 Kathryn L. Howe
Kathryn L. Howe Myron Cybulsky
Myron Cybulsky Jason E. Fish
Jason E. Fish- 1Toronto General Hospital Research Institute, University Health Network, Toronto, ON, Canada
- 2Institute of Medical Science, University of Toronto, Toronto, ON, Canada
- 3Peter Munk Cardiac Centre, University Health Network, Toronto, ON, Canada
- 4Division of Vascular Surgery, Department of Surgery, University of Toronto, Toronto, ON, Canada
- 5Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
Endothelial cells line every blood vessel and thereby serve as an interface between the blood and the vessel wall. They have critical functions for maintaining homeostasis and orchestrating vascular pathogenesis. Atherosclerosis is a chronic disease where cholesterol and inflammatory cells accumulate in the artery wall below the endothelial layer and ultimately form plaques that can either progress to occlude the lumen or rupture with thromboembolic consequences – common outcomes being myocardial infarction and stroke. Cellular communication lies at the core of this process. In this review, we discuss traditional (e.g., cytokines, chemokines, nitric oxide) and novel (e.g., extracellular vesicles) modes of endothelial communication with other endothelial cells as well as circulating and vessel wall cells, including monocytes, macrophages, neutrophils, vascular smooth muscle cells and other immune cells, in the context of atherosclerosis. More recently, the growing appreciation of endothelial cell plasticity during atherogenesis suggests that communication strategies are not static. Here, emerging data on transcriptomics in cells during the development of atherosclerosis are considered in the context of how this might inform altered cell-cell communication. Given the unique position of the endothelium as a boundary layer that is activated in regions overlying vascular inflammation and atherosclerotic plaque, there is a potential to exploit the unique features of this group of cells to deliver therapeutics that target the cellular crosstalk at the core of atherosclerotic disease. Data are discussed supporting this concept, as well as inherent pitfalls. Finally, we briefly review the literature for other regions of the body (e.g., gut epithelium) where cells similarly exist as a boundary layer but provide discrete messages to each compartment to govern homeostasis and disease. In this light, the potential for endothelial cells to communicate in a directional manner is explored, along with the implications of this concept – from fundamental experimental design to biomarker potential and therapeutic targets.
Introduction: Endothelial Cells as Gatekeepers
Endothelial cells (ECs) lie at a critical interface between the circulating blood and cellular milieu below, and hence serve as gatekeepers of vascular biology (1). Lining every blood vessel throughout the human body, this linked network of cells performs key roles unique to the local environment to maintain organ homeostasis. In large and medium-sized arteries, endothelial cells function as an anti-thrombotic physiological barrier, are sensors of mechanical forces, oxygen tension and nutrient availability, serve as sentinels of infectious threats, and mount appropriate responses to distortions in normal physiology to ensure tissue viability (2). Intercellular communication is at the core of this homeostatic process. Through soluble mediators such as nitric oxide and other small molecules, release of growth factors and cytokines, production or degradation of extracellular matrix, and secretion of extracellular vesicles (EVs) containing nucleic acids, proteins, and lipids, endothelial cells can govern one another, nearby cells in the local milieu, and cells located in remote tissues (1, 3, 4). Control of permeability (e.g., glycocalyx and junctional proteins), transcytosis (e.g., transcellular transport), and basement membrane composition, are additional signaling hubs where endothelial cells govern communication between the blood and tissue (5–7). Completing this communication loop, the endothelium is also exquisitely sensitive to mechanical and biochemical signals in their microenvironment. Regardless of whether an EC communication strategy is just being uncovered (e.g., EVs) or has been investigated for decades (e.g., nitric oxide), they each play a critical role in conditions such as atherosclerosis where vascular homeostasis is dysregulated. In the following sections, the nature of endothelial communication – both autocrine and paracrine – is explored, with a specific focus on novel modes of communication. Additionally, we will discuss the impact of cellular plasticity on cell communication and will consider approaches to exploit these lines of communication therapeutically. Finally, we will determine whether there is evidence for directionality of the messages that are sent and received (Figure 1). Together, this review will provide the reader with a highly focused update on the dynamic role of the endothelium as a maestro in cellular communication in atherosclerosis and provide provocative insights into how these signals might be intercepted or modulated to mitigate disease.
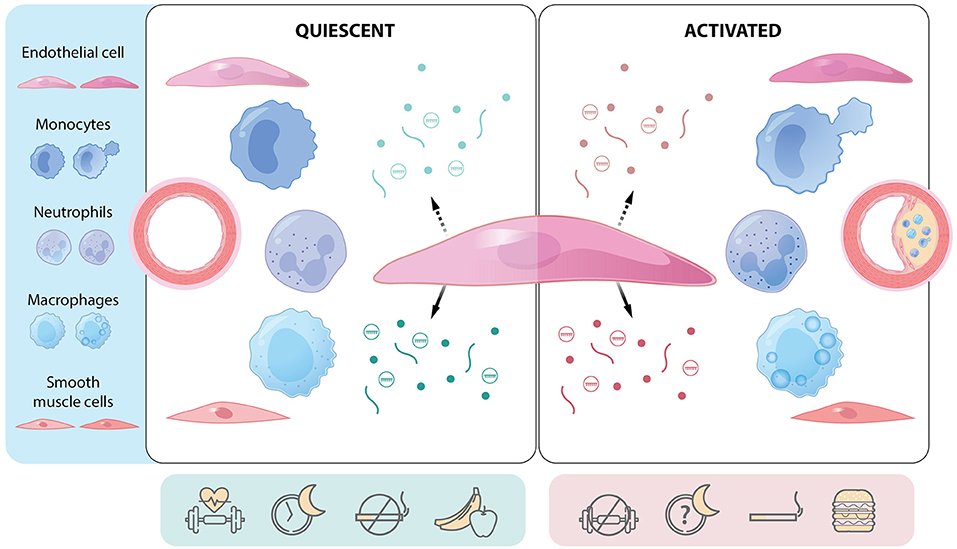
Figure 1. The endothelial cell as a communication hub in quiescent and activated states. Central endothelial cell with apical (hatched arrow) and basolateral (solid arrow) release of mediators [e.g., proteins (lines), gases and small molecules (dots), extracellular vesicles (circles containing cargo)]. Communication differs depending on whether the endothelium is quiescent or activated (e.g., regions prone to atherosclerosis) and can affect several cell types. Endothelial function and communication are affected by several lifestyle factors such as physical activity, sleep hygiene, smoking, and diet. Atherosclerotic plaque development is a function of cellular communication and represents a multicellular response to environmental, genetic, and epigenetic cues. Green: mediators and factors that actively maintain endothelial quiescence. Red: mediators and factors that promote endothelial activation.
Role of the Endothelium and Cell-Cell Crosstalk in Health and Atherosclerosis
In its healthy state, the endothelium is quiescent and autocrine and paracrine communication emanating from ECs preserves organ function [reviewed in (8)]. Endothelial quiescence is a process that must be actively maintained – it is not a “default” setting. As Ricard et al. elegantly discuss in their review (8), single cell sequencing is highlighting the regional- and organ-specific heterogeneity of ECs (i.e., lymphatic vs. venous vs. arterial and large-vessel vs. capillary) (9) and is uncovering the distinct signaling pathways required for EC quiescence. For example, in the vasculature, endothelial barrier function and cell fate is maintained by the Angiopoietin/Tie2 pathway and limitation of TGF-β signaling, respectively, among others (10, 11). The importance of preserving endothelial quiescence has been well-illustrated: loss of endothelial nitric oxide synthase accelerates murine atherosclerosis and aortic aneurysm formation (12), while endothelial-specific MAPK1/2 (ERK1/2) knockout in mice has a catastrophic impact, with hypertension, decreased endothelial nitric oxide synthase expression, increased endothelin-1 expression, and death within 5 weeks (13). Bi-directional cellular communication also participates in endothelial homeostasis. Beyond autocrine signals from the endothelium, monocytes patrolling the apical surface of the endothelium have been shown to protect against endothelial death (i.e., apoptosis) and limit inflammation – with a large artery model suggesting these cells possibly serve a housekeeping function to preserve endothelial integrity in the face of hyperlipidemia and atherosclerosis (14). Conversely, loss of endothelial quiescence causes a shift in metabolism and loss of critical protective communication via soluble mediators, including nitric oxide (15, 16). Equally important to endothelial homeostasis is physiologic high shear stress from laminar blood flow, modulated in part by the anti-inflammatory and cytoprotective actions of the transcription factor, Kruppel-like factor 2. Reduced or oscillatory shear stress, as found in arterial branch points and curvatures, renders the artery prone to atherosclerotic plaque development. Recent systems biology and omics approaches are revealing that endothelial responses to low shear stress involves activation of developmental pathways such as WNT, Notch, HIF1-α, and Hippo-YAP-Taz [reviewed in (17)] – consequently, these once dormant embryonic programs lead to altered endothelial signaling and undoubtedly contribute to the altered EC communication prevalent in atherosclerotic plaques.
Atherosclerosis is a disease of aging and cellular senescence (18). Atherosclerotic plaques are a complex collection of lipids, extracellular matrix, cells, and cellular debris that accumulate in the vessel wall (19). Plaque formation has a predilection for vascular branch points, where ECs are exposed to disturbed laminar blood flow, resulting in activated endothelial phenotypes (20). Activation of the endothelium can include the following core changes: (1) upregulation of proinflammatory cytokines (e.g., IL-1β, TNF-α) and chemokine production; (2) expression of adhesion molecules such as VCAM-1 and ICAM-1, which are critical for the recruitment of circulating immune cells; (3) loss of an anti-thrombotic surface; (4) upregulation of class II HLA molecules with consequent function in antigen presentation; and (5), loss of barrier integrity (21). The dysfunctional vascular endothelium as a driver of atherogenesis has been recently reviewed (22). In contrast to classic dogma citing loss of endothelial barrier integrity and passive movement of low-density lipoprotein (LDL) into the vessel wall (23, 24), transport of LDL cholesterol (a critical event in atherogenesis) requires active endothelial transcellular transport via expression of scavenger receptor class B type 1 (SR-B1) (25–27) and potentially other molecules such as activin-like kinase 1 (28). In regions of endothelial activation, cellular communication mediated by expression of adhesion molecules and chemokines leads to recruitment of inflammatory cells, including neutrophils (29) and monocytes (30), and accumulation and maintenance of macrophage populations through survival/proliferation signals (31, 32). New data is emerging however, that beyond the prototypic adhesion molecules, ECs have the intrinsic capacity to form “hotspots” for transendothelial neutrophil migration through the formation of junctional membrane protrusions in regions of high Rac1 activity (33). The process is asymmetric within the endothelial luminal and abluminal surfaces and heterogeneous between ECs, with more comprehensive mechanistic understanding still being unraveled (34). While beyond the scope of this review, the reader is referred to work from the Muller laboratory delineating the role of the lateral border recycling compartment – a key region communicating transmigration of leukocytes across ECs (35).
While there are well-described roles for endothelial-monocyte/macrophage communication through cell adhesion molecules, cytokines and chemokines in atherosclerotic plaques (36), more recently, it has been shown that local, but not systemic, production of colony-stimulating factor 1 from endothelial and smooth muscle cells provide macrophage survival signals that drive macrophage proliferation and atherosclerotic plaque progression (37). Atherosclerotic plaque progression is a complex process and develops in response to local and systemic intercellular communication. For many diseases – atherosclerosis included – emerging studies have focused on nanoparticle sized packages of information (EVs) and how intercellular communication is regulated through secretion and uptake of EVs and their contents (4). Endothelial-derived EV communication is beginning to be understood in the context of models of atherosclerosis (38). EVs are heterogeneous and include exosomes, derived from the multivesicular body, and microparticles, derived from budding and excision of cell membrane. EV cargo contains a range of biologically relevant material, including lipids, nucleic acids (e.g., microRNA), and proteins. Of these, microRNA has been studied extensively. These short sequences of non-coding RNA bind to the 3′ untranslated regions of mRNAs via their seed sequences and destabilize mRNAs or repress their translation into protein. Each microRNA typically has multiple mRNA targets, and thus microRNAs are considered as microregulators of health and disease (39, 40). The microRNA content of EVs has begun to be explored in the context of atherogenesis, and comprehensive reviews of their role in atherosclerosis (41) and carotid artery atherosclerotic disease (42) are available. This form of cellular crosstalk is also beginning to explain gaps in our understanding of acute myocardial infarction pathogenesis. For example, VCAM-1 positive endothelial-derived EVs enriched in microRNA-126 are responsible for the rapid neutrophil mobilization from the spleen, making these EVs a potential therapeutic target to limit infarct size (43).
In vitro models are the simplest way to examine EV communication. This reductionist approach has revealed that EVs derived from quiescent ECs have different effects on cells than those derived from inflamed (e.g., TNF-α stimulated) ECs (44). For example, under healthy quiescent conditions, ECs suppress monocyte activation by release of EVs containing anti-inflammatory microRNAs (45, 46). Similarly, ECs can communicate atheroprotective microRNAs to vascular smooth muscle cells via EVs (47). When ECs are activated with pro-inflammatory cytokines or oscillatory shear stress, they release EVs containing miR-92a, which cause macrophage activation, increased lipoprotein uptake, and decreased migration (48). In response to stress from serum-starvation, ECs release EVs containing the inflammatory adhesion molecule VCAM-1, which drive inflammatory and senescent pathways in vascular smooth muscle cells (49). Although in vitro studies suggest that endothelial-derived EVs may serve as an important mode of intercellular communication, they lack the complexity of animal models. To that end, reporter systems in mice and zebrafish may provide new opportunities to track endothelial-derived EVs and better evaluate intercellular communication in complex disease phenotypes such as atherosclerosis (50–52).
Endothelial Plasticity in Atherosclerosis: Altered Cell-Cell Communication?
Cellular plasticity and diversity within atherosclerotic plaques has been increasingly appreciated in recent years (53). Single cell RNA sequencing is a robust tool that has revealed in intricate detail the complexity of the atherosclerotic plaques [reviewed in (54)]. Lineage-tracing studies are supporting the notion that cellular identities are on a continuum and have the potential to “transform”, where cells acquire the phenotype and properties typically attributed to another cell [e.g., macrophage subsets (55, 56) and vascular smooth muscle cell plasticity and plaque development (57, 58)]. Notably, ECs can undergo partial or complete endothelial-to-mesenchymal transition (EndMT), where classic cell-cell junction proteins are lost, and mesenchymal markers are gained. This is often, but not always, associated with enhanced migratory capabilities and delamination from the endothelial monolayer, allowing cells to migrate into the plaque. Ligand-receptor interactions from transcriptomics are likewise informing our understanding of cellular communication in states of health and disease in the cardiovascular system (59). The Giannarelli laboratory studied the immune landscape of human carotid atherosclerotic plaques and found distinct T cell and macrophage populations that differed from the blood and importantly, the putative cell-cell interactions occurring in plaques from patients that had clinical cerebrovascular events (i.e., strokes) (60). The Lutgens laboratory observed the diversity of the endothelium overlying atherosclerotic plaques in both mice and humans, with electron microscopic images showing greater abnormalities in early plaques and at the shoulder regions (61). The junctional disruptions and large transcellular endothelial pores seen in early plaques and aggravated on the shoulders of advanced plaques are very likely to influence communication between the blood and the medial layer of the vessel wall (61). Moreover, these locations for endothelial transformations may correspond to the extensive proatherogenic endothelial reprogramming in response to disturbed flow that has been revealed by single-cell RNA and chromatin accessibility studies (62). Indeed, Andueza et al. show EC responses to disturbed flow are highly plastic, with evidence that disturbed laminar flow leads to a transition to pro-inflammatory cells, endothelial progenitor cells, EndMT, and even a novel immune-like cell (EndICLT) (62). While lineage tracing studies will be needed to confirm the origin of EndICLT cells, prior lineage tracing studies have shown that EndMT occurs during atherosclerotic plaque development in mice (63). Moreover, analysis of human tissue has shown that EndMT correlates with an unstable plaque phenotype, which is likely driven by altered collagen and matrix metalloproteinase expression causing decreased cap thickness (63). However, EndMT also contributes to formation of the fibrous cap and may therefore have protective, as well as detrimental roles (64). As TGF-β is a key driver of EndMT, there is work showing that limiting endothelial responses to TGF-β (i.e., by endothelial-specific Tgfbr1/2 knock out in mouse models of atherosclerosis) limits EndMT, decreases inflammation and plaque progression, and even facilitates plaque regression (65). However, TGF-β is also a potent anti-inflammatory cytokine, and a more nuanced therapeutic target will therefore be required (66). What is clear, however, is that endothelial activation and cellular plasticity are drivers of atherosclerotic disease. Whether this cell identity shift is truly “fluid” and reversible is unknown – it is certainly conceivable, given the observation that mesenchymal to epithelial transition occurs in development and reprogramming (67). Regardless, given ongoing cell-cell communication within the atherosclerotic plaque environment, ECs transitioning to other functional cell phenotypes will undoubtedly send and receive different messages to/from their neighboring cells. As seen in in vitro models, ECs induced to undergo EndMT produce a different EV cargo that affect the metabolism and angiogenic potential of naïve recipient ECs (68). We must now look at more complex tissue to understand how altered endothelial signals can be harnessed to detect disease earlier, intervene, and/or identify targets for plaque stabilization or regression. To that end, Depuydt et al. performed single cell transcriptomics and chromatin accessibility on human carotid atherosclerotic plaques and not only identified 4 distinct endothelial populations, but also utilized ligand-receptor interactions between cell types to predict endothelial communication with myeloid and smooth muscle populations (53). Together, these studies make the case for continued work to focus on EC plasticity and consequent EC-myeloid and EC-smooth muscle cell communication in atherosclerotic disease. Moreover, there is emerging evidence that EVs act on ECs to govern cell fate/plasticity – this will undoubtedly affect the communication loop between the endothelium in both a local and systemic manner (68, 69).
Directionality of Endothelial Communication – a Provocative Idea With Implications for Cell-Cell Crosstalk?
Cellular polarity is common across tissues, particularly for cells that serve as boundary layers. In the vasculature, endothelial cells have apical-basal polarity to establish a barrier between the circulation (i.e., blood) and tissues (70). Key functions are performed by each surface, with polarized expression of anticoagulant factors, receptors, ion channels, mechanosensors, and junctional proteins conferring regional properties (71). Importantly, uniform laminar and disturbed laminar blood flow have distinct spatio-temporal shear stress profiles and impart unique frictional forces that are sensed by ECs through multiple mechanosensors, including Peizo1 (72, 73). Endothelial mechanosensing contributes to the regulation of many aspects of EC biology, including shape, orientation, and polarity. Recent work has shown that PAR3 expression by ECs governs endothelial polarity relative to the direction of blood flow but not apical-basal polarity (74). Conversely, Scrib is a polarity protein that helps to maintain apical-basolateral polarity, endothelial quiescence, and confers atheroprotection (75). Notably, the endothelial secretome is also polarized, with proteomic analysis showing that extracellular matrix components are predominantly secreted basally while the apical proteome contains a significant proportion of EV proteins (76). Further, ECs respond to inflammatory stimuli (TNF-α) differentially depending on whether the apical vs. basolateral surface is exposed (77). Endothelial-derived EVs contribute to the circulating secretome and carry cargo capable of governing endothelial function in a paracrine and perhaps endocrine manner, as well as other cells: truly, EC-EVs have been referred to as “keepers of health” as well as “messengers of disease” (50). As ECs are situated at the interface of the blood vessel lumen and wall, it is conceivable they selectively load and directionally release EVs containing different cargo depending on the stimulus (Figure 2). Other cells in similar physiological environments, such as retinal pigmented epithelium and intestinal epithelial cells, demonstrate polarized release of exosomes containing differential protein content (80, 81). Support for the concept that ECs communicate with directional particle release is further supported by Yun et al. who showed human brain microvascular endothelial cells release microparticles (microvesicles) constitutively and in response to pro-inflammatory cytokines and critically, that protein content and functional effects (permeability) were different between apical and basolateral sources (82). Given the unique location of ECs, communication from activated cells might reflect critical loco-regional information and this information may be shared through distinct messages via apical and basolateral EV release – providing unique candidates for diagnostic and therapeutic targets to identify at-risk patients and intervene at different stages of atherosclerotic plaque development.
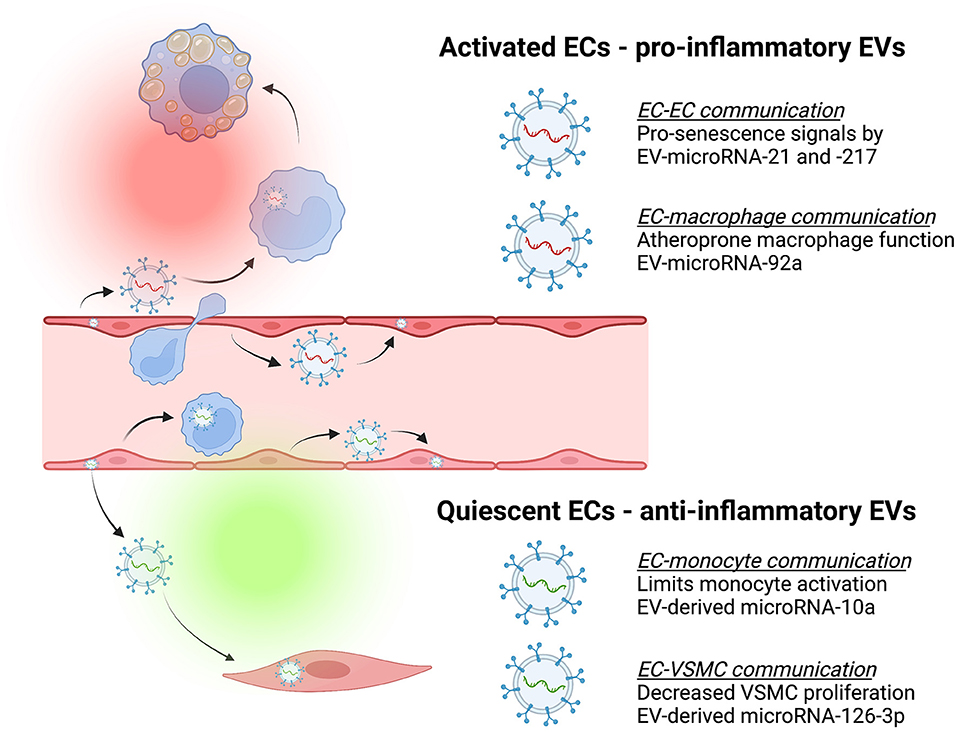
Figure 2. Endothelial-derived extracellular vesicles (EVs) contribute to cell-cell communication that can be atheroprotective (green) or atheroprone (red). Endothelial cells (ECs) release EVs containing different cargo depending on whether they are activated or quiescent, and these have effects on recipient cells [e.g., monocytes, macrophages, and vascular smooth muscle cells (VSMC)]. For example, activated ECs release EVs that deliver pro-senescence signals such as microRNA-21 and microRNA-217 to other ECs (78) or separately, release EVs containing microRNA-92a capable of inducing macrophage inflammation and impaired migration (48). In contrast, quiescent ECs release EVs containing cargo such as microRNA-10a that limit monocyte activation (45) or EVs containing microRNA-126-3p to decrease VSMC proliferation and neointima formation (79).
The Endothelium as a Therapeutic Target in Atherosclerosis: can we Exploit EV-Based Cell-Cell Crosstalk?
It is clear the endothelium actively governs cardiovascular health. From pro-homeostatic signals such as nitric oxide release (1, 15), to atheroprotective EVs (45, 47), to molecules that facilitate resolution of inflammation (83–85), there are several means by which endothelial cross-talk is beneficial. As the endothelium becomes activated with transition to distinct phenotypes, cellular communication is altered, such that these ECs now have the capacity to mediate atherosclerotic disease through cell-cell crosstalk. Strategies should therefore be designed to preserve or “boost” homeostatic roles of the endothelium or alternatively, target dysregulated endothelium to modulate atherosclerotic plaque development. The concept that EVs participate in intercellular communication is firmly established and multiple groups are exploring their diagnostic and therapeutic potential. As these lipid bilayers are ubiquitously released throughout the body, protect their molecular cargo from degradation, carry surface markers identifying the parent cell, and can be employed as delivery systems, they have tremendous potential in multiple disease applications (86, 87). If we consider the profound significance of the endothelium located at the interface of the vessel wall where it participates in systemic (i.e., blood) and local (i.e., vessel wall) communication, it may be that modifying endothelial function is a viable target in cardiovascular disease. Here, we consider a few novel endothelial-specific approaches in the context of EV communication and atherosclerosis: (1) designing treatments that preserve endothelial quiescence and limit senescence; (2) interrupting EC communication promoting early atherosclerotic plaques; and (3), selectively targeting regions of activated endothelium to deliver plaque-stabilizing/regression therapies to the atherosclerotic plaque lying below.
Preserving Endothelial Quiescence Through EV-Mediated Delivery of Bioactive Molecules
Identifying EV components that govern endothelial health may be difficult as few studies have focused on this process. However, we can look to recent work on the protective effects of a healthy lifestyle on atherosclerotic disease, such as nutrition, sleep, stress reduction and exercise, to provide some clues on where to start (88). To date, a prototypic functional measure of endothelial health is flow-mediated vasodilatation (FMD). Human studies exploring Mediterranean diets have demonstrated the benefits on EC function when assessed by FMD (89). More recently, the benefits of carbohydrate restriction improved FMD, markers of endothelial activation, and decreased endothelial microparticles in subjects with type 2 diabetes (90). Sleep is also critical for health. Key research by the Swirski laboratory has emerged showcasing the importance of sleep quality in the control of hematopoiesis and atheroprotection (91). Endothelial health is specifically affected by altered circadian rhythms and sleep deprivation, with reduced FMD observed by several investigators (92, 93). Notably, aerobic exercise is considered a particularly effective counterstrategy through its ability to limit endothelial inflammation and bolster nitric oxide production (94). Clearly, exercise affects multiple cellular populations, with cardiovascular health depending upon orchestrated crosstalk between them (95). A comprehensive evaluation by Brahmer et al. has demonstrated leukocyte, platelet, and endothelial-derived EVs increase in response to exercise and represent a complex communication network that could be exploited to preserve endothelial health (96). This concept of “exerkines” has been recently established in models of cardioprotection, which showed that both exercise training or laminar shear stress directly enhanced EV-microRNA-342-5p in ECs to provide cardiomyocyte protection (97). Whether or not there is a similar beneficial autocrine effect on ECs is unknown (97). As we seek to understand how cardiovascular fitness can be preserved through maintenance of endothelial health and potentially EV-based therapeutics, it would be prudent to discern whether novel mediators act on ECs alongside other cellular targets. In this light, some of the most promising work might evolve from studies determining EV cargo produced by senescent ECs – key insights precisely because of the paracrine induction of senescence on younger cells through a “bystander effect” from aged cells (98). Reversing or preventing effectors of senescence would thereby limit “spread” and preserve endothelial health. Indeed, recent work by Mensa et al. determined that senescent endothelial EVs spread pro-senescence signals through microRNA-21 and−217, affecting DNA methylation and cell replication, and these have an age-specific human biomarker correlate when studied across subjects 40–100 years of age (78). The therapeutic potential of using EVs to program target cells with anti-senescence or rejuvenation strategies with a range of natural EV sources (e.g., young donor-derived) and artificial (e.g., nanomedicines) is an area of active exploration and promise (99). Taken together, there are multiple early insights that can be further explored from work being done on circadian rhythms, exercise, nutrition and aging to target endothelial health and prevent atherosclerosis.
Interrupting Early Atherosclerosis
An additional target to limit atherosclerotic disease would be honing endothelial-directed strategies to interrupt early plaque development. The accumulation of LDL in the subendothelial space would be a reasonable target. Antagonism of endothelial SR-B1 (25–27) or downstream regulators of LDL transcytosis and diverting LDL transcytosis to endolysosomal degradation (100) could be promising approaches, but will require teasing apart the pathways used by SR-B1 to effect the anti-atherogenic process of reverse cholesterol transport (6). Alternatively, immunotargeting activated ECs provides a strategy for delivering therapies to atheroprone regions. While not new (101), this concept has renewed potential in the era of EVs and nanomedicines. Recent work by Distasio and colleagues demonstrated VCAM-1-targeted gene delivery of nanoparticles (NPs) containing the anti-inflammatory cytokine interleukin-10, localized to inflamed ECs and atherosclerotic plaques (102). The Fang laboratory and others have done elegant work over the past decade to not only identify that disturbed flow-induced microRNA-92a causes endothelial dysfunction (103, 104), but also recently reported VCAM-1 targeted NPs can preferentially deliver microRNA-92a inhibitors to inflamed ECs and reduce aortic atherosclerotic plaque development in a murine model (105). Considering endogenous sources, there is certainly promise that endothelial-derived EVs play roles in the progression of atherosclerosis (106); by focusing on those that are considered beneficial such as microRNA-10a (45) or microRNA-126 (79, 107) and what drives their expression, we may likewise find strategies to interrupt early atherogenesis. Early work from the Fish laboratory revealed that microRNA-146 repressed endothelial activation (108), with later in vivo work showing its importance in restraining EC activation and atherosclerosis (109). As highlighted, targeting the activated endothelium with site-directed approaches to limit atherogenesis has some clear promise – at least in pre-clinical models. Selectively targeting advanced atherosclerotic plaques will have different, but perhaps more clinically relevant, objectives; namely, to stabilize and protect from rupture.
Selective Targeting Over Advanced Plaque
The advanced atherosclerotic plaque has unique biology with distinct therapeutic targets that are emerging. One heavily studied target is microRNA-33 (110, 111). Studies using non-human primates have demonstrated that pharmacological inhibition of microRNA-33 can favorably improve cholesterol profiles, with the potential for reducing cardiovascular disease (112). Further insights from the Moore laboratory have shown that microRNA-33 silencing reprograms the immune cell landscape in atherosclerotic plaques to promote regression (113). Intriguingly, ECs might serve as a “trojan horse” for anti-microRNA-33 therapy: using adenoviral delivery, ECs can be steered to release exosomes containing anti-microRNA-33a-5p that transfer their contents to macrophages and vascular smooth muscle cells to enhance cholesterol efflux (114). Beyond the microRNA-33 family, additional novel targets exist within resolution biology pathways. Defective efferocytosis (clearing of the dead cells) has been associated with advanced atherosclerotic plaques and is considered the linchpin in plaque vulnerability (115). EVs from cardiac progenitor-derived cells enhance macrophage efferocytosis through microRNA-26 to sustain expression of a key efferocytosis receptor (MerTK) (116). NPs containing small interfering RNA targeting a macrophage molecule (CaMKIIy) can improve efferocytosis, decrease necrotic core area, and increase fibrous cap thickness – signs of atherosclerotic plaque stability – in a preclinical model of atherosclerosis (117). Certainly, macrophage-targeted nanomedicine holds unique promise (118). Work from the Leeper laboratory has demonstrated that anti-CD47 (pro-efferocytic therapy), delivered systemically (119, 120) or in a macrophage-specific nanotube therapy (121), holds promise in animal models of atherosclerosis, while human data has suggested anti-CD47 therapy reduces carotid artery inflammation (122). Whether the endothelium can or should be exploited for these delivery methods remains to be determined. Within the sphere of resolution biology, pro-resolving lipid mediators such as the resolvins and maresin, have also shown promise (85, 123, 124). While several papers have focused on the effect these mediators have on macrophage function (125–127), it is notable that endothelial cells release resolvin-D1 as a pro-resolution response to a range of low-density lipoproteins (84). Given the ongoing intercellular communication regulated by the endothelium over the lifetime of an atherosclerotic plaque, it behooves us to determine how ECs govern efferocytosis and resolution of inflammation. Lastly, understanding how the endothelium governs plaque stabilizing features will likewise provide new therapeutic targets. Stable plaque phenotypes can be induced through endothelial-specific targets, with EC-deletion of the P2Y2 receptor (regulates VCAM-1 expression and vascular inflammation) (128) and EC-deletion of CD40 (leukocyte adhesion) (129). MicroRNA have also been implicated, with microRNA-210 and−21 both showing the potential to stabilize fibrous caps in advanced atherosclerotic plaques (130, 131). Lastly, sophisticated work from the Owens laboratory has shown that endothelial cells can undergo EndMT and contribute to atherosclerotic fibrous cap stability as a means to compensate, at least for a time, the role usually played by smooth muscle cells (64). Whether we can use ECs as the “trojan horse” to drive any of these processes through specific EV cargo requires exploration. Importantly, understanding the nature of distinct endothelial crosstalk with cells in both the luminal and abluminal space may yield additional critical insights and new targets.
Discussion: Challenging Dogma for Novel Insights
As discussed in the preceding paragraphs, ECs are dynamic, they anchor polarized communication hubs (e.g., “hot spots” for leukocyte transmigration) and have the capacity to shift identity and transform their regional ultrastructure. In the context of atherosclerotic plaque progression, where the microenvironment is continuously evolving, the endothelium likewise can provide dynamic responses both locally and systemically. One challenge will be to improve upon insights from single-cell transcriptomics to distinguish between the endothelium within the vessel lumen vs. that within the adventitia or vasa vasorum (58, 132). For this, we will need emerging techniques such as microdissection, spatial transcriptomics (133) and imaging mass spectrometry (134) to provide loco-regional information on endothelial phenotype and function in atherosclerosis. Being able to target the endothelium in a site-specific manner will be a critical advance for any therapeutic target. In this way, NPs that can target ECs for robust genome editing holds promise (135). Ongoing work will be required to better understand the mechanisms of EC-enriched NP or EV uptake, followed by evaluation in large animal models. In this light, it is encouraging that endothelial small interfering RNA delivery in non-human primates can be performed using NPs that target multiple organs (136). EV- or NP-based approaches will ultimately need to target regions of activated endothelium, and in this way, should capitalize on VCAM-1 expression for therapies or molecular imaging (105, 137–139). More recent work on the endothelial transcriptome suggests that epigenetic targets exist for anti-atherogenic therapy such as EZH2 antagonism and SIRT1 agonism – either through epigenetic drugs or epigenetic editing [reviewed in (140)]. If endothelial communication is distinct between the luminal (i.e., circulation) and abluminal (i.e., vascular wall) environments, then it is conceivable that therapies will need to consider how to exploit directional communication. Moreover, with respect to EVs, it will be imperative to improve our rudimentary understanding of EV biogenesis, cargo loading, cellular uptake, and processing (3, 4, 141), done in accordance with standards set by the research field (e.g., MISEV2018 guidelines) (142). Reductionist approaches used to study endothelial function have historically collected supernatants from plate-grown cells. As we look to better understand EC communication in distinct environments, it will be important to determine whether and how mediators are released in a polarized fashion. More sophisticated approaches will need to incorporate other cell types, extracellular matrix proteins, and flow. In this way, we may need to move to organ-on-a-chip methods (143) to better elucidate the ideal endothelial communication strategy we wish to either boost or interrupt. Likewise, more complex animal models will be required to track endothelial communication and target endothelial gene editing accordingly (135, 144). While these suggestions add inherent complexity to our research paradigms, they will serve useful as we strive to identify circulating biomarkers of endothelial health and disease or novel targets that directly affect the endothelium and/or can be conveyed across this strategic layer to target cells below.
Author Contributions
KH conceived the idea, synthesized the literature, and drafted the review article. MC and JF provided conceptual insight and manuscript editing. All authors have read and approved the published version of the manuscript.
Funding
KH was supported by a Canadian Institutes of Health Research (CIHR) Project Grant (PJT178006) and the Wylie Scholar Award (Vascular Cures), Blair Early Career Professorship in Vascular Surgery (University of Toronto), Peter Munk Cardiac Centre, and University Health Network. MC holds a Tier I Canada Research Chair and a CIHR Foundation Grant (FDN-154299). JF was supported by project grants from the CIHR (PJT148487 and PJT173489). MC and JF are supported by a team grant from the Medicine by Design program, which received funding from the Canada First Research Excellence Fund. JF received infrastructure funding from the John R. Evans Leaders Fund and the Ontario Research Fund (Canada Foundation for Innovation) and is the recipient of a Tier II Canada Research Chair from CIHR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
BioRender.com was used in the creation of the figures for this review, with final illustration of Figure 1 completed by BioHues Digital©.
Abbreviations
CD, cluster of differentiation; EC, endothelial cell; NP, nanoparticle; EndMT, endothelial-to-mesenchymal transition; EV, extracellular vesicle; EZH2, enhancer of zeste homolog 2; SIRT1, sirtuin 1; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; PAR3, partitioning defective 3; KLF2, Kruppel-like factor 2; LDL, low-density lipoprotein; TAZ, tafazzin; VCAM-1, vascular cell adhesion molecule 1; VE-cadherin, vascular endothelial-cadherin; YAP, yes-associated protein; MerTK, MER proto-oncogene tyrosine kinase; CaMKIIy, Ca2+/calmodulin-dependent protein kinase II; SR-B1, scavenger receptor class B type 1; FMD, flow-mediated dilatation; TGF, transforming growth factor; TNF, tumor necrosis factor; LDL, low density lipoprotein; EndICLT, endothelial-to-immune cell like transition; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase; HIF-1, hypoxia-inducible factor.
References
1. Gimbrone MA, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
2. Kruger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20184411
3. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
4. Mathieu M, Martin-Jaular L, Lavieu G, Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. (2019) 21:9–17. doi: 10.1038/s41556-018-0250-9
5. Potje SR, Paula TD, Paulo M, Bendhack LM. The role of glycocalyx and caveolae in vascular homeostasis and diseases. Front Physiol. (2020) 11:620840. doi: 10.3389/fphys.2020.620840
6. Zhang X, Sessa WC, Fernandez-Hernando C. Endothelial transcytosis of lipoproteins in atherosclerosis. Front Cardiovasc Med. (2018) 5:130. doi: 10.3389/fcvm.2018.00130
7. Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, et al. Endothelial basement membrane laminin 511 contributes to endothelial junctional tightness and thereby inhibits leukocyte transmigration. Cell Rep. (2017) 18:1256–69. doi: 10.1016/j.celrep.2016.12.092
8. Ricard N, Bailly S, Guignabert C, Simons M. The quiescent endothelium: signalling pathways regulating organ-specific endothelial normalcy. Nat Rev Cardiol. (2021) 18:565–80. doi: 10.1038/s41569-021-00517-4
9. Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. (2020) 180:764–79 e20. doi: 10.1016/j.cell.2020.01.015
10. Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun. (2017) 8:14361. doi: 10.1038/ncomms14361
11. Mack JJ, Iruela-Arispe ML. NOTCH regulation of the endothelial cell phenotype. Curr Opin Hematol. (2018) 25:212–8. doi: 10.1097/MOH.0000000000000425
12. Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. (2001) 104:448–54. doi: 10.1161/hc2901.091399
13. Ricard N, Scott RP, Booth CJ, Velazquez H, Cilfone NA, Baylon JL, et al. Endothelial ERK1/2 signaling maintains integrity of the quiescent endothelium. J Exp Med. (2019) 216:1874–90. doi: 10.1084/jem.20182151
14. Quintar A, McArdle S, Wolf D, Marki A, Ehinger E, Vassallo M, et al. Endothelial protective monocyte patrolling in large arteries intensified by western diet and atherosclerosis. Circ Res. (2017) 120:1789–99. doi: 10.1161/CIRCRESAHA.117.310739
15. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. (2015) 116:1231–44. doi: 10.1161/CIRCRESAHA.116.302855
16. Wong BW, Marsch E, Treps L, Baes M, Carmeliet P. Endothelial cell metabolism in health and disease: impact of hypoxia. EMBO J. (2017) 36:2187–203. doi: 10.15252/embj.201696150
17. Souilhol C, Serbanovic-Canic J, Fragiadaki M, Chico TJ, Ridger V, Roddie H, et al. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat Rev Cardiol. (2020) 17:52–63. doi: 10.1038/s41569-019-0239-5
18. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012) 111:245–59. doi: 10.1161/CIRCRESAHA.111.261388
19. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
20. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Nat Acad Sci. (2000) 97:9052–7. doi: 10.1073/pnas.97.16.9052
21. Hunt BJ, Jurd KM. Endothelial cell activation. A central pathophysiological process. BMJ. (1998) 316:1328–9. doi: 10.1136/bmj.316.7141.1328
22. Botts SR, Fish JE, Howe KL. Dysfunctional vascular endothelium as a driver of atherosclerosis: emerging insights into pathogenesis and treatment. Front Pharmacol. (2021) 12:787541. doi: 10.3389/fphar.2021.787541
23. Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
24. Stocker R, Keaney JF. Role of oxidative modifications in atherosclerosis. Physiol Rev. (2004) 84:1381–478. doi: 10.1152/physrev.00047.2003
25. Armstrong SM, Sugiyama MG, Fung KY, Gao Y, Wang C, Levy AS, et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res. (2015) 108:268–77. doi: 10.1093/cvr/cvv218
26. Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. (2019) 569:565–9. doi: 10.1038/s41586-019-1140-4
27. Ghaffari S, Jang E, Naderinabi F, Sanwal R, Khosraviani N, Wang C, et al. Endothelial HMGB1 is a critical regulator of LDL transcytosis via an SREBP2-SR-BI axis. Arterioscler Thromb Vasc Biol. (2021) 41:200–16. doi: 10.1161/ATVBAHA.120.314557
28. Kraehling JR, Chidlow JH, Rajagopal C, Sugiyama MG, Fowler JW, Lee MY, et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat Commun. (2016) 7:13516. doi: 10.1038/ncomms13516
29. Luscinskas FW, Cybulsky MI, Kiely JM, Peckins CS, Davis VM, Gimbrone MA. Cytokine-activated human endothelial monolayers support enhanced neutrophil transmigration via a mechanism involving both endothelial-leukocyte adhesion molecule-1 and intercellular adhesion molecule-1. J Immunol. (1991) 146:1617–25.
30. Cybulsky MI, Gimbrone MA. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. (1991) 251:788–91. doi: 10.1126/science.1990440
31. Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. (1992) 140:301–16.
32. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. (2013) 19:1166–72. doi: 10.1038/nm.3258
33. Arts JJ, Mahlandt EK, Gronloh ML, Schimmel L, Noordstra I, Gordon E, et al. Endothelial junctional membrane protrusions serve as hotspots for neutrophil transmigration. Elife. (2021) 10. doi: 10.7554/eLife.66074
34. Gronloh MLB, Arts JJG, van Buul JD. Neutrophil transendothelial migration hotspots - mechanisms and implications. J Cell Sci. (2021) 134. doi: 10.1242/jcs.255653
35. Muller WA. Transendothelial migration: unifying principles from the endothelial perspective. Immunol Rev. (2016) 273:61–75. doi: 10.1111/imr.12443
36. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. (2008) 18:228–32. doi: 10.1016/j.tcm.2008.11.004
37. Sinha SK, Miikeda A, Fouladian Z, Mehrabian M, Edillor C, Shih D, et al. Local M-CSF (Macrophage Colony-Stimulating Factor) expression regulates macrophage proliferation and apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. (2021) 41:220–33. doi: 10.1161/ATVBAHA.120.315255
38. Jansen F, Li Q, Pfeifer A, Werner N. Endothelial- and immune cell-derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl Sci. (2017) 2:790–807. doi: 10.1016/j.jacbts.2017.08.004
39. Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. (2011) 469:336–42. doi: 10.1038/nature09783
40. Laffont B, Rayner KJ. MicroRNAs in the pathobiology and therapy of atherosclerosis. Can J Cardiol. (2017) 33:313–24. doi: 10.1016/j.cjca.2017.01.001
41. Feinberg MW, Moore KJ. MicroRNA Regulation of atherosclerosis. Circ Res. (2016) 118:703–20. doi: 10.1161/CIRCRESAHA.115.306300
42. Raju S, Fish JE, Howe KL. MicroRNAs as sentinels and protagonists of carotid artery thromboembolism. Clin Sci. (2020) 134:169–92. doi: 10.1042/CS20190651
43. Akbar N, Braithwaite AT, Corr EM, Koelwyn GJ, van Solingen C, Cochain C, et al. Rapid neutrophil mobilisation by VCAM-1+ endothelial extracellular vesicles. Cardiovasc Res. (2022). doi: 10.1093/cvr/cvac012 [Epub ahead of print].
44. Hosseinkhani B, Kuypers S, van den Akker NMS, Molin DGM, Michiels L. Extracellular vesicles work as a functional inflammatory mediator between vascular endothelial cells and immune cells. Front Immunol. (2018) 9:1789. doi: 10.3389/fimmu.2018.01789
45. Njock MS, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, et al. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood. (2015) 125:3202–12. doi: 10.1182/blood-2014-11-611046
46. Das K, Keshava S, Pendurthi UR, Rao LVM. Factor VIIa suppresses inflammation and barrier disruption through the release of EEVs and transfer of microRNA 10a. Blood. (2022) 139:118–33. doi: 10.1182/blood.2021012358
47. Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56. doi: 10.1038/ncb2441
48. Chang YJ Li YS, Wu CC, Wang KC, Huang TC, Chen Z, et al. Extracellular MicroRNA-92a mediates endothelial cell-macrophage communication. Arterioscler Thromb Vasc Biol. (2019) 39:2492–504. doi: 10.1161/ATVBAHA.119.312707
49. Boyer MJ, Kimura Y, Akiyama T, Baggett AY, Preston KJ, Scalia R, et al. Endothelial cell-derived extracellular vesicles alter vascular smooth muscle cell phenotype through high-mobility group box proteins. J Extracell Vesicles. (2020) 9:1781427. doi: 10.1080/20013078.2020.1781427
50. Mathiesen A, Hamilton T, Carter N, Brown M, McPheat W, Dobrian A. Endothelial extracellular vesicles: from keepers of health to messengers of disease. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22094640
51. Verweij FJ, Revenu C, Arras G, Dingli F, Loew D, Pegtel DM, et al. Live tracking of inter-organ communication by endogenous exosomes in vivo. Dev Cell. (2019) 48:573–89 e4. doi: 10.1016/j.devcel.2019.01.004
52. Verweij FJ, Balaj L, Boulanger CM, Carter DRF, Compeer EB, D'Angelo G, et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat Methods. (2021) 18:1013–26. doi: 10.1038/s41592-021-01206-3
53. Depuydt MA, Prange KH, Slenders L, Ord T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1016/j.atherosclerosis.2019.06.013
54. Williams JW, Winkels H, Durant CP, Zaitsev K, Ghosheh Y, Ley K. Single cell RNA sequencing in atherosclerosis research. Circ Res. (2020) 126:1112–26. doi: 10.1161/CIRCRESAHA.119.315940
55. Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.124574
56. Sharma M, Schlegel M, Brown EJ, Sansbury BE, Weinstock A, Afonso MS, et al. Netrin-1 alters adipose tissue macrophage fate and function in obesity. Immunometabolism. (2019) 1:e190010. doi: 10.20900/immunometab20190010
57. Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT, et al. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation. (2020) 142:2045–59. doi: 10.1161/CIRCULATIONAHA.120.046672
58. Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. (2020) 142:2060–75. doi: 10.1161/CIRCULATIONAHA.120.048378
59. Nicin L, Schroeter SM, Glaser SF, Schulze-Bruning R, Pham M-D, Hille SS, et al. A human cell atlas of the pressure-induced hypertrophic heart. Nat Cardiovasc Res. (2022) 1:174–85. doi: 10.1038/s44161-022-00019-7
60. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
61. Kluza E, Beldman TJ, Shami A, Scholl ER, Malinova TS, Grootemaat AE, et al. Diverse ultrastructural landscape of atherosclerotic endothelium. Atherosclerosis. (2021) 339:35–45. doi: 10.1016/j.atherosclerosis.2021.11.017
62. Andueza A, Kumar S, Kim J, Kang DW, Mumme HL, Perez JI, et al. Endothelial reprogramming by disturbed flow revealed by single-cell RNA and chromatin accessibility study. Cell Rep. (2020) 33:108491. doi: 10.1016/j.celrep.2020.108491
63. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2016) 7:11853. doi: 10.1038/ncomms11853
64. Newman AAC, Serbulea V, Baylis RA, Shankman LS, Bradley X, Alencar GF, et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRbeta and bioenergetic mechanisms. Nat Metab. (2021) 3:166–81. doi: 10.1038/s42255-020-00338-8
65. Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat Metab. (2019) 1:912–26. doi: 10.1038/s42255-019-0102-3
66. Howe KL, Fish JE. Transforming endothelial cells in atherosclerosis. Nat Metab. (2019) 1:856–7. doi: 10.1038/s42255-019-0100-5
67. Pei D, Shu X, Gassama-Diagne A, Thiery JP. Mesenchymal-epithelial transition in development and reprogramming. Nat Cell Biol. (2019) 21:44–53. doi: 10.1038/s41556-018-0195-z
68. Haynes BA, Yang LF, Huyck RW, Lehrer EJ, Turner JM, Barabutis N, et al. Endothelial-to-mesenchymal transition in human adipose tissue vasculature alters the particulate secretome and induces endothelial dysfunction. Arterioscler Thromb Vasc Biol. (2019) 39:2168–91. doi: 10.1161/ATVBAHA.119.312826
69. Kuriyama N, Yoshioka Y, Kikuchi S, Azuma N, Ochiya T. Extracellular vesicles are key regulators of tumor neovasculature. Front Cell Dev Biol. (2020) 8:611039. doi: 10.3389/fcell.2020.611039
70. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. (2011) 146:873–87. doi: 10.1016/j.cell.2011.08.039
71. Wolpe AG, Ruddiman CA, Hall PJ, Isakson BE. Polarized proteins in endothelium and their contribution to function. J Vasc Res. (2021) 58:65–91. doi: 10.1159/000512618
72. Beech DJ, Kalli AC. Force sensing by piezo channels in cardiovascular health and disease. Arterioscler Thromb Vasc Biol. (2019) 39:2228–39. doi: 10.1161/ATVBAHA.119.313348
73. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. (2010) 330:55–60. doi: 10.1126/science.1193270
74. Hikita T, Mirzapourshafiyi F, Barbacena P, Riddell M, Pasha A, Li M, et al. PAR-3 controls endothelial planar polarity and vascular inflammation under laminar flow. EMBO Rep. (2018) 19. doi: 10.15252/embr.201745253
75. Schurmann C, Dienst FL, Palfi K, Vasconez AE, Oo JA, Wang S, et al. The polarity protein Scrib limits atherosclerosis development in mice. Cardiovasc Res. (2019) 115:1963–74. doi: 10.1093/cvr/cvz093
76. Wei H, Sundararaman A, Dickson E, Rennie-Campbell L, Cross E, Heesom KJ, et al. Characterization of the polarized endothelial secretome. FASEB J. (2019) 33:12277–87. doi: 10.1096/fj.201900262R
77. Salminen AT, Tithof J, Izhiman Y, Masters EA, McCloskey MC, Gaborski TR, et al. Endothelial cell apicobasal polarity coordinates distinct responses to luminally versus abluminally delivered TNF-alpha in a microvascular mimetic. Integr Biol (Camb). (2020) 12:275–89. doi: 10.1093/intbio/zyaa022
78. Mensa E, Guescini M, Giuliani A, Bacalini MG, Ramini D, Corleone G, et al. Small extracellular vesicles deliver miR-21 and miR-217 as pro-senescence effectors to endothelial cells. J Extracell Vesicles. (2020) 9:1725285. doi: 10.1080/20013078.2020.1725285
79. Jansen F, Stumpf T, Proebsting S, Franklin BS, Wenzel D, Pfeifer P, et al. Intercellular transfer of miR-126-3p by endothelial microparticles reduces vascular smooth muscle cell proliferation and limits neointima formation by inhibiting LRP6. J Mol Cell Cardiol. (2017) 104:43–52. doi: 10.1016/j.yjmcc.2016.12.005
80. Klingeborn M, Dismuke WM, Skiba NP, Kelly U, Stamer WD, Bowes Rickman C. Directional Exosome Proteomes Reflect Polarity-Specific Functions in Retinal Pigmented Epithelium Monolayers. Sci Rep. (2017) 7:4901. doi: 10.1038/s41598-017-05102-9
81. van Niel G, Raposo G, Candalh C, Boussac M, Hershberg R, Cerf-Bensussan N, et al. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology. (2001) 121:337–49. doi: 10.1053/gast.2001.26263
82. Yun JW, Barzegar M, Boyer CJ, Minagar A, Couraud PO, Alexander JS. Brain endothelial cells release apical and basolateral microparticles in response to inflammatory cytokine stimulation: relevance to neuroinflammatory stress? Front Immunol. (2019) 10:1455. doi: 10.3389/fimmu.2019.01455
83. Xu S, Ilyas I, Little PJ Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. (2021) 73:924–67. doi: 10.1124/pharmrev.120.000096
84. Dufour D, Khalil A, Nuyens V, Rousseau A, Delporte C, Noyon C, et al. Native and myeloperoxidase-oxidized low-density lipoproteins act in synergy to induce release of resolvin-D1 from endothelial cells. Atherosclerosis. (2018) 272:108–17. doi: 10.1016/j.atherosclerosis.2018.03.012
85. Fredman G, Tabas I. Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol. (2017) 187:1211–21. doi: 10.1016/j.ajpath.2017.01.018
86. Sluijter JPG, Davidson SM, Boulanger CM, Buzas EI, de Kleijn DPV, Engel FB, et al. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. (2018) 114:19–34. doi: 10.1093/cvr/cvx211
87. Wiklander OPB, Brennan MA, Lotvall J, Breakefield XO, El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aav8521
88. Schloss MJ, Swirski FK, Nahrendorf M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ Res. (2020) 126:1242–59. doi: 10.1161/CIRCRESAHA.120.315936
89. Davis CR, Hodgson JM, Woodman R, Bryan J, Wilson C, Murphy KJ, et al. Mediterranean diet lowers blood pressure and improves endothelial function: results from the MedLey randomized intervention trial. Am J Clin Nutr. (2017) 105:1305–13. doi: 10.3945/ajcn.116.146803
90. Francois ME, Myette-Cote E, Bammert TD, Durrer C, Neudorf H, DeSouza CA, et al. Carbohydrate restriction with postmeal walking effectively mitigates postprandial hyperglycemia and improves endothelial function in type 2 diabetes. Am J Physiol Heart Circ Physiol. (2018) 314:H105–H13. doi: 10.1152/ajpheart.00524.2017
91. McAlpine CS, Kiss MG, Rattik S, He S, Vassalli A, Valet C, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. (2019) 566:383–7. doi: 10.1038/s41586-019-0948-2
92. Bain AR, Weil BR, Diehl KJ, Greiner JJ, Stauffer BL, DeSouza CA. Insufficient sleep is associated with impaired nitric oxide-mediated endothelium-dependent vasodilation. Atherosclerosis. (2017) 265:41–6. doi: 10.1016/j.atherosclerosis.2017.08.001
93. Calvin AD, Covassin N, Kremers WK, Adachi T, Macedo P, Albuquerque FN, et al. Experimental sleep restriction causes endothelial dysfunction in healthy humans. J Am Heart Assoc. (2014) 3:e001143. doi: 10.1161/JAHA.114.001143
94. Cherubini JM, Cheng JL, Williams JS, MacDonald MJ. Sleep deprivation and endothelial function: reconciling seminal evidence with recent perspectives. Am J Physiol Heart Circ Physiol. (2021) 320:H29–35. doi: 10.1152/ajpheart.00607.2020
95. Heinonen I, Kalliokoski KK, Hannukainen JC, Duncker DJ, Nuutila P, Knuuti J. Organ-specific physiological responses to acute physical exercise and long-term training in humans. Physiology (Bethesda). (2014) 29:421–36. doi: 10.1152/physiol.00067.2013
96. Brahmer A, Neuberger E, Esch-Heisser L, Haller N, Jorgensen MM, Baek R, et al. Platelets, endothelial cells and leukocytes contribute to the exercise-triggered release of extracellular vesicles into the circulation. J Extracell Vesicles. (2019) 8:1615820. doi: 10.1080/20013078.2019.1615820
97. Hou Z, Qin X, Hu Y, Zhang X, Li G, Wu J, et al. Longterm exercise-derived exosomal miR-342-5p: a novel exerkine for cardioprotection. Circ Res. (2019) 124:1386–400. doi: 10.1161/CIRCRESAHA.118.314635
98. McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. (2018) 217:65–77. doi: 10.1083/jcb.201708092
99. Yin Y, Chen H, Wang Y, Zhang L, Wang X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J Extracell Vesicles. (2021) 10:e12154. doi: 10.1002/jev2.12154
100. Ikeda J, Scipione CA, Hyduk SJ, Althagafi MG, Atif J, Dick SA, et al. Radiation impacts early atherosclerosis by suppressing intimal LDL accumulation. Circ Res. (2021) 128:530–43. doi: 10.1161/CIRCRESAHA.119.316539
101. Spragg DD, Alford DR, Greferath R, Larsen CE, Lee KD, Gurtner GC, et al. Immunotargeting of liposomes to activated vascular endothelial cells: a strategy for site-selective delivery in the cardiovascular system. Proc Natl Acad Sci U S A. (1997) 94:8795–800. doi: 10.1073/pnas.94.16.8795
102. Distasio N, Salmon H, Dierick F, Ebrahimian T, Tabrizian M, Lehoux S. VCAM-1 targeted gene delivery nanoparticles localize to inflamed endothelial cells and atherosclerotic plaques. Adv Therap. (2021) 4:1–11. doi: 10.1002/adtp.202000196
103. Fang Y, Davies PF. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Biol. (2012) 32:979–87. doi: 10.1161/ATVBAHA.111.244053
104. Chen Z, Wen L, Martin M, Hsu CY, Fang L, Lin FM, et al. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation. (2015) 131:805–14. doi: 10.1161/CIRCULATIONAHA.114.013675
105. Zhou Z, Yeh CF, Mellas M, Oh MJ, Zhu J, Li J, et al. Targeted polyelectrolyte complex micelles treat vascular complications in vivo. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2114842118
106. Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci. (2019) 76:1093–106. doi: 10.1007/s00018-018-2983-9
107. Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. (2013) 128:2026–38. doi: 10.1161/CIRCULATIONAHA.113.001720
108. Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, et al. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med. (2013) 5:1017–34. doi: 10.1002/emmm.201202318
109. Cheng HS, Besla R, Li A, Chen Z, Shikatani EA, Nazari-Jahantigh M, et al. Paradoxical suppression of atherosclerosis in the absence of microRNA-146a. Circ Res. (2017) 121:354–67. doi: 10.1161/CIRCRESAHA.116.310529
110. Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. (2011) 121:2921–31. doi: 10.1172/JCI57275
111. Ouimet M, Ediriweera H, Afonso MS, Ramkhelawon B, Singaravelu R, Liao X, et al. microRNA-33 regulates macrophage autophagy in atherosclerosis. Arterioscler Thromb Vasc Biol. (2017) 37:1058–67. doi: 10.1161/ATVBAHA.116.308916
112. Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. (2011) 478:404–7. doi: 10.1038/nature10486
113. Afonso MS, Sharma M, Schlegel M, van Solingen C, Koelwyn GJ, Shanley LC, et al. miR-33 silencing reprograms the immune cell landscape in atherosclerotic plaques. Circ Res. (2021) 128:1122–38. doi: 10.1161/CIRCRESAHA.120.317914
114. Stamatikos A, Knight E, Vojtech L, Bi L, Wacker BK, Tang C, et al. Exosome-mediated transfer of anti-miR-33a-5p from transduced endothelial cells enhances macrophage and vascular smooth muscle cell cholesterol efflux. Hum Gene Ther. (2020) 31:219–32. doi: 10.1089/hum.2019.245
115. Tabas I. 2016 russell ross memorial lecture in vascular biology: molecular-cellular mechanisms in the progression of atherosclerosis. Arterioscler Thromb Vasc Biol. (2017) 37:183–9. doi: 10.1161/ATVBAHA.116.308036
116. de Couto G, Jaghatspanyan E, DeBerge M, Liu W, Luther K, Wang Y, et al. Mechanism of enhanced MerTK-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler Thromb Vasc Biol. (2019) 39:2082–96. doi: 10.1161/ATVBAHA.119.313115
117. Tao W, Yurdagul A, Kong N, Li W, Wang X, Doran AC, et al. siRNA nanoparticles targeting CaMKIIgamma in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.aay1063
118. Chen W, Schilperoort M, Cao Y, Shi J, Tabas I, Tao W. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat Rev Cardiol. (2021) 19:228–49. doi: 10.1038/s41569-021-00629-x
119. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. (2016) 536:86–90. doi: 10.1038/nature18935
120. Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci U S A. (2020) 117:15818–26. doi: 10.1073/pnas.2006348117
121. Flores AM, Hosseini-Nassab N, Jarr KU, Ye J, Zhu X, Wirka R, et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat Nanotechnol. (2020) 15:154–61. doi: 10.1038/s41565-019-0619-3
122. Jarr KU, Nakamoto R, Doan BH, Kojima Y, Weissman IL, Advani RH, et al. Effect of CD47 blockade on vascular inflammation. N Engl J Med. (2021) 384:382–3. doi: 10.1056/NEJMc2029834
123. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. (2016) 119:1030–8. doi: 10.1161/CIRCRESAHA.116.309492
124. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92–101. doi: 10.1038/nature13479
125. Gerlach BD, Marinello M, Heinz J, Rymut N, Sansbury BE, Riley CO, et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. (2020) 27:525–39. doi: 10.1038/s41418-019-0370-1
126. Hosseini Z, Marinello M, Decker C, Sansbury BE, Sadhu S, Gerlach BD, et al. Resolvin D1 enhances necroptotic cell clearance through promoting macrophage fatty acid oxidation and oxidative phosphorylation. Arterioscler Thromb Vasc Biol. (2021) 41:1062–75. doi: 10.1161/ATVBAHA.120.315758
127. Arnardottir H, Thul S, Pawelzik SC, Karadimou G, Artiach G, Gallina AL, et al. The resolvin D1 receptor GPR32 transduces inflammation resolution and atheroprotection. J Clin Invest. (2021) 131. doi: 10.1172/JCI142883
128. Chen X, Qian S, Hoggatt A, Tang H, Hacker TA, Obukhov AG, et al. Endothelial cell-specific deletion of P2Y2 receptor promotes plaque stability in atherosclerosis-susceptible ApoE-null mice. Arterioscler Thromb Vasc Biol. (2017) 37:75–83. doi: 10.1161/ATVBAHA.116.308561
129. Gissler MC, Scherrer P, Anto-Michel N, Pennig J, Hoppe N, Funer L, et al. Deficiency of endothelial CD40 induces a stable plaque phenotype and limits inflammatory cell recruitment to atherosclerotic lesions in mice. Thromb Haemost. (2021) 121:1530–40. doi: 10.1055/a-1397-1858
130. Eken SM, Jin H, Chernogubova E, Li Y, Simon N, Sun C, et al. MicroRNA-210 enhances fibrous cap stability in advanced atherosclerotic lesions. Circ Res. (2017) 120:633–44. doi: 10.1161/CIRCRESAHA.116.309318
131. Jin H, Li DY, Chernogubova E, Sun C, Busch A, Eken SM, et al. Local delivery of miR-21 stabilizes fibrous caps in vulnerable atherosclerotic lesions. Mol Ther. (2018) 26:1040–55. doi: 10.1016/j.ymthe.2018.01.011
132. Gu W, Ni Z, Tan YQ, Deng J, Zhang SJ, Lv ZC, et al. Adventitial cell atlas of wt (Wild Type) and ApoE (Apolipoprotein E)-deficient mice defined by single-Cell RNA sequencing. Arterioscler Thromb Vasc Biol. (2019) 39:1055–71. doi: 10.1161/ATVBAHA.119.312399
133. Hill CA, Fernandez DM, Giannarelli C. Single cell analyses to understand the immune continuum in atherosclerosis. Atherosclerosis. (2021) 330:85–94. doi: 10.1016/j.atherosclerosis.2021.04.003
134. Ali HR, Jackson HW, Zanotelli VRT, Danenberg E, Fischer JR, Bardwell H, et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat Cancer. (2020) 1:163–75. doi: 10.1038/s43018-020-0026-6
135. Zhang X, Jin H, Huang X, Chaurasiya B, Dong D, Shanley TP, et al. Robust genome editing in adult vascular endothelium by nanoparticle delivery of CRISPR-Cas9 plasmid DNA. Cell Rep. (2022) 38:110196. doi: 10.1016/j.celrep.2021.110196
136. Khan OF, Kowalski PS, Doloff JC, Tsosie JK, Bakthavatchalu V, Winn CB, et al. Endothelial siRNA delivery in nonhuman primates using ionizable low-molecular weight polymeric nanoparticles. Sci Adv. (2018) 4:eaar8409. doi: 10.1126/sciadv.aar8409
137. Qiu P, Xu Y. The construction of multifunctional nanoparticles system for dual-modal imaging and arteriosclerosis targeted therapy. Am J Transl Res. (2021) 13:4026–39.
138. Atukorale PU, Covarrubias G, Bauer L, Karathanasis E. Vascular targeting of nanoparticles for molecular imaging of diseased endothelium. Adv Drug Deliv Rev. (2017) 113:141–56. doi: 10.1016/j.addr.2016.09.006
139. Thayse K, Kindt N, Laurent S, Carlier S. VCAM-1 target in non-invasive imaging for the detection of atherosclerotic plaques. Biology. (2020) 9. doi: 10.3390/biology9110368
140. Fledderus J, Vanchin B, Rots MG, Krenning G. The endothelium as a target for anti-atherogenic therapy: a focus on the epigenetic enzymes EZH2 and SIRT1. J Pers Med. (2021) 11. doi: 10.3390/jpm11020103
141. Margolis L, Sadovsky Y. The biology of extracellular vesicles: the known unknowns. PLoS Biol. (2019) 17:e3000363. doi: 10.1371/journal.pbio.3000363
142. Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013708.2018.1535750
143. Vunjak-Novakovic G, Ronaldson-Bouchard K, Radisic M. Organs-on-a-chip models for biological research. Cell. (2021) 184:4597–611. doi: 10.1016/j.cell.2021.08.005
Keywords: endothelium, crosstalk, extracellular vesicles, atherosclerosis, polarity, directionality, inflammation, microRNA
Citation: Howe KL, Cybulsky M and Fish JE (2022) The Endothelium as a Hub for Cellular Communication in Atherogenesis: Is There Directionality to the Message? Front. Cardiovasc. Med. 9:888390. doi: 10.3389/fcvm.2022.888390
Received: 02 March 2022; Accepted: 21 March 2022;
Published: 15 April 2022.
Edited by:
Elena Aikawa, Harvard Medical School, United StatesReviewed by:
Elena V. Galkina, Eastern Virginia Medical School, United StatesHong Chen, Harvard Medical School, United States
Copyright © 2022 Howe, Cybulsky and Fish. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn L. Howe, a2F0aHJ5bi5ob3dlQHVobi5jYQ==
†ORCID: Kathryn L. Howe orcid.org/0000-0002-6519-2280
Jason E. Fish orcid.org/0000-0003-0640-7277