglin and betaglycan. Upon activation the complex results in initiation of canonical or non-canonical signaling cascades.
The Role of Mediators in Canonical Transforming Growth Factor-β Initiated Myxomatous Mitral Valve Degeneration
While Smad2/3-mediated signaling probably is a major contributor to the initiating of MMVD, other molecular mediators related to canonical signaling pathways have been shown to play a significant role in the modulation of MMVD progression in animal models in vivo and in vitro in VICs. These regulatory mechanisms include molecules exerting their effects on MMVD by means of canonical TGF-β signaling pathways or through activation by TGF-β itself. Examples include fibrillin-1 (Fbn-1), filamin A (FLN-A), follistatin-like 1 (Fstl1), fibroblast growth factor (FGF)-2 and angiotensin (Ang) II (32, 84, 89, 100, 101).
Fbn-1 beyond being a structural component of ECM microfibrils has an important role in regulating TGF-β activity and signaling through interaction with LTBP in the myxomatous mitral valve of MFS patients (32). Ng and colleagues showed that the intense immunohistochemical signal of both active TGF-β and its intracellular responder pSmad2 were significantly increased in mitral valves in a MFS mouse model. These observations were further validated by TGF-β antagonism by neutralizing antibodies in vivo with rescuing of the myxomatous mitral valve phenotype. Additionally, the expression of the downstream TGF-β-related effectors, which are either members of the TGF-β superfamily or directly mediated by TGF-βs, such as βIGH3, endothelin-1 (EDN1) and BMPs, are upregulated in the mitral valves of MFS mice. This suggests a promoter role of the mutation in Fbn-1 in initiation of canonical TGF-β signaling in MFS, and perhaps more commonly non-syndromic variants of human MMVD (32). Evidence for the extracellular control by Fbn-1 of TGF-β signaling activation in other MFS-induced systemic disorders has been shown in other studies (102–108). Recent work on the age-dependent cardiac remodeling in a mouse model of MFS revealed a comparable expression pattern of the myofibroblast marker α-smooth muscle actin (α-SMA) in cardiac tissues of Fbn-1 transgenic mice and their wild-type littermates.
FLN-A is highly expressed in the mitral valve during development and is significantly diminished after birth, suggesting an important role for FLN-A during valve development (109). Mutations in the FLNA gene had been identified as a cause to a rare X-linked myxomatous valvular disease (17). FLN-A might act as a contributor to the initiation and development of myxomatous cardiac valves by its interaction with activated Smads to regulate TGF-β signaling (110, 111). In contrast, a recent study showed that FLN-A might not be involved in the pathogenesis of non-syndromic MVP, since the mRNA expression level of FLN-A in diseased valves is comparable to control leaflets, despite upregulation of TGF-β1 and pSmad2 signaling in diseased mitral valves (86). Another study also suggested that there were interactions between FLN-A and TGF-β since FLN-A acted as a promoter of TGF-β induced ECM remodeling in Fstl1-deficient mice (101). In a FLN-A knockout mouse the expression of the canonical TGF-β-dependent effector pSmad3 and its downstream target molecule collagen IαI are markedly increased in mitral valves suggesting deficiency in FLN-A positively influences Smad activation and correlates with increased collagen expression (89).
In Fstl1-knockout mice there is a sustained increase in TGF-β signaling after birth, while deletion of Fstl1 from the endocardial lineage results in myxomatous mitral valves with cell proliferation and endocardial-to-mesenchymal transition. Fstl1-deficient mitral valves show significantly upregulated expression of TGF-β in mitral valve VICs. Immunofluorescent analysis has shown that the positive signal for pSmad2/3 is mainly detected in the nuclei of VECs and VICs in diseased mitral leaflets, indicating active TGF-β signaling during postnatal development (101). Additional evidence that Fstl1 could regulate MMVD is shown by increased upregulation of a series of unique makers of ECM remodeling and EndoMT mediated by TGF-β signaling in Fst1-KO mice, including Fbn-1, FLN-A, vimentin and α-SMA. These data suggest important roles of Fstl1 in promoting homeostasis of the mitral valve undergoing embryonic development, postnatal maturation, and even into adulthood (101).
Several studies have shown that fibroblast growth factor (FGF)-2 is able to potentiate canonical pSmad2/3-dependent TGF-β signaling by binding to TGF-β type III receptor beta-glycan, which both possess the binding site of endogenous TGF-β and FGF-2, and thereby result in increased TGF-β availability for the activation of the canonical pathway. A significant role for FGF-2/AKT-1 signaling in mitral VICs in response to experimental wounding and remodeling, independent of the TGF-β/Smad signaling, has been identified (100). Ang II has also been shown to play a significant role in canonical TGF-β-dependent ECM remodeling causing myxomatous degeneration in murine mitral valves. Infusion of Ang II into mice triggers activation of a canonical Smad-mediated TGF-β2 cascade and transcriptional activity of TGF-β-responsive genes (84). There is increased mRNA levels of TGF-β1 and TGF-β2 as well as downstream pSmad2, detected by both qRT-PCR and IHC, alongside an increase in MMP2 and CTGF expression, respectively (84). As previously mentioned canonical TGF-β-induced SMAD2/3-dependent ECM production in cultured human VICs can be effectively inhibited by the Ang II receptor blocker losartan (34).
Non-canonical Transforming Growth Factor-β Signaling in Myxomatous Mitral Valve Degeneration
In addition to the canonical signaling, TGF-β family members contribute to MMVD progression by via several non-Smad/non-canonical pathways. Similar to the Smad-dependent signaling, the non-canonical cascades are initially amplified by the phosphorylation of TGF-βRI/II through binding with activated TGF-β ligands regulating downstream cellular activities. These pathways comprise the kinases members from the mitogen-activated protein kinase (MAPK) family and other kinases such as phosphatidylinositol 3-kinase (PI3K). TGF-β can activate all three known MAPK pathways, and these are also known as TGF-β activated kinase 1 (TAK1)-mediated p38 activation, c-Jun amino-terminal kinase (JNK) MAPK, and extracellular signal-regulated kinase (ERK). Signal transduction through these pathways modulates non-Smad-mediated TGF-β-responsive cellular activities. However, these pathways can also modify canonical pathways with complex interactions between non-Smad- or Smad-mediated components occurring in most TGF-β-mediated biological effects. This can result in up- or down-regulation of TGF-β signaling and complex regulation of biological responses (112).
Although Smad-independent TGF-β signaling is implicated in a wide spectrum of intracellular transduction cascades and diverse cellular responses only a few studies have examined its involvement in MMVD (34, 41, 89, 113, 114). Nevertheless, these studies show that non-Smad signaling, including ERK, p38 MAPK, and PI3K, can contribute to MMVD pathogenesis affecting the function of both VICs and VECs by interacting with canonical TGF-β signaling or directly. Recent transcriptomic profiling of canine valve tissues has shown that positively regulated gene expression of the ERK1/2 cascade is most noticeably in end-stage valves (37). In FLNA conditional knockout mice, as found in the human FLNA mutation X-linked MMVD, progression can be attributed to a balance between the opposing regulatory effect of the non-canonical Ras/Mek/Erk and canonical TGF-β-dependent effector pSmad3 (89). In human mitral VICs exposed to cyclic mechanical strain, expression of TGF-β2 and α-SMA are significantly increased, which is partly dependent on the activation of RhoC/ROCK in tandem with the non-canonical MEK/ERK1/2 pathway (114). The expression of α-SMA induced by activation of the TGF-β-dependent PI3K/AKT signaling is also observed in bone marrow-derived mesenchymal stem cells (MSCs) modified to generate VICs for tissue-engineered heart valves (113). In human mitral VECs EndoMT can be induced via TGF-β-mediated activation of Ras/Mek/Erk rather than the canonical pSmad3 signaling with increased expression of the EndoMT markers Slug, Snai1 and MMP-2 (41). TGF-β-mediated ECM production has also been shown to be dependent on non-Smad p38 MAPK pathways, working in combination with the canonical Smad2/3 signaling in diseased VICs in sporadic non-syndromic mitral valve prolapse (34).
The potential interaction between Ang II and TGF-β in the pathogenesis of MMVD should also be considered as this is well recognized in fibrosis and ECM deposition, involving both canonical and non-canonical parts of the TGF-β signaling pathway, and these effects can be inhibited by angiotensin II receptor blockers (ARBs) (34). One consideration, as previously mentioned, is the disparity in the level of fibrosis comparing end-stage canine and human MMVD (BD), and to what extent signaling pathways more important in fibrosis might actually contribute to the ECM remodeling seen with myxomatous degeneration. Nevertheless, activation of Smad-mediated signaling and TGF-β2-responsive gene expression has been observed in the mitral valves of mice treated with Ang II, with a relatively moderate change in the activity of BMP and Wnt-β-catenin signaling, both of which are suggested to contribute to human MMVD (84). The ARB antagonist losartan competitively antagonizes binding of Ang II to the AT1 receptor, but also inhibits progression of aortic root aneurysms in Fbn-1 mutant mice and Marfan disease patients through anti-TGF-β effects (104, 115). It has been proposed that losartan shunts Ang II signaling toward the Type II receptor involving both SMAD and ERK (116). In human mitral VICs treated with losartan, phosphorylation of SMAD2/3 and p38 is inhibited, but not ERK, suggesting SMAD2/3-dependent canonical TGF-β and ERK-independent signaling contribute to MMVD progression. Furthermore, in a mouse model of Loeys-Dietz syndrome (LDS), which exhibits phenotypic features overlap with MMVD and is a consequence of dysregulated TGF-β signaling, losartan normalizes growth of aortic roots and protects the aortic wall from the damage associated with decreased expression of pSmad2, pERK and TGF-β1 ligands (117). Losartan antagonism of TGF-β-dependent pSmad and ERK signaling also prevents EndoMT in ovine VECs and reduces expression of TGF-β and pERK in sheep mitral valve tissues (43, 118). In transgenic Runx2± mice treated with AngII there is increased expression levels for the COL3A1 gene and immunostaining for pAKT proteins despite the lack of expression of TGF-β3 and pSMAD2/3 in mitral valve tissues. Conversely, there is increased expression of TGF-β3, COL3A1, p-SMAD2/3 and p-AKT in Runx2+/+ mice, suggesting that Runx2 may contribute to trigger tissue ECM responses and cellular proliferation, similar to that seen in MMVD (84). However, a large-scale clinical trial for children and young adults with MFS showed that there were no significant difference in the rate of aortic-root dilatation between the two groups treated with losartan and atenolol, the current standard therapy in most hospitals (119).
Bone Morphogenetic Proteins Signaling in Myxomatous Mitral Valve Degeneration
BMPs are important members of the TGF-β superfamily and may also be involved in MMVD progression. While they were originally discovered because of their capacity to mediate bone and cartilage formation, there is an increasing awareness of their role in non-osteogenic processes, such as heart development, circulation homeostasis and several cardiovascular diseases (120). Similar to TGF-βs BMPs ligands can bind to the heterotetrameric transmembrane receptor complexes comprised of two serine-threonine kinase type I and type II receptors, resulting in initiation of canonical or non-canonical signaling cascades, including the downstream phosphorylation of R-SMAD effector proteins and the three well-characterized ERK, JNK and p38 MAPK pathways (121–123). Following activation of R-SMADs, also known as SMADs 1, 5, and 8, BMPs form heteromeric complexes by binding with the co-Smad mediator, Smad4, and translocate into the nucleus to mediate transcription of BMP-responsive genes (Figure 3) (124).
The potential role of BMP signaling was first recognized in MMVD degeneration in a Fbn-1 mutant murine model of MFS. Several studies have shown that BMP 2, 4, and 6 can contribute to cardiac valve development and EndoMT involving a key contribution of Smad1/4/5-dependent BMP signaling (125–129). Although the molecular mechanisms in which the BMPs might impact MMVD pathogenesis have not been clarified some studies suggest that non-canonical BMP4/SOX9 signaling regulates the phenotypic change in VICs and ECM remodeling in human myxomatous mitral valve tissue, with increased gene and protein expression of BMP4, Sox9, CRTAC1, CTGF, α-SMA, vimentin and desmin (67). BMP4 treatment itself results in increased expression of Sox9 and other markers of ECM reorganization and VIC activation in human MMVD valves (67). BMP2 signaling in the human endocardial lineage is essential for remodeling of atrioventricular valves since BMP2 knockout mice show reduced Sox9 expression and mitral valve malformation deficiencies (130). BMP2 and TGF-β1 synergistically stimulate the expression of the transcriptional factors SOX9, Twist1, and Snail1/2 and initiate EndoMT via canonical Smad1/5- and Smad2/3-dependent pathways (101). TGF-β induces quiescent VIC activation by BMP2 stimulation in deformed mitral valves in Fstl1-deficient transgenic mouse model indicating a potential molecular target for myxomatous mitral valve disease (101). Comparable activity of canonical TGF-β and BMP signaling has been detected in surgically excised human MMVD tissue (84).
Other Important Pathways in Myxomatous Mitral Valve Degeneration
Several important regulatory pathways besides TGF-β signaling have also been shown to participate in the development of heart valves and progression of MMVD under certain specific cellular contexts. These molecular pathways include Wnt/β-catenin and Notch pathways (42, 84, 131). A microarray pathway analyses showed that the expression level of Wnt ligand (Wnt9A) and its receptor (frizzled 8), accompanied by upregulation of the extracellular positive modulator R-Spondin 2 and the target gene runt-related transcription factor 2 (Runx2), were increased in human MMVD tissues. These observations were further supported by the increased expression of Wnt9A, β-catenin and Wnt-target gene WISP1 in TGF-β2-treated human mitral VICs and Ang II-induced myxomatous degeneration in murine mitral valves (84). The Notch pathway also plays an important role in the development of outflow tract of the heart which starts with EndoMT in the endocardial cells leading to the formation of cardiac valves (132). However, there lacks enough evidence that Notch signaling is directly involved in the onset and progression of MMVD in human and dogs. To what extent TGF-β signaling might interact with these pathways is beyond the scope of this review.
Conclusion
MMVD is the most important acquired mitral valve disease in both dogs and humans and its natural history, pathogenesis and progression, pathology, cell and molecular changes are reasonably well characterized. Current areas of investigation are focused on genetic analysis, small and large animal models, the role of TGF-β dysregulation, abnormal EndoMT, and the impact of biomechanical strain in the pathogenesis of MMVD. There is also parallel interest in monocyte infiltration, particularly macrophages, as potential contributors to MMVD pathogenesis, at least in human mitral valvulopathies. While transgenic rodent models and surgically resected human valve tissues give insight into many molecular aspects of MMVD they are limited in modeling the chronicity of this disease and the extensive secondary fibrosis in patient-derived human valve tissues hampers examination of molecular events controlling myxomatous initiation and progression. Interestingly, much more is known about the earlier stages of the disease in the dog, and the naturally occurring disease in the dog might be the best large animal model to study human MMVD considering the shared cellular and molecular events in the two species. The important role of TGF-β signaling in the onset and progression of MMVD has been emphasized by studies using mouse models, valve samples of human and canine myxomatous mitral valves and in in vitro canine aVIC cell culture models. Phenotypic transition of qVICs to aVICs induced by dysregulated TGF-β signaling appears to be a key contributor to valve myxomatous degeneration though aberrant matrix remodeling, exerting control through complex canonical and non-canonical signaling pathways interactions and effects, which can conceivably affect the disease phenotype alone or in combinations. To what extent one of these might be a dominant pathway for the diseased is still unknown. Furthermore, what abnormal signaling contributes to the survival and persistence of aVICs in diseased valves remains unanswered. Understanding the mechanisms that control cell persistence in this disease likely will give clues to the pathogenesis and identify potential therapeutic targets in both the dog and human.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
QT: major contribution to writing and preparation. AM, KP, and VEM: additional writing and editing. BMC: initiator of review, major contribution to writing and final editing. All authors contributed to the article and approved the submitted version.
Funding
QT was funded by the China Scholarship Council (Ph.D.). KP and VEM were supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) in the form of an Institute Strategic Programme Grant (BB/J004316/1). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
BarlowJBPocockWA. The significance of late systolic murmurs and mid-late systolic clicks.Md State Med J. (1963) 12:76–7.
2.
PensingerRR. Comparative aspects of mitral valve disease in dogs.Ann N Y Acad Sci. (1965) 118:525–34. 10.1111/j.1749-6632.1965.tb33974.x
3.
DellingFNVasanRS. Epidemiology and pathophysiology of mitral valve prolapse new insights into disease progression, genetics, and molecular basis.Circulation. (2014) 129:2158–70. 10.1161/CIRCULATIONAHA.113.006702
4.
LevineRAHagégeAAJudgeDPPadalaMDal-BiancoJPAikawaEet alMitral valve disease–morphology and mechanisms.Nat Rev Cardiol. (2015) 12:689–710. 10.1038/nrcardio.2015.161
5.
MarkbyGRSummersKMMacRaeVECorcoranBM. Comparative transcriptomic profiling and gene expression for myxomatous mitral valve disease in the dog and human.Vet Sci. (2017) 4:34. 10.3390/vetsci4030034
6.
KimAJXuNYutzeyKE. Macrophage lineages in heart valve development and disease.Cardiovasc Res. (2021) 117:663–73. 10.1093/cvr/cvaa062
7.
FreedLALevyDLevineRALarsonMGEvansJCFullerDLet alPrevalence and clinical outcome of mitral-valve prolapse.N Engl J Med. (1999) 341:1–7.
8.
DevereuxRBJonesECRomanMJHowardBVFabsitzRRLiuJEet alPrevalence and correlates of mitral valve prolapse in a population-based sample of American Indians: the strong heart study.Am J Med. (2001) 111:679–85. 10.1016/s0002-9343(01)00981-0
9.
DetweilerDKPattersonDF. The prevalence and types of cardiovascular disease in dogs.Ann N Y Acad Sci. (1965) 127:481–516. 10.1111/j.1749-6632.1965.tb49421.x
10.
BonnettBEgenvallAOlsonPHedhammarA. Mortality in insured Swedish dogs: rates and causes of death in various breeds.Vet Rec. (1997) 141:40–4. 10.1136/vr.141.2.40
11.
ParkerHGKilroy-GlynnP. Myxomatous mitral valve disease in dogs: does size matter?J Vet Cardiol. (2012) 14:19–29. 10.1016/j.jvc.2012.01.006
12.
MaronBJEpsteinSE. Hypertrophic cardiomyopathy. Recent observations regarding the specificity of three hallmarks of the disease: asymmetric septal hypertrophy, septal disorganization and systolic anterior motion of the anterior mitral leaflet.Am J Cardiol. (1980) 45:141–54. 10.1016/0002-9149(80)90232-5
13.
JudgeDPRoufRHabashiJDietzHC. Mitral valve disease in Marfan syndrome and related disorders.J Cardiovasc Transl Res. (2011) 4:741–7. 10.1007/s12265-011-9314-y
14.
AtzingerCLMeyerRAKhouryPRGaoZTinkleBT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic ehlers-danlos syndrome.J Pediatr. (2011) 158:826–830.e1. 10.1016/j.jpeds.2010.11.023
15.
LardeuxAKyndtFLecointeSMarecHLMerotJSchottJJet alFilamin-a-related myxomatous mitral valve dystrophy: genetic, echocardiographic and functional aspects.J Cardiovasc Transl Res. (2011) 4:748–56. 10.1007/s12265-011-9308-9
16.
SaulsKde VlamingAHarrisBSWilliamsKWesselsALevineRAet alDevelopmental basis for filamin-A-associated myxomatous mitral valve disease.Cardiovasc Res. (2012) 96:109–19. 10.1093/cvr/cvs238
17.
Le TourneauTLe ScouarnecSCueffCBernsteinDAalbertsJLecointeSet alNew insights into mitral valve dystrophy: a filamin-A genotype-phenotype and outcome study.Eur Heart J. (2018) 39:1269–77. 10.1093/eurheartj/ehx505
18.
FornesPHeudesDFuzellierJFTixierDBrunevalPCarpentierA. Correlation between clinical and histologic patterns of degenerative mitral valve insufficiency: a histomorphometric study of 130 excised segments.Cardiovasc Pathol. (1999) 8:81–92. 10.1016/s1054-8807(98)00021-0
19.
PedersenHDHäggströmJ. Mitral valve prolapse in the dog: a model of mitral valve prolapse in man.Cardiovasc Res. (2000) 47:234–43. 10.1016/s0008-6363(00)00113-9
20.
RauschMKBotheWKvittingJPGöktepeSMillerDCKuhlE. In vivo dynamic strains of the ovine anterior mitral valve leaflet.J Biomech. (2011) 44:1149–57. 10.1016/j.jbiomech.2011.01.020
21.
CuiYCLiKTianYYuanWMPengPYangJZet alA pig model of ischemic mitral regurgitation induced by mitral chordae tendinae rupture and implantation of an ameroid constrictor.PLoS One. (2014) 9:e111689. 10.1371/journal.pone.0111689
22.
RabkinEAikawaMStoneJRFukumotoYLibbyPSchoenFJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves.Circulation. (2001) 104:2525–32. 10.1161/hc4601.099489
23.
CorcoranBMBlackAAndersonHMcEwanJDFrenchASmithPet alIdentification of surface morphologic changes in the mitral valve leaflets and chordae tendineae of dogs with myxomatous degeneration.Am J Vet Res. (2004) 65:198–206. 10.2460/ajvr.2004.65.198
24.
HanRIBlackACulshawGJFrenchATElseRWCorcoranBM. Distribution of myofibroblasts, smooth muscle like cells, macrophages, and mast cells in mitral valve leaflets of dogs with myxomatous mitral valve disease.Am J Vet Res. (2008) 69:763–9. 10.2460/ajvr.69.6.763
25.
KimAJXuNUmeyamaKHulinAPonnySRVagnozziRJet alDeficiency of circulating monocytes ameliorates the progression of myxomatous valve degeneration in Marfan syndrome.Circulation. (2020) 141:132–46. 10.1161/CIRCULATIONAHA.119.042391
26.
McDonaldPCWilsonJEMcNeillSGaoMSpinelliJJRosenbergFet alThe challenge of defining normality for human mitral and aortic valves: geometrical and compositional analysis.Cardiovasc Pathol. (2002) 11:193–209. 10.1016/s1054-8807(01)00102-8
27.
RobertsWCVowelsTJKoJMHebelerRFJr.Gross and histological features of excised portions of posterior mitral leaflet in patients having operative repair of mitral valve prolapse and comments on the concept of missing (= ruptured) chordae tendineae.J Am Coll Cardiol. (2014) 63:1667–74. 10.1016/j.jacc.2013.11.017
28.
SainiNSaikiaUNSahniDSinghRS. A comparative analysis of mitral valve changes in different age groups by histochemical, immunohistochemical and ultrastructural study.J Anat Soc India. (2014) 63:103–9. 10.1016/j.jasi.2014.11.012
29.
KruithofBPPaardekooperLHiemstraYLGoumansMJPalmenMDelgadoVet alStress-induced remodelling of the mitral valve: a model for leaflet thickening and superimposed tissue formation in mitral valve disease.Cardiovasc Res. (2020) 116:931–43. 10.1093/cvr/cvz204
30.
GuptaVBarzillaJEMendezJSStephensEHLeeELCollardCDet alAbundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves.Cardiovasc Pathol. (2009) 18:191–7. 10.1016/j.carpath.2008.05.001
31.
KingBDClarkMABabaNKilmanJWWooleyCF. ‘Myxomatous’ mitral valves: collagen dissolution as the primary defect.Circulation. (1982) 66:288–96. 10.1161/01.cir.66.2.288
32.
NgCMChengAMyersLAMartinez-MurilloFJieCBedjaDet alTGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome.J Clin Invest. (2004) 114:1586–92. 10.1172/JCI22715
33.
ObayashiKMiyagawa-TomitaSMatsumotoHKoyamaHNakanishiTHiroseH. Effects of transforming growth factor-β3 and matrix metalloproteinase-3 on the pathogenesis of chronic mitral valvular disease in dogs.Am J Vet Res. (2011) 72:194–202. 10.2460/ajvr.72.2.194
34.
GeirssonASinghMAliRAbbasHLiWSanchezJAet alModulation of transforming growth factor-β signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers.Circulation. (2012) 126:S189–97. 10.1161/CIRCULATIONAHA.111.082610
35.
HaglerMAHadleyTMZhangHMehraKRoosCMSchaffHVet alTGF-β signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves.Cardiovasc Res. (2013) 99:175–84. 10.1093/cvr/cvt083
36.
TanKMarkbyGMuirheadRBlakeRBergeronLFiciGet alEvaluation of canine 2D cell cultures as models of myxomatous mitral valve degeneration.PLoS One. (2019) 14:e0221126. 10.1371/journal.pone.0221126
37.
MarkbyGRMacraeVESummersKMCorcoranBM. Disease severity-associated gene expression in canine myxomatous mitral valve disease is dominated by TGFβ signaling.Front Genet. (2020) 11:372. 10.3389/fgene.2020.00372
38.
GoumansMJvan ZonneveldAJTen DijkeP. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis?Trends Cardiovasc Med. (2008) 18:293–8. 10.1016/j.tcm.2009.01.001
39.
GoumansMJTen DijkeP. TGF-β signaling in control of cardiovascular function.Cold Spring Harb Perspect Biol. (2018) 10:a022210. 10.1101/cshperspect.a022210
40.
Garcia-PenaAIbarrolaJNavarroASadabaATirapleguiCGaraikoetxeaMet alActivation of the interleukin-33/ST2 pathway exerts deleterious effects in myxomatous mitral valve disease.Int J Mol Sci. (2021) 22:2310. 10.3390/ijms22052310
41.
SaulsKToomerKWilliamsKJohnsonAJMarkwaldRRHajduZet alIncreased infiltration of extra-cardiac cells in myxomatous valve disease.J Cardiovasc Dev Dis. (2015) 2:200–13. 10.3390/jcdd2030200
42.
HulinAMooreVJamesJMYutzeyKE. Loss of axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease.Cardiovasc Res. (2017) 113:40–51. 10.1093/cvr/cvw229
43.
BartkoPEDal-BiancoJPGuerreroJLBeaudoinJSzymanskiCKimDHet alEffect of losartan on mitral valve changes after myocardial infarction.J Am Coll Cardiol. (2017) 70:1232–44. 10.1016/j.jacc.2017.07.734
44.
KimAJAlfieriCMYutzeyKE. Endothelial cell lineage analysis does not provide evidence for EMT in adult valve homeostasis and disease.Anat Rec (Hoboken). (2019) 302:125–35. 10.1002/ar.23916
45.
BischoffJCasanovasGWylie-SearsJKimDHBartkoPEGuerreroJLet alCD45 expression in mitral valve endothelial cells after myocardial infarction. Circ Res. (2016) 119:1215–25.
46.
LuCCLiuMMClintonMCulshawGArgyleDJCorcoranBM. Developmental pathways and endothelial to mesenchymal transition in canine myxomatous mitral valve disease.Vet J. (2015) 206:377–84. 10.1016/j.tvjl.2015.08.011
47.
HulinADeroanneCFLambertCADumontBCastronovoVDefraigneJOet alMetallothionein-dependent up-regulation of TGF-beta2 participates in the remodelling of the myxomatous mitral valve.Cardiovasc Res. (2012) 93:480–9. 10.1093/cvr/cvr337
48.
DisatianSEhrhartEJIIIZimmermanSOrtonEC. Interstitial cells from dogs with naturally occurring myxomatous mitral valve disease undergo phenotype transformation.J Heart Valve Dis. (2008) 17:402–11.
49.
DisatianSOrtonEC. Autocrine serotonin and transforming growth factor-b1 signaling mediates spontaneous myxomatous mitral valve disease.J Heart Valve Dis. (2009) 18:44–51.
50.
von GiseAPuWT. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease.Circ Res. (2012) 110:1628–45. 10.1161/CIRCRESAHA.111.259960
51.
TaoGKotickJDLincolnJ. Heart valve development, maintenance, and disease: the role of endothelial cells.Curr Top Dev Biol. (2012) 100:203–32. 10.1016/B978-0-12-387786-4.00006-3
52.
HintonRBYutzeyKE. Heart valve structure and function in development and disease.Annu Rev Physiol. (2011) 73:29–46. 10.1146/annurev-physiol-012110-142145
53.
GuthrieRBEdwardsJE. Pathology of the myxomatous mitral value. Nature, secondary changes and complications.Minn Med. (1976) 59:637–47.
54.
CulshawGJFrenchATHanRIBlackAPearsonGTCorcoranBM. Evaluation of innervation of the mitral valves and the effects of myxomatous degeneration in dogs.Am J Vet Res. (2010) 71:194–202. 10.2460/ajvr.71.2.194
55.
HanRIClarkCHBlackAFrenchACulshawGJKempsonSAet alMorphological changes to endothelial and interstitial cells and to the extra-cellular matrix in canine myxomatous mitral valve disease (endocardiosis).Vet J. (2013) 197:388–94. 10.1016/j.tvjl.2013.01.027
56.
TamuraKFukudaYIshizakiMMasudaYYamanakaNFerransVJ. Abnormalities in elastic fibers and other connective-tissue components of floppy mitral valve.Am Heart J. (1995) 129:1149–58. 10.1016/0002-8703(95)90397-6
57.
Rabkin-AikawaEFarberMAikawaMSchoenFJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves.J Heart Valve Dis. (2004) 13:841–7.
58.
BischoffJAikawaE. Progenitor cells confer plasticity to cardiac valve endothelium.J Cardiovasc Transl Res. (2011) 4:710–9. 10.1007/s12265-011-9312-0
59.
HanRIBlackACulshawGFrenchATCorcoranBMStructural and cellular changes in canine myxomatous mitral valve disease: an image analysis study. J Heart Valve Dis. (2010) 19:60–70.
60.
HadianMCorcoranBMBradshawJP. Molecular changes in fibrillar collagen in myxomatous mitral valve disease.Cardiovasc Pathol. (2010) 19:e141–8. 10.1016/j.carpath.2009.05.001
61.
LiuMMFlanaganTCLuCCFrenchATArgyleDJCorcoranBMCulture and characterisation of canine mitral valve interstitial and endothelial cells. Vet J. (2015) 204:32–9.
62.
BlakeRRMarkbyGRCulshawGJMartinez-PereiraYLuCCCorcoranBM. Survival of activated myofibroblasts in canine myxomatous mitral valve disease and the role of apoptosis.Res Vet Sci. (2020) 128:99–106. 10.1016/j.rvsc.2019.11.004
63.
SalhiyyahKYacoubMHChesterAH. Cellular mechanisms in mitral valve disease.J Cardiovasc Transl Res. (2011) 4:702–9. 10.1007/s12265-011-9318-7
64.
LiuACJoagVRGotliebAI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology.Am J Pathol. (2007) 171:1407–18. 10.2353/ajpath.2007.070251
65.
ArmstrongEJBischoffJ. Heart valve development: endothelial cell signaling and differentiation.Circ Res. (2004) 95:459–70. 10.1161/01.RES.0000141146.95728.da
66.
HakunoDKimuraNYoshiokaMMukaiMKimuraTOkadaYet alPeriostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents.J Clin Invest. (2010) 120:2292–306. 10.1172/JCI40973
67.
SaingerRGrauJBBranchettiEPoggioPSeefriedWFFieldBCet alHuman myxomatous mitral valve prolapse: role of bone morphogenetic protein 4 in valvular interstitial cell activation.J Cell Physiol. (2012) 227:2595–604. 10.1002/jcp.22999
68.
HulinADeroanneCLambertCDefraigneJONusgensBRadermeckerMet alEmerging pathogenic mechanisms in human myxomatous mitral valve: lessons from past and novel data. Cardiovasc Pathol. (2013) 22:245–50.
69.
ButcherJTMarkwaldRR. Valvulogenesis: the moving target.Philos Trans R Soc Lond B Biol Sci. (2007) 362:1489–503. 10.1098/rstb.2007.2130
70.
CombsMDYutzeyKE. Heart valve development: regulatory networks in development and disease.Circ Res. (2009) 105:408–21. 10.1161/CIRCRESAHA.109.201566
71.
ChakrabortySCombsMDYutzeyKE. Transcriptional regulation of heart valve progenitor cells.Pediatr Cardiol. (2010) 31:414–21. 10.1007/s00246-009-9616-x
72.
AkiyamaHChaboissierMCBehringerRRRowitchDHSchedlAEpsteinJAet alEssential role of Sox9 in the pathway that controls formation of cardiac valves and septa.Proc Natl Acad Sci USA. (2004) 101:6502–7. 10.1073/pnas.0401711101
73.
ParanyaGVinebergSDvorinEKaushalSRothSJRabkinEet alAortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro.Am J Pathol. (2001) 159:1335–43. 10.1016/s0002-9440(10)62520-5
74.
ParuchuriSYangJHAikawaEMelero-MartinJMKhanZALoukogeorgakisSet alHuman pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-beta2.Circ Res. (2006) 99:861–9. 10.1161/01.RES.0000245188.41002.2c
75.
ShaperoKWylie-SearsJLevineRAMayerJEJrBischoffJ. Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelialto-mesenchymal transition and myofibroblastic activation.J Mol Cell Cardiol. (2015) 80:175–85. 10.1016/j.yjmcc.2015.01.006
76.
BorderWANobleNA. Transforming growth factor beta in tissue fibrosis.N Engl J Med. (1994) 331:1286–92.
77.
RobertsABSpornMBAssoianRKSmithJMRocheNSWakefieldLMet alTransforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro.Proc Natl Acad Sci USA. (1986) 83:4167–71. 10.1073/pnas.83.12.4167
78.
VargaJJimenezSA. Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta.Biochem Biophys Res Commun. (1986) 138:974–80. 10.1016/s0006-291x(86)80591-5
79.
KyndtFGueffetJPProbstVJaafarPLegendreABouffantFLet alMutations in the gene encoding filamin a as a cause for familial cardiac valvular dystrophy.Circulation. (2007) 115:40–9. 10.1161/CIRCULATIONAHA.106.622621
80.
MediciDPotentaSKalluriR. Transforming growth factor-β2 promotes snail-mediated endothelial-mesenchymal transition through convergence of SMAD-dependent and SMAD-independent signalling.Biochem J. (2011) 437:515–20. 10.1042/BJ20101500
81.
MihiraHSuzukiHIAkatsuYYoshimatsuYIgarashiTMiyazonoKet alTGF-β-induced mesenchymal transition of MS-1 endothelial cells requires SMAD-dependent cooperative activation of Rho signals and MRTF-A.J Biochem. (2012) 151:145–56. 10.1093/jb/mvr121
82.
PardaliESanchez-DuffhuesGGomez-PuertoMCTen DijkeP. TGF-β-induced endothelial-mesenchymal transition in fibrotic diseases.Int J Mol Sci. (2017) 18:2157. 10.3390/ijms18102157
83.
Van MeeterenLATen DijkeP. Regulation of endothelial cell plasticity by TGF-β.Cell Tissue Res. (2012) 347:177–86. 10.1007/s00441-011-1222-6
84.
ThaljiNMHaglerMAZhangBCasaclang-VerzosaGNairAASuriRMet alNonbiased molecular screening identifies novel molecular regulators of fibrogenic and proliferative signaling in myxomatous mitral valve disease.Circ Cardiovasc Genet. (2015) 8:516–28. 10.1161/CIRCGENETICS.114.000921
85.
AupperleHMärzIThielebeinJSchoonHA. Expression of transforming growth factor-beta1, -beta2 and -beta3 in normal and diseased canine mitral valves.J Comp Pathol. (2008) 139:97–107. 10.1016/j.jcpa.2008.05.007
86.
RizzoSBassoCLazzariniECeleghinRPaolinAGerosaGet alTGF-beta1 pathway activation and adherens junction molecular pattern in nonsyndromic mitral valve prolapse.Cardiovasc Pathol. (2015) 24:359–67. 10.1016/j.carpath.2015.07.009
87.
DietzHCLoeysBCartaLRamirezF. Recent progress towards a molecular understanding of Marfan syndrome.Am J Med Genet C Semin Med Genet. (2005) 139:4–9. 10.1002/ajmg.c.30068
88.
ChaudhrySSCainSAMorganADallasSLShuttleworthCAKieltyCM. Fibrillin-1 regulates the bioavailability of TGFbeta1.J Cell Biol. (2007) 176:355–67. 10.1083/jcb.200608167
89.
ToomerKSaulsKFulmerDGuoLMooreKGloverJet alFilamin-A as a balance BETWEEN Erk/Smad activities during cardiac valve development.Anat Rec. (2019) 302:117–24. 10.1002/ar.23911
90.
SurachetpongSJiranantasakTRungsipipatAOrtonEC. Apoptosis and abundance of Bcl-2 family and transforming growth factor beta1 signaling proteins in canine myxomatous mitral valves.J Vet Cardiol. (2013) 15:171–80. 10.1016/j.jvc.2013.02.005
91.
MoesgaardSGAupperleHRajamakiMMFalkTRasmussenCEZoisNEet alMatrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs) and transforming growth factor-beta (TGF-beta) in advanced canine myxomatous mitral valve disease.Res Vet Sci. (2014) 97:560–7. 10.1016/j.rvsc.2014.10.003
92.
AnnesJPChenYMungerJSRifkinDB. Integrin alphaVbeta6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1.J Cell Biol. (2004) 165:723–34. 10.1083/jcb.200312172
93.
ArciniegasEFridMGDouglasISStenmarkKR. Perspectives on endothelial-tomesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension.Am J Physiol Lung Cell Mol Physiol. (2007) 293:L1–8. 10.1152/ajplung.00378.2006
94.
ArciniegasECarrilloLMDe SanctisJBCandelleD. Possible role of NFkappaB in the embryonic vascular remodeling and the endothelial mesenchymal transition process.Cell Adhes Migr. (2008) 2:17–29. 10.4161/cam.2.1.5789
95.
RobertsonIBHoriguchiMZilberbergLDabovicBHadjiolovaKRifkinDBLatent TGF-β-binding proteins. Matrix Biol. (2015) 47:44–53.
96.
WranaJLAttisanoLWieserRVenturaFMassaguéJ. Mechanism of activation of the TGF-β receptor.Nature. (1994) 370:341–7. 10.1038/370341a0
97.
HataAChenYG. TGF-β signaling from receptors to smads.Cold Spring Harb Perspect Biol. (2016) 8:a022061. 10.1101/cshperspect.a022061
98.
HeldinCHMoustakasA. Signaling receptors for TGF-β family members.Cold Spring Harb Perspect Biol. (2016) 8:a022053. 10.1101/cshperspect.a022053
99.
Euler-TaimorGHegerJ. The complex pattern of SMAD signaling in the cardiovascular system.Cardiovasc Res. (2006) 69:15–25. 10.1016/j.cardiores.2005.07.007
100.
HanLGotliebAI. Fibroblast growth factor-2 promotes in vitro heart valve interstitial cell repair through the Akt1 pathway.Cardiovasc Pathol. (2012) 21:382–9. 10.1016/j.carpath.2011.12.001
101.
PrakashSBorregueroLSylvaMFlores RuizLRezaiFGunstQDet alDeletion of Fstl1 (follistatin-like 1) from the endocardial/endothelial lineage causes mitral valve disease.Arterioscler Thromb Vasc Biol. (2017) 37:e116–30. 10.1161/ATVBAHA.117.309089
102.
BoileauCGuoDCHannaNRegaladoESDetaintDGongLet alTGF2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome.Nat Genet. (2012) 44:916–21. 10.1038/ng.2348
103.
CañadasVVilacostaIBrunaIFusterV. Marfan syndrome. Part 2: treatment and management of patients.Nat Rev Cardiol. (2010) 7:266–76. 10.1038/nrcardio.2010.31
104.
HabashiJPJudgeDPHolmTMCohnRDLoeysBLCooperTKet alLosartan, an AT1 antagonist, prevents aortic aneurism in a mouse model of Marfan syndrome.Science. (2006) 312:117–21. 10.1126/science.1124287
105.
IsogaiZOnoRNUshiroSKeeneDRChenYMazzieriRet alLatent transforming growth factor-beta binding protein 1 interacts with fibrillin and is micro-fibril-associated protein.J Biol Chem. (2003) 278:2750–7. 10.1074/jbc.m209256200
106.
JudgeDPDietzHC. Marfan syndrome.Lancet. (2005) 366:1965–76.
107.
LindsayMESchepersDBolarNADoyleJJGalloEFert-BoberJet alLoss-of-function mutations in TGF-2 cause a syndromic presentation of thoracic aortic aneurism.Nat Genet. (2012) 44:922–7. 10.1038/ng.2349
108.
NeptuneERFrischmeyerPAArkingDEMyersLBuntonTEGayraudBet alDysregulation of TGFbeta activation contributes to pathogenesis in Marfan syndrome.Nat Genet. (2003) 33:407–11. 10.1038/ng1116
109.
NorrisRAMoreno-RodriguezRWesselsAMerotJBrunevalPChesterAHet alExpression of the familial cardiac valvular dystrophy gene, filamin-A, during heart morphogenesis.Dev Dyn. (2010) 239:2118–27. 10.1002/dvdy.22346
110.
SasakiAMasudaYOhtaYIkedaKWatanabeK. Filamin associates with SMADS and regulates transforming growth factor-β signaling.J Biol Chem. (2001) 276:17871–7. 10.1074/jbc.M008422200
111.
DerynckRZhangYE. Smad-dependent and smad-independent pathways in TGF-beta family signalling.Nature. (2003) 425:577–84. 10.1038/nature02006
112.
ZhangYE. Non-SMAD signaling pathways of the TGF-β family.Cold Spring Harb Perspect Biol. (2017) 9:a022129. 10.1101/cshperspect.a022129
113.
HuangWXiaoDZWangYShanZXLiuXYLinQXet alFn14 promotes differentiation of human mesenchymal stem cells into heart valvular interstitial cells by phenotypic characterization.J Cell Physiol. (2014) 229:580–7. 10.1002/jcp.24480
114.
BlommeBDeroanneCHulinALambertCDefraigneJONusgensBet alMechanical strain induces a pro-fibrotic phenotype in human mitral valvular interstitial cells through RhoC/ROCK/MRTF-A and Erk1/2 signaling pathways.J Mol Cell Cardiol. (2019) 135:149–59. 10.1016/j.yjmcc.2019.08.008
115.
BrookeBSHabashiJPJudgeDPPatelNLoeysBDietzHC. Angiotensin II blockade and aortic-root dilatation in marfan’s syndrome.N Engl J Med. (2008) 358:2787–95.
116.
HabashiJPDoyleJJHolmTMAzizHSchoenhoffFBedjaDet alAngiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism.Science. (2011) 332:361–5. 10.1126/science.1192152
117.
GalloEMLochDCHabashiJPCalderonJFChenYBedjaDet alAngiotensin II-dependent TGF-β signaling contributes to loeys-dietz syndrome vascular pathogenesis.J Clin Invest. (2014) 124:448–60. 10.1172/JCI69666
118.
Wylie-SearsJLevineRABischoffJ. Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-β-induced phosphorylation of ERK.Biochem Biophys Res Commun. (2014) 446:870–5. 10.1016/j.bbrc.2014.03.014
119.
LacroRVDietzHCSleeperLAYetmanATBradleyTJColanSDet alAtenolol versus losartan in children and young adults with Marfan’s syndrome.N Engl J Med. (2014) 371:2061–71. 10.1056/NEJMoa1404731
120.
MorrellNWBlochDBten DijkePGoumansMJHataASmithJet alTargeting BMP signalling in cardiovascular disease and anaemi.Nat Rev Cardiol. (2016) 13:106–20. 10.1038/nrcardio.2015.156
121.
ScharpfeneckerMvan DintherMLiuZvan BezooijenRLZhaoQPukacLet alBMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis.J Cell Sci. (2007) 120:964–72. 10.1242/jcs.002949
122.
KirkbrideKCTownsendTABruinsmaMWBarnettJVBlobeGC. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptor.J Biol Chem. (2008) 283:7628–37. 10.1074/jbc.M704883200
123.
CorradiniEttJLLinHY. The RGM/DRAGON family of BMP co-receptors.Cytokine Growth Factor Rev. (2009) 20:389–98. 10.1016/j.cytogfr.2009.10.008
124.
LuoJYZhangYWangLHuangY. Regulators and effectors of bone morphogenetic protein signalling in the cardiovascular system.J Physiol. (2015) 593:2995–3011. 10.1113/JP270207
125.
SugiYYamamuraHOkagawaHMarkwaldRR. Bone morphogenetic protein-2 can mediate myocardial regulation of atriventricular cushion mesenchymal cell formation in mice.Dev Biol. (2004) 269:505–18. 10.1016/j.ydbio.2004.01.045
126.
NakajimaYYamagishiTHokariSNakamuraH. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-β and bone morphogenetic protein (BMP).Anat Rec. (2000) 258:119–27. 10.1002/(SICI)1097-0185(20000201)258:2<119::AID-AR1>3.0.CO;2-U
127.
JiaoKKulessaHTompkinsKZhouYBattsLBaldwinHSet alAn essential role of Bmp4 in the atrioventricular septation of the mouse heart.Genes Dev. (2003) 17:2362–7. 10.1101/gad.1124803
128.
KimRYRobertsonEJSollowayMJ. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart.Dev Biol. (2001) 235:449–66. 10.1006/dbio.2001.0284
129.
JacksonLFQiuTHSunnarborgSWChangAZhangCPattersonCet alDefective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling.EMBO J. (2003) 2:2704–16. 10.1093/emboj/cdg264
130.
SaxonJGBaerDRBartonJAHawkinsTWuBTruskTCet alBMP2 expression in the endocardial lineage is required for AV endocardial cushion maturation and remodeling.Dev Biol. (2017) 430:113–28. 10.1016/j.ydbio.2017.08.008
131.
AquilaGKostinaAVieceli Dalla SegaFShlyakhtoEKostarevaAMarracinoLet alThe notch pathway: a novel therapeutic target for cardiovascular diseases?Expert Opin Ther Targets. (2019) 23:695–710. 10.1080/14728222.2019.1641198
132.
Del MonteGGrego-BessaJGonzález-RajalABolósVDe La PompaJL. Monitoring notch1 activity in development: evidence for a feedback regulatory loop.Dev Dyn. (2007) 236:2594–614. 10.1002/dvdy.21246
133.
Ayme-DietrichELawsonRC\̂text{o}téFde TapiaCDa SilvaSEbelCet alThe role of 5-HT2B receptors in mitral valvulopathy: bone marrow mobilization of endothelial progenitors.Br J Pharmacol. (2017) 174:4123–39. 10.1111/bph.13981
134.
IbarrolaJGaraikoetxeaMGarcia-PeñaAMatillaLJoverEBonnardBet alBeneficial effects of mineralocorticoid receptor antagonism on myocardial fibrosis in an experimental model of the myxomatous degeneration of the mitral valve.Int J Mol Sci. (2020) 21:5372. 10.3390/ijms21155372
135.
Ayme-DietrichEDa SilvaSBouaboutGAArnouxAGuyonnetJBeckerGet alCharacterization of the spontaneous degenerative mitral valve disease in FVB mice.PLoS One. (2021) 16:e0257022. 10.1371/journal.pone.0257022
136.
ZhuASMustafaTConnellJPGrande-AllenKJ. Tumor necrosis factor alpha and interleukin 1 beta suppress myofibroblast activation via nuclear factor kappa B signaling in 3D-cultured mitral valve interstitial cells.Acta Biomater. (2021) 127:159–68. 10.1016/j.actbio.2021.03.075
137.
LiuMMFlanaganTCJockenhovelSBlackALuCCFrenchATet alDevelopment and evaluation of a tissue-engineered fibrin-based canine mitral valve three-dimensional cell culture system.J Comp Pathol. (2018) 160:23–33. 10.1016/j.jcpa.2018.02.001
138.
SyxDMillerREObeidatAMTranPBVromanRMalfaitZet alPain-related behaviors and abnormal cutaneous innervation in a murine model of classical Ehlers-Danlos syndrome.Pain. (2020) 161:2274–83. 10.1097/j.pain.0000000000001935
Summary
Keywords
MVP, MMVD, valve interstitial cell, valve endothelial cell, TGF-β, BMP
Citation
Tang Q, McNair AJ, Phadwal K, Macrae VE and Corcoran BM (2022) The Role of Transforming Growth Factor-β Signaling in Myxomatous Mitral Valve Degeneration. Front. Cardiovasc. Med. 9:872288. doi: 10.3389/fcvm.2022.872288
Received
09 February 2022
Accepted
12 April 2022
Published
17 May 2022
Volume
9 - 2022
Edited by
Elena Aikawa, Brigham and Women’s Hospital and Harvard Medical School, United States
Reviewed by
Katherine Yutzey, Cincinnati Children’s Hospital Medical Center, United States; Anna Malashicheva, Institute of Cytology, Russia
Updates
Copyright
© 2022 Tang, McNair, Phadwal, Macrae and Corcoran.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brendan M. Corcoran, Brendan.Corcoran@ed.ac.uk
This article was submitted to Heart Valve Disease, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.