Abstract
The ANK2 gene encodes for ankyrin-B (ANKB), one of 3 members of the ankyrin family of proteins, whose name is derived from the Greek word for anchor. ANKB was originally identified in the brain (B denotes “brain”) but has become most widely known for its role in cardiomyocytes as a scaffolding protein for ion channels and transporters, as well as an interacting protein for structural and signaling proteins. Certain loss-of-function ANK2 variants are associated with a primarily cardiac-presenting autosomal-dominant condition with incomplete penetrance and variable expressivity characterized by a predisposition to supraventricular and ventricular arrhythmias, arrhythmogenic cardiomyopathy, congenital and adult-onset structural heart disease, and sudden death. Another independent group of ANK2 variants are associated with increased risk for distinct neurological phenotypes, including epilepsy and autism spectrum disorders. The mechanisms underlying ANKB's roles in cells in health and disease are not fully understood; however, several clues from a range of molecular and cell biological studies have emerged. Notably, ANKB exhibits several isoforms that have different cell-type–, tissue–, and developmental stage– expression profiles. Given the conservation within ankyrins across evolution, model organism studies have enabled the discovery of several ankyrin roles that could shed important light on ANKB protein-protein interactions in heart and brain cells related to the regulation of cellular polarity, organization, calcium homeostasis, and glucose and fat metabolism. Along with this accumulation of evidence suggesting a diversity of important ANKB cellular functions, there is an on-going debate on the role of ANKB in disease. We currently have limited understanding of how these cellular functions link to disease risk. To this end, this review will examine evidence for the cellular roles of ANKB and the potential contribution of ANKB functional variants to disease risk and presentation. This contribution will highlight the impact of ANKB dysfunction on cardiac and neuronal cells and the significance of understanding the role of ANKB variants in disease.
Introduction
Loss-of-function variants in the ANK2 gene are associated with a wide range of electrical and structural heart disease. Reported cardiac phenotypes include arrhythmia, corrected QT interval prolongation (sometimes referred to as long QT type 4), and sudden cardiac death. A prolonged QT interval on an electrocardiogram corrected for heart rate (QTc) is a predictor of ventricular arrythmias and sudden cardiac death (1, 2). At least 15 congenital long QT syndromes (LQTS) have been described, associated with genes encoding for ion channels, ion channel modulatory subunits, signaling proteins, and cytoskeleton-associated proteins (3). One of the first identified ANK2 variants, p.E1458G, was associated with prolonged QTc, and this QTc prolongation has since been associated with other ANK2 variants (4–7). Notably, a prolonged QTc is not observed in all patients harboring cardiac phenotype- associated ANK2 variants. In fact, there is minimal evidence of a prolonged QTc in individuals under the age of 25 (7). Additional reported cardiac manifestations include bradycardia, sinus arrhythmia, idiopathic ventricular fibrillation, and catecholaminergic polymorphic ventricular tachycardia (5, 8). Separately, ANK2 is also emerging as a gene of interest in neurological disorders. ANK2 has been identified as a key risk gene for autism spectrum disorders (ASD) (9, 10) and as a candidate gene for epilepsy (11).
The protein produced from the ANK2 gene, ankyrin-B (ANKB), is a large scaffolding protein that has become known as a key regulator of cardiac physiology (4, 12). There are three mammalian ankyrin protein family members, including ANKB, ankyrin-R (ANKR, ANK1 gene), and ankyrin-G (ANKG, ANK3 gene). ANKR is primarily expressed in erythrocytes (13) while ANKB and ANKG are co-expressed in a variety of cell types and tissues (14–16). Ankyrins, including ANKB, are composed of four domains: a membrane binding domain comprised of 24 ANK repeats that interacts with membrane proteins such as ion channels and transporters, a spectrin binding domain responsible for interacting with βII spectrin, a death domain of which the function has not yet been identified but in other proteins is key for signal transduction cascades resulting in apoptosis and inflammation (17), and a C-terminal domain. The death domain and C-terminal domain comprise the regulatory domain which is named due to its ability to directly bind the membrane binding domain and play a role in inhibition (15). As this review is focused on ANKB, the following information is specific to ANKB, except where information about other ankyrin family members provides key insight.
ANK2 has critical roles for cardiac and neuronal physiology as indicated by loss-of-function variants and studies using model organisms. ANKB's structure and different isoforms allow for a diverse array of protein-protein interactions within a variety of different cell types. As such, dysfunction in ANKB can lead to a wide range of cellular impacts. There are different groups of variants associated with different phenotypes; one group of ANK2 variants is primarily associated with a broad cardiac phenotype, another is associated with neurological diseases including ASD and epilepsy, and others are linked to metabolic perturbations. The ANK2 variant-associated clinical phenotypes inform investigation of ANKB cellular roles, including key potential protein-protein interactions and cellular processes that could, in turn, help to develop new therapeutic strategies. To this end, we first highlight certain ANK2 variants associated with disease and then discuss the potential underlying mechanisms garnered from cell biological studies using a variety of model organisms. These studies have revealed key cellular roles for ANKB in the localization and spatial organization of ion channels and transporters, signaling molecules, and structural proteins involved in variety of cellular processes, including development of cellular morphology, calcium homeostasis, and glucose and fat metabolism. By linking ANKB's emergent cellular roles with phenotypes associated with ANK2 variants, a picture of ANKB's many contributions to cardiac, neurological, and metabolic health and disease begins to emerge. Making these links is key to translating this knowledge into the clinical setting and helps understand disease risk and presentation.
Tissue- and cell-type-specific expression of ANKB isoforms across development
There are several ANKB isoforms which exhibit cell-type–, tissue–, and developmental stage–specific expression patterns. While the 220 kDa ANKB isoform is the primary isoform in both the heart and brain [as well as other cells and tissues, such as skeletal muscle, thymus, pancreas, and adipose tissue (18, 19) certain isoforms exhibit tissue-specific expression. The initial discovery of ANK2 (and its product ANKB) resulted from a series of studies characterizing ankyrin cDNA enriched in non-erythroid cells (20, 21). After the identification of a 440 kDa isoform, consisting of a large insertion (exon 40) between the regions encoding for the spectrin binding domain and death domain (20, 22), transcript and protein level characterization showed that 440 kDa ANKB was detectable at birth, with expression levels peaking at postnatal day 10 and decreasing progressively in the adult rat brain (down to 30% of peak levels) (22). Meanwhile, the 220 kDa ANKB transcript and protein levels were found to increase progressively through development into adulthood (20, 23). In addition to the 220 kDa isoform, additional ANKB isoforms have been detected in the heart: a 188 kDa isoform that, similarly to 220 kDa ANKB isoform, when knocked down results in altered expression and localization of the sodium calcium exchanger, a 212 kDa isoform which is localized to striated muscle and the cardiac M-line (24), and a 160 kDa isoform that is highly expressed in mouse hearts along with the 220 kDa isoform (25).
Newer transcriptomics studies and databases of the developing human heart show that ANK2 is differentially expressed in human embryonic ventricular and atrial cardiomyocytes, with high transcript levels also detected in fibroblast-like cells associated with vascular development and cardiac neural crest cells (https://spatialtranscriptomics3d.shinyapps.io/Developmental_heart_explorer/) (26). ANK2 transcript levels peak during early mid-fetal human development (and mouse Ank2 transcript levels peak during late embryonic development) to eventually plateau during later developmental stages (https://hbatlas.org/mouseNCXtranscriptome/, https://hbatlas.org/pages/hbtd) (26–29). In the 1-week-old mouse brain, Ank2 transcript levels are enriched in astrocytes, neurons, and oligodendrocyte progenitors (https://www.brainrnaseq.org/) (30). At the single RNA-seq level, ANK2 transcript levels are slightly enriched in inhibitory and excitatory neuron populations [Allen Cell Type Database – “M1 - 10X GENOMICS (2020)”; https://portal.brain-map.org/atlases-and-data/rnaseq] (31). Consideration of this ANKB enrichment in select cell types, tissues, and developmental stages could help provide important clues to the clinical impacts of ANK2 variants. In the next section, we will highlight several ANK2 variants and associated phenotypes that provide important areas of focus for investigation of ANKB's cellular roles.
ANK2 variants and risk for disease
Consistent with enriched ANKB expression in heart and brain, a number of ANK2 variants have been associated with a range of cardiac phenotypes while others are associated with neurological or metabolic phenotypes.
ANK2 variants associated with (primarily) cardiac phenotypes
Certain ANK2 loss-of-function variants are associated with a broad spectrum of cardiac phenotypes including arrhythmia, conduction abnormalities, and cardiomyopathy (Table 1) (4, 8, 32). Amino acid changes produced by these variants are present in all four domains of ANKB and are associated with autosomal dominant inheritance, reduced penetrance, and variable expressivity (Figure 1) (32). Initially described as LQTS type 4, QTc prolongation is commonly linked with cardiac-phenotype associated ANK2 variants, although the role in QT prolongation has been since debated (33) (Table 1). ANKB p.E1458G (previously p.E1425G), the result of an amino acid substitution in the spectrin binding domain, was among the first ANK2 variants identified. It was found in a French kindred with LQTS associated with atrial fibrillation and sinus node dysfunction (4). There was a family history of sudden death, including an 18 and 12 y.o. The variant demonstrated incomplete penetrance in one out of 23 carriers. Age related effects were also observed, affected children had sinus node abnormalities (diagnosed in utero) whereas atrial fibrillation was present only in adults (4). Of note, the p.E1458G variant has also been identified in a healthy Danish exome cohort without evidence of QTc prolongation and has a frequency of 0.11% (41/35360) in the Latino population according to the Genome Aggregation Database (gnomAD) (34, 35). Similarly, while two ANK2 variants p.E1458G and p.V3634D (initially reported as p.V1516D) were over-represented in a private cohort from an inherited heart rhythm clinic, most patients carrying ANK2 variants that were referred to this clinic showed no symptoms or had electrocardiographic findings of unknown significance; however, their genetic ancestry composition and clinical and epidemiological information is not publicly available (36). Another variant associated with prolonged QTc and ventricular tachyarrhythmias is the ANK2 p.L1622I variant, found with higher frequency in individuals of African ancestry (minor allele frequency: 0.03, 850/24964, gnomAD) (5, 37).
Table 1
| E1458Ga N = 25 | R990Q N = 2 | V3634Db N = 4 | S646F N = 15 | E1813Kc N = 3 | Q1283H N = 1 | T1404I N = 1 | M1988T N = 5 | T3744Nd N = 10 | R3906We N = 2 | I3437T N = 1 | W1535R N = 6 | 46,XX,t (4;9) (q25;q31.1) N = 5 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant type | Missense | Missense | Missense | Missense | Missense | Missense | Missense | Missense | Missense | Missense | Missense | Missense | LOF | |
| Location | SBD | SBD | DD | MBD | CTD | SBD | SBD | CTD | CTD | CTD | Disordered | DD | n/a | |
| Arrhythmia | LQTS | X | X | X | X | X | X | |||||||
| Drug-induced LQTS | X | X | X | X | ||||||||||
| CPVT | X | X | ||||||||||||
| Atrial Fibrillation | X | X | X | |||||||||||
| Cardiac arrest | X | X | X | |||||||||||
| SCD | X | X | X | X | ||||||||||
| Bradycardia | X | X | X | X | X | X | ||||||||
| VT | X | X | X | X | ||||||||||
| Other arrhythmia | Recurrent VF | Type 1 brugada pattern | SVT | Late potential on SAECG | Torsades de pointes | Torsades de pointes, VF, BrS | ||||||||
| Conduction abnormalities | WPW | X | ||||||||||||
| SND | X | X | ||||||||||||
| Other | Heart block | |||||||||||||
| Symptoms | Syncope | X | X | X | X | X | X | X | X | X | X | X | ||
| Palpitations | X | |||||||||||||
| Structural | HCM | X | X | X | ||||||||||
| DCM | X | |||||||||||||
| ARVC | X | X | ||||||||||||
| Other structural | LV dysfunction, cardiomegaly | |||||||||||||
| Congenital Heart Defect | X | X | ||||||||||||
| Other | Seizures | X | X | |||||||||||
| GnomAD | 0.0005346 | 0.00001971 | 0.002051 | Not observed | 0.002286 | Not observed | Not observed | Not observed | 0.0007363 | 0.0009337 | Not observed | 0.00001314 | N/a | |
| Clinvar Classification (# labs) | LP (3) VUS (4) LB (2) | VUS (3) | VUS (2) LB (5) B (1) | P (1) | VUS (1) LB (5) B (5) | VUS (1) | VUS (4) LB (7) | VUS (1) LB (6) B (1) | LP (1) VUS (1) | |||||
| ClinVar ID | VCV0000 18056.18 | VCV000 190560.9 | VCV0000 67599.12 | VCV000 190552.2 | VCV0000 18060.14 | VCV00 1341732.1 | VCV0000 18057.26 | VCV0000 18059.23 | VCV0000 67596.4 | |||||
| Reference | (4, 44) | (6, 43) | (8) | (7) | (130) | (95) | (8) | (44) | (5) | (5) | (40) | (46) | (39) |
Spectrum of cardiac features associated with ANK2 variants in humans.
SBD, Spectrin Binding Domain; DD, Death Domain; MBD, Membrane Binding Domain; CTD, C-Terminal Domain; LQTS, Long QT Syndrome; CPVT, Catecholaminergic Polymorphic Ventricular Tachycardia; SCD, Sudden Cardiac Death; VT, Ventricular Tachycardia; VF, Ventricular Fibrillation; SVT, Supraventricular Tachycardia; SAECG, Signal Averaged ECG; BrS, Brugada Syndrome; WPW, Wolff Parkinson White; SND, Sinus Node Dysfunction; HCM, Hypertrophic Cardiomyopathy; DCM, Dilated Cardiomyopathy; ARVC, Arrhythmogenic Right Ventricular Cardiomyopathy; P, Pathogenic; LP, Likely pathogenic; VUS, Variant of Uncertain Significance; LB, Likely benign; B, Benign.
Previously E1425G;
Previously V1516D;
Proband also carried variant in KCNH2-H562R, only the phenotype in those carrying the ANK2 E1813K variant alone described in table. See text for description of the combined KCNH2 and ANK2 phenotype;
Previously T1626N;
Previously R1788W.
Figure 1
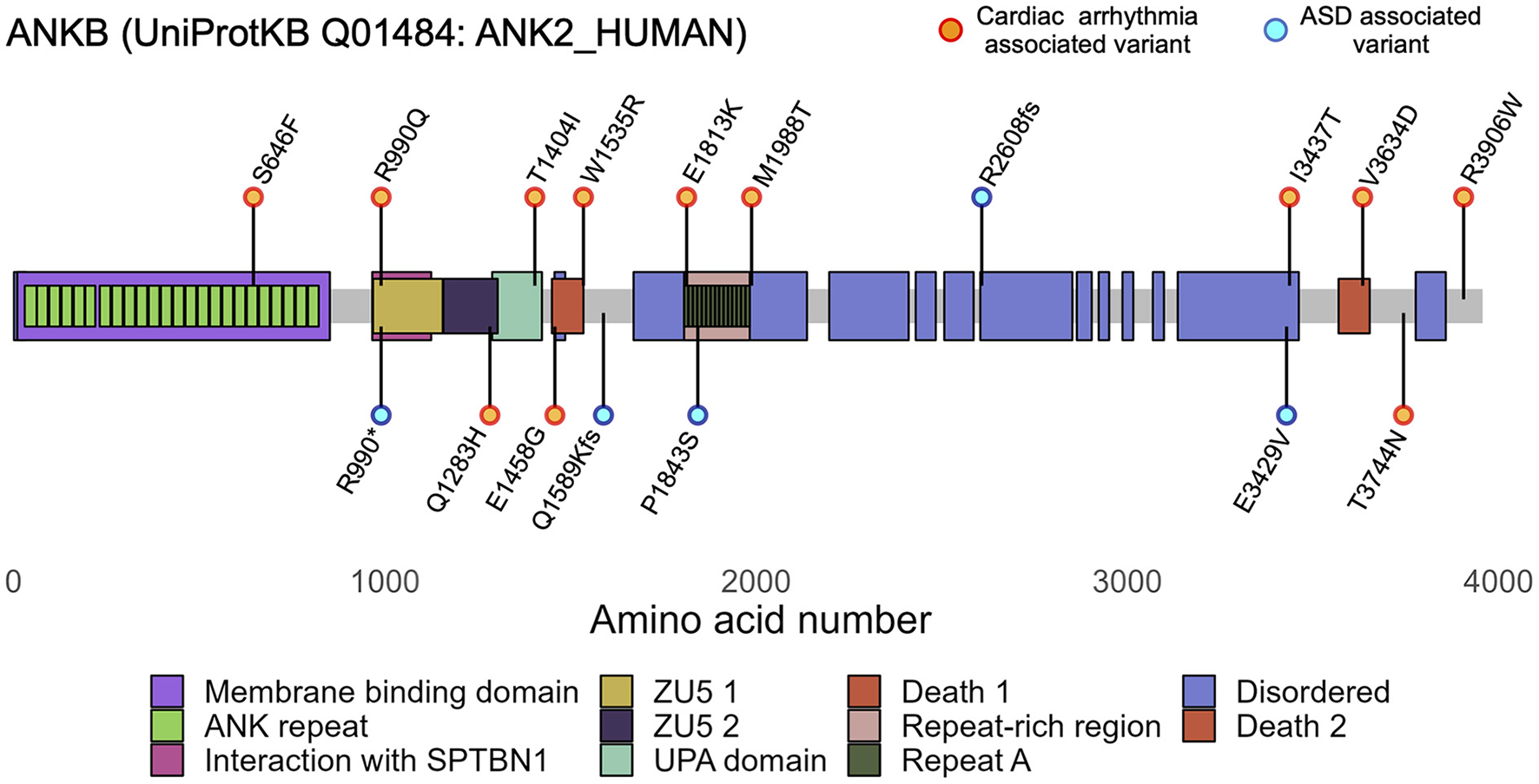
ANK2 variants are associated to cardiac arrhythmias and autism spectrum disorders (ASD). Diagram of canonical human ANKB (UniProtKB Q01484-4, 3,957 aa) showing ANKB regions and domains. Canonically, ANKB can be divided into 4 main domains: an initial membrane binding domain, a spectrin binding domain, a death domain, and a C-terminus domain. The ANKB membrane binding domain contains 24 consecutive ankyrin (ANK) repeats and associates with ion channels and transporters; the spectrin binding domain contains highly conserved ZU5-ZU5-UPA domains and associates with βII-spectrin and the B56α subunit of PP2A; and the regulatory domain comprised of the death domain and C-terminus domain comprised is unstructured and highly variable between species. Orange and cyan dots represent the main cardiac and ASD variants discussed in the main text. Functional variants have been identified in each of ANKB's domains.
ANKB p.S646F, the first identified variant located in the membrane binding domain, also came to attention due to LQTS. This variant was found in two large multigenerational Gitxsan families identified because of LQTS in the context of a known high community prevalence of KCNQ1-mediated LQTS. The probands in each family did not carry the known KCNQ1 variant (38), but instead, carried the p.S646F variant (7). As with the p.E1458G variant, QTc prolongation was not the only associated feature. The variant was identified in one individual who died suddenly due to dilated cardiomyopathy, another carrier had a history of Wolf-Parkinson-White (WPW) syndrome, and this individual's daughter was born with a congenital heart defect (total anomalous pulmonary venous return). Age related effects were observed, with limited evidence of QTc prolongation in those under 25 years (7). Congenital heart defects were also reported in a fetus carrying a duplication of 4q25-ter and 9pter-q31.1 with breakpoints in chromosome four transecting ANK2; the fetus was born with multiple cardiac malformations including a large atrioventricular septal defect (39). However, it is unclear whether the congenital heart defects may be related to the duplications or whether ANK2 haploinsufficiency played a role. Carriers of the balanced translocation, which includes breakpoints transecting ANK2, did not have congenital heart defects but other cardiac features including bradycardia, ventricular ectopy, sinus node dysfunction, and mild left ventricular dysfunction (Table 1).
LQTS is not the only phenotype associated with loss of ANKB function. Over time, several other arrhythmias and conduction anomalies have been associated with ANK2 variants including catecholaminergic polymorphic ventricular tachycardia (CPVT), bradycardia, and WPW. An occurrence of CPVT in carriers of an ANK2 variant has been reported in a small number of cases, with a recent report by Song et al. (40) of a 20 y.o. man with diagnosis of CPVT and non-ischemic cardiomyopathy who was found to carry the p.I3437T variant located in the disordered domain of ANKB (8, 41). WPW has been suggested to be another feature of Ankyrin-B syndrome. In addition to the one individual with WPW carrying the p.S646F variant previously mentioned, two rare de novo and one inherited variant in ANK2 were identified in a cohort of patients with WPW (7, 42).
Beyond inherited arrhythmias, loss-of-function variants in ANK2 have also been associated with cardiomyopathy, such as hypertrophic cardiomyopathy, dilated cardiomyopathy, and LV dysfunction (7, 39, 43). In a cohort of patients with HCM, rare variants in ANK2 showed a statistically significant association with greater maximal mean left ventricular wall thickness, contributing to more severe LV hypertrophy (43). Recently, the role of ANK2 in arrhythmogenic right ventricular cardiomyopathy (ARVC), a condition characterized by fibrofatty replacement of the myocardium, ventricular arrhythmias, and sudden cardiac death has come to attention. The previously reported p.E1458G variant was identified in an individual who died suddenly while running and was found to have ARVC on autopsy. A second ANK2 variant was identified in a family where the proband died suddenly during exercise and was also found to have ARVC on autopsy. Post-mortem genetic testing was carried out and identified a novel p. M1988T variant, which is located in the C-terminal domain. Additional family members were identified through cascade screening to have definite or borderline diagnoses of ARVC (44).
With the emergence of a broad spectrum of features linked to ANK2 variants came the term “Ankyrin-B syndrome,” which at the time more fully captured the complexity of the range of associated phenotypes (8, 32). Although the origin of Ankyrin-B syndrome is associated solely with cardiac phenotypes (8, 45), through the investigation and identification of new variants, it has become apparent that ANKB dysfunction is not exclusive to cardiac phenotypes but underlies neurological ones as well. Thus the term, Ankyrin-B syndrome, does not fully capture the broad spectrum of ANKB dysfunction across all cell types and variants. The pleiotropic nature of ANK2 is highlighted by individuals that experience seizures in combination to the cardiac manifestations (7, 46), as well as unique ANK2 variants associated with ASD (47–49) of which we will discuss in the next section.
ANK2 variants associated with neurological phenotypes
Beyond the heart, ANK2 is emerging as an important gene in neurological conditions, including ASD and epilepsy. It is important to note that the variants associated with Ankyrin-B syndrome (cardiac-phenotype associated ANKB variants) are distinct from those reported in association with ASD, and a combination of cardiac and ASD phenotypes has not been reported. Rare variants in ANK2 including missense, frameshift, non-sense, and copy number variants have been identified in individuals with ASD (Table 2) (10, 47–50). ANK2 is classified as a high-confidence gene clearly implicated in ASD by the Simons Foundation Autism Research initiative due to the reports of at least three de-novo loss-of-function variants in the literature and meeting the threshold false discovery rate of <0.1 (https://gene.sfari.org/database/human-gene/ANK2). ASD-associated ANK2 variants are largely non-syndromic and typically not associated with intellectual disability (Table 2) (51). While some variants are present within both the 220-kDa and 440 kDa ANKB proteins, certain variants are unique to the 440-kDa giant ANKB isoform. For instance, a knock-in mouse model carrying ANKB p.P2580fs (analogous to the human p.R2608fs), which expresses a truncated giant ANKB polypeptide, demonstrated ASD-like behaviors including repetitive behavior, decreased ultrasonic vocalization, reduced territory marking, and superior executive functioning. Of note, mice homozygous and heterozygous for the p.P2580fs variant exhibited the same behaviors, supporting that haploinsufficiency of ANK2 could contribute to risk for ASD (51). Using a multiplex network that characterized modules of epilepsy and ASD genes sharing similar phenotypes and protein-protein interactions, ANK2 has also been identified as a novel candidate gene for epilepsy (11). Similarly, in a workflow using the random walk with restart algorithm in addition to permutation and functional association tests ANK2 was also predicted as a novel gene for epilepsy (52).
Table 2
| Variant | Type | Location | ASD | Intellectual disability | Other | gnomAD | Clinvar ID | Reference |
|---|---|---|---|---|---|---|---|---|
| Affect only giant ANKB(440 kDa)isoform | ||||||||
| P1843S | Missense | Disordered | X | 0.000003979 | (134) | |||
| E3429V | Missense | Disordered | X | Absent | (48) | |||
| R2608fs | Frameshift | Disordered | Pervasive developmental disorder | Absent | (135) | |||
| Affect giant ANKB(440 kDa)and220 kDaisoform | ||||||||
| R990* | Nonsense | ZU5-1 | Asperger's disorder | Absent | VCV000450028.2 | (10) | ||
| Q1589Kfs | Frameshift | X | X | Sensorimotor neuropathy, facial dysmorphism | Absent | VCV000235896.1 | (50) | |
| 4:113593803_113967887dup | Duplication | X | n/a | VCV000236353.1 | (47) | |||
| 4:114077690_118094709dup | Duplication | X | n/a | VCV000236354.1 | (47) | |||
| 4: 114225715-114429181del | Deletion | X | n/a | VCV000236355.1 | (47) | |||
ANK2 variants associated with autism spectrum disorder.
Table adapted from Yang et al. (51); ASD, Autism Spectrum Disorder.
Notably, independent of the connection between ANK2 variants and risk for epilepsy, seizures were reported in association with cardiac-phenotype associated ANK2 variants. A history of seizures was reported in eight of eighteen carriers of the ANKB p.S646F variant, and in two out of six patients carrying the ANKB p.W1535R variant (7, 46). In a study which sequenced cases of epilepsy-related sudden unexpected death for inherited heart disease related genes, one individual was found to carry two variants in ANK2 (p.Ser105Thr, p.Glu1934Val). Of note, this death occurred by drowning, and the individual was reported to have mildly prolonged QTc (53). Given that seizures can be linked to cardiac arrhythmias (54) and the fact that some cardiac-associated ANK2 variants are linked with seizures (7, 46) it would be worth investigation to determine if the seizures are a result of the arrhythmia or independent and owed to dysfunction in the brain. Furthermore, with the link to epilepsy in neurological-associated ANK2 variants (11) it raises the question of how ANKB dysfunction is impacting neuronal mechanisms.
ANK2 variants associated with metabolic phenotypes
ANK2 variants have also been implicated in the regulation of fat and glucose metabolism. In particular, the ANK2 p.R1788W variant, which is associated to cardiac phenotypes (Table 1), was enriched in individuals of white and Hispanic descent diagnosed with type 2 diabetes in the American Diabetes Association GENNID cohort. Moreover, the ANK2 p.L1622I variant, associated with a less severe cardiac phenotype, is the most frequent ANK2 variant (7.5%) in African Americans who carry up to a 2-fold increased risk for type 2 diabetes (19, 55, 56). Whether other primarily cardiac or neuronal ANK2 variants also result in global or local metabolic disturbances remains to be investigated.
Fundamental research studies revealing the many roles of ANKB within cells have provided insights into possible mechanisms behind the various phenotypes associated with ANK2 variants. ANKB is implicated in different pathways, as it is a scaffolding protein for ion channels and transporters as well as a link for structural and signaling proteins, some of which are outlined below and summarized in Table 3.
Table 3
| Domain | Classification | Interacting partner | Cell type interaction confirmed in | References |
|---|---|---|---|---|
| MBD | Ion channels/ Transporters | Inositol 1,4,5-trisphosphate receptor | Cardiac and Neuronal | (4, 5, 14, 60, 103, 136) |
| CaV1.3 | Cardiac | (136, 137) | ||
| CaV2.1 | Neuronal | (101, 102) | ||
| CaV2.2 | Neuronal | (101) | ||
| CaV3.1 | Neuronal | (138) | ||
| CaV3.2 | Neuronal | (138) | ||
| Kir6.2 | Cardiac | (139, 140) | ||
| Sodium Calcium Exchanger | Cardiac and Neuronal | (4, 5, 24, 60, 103, 137) | ||
| Sodium Potassium ATPase (a1 and a2) | Cardiac and Neuronal | (4, 5, 60, 103, 137, 140, 141) | ||
| Erythrocyte anion channel | Neuronal | (12) | ||
| Structural | EHD1-4 | Cardiac | (142) | |
| Structural/ Signaling | Beta-catenin | Cardiac | (44) | |
| Cell adhesion | L1CAM | Neuronal | (51, 120, 143) | |
| Dystrophin | Cardiac | (144, 145) | ||
| SBD | Motor movement | Dynactin-4 | Cardiac and Neuronal | (144–146) |
| Structural | βII-spectrin | Cardiac and Neuronal | (6, 59, 103, 147) | |
| Signaling | Phosphatidylinositol 3-phosphate | Neuronal and Fibroblast | (146, 148) | |
| PP2A B56α | Cardiac | (95, 149, 150) | ||
| Giant insertion | Enzyme | Ndel1 | Neuronal | (151) |
| DD | Signaling | RABGAP1L | Fibroblast | (148) |
| CTD | Chaperone | HSP40 | Cardiac | (69) |
| Signaling | Obscurin | Cardiac | (24, 152) | |
| Regulatory | Ankyrin-B MBD | Cardiac | (15) | |
| Unknown | Signaling | SadA/SadB | Neuronal | (153) |
| Chaperone | UNC-119 | Neuronal | (80) |
ANKB interacting partners.
Insights on the cellular role(s) of ANKB from model organism studies
Given their sequence similarity, it is possible to understand the biological role of ANK2 (and its homologs) through studying model organisms. Mouse Ank2 is comprised of exons exhibiting considerable homology to those found in human ANK2 and exhibits similar tissue-specific isoform expression patterns (24, 25). In mice, global Ank2 knockout causes neonatal death (57), while conditional Ank2 knockout in the heart and brain results in significant electrical and structural impairments and death (44, 51, 58, 59). Heterozygous Ank2 knockout (Ank2+/−) mice model haploinsufficiency (i.e., expression of a single wildtype Ank2 allele fails to produce a wildtype phenotype), are relatively viable, and therefore used in many preclinical studies. Ank2+/− mice display increased susceptibility to atrial and catecholamine-induced ventricular arrhythmias and sudden death, as well as, premature senescence and reduced lifespan (4, 8, 45). These cardiac manifestations have been associated with decreased presence of the sodium calcium exchanger, the sodium potassium ATPase subunits 1 and 2, and the inositol 1,4,5-trisphosphate receptor at the T-tubules of cultured primary cardiomyocytes (4, 60) (Figure 2). Mice with complete global loss of Ank2 (Ank2−/−) display severe structural brain defects, such as hypoplasia of white matter tracts, dilated ventricles, and degeneration of the optic nerve (57). As several developmental signaling pathways are strongly intertwined with the homeostasis of ions, such as calcium (61–63), the severe structural phenotypes observed in the context of ANKB dysfunction (or haploinsufficiency) may be related to ANKB regulatory roles in ion homeostasis and cytoskeletal proteins.
Figure 2
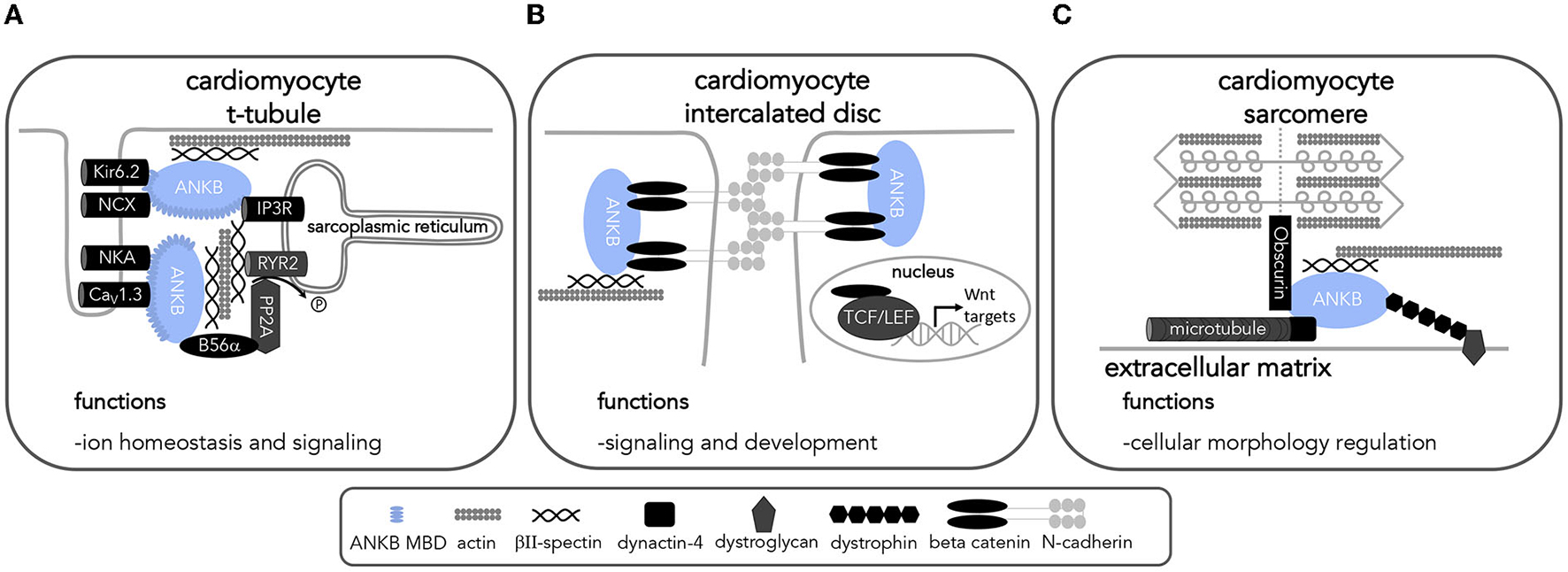
ANKB interactions in cardiomyocytes implicates ANKB in a variety of cellular processes. Diagram of ANKB interacting partners and their localization allowing for proper cardiac function. ANKB interactions at the (A) t-tubule (B) intercalated disc and (C) sarcomere allow for proper cell functions. Kir6.2, inward rectifier potassium channel; NCX, sodium calcium exchanger; NKA, sodium potassium ATPase; CaV1.3, voltage-gated calcium channel; IP3R, Inositol 1,4,5-trisphosphate receptor; PP2A, protein phosphatase 2A; RYR2, ryanodine receptor 2; L1CAM, L1 cell adhesion molecule; PI3P, phosphatidylinositol 3-phosphate; CaV1.3, voltage-gated calcium channel; TCF/LEF, T cell factor/lymphoid enhancer factor transcription factors.
Analysis of the molecular evolution of ankyrins has revealed a single ankyrin gene in Caenorhabditis elegans (unc-44), two ankyrin genes in Drosophila melanogaster (Dank1, Dank2), and three mammalian ankyrin genes (ANK1, ANK2, ANK3) that likely originated from a single ankyrin ancestor gene in Ciona intestinalis (64, 65). Moreover, these analyses have also demonstrated a closer evolutionary relationship between ANK2 and ANK3, which despite their high sequence and structural similarity localize to different cellular compartments and associate with different proteins (66, 67). In most cases, ankyrins do not have the ability to compensate for each other (68, 69). However, previous studies from the Rasband lab have shown that in the central nervous system ANKB can partially compensate for loss of paranodal ANKG and ANKR can compensate ANKG's role in sodium channel clustering at nodes of Ranvier (70, 71). While each ankyrin protein appears to have different roles they share some protein-protein interactions and can provide insight into each other's roles in cells.
The use of model organisms can help unravel the mechanisms behind the clinical phenotypes associated with ANK2 variants. Insights from model organisms have elucidated ANKB's essential roles in regulating cellular morphology, polarization, calcium homeostasis, and glucose and fat metabolism, as outlined below and summarized in Tables 3–5 and Figures 2, 3.
Table 4
| Model organism | Biological process | Elucidated roles and implications | Reference | |
|---|---|---|---|---|
| Mus | Ank2−/− (Ank2 null) | N/a | Global knockout is deadly | (57) |
| musculus | Ank2+/− (Models haploinsufficiency) | Cardiomyocyte structural development | Cardiac malformations imply role in structural development | (4, 8, 45) |
| Calcium homeostasis and signaling | Localization and expression of the sodium/calcium exchanger, inositol trisphosphate receptor, and voltage-gated calcium channels L-type channels; Cav1.3 expression (SAN isolated cells and atrial cardiomyocytes) P/Q-type channels; Cav2.1 and Cav2.2 expression (cortex, cerebellum, and brainstem) | (4, 60, 101, 136, 137) | ||
| Regulation of RYR2-mediated sarcoplasmic reticulum calcium leak via PP2A (cardiomyocytes) | (104) | |||
| Regulation of calcium homeostasis affects calcium cycling dynamics (calcium transients, sparks) and delayed afterdepolarizations | (4, 5, 95, 104) | |||
| Glucose and fat metabolism | Downstream effects on oral glucose tolerance | (114) | ||
| shAnkB knockdown | Calcium homeostasis and signaling | Localization and expression of T-type channels Cav3.2 expression (hippocampal neurons) | (138) | |
| Cardiac-specific conditional knockout | Cardiomyocyte structural development | Cardiac remodeling implies structural role | (44) | |
| Involved in Beta-catenin localization and expression; possible implications on beta-catenin/Wnt signaling | ||||
| Brain-specific knockout (brain-specific ANKB 440-kDa isoform not expressed) | Neuronal structural development | Synaptic signaling and synapse excitability | (51) | |
| Axon branching and connectivity (linked to Ank2 involvement in microtubule bundle formation) | ||||
| Abnormal social behavior. Impaired communicative behavior. Enhanced executive function. | ||||
| Excitatory neuron-specific knockout (ANKB 220-kDa and 440-kDa are not expressed in excitatory neurons) | Calcium homeostasis and signaling | Regulation of Cav2.1 expression (decreased Cav2.1 expression in whole cortex homogenates) | (102) | |
| Adipose tissue-specific conditional knockout | Glucose and fat metabolism | Adiposity | (117) | |
| Pancreatic islet size | ||||
| Insulin resistance | ||||
| ANK2 p.R1788W knock-in | Glucose and fat metabolism | Abnormal insulin secretion. Insulin resistance | (19) | |
| Increased peripheral glucose uptake (increased cell surface GLUT4) | ||||
| Adiposity | ||||
| Caenorhabditis elegans: unc-44 | Neuronal development and polarization | Regulating organization/ polarization neurite microtubule networks | (79, 81) | |
| Drosophila melanogaster: Dank2 (Dmel\ank2) | Neuronal development and polarization | Supporting stabilization and remodeling of synaptic microtubule network | (87, 88) | |
Summary of ANKB's cellular roles identified using model organisms.
Table 5
| Biological function | Level | Heart | Brain |
|---|---|---|---|
| Structural development and cell polarization | Cellular | Trafficking and distribution of ion channels and exchangers along T-tubules and beta-catenin at the intercalated disc | Definition of axonal and dendritic compartments in neurons Trafficking of proteins to axonal and dendritic compartments |
| Tissue/Organ | Dilated cardiomyopathy Ventricular wall fibrosis | White matter tract defects Increased axonal connectivity | |
| Calcium homeostasis and signaling | Cellular | Increased calcium transient amplitude (putatively, increased intracellular calcium concentration) Increased calcium sparks (calcium release events from the sarcoplasmic reticulum) Decreased calcium transient frequency Decreased spontaneous contraction rate | Increased miniature excitatory postsynaptic potentials Decreased excitability Decreased action potential firing rate |
| Tissue/Organ | Increased contractility Increased rate of delayed afterdepolarizations | Decreased expression of calcium voltage gated channels (CaV2.1 and CaV2.2) |
Summary of primary biological functions affected by ANKB dysfunction/loss-of-function.
Figure 3
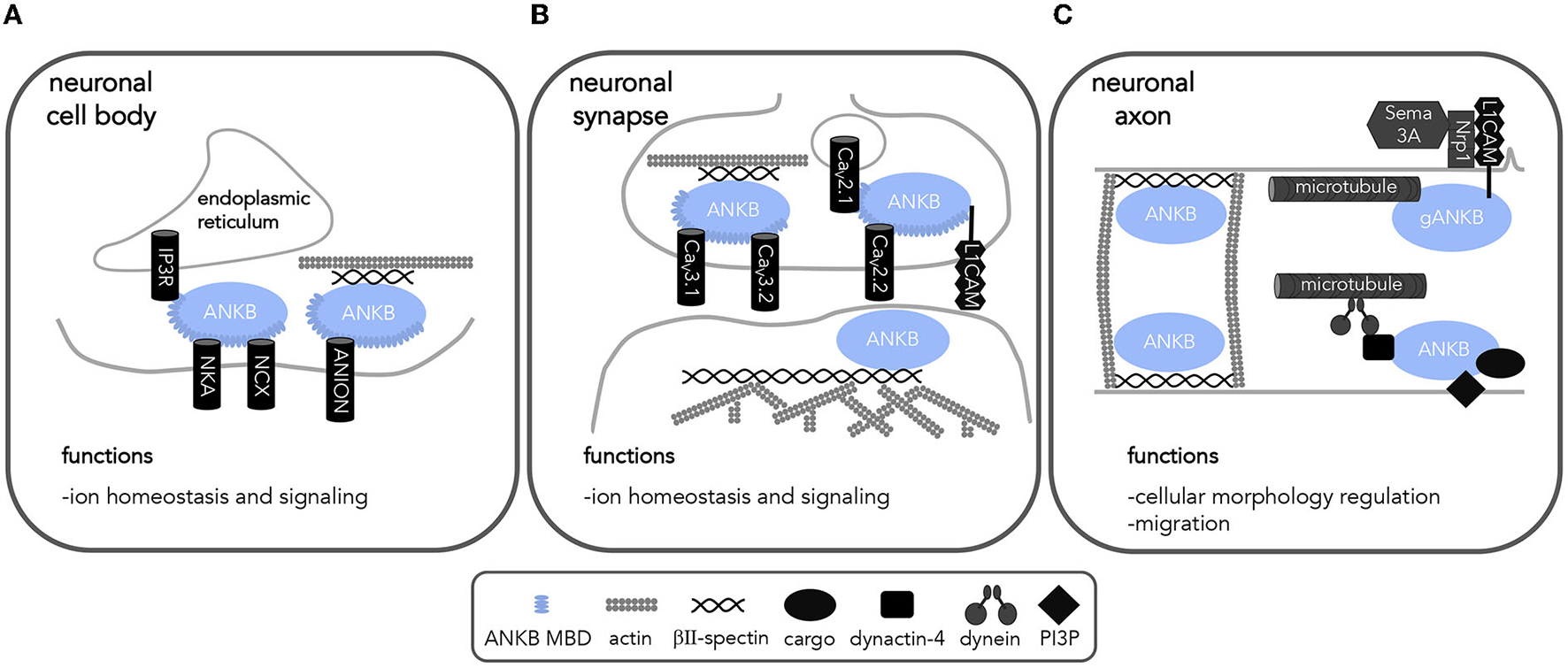
ANKB interactions in neurons implicates ANKB in a variety of cellular processes. Diagram of ANKB interacting partners and their localization allowing for proper neuronal function. (A) cell body (B) synapse and (C) axon allow for proper cell functions. NCX, sodium calcium exchanger; NKA, sodium potassium ATPase; IP3R, Inositol 1,4,5-trisphosphate receptor; CaV, voltage-gated calcium channels; PP2A, protein phosphatase 2A; RYR2, ryanodine receptor 2; L1CAM, L1 cell adhesion molecule; PI3P, phosphatidylinositol 3-phospate; Sema 3A, semaphorin 3A; Nrp1, semaphorin 3A receptor neuropilin 1.
ANKB regulates cellular morphology and polarization
As cells develop, migrate, and mature, cytoskeletal rearrangements lead to the specification of a directionality axis resulting in well-organized regions that support motility, cell-to-cell contacts, and surfaces for secretion or absorption (72). This spatiotemporal phenomenon is known as cell polarity and it is what influences the shape, motility, and trafficking and signaling domains in cells, as well as their ability to respond and adapt to extracellular and intracellular signals (72, 73). In the mouse heart, for example, cell polarization allows round embryonic ventricular cardiomyocytes to postnatally adopt the shape of a rod and direct their junctional proteins to the ends of the now elongated cells to form the intercalated disc, a specialization for cell-to-cell communication (74, 75). In other cell types, such as neurons, cell polarization defines specialized compartments for receiving (dendrites) and sending (axons) electrochemical signals (76). Insights from studies using model organisms have shown ANKB's essential role in neuronal development which raises the question of how ANKB may play a role in cardiomyocyte development as well.
Although it is yet unclear whether ANKB plays a role in the morphological development of cardiomyocytes and other cardiac cell-types in which it is expressed, there is evidence of ANKB's roles in neuronal development. Studies using the model organism C. elegans have demonstrated that ANK2's ortholog unc-44/ankyrin is a known master regulator cell polarization and axonal neurite outgrowth in this roundworm's sensory neurons (77–80). Mutations affecting unc-44/ankyrin function result in abnormal neural development, locomotor defects, and microtubule networks with mixed polarity in axons and dendrites leading to abnormal protein sorting and trafficking into these compartments (77–79, 81–83). Unc-44/ankyrin, along with unc-33/crmp (an actin and microtubule associating protein) and unc-116/kinesin-1 (a motor protein) help establish neuronal polarity by regulating the organization of dendritic and axonal microtubule networks (79, 81). Furthermore, unc-44/ankyrin acts upstream of unc-33/crmp and vab-8/kinesin-like protein to regulate the removal of gap junction channels (84) which allow for the direct electrical communication between cells and play a key role in development (85, 86). In Drosophila melanogaster, Dmel\ank2, which has a short and long/ giant isoform localized to different sub-cellular compartments (cell body and axon, respectively) supports the stabilization and remodeling of the synaptic microtubule network (87, 88). Loss of Dmel\ank2 results in retraction of synaptic boutons, collapse of the pre-synaptic active zones, reduction of the terminal size, and altered neuromuscular junction morphology (88, 89). While the role of ANK2 in cardiomyocyte polarization during heart development remains to be investigated, some of the ANK2 variants listed above have been associated with cardiac malformations suggesting that ANKB dysfunction results in an impact to the structural development of these cells (7, 39).
Recent organ-specific ANK2 conditional knockouts further underscore the important role of ANK2 in the structural development of cardiomyocytes and neurons. Specifically, the beta-catenin/Wnt signaling pathway is important in both cardiac and neuronal cell fate determination, axis patterning and polarity, and proliferation (90–92). This pathway is initiated by the accumulation of beta-catenin in the nucleus leading to the transcription of Wnt responsive genes (92). Evidence underlying ANKB's role in cell proliferation and survival has been highlighted by the p.S646F variant. In H9c2 cells, a cell model with similar traits of primary cardiomyocytes (93), expression of the p.S646F variant resulted in decreased cellular viability and proliferation (94). Using a cardiac-specific conditional knockout model, Roberts et al. found that loss of Ank2 in the heart leads to severe cardiac remodeling resulting in ventricular dilation, fibrosis, bradycardia, QTc prolongation, and increased susceptibility to catecholamine-induced ventricular arrythmias (44). Associated with decreased protein expression and altered localization of beta-catenin away from the intercalated disk, this cardiac-specific ANK2 knockout phenotype recapitulates what has been observed in arrhythmogenic cardiomyopathy phenotypes as well as in patients carrying predicted loss-of-function ANK2 variants and their respective knock-in mouse models (Figure 2) (4, 37, 95). It is worth noting that cardiomyocytes with Ank2 loss do not show altered expression nor mislocalization of intercalated disc proteins such as plakoglobin, plakophilin 2, connexin 43, N-cadherin, desmoplakin, and desmoglein 2 (44). Further insights regarding the involvement of ankyrin proteins in this context may be drawn from ANKG, which also interacts with beta-catenin. Loss of ANKG results in a comparable decrease in beta-catenin localization at the membrane and increased nuclear levels leading to an increase in neural progenitor proliferation in mice via Wnt signaling (96). Given ANKB also plays a role in organizing beta-catenin localization and expression, it is worth future investigations to determine if ANKB leads to any effects on Wnt signaling. In parallel, another ANKB interacting partner, protein phosphatase 2A (PP2A), is also a regulator of Wnt signaling (97). With ANKB's potential involvement at two stages of the Wnt signaling pathway, future studies should explore the implications of ANKB dysfunction on the latter as well as concomitant developmental processes.
In the case of loss of brain-specific giant ANKB 440-kDa, which primarily localizes to axons, mice display ectopic axon branching and connectivity, transient increase in excitatory synapses, and neurodevelopmental disorder-like behaviors such as stereotype movements and impaired social behavior (51). The impairment on axonal connectivity has been linked to ANK2's role in regulating the formation of microtubule bundles in the axon and reducing branching points enriched with F-actin by promoting growth cone collapse in response to semaphorin 3A signaling (58, 98) (Figure 3). While brain specific Ank2 knockout mice do not exhibit impairments in memory and learning (51), the identified structural and connectivity changes recapitulate some of the morphological features observed in neurodevelopmental disorders, such as ASD (51, 99, 100). The identification of giant ANKB-specific roles in critical aspects of neuronal structural development warrants further exploration in the heart and its intrinsic nervous system. Furthermore, given that ANKB is associated with several critical steps in the development of cells and the establishment of their polarity, studies aiming to elucidate the role that ANK2 plays during early heart and cardiac conduction system development will be crucial to understand the various phenotypes associated with ANK2 variants.
ANKB regulates calcium homeostasis
With its role in proper localization of the calcium the sodium/calcium exchanger, the inositol trisphosphate receptor, and calcium voltage-gated channels, ANKB is a key hub for regulation of calcium homeostasis in excitable cells (14, 101–103). In mouse cardiomyocytes, complete and partial loss of Ank2 leads to abnormal calcium dynamics as summarized in Tables 4, 5 (4, 5, 104). Using global and partial loss of Ank2 knockout mouse models, it has been demonstrated that ANK2 variants identified in cardiogenetic studies have differential effects on cardiomyocyte calcium dynamics in vitro, with some variants (namely: p.G1406C, p.R1450W, p.L1503V) rescuing calcium transient amplitude defects, while others (namely: p.E1425G, p.L1622I, p.T1626N, p.R1788W, p.T1404I, p.V1516D, p.T1552N, p.V1777M, and p.E1813K) fail to rescue calcium and spontaneous activity abnormalities (8). These in vitro experimental findings are in line the variable expressivity and penetrance observed in individuals carrying ANK2 variants (4, 7).
ANK2 regulates calcium homeostasis in excitable cells through various potential mechanisms, some of which still require additional in-depth characterization. Loss of ANK2 or ANK2 dysfunction (as in the case of pathogenic ANK2 variants) leads to the mis-localization of channels and transporters involved in calcium handling (sodium/calcium exchanger, inositol trisphosphate receptor, and calcium voltage-gated channels) (4, 7, 95, 105). Furthermore, lack of ANK2 (and in some cases, ANK2 dysfunction) leads to decreased protein expression of the sodium/calcium exchanger and L-type, T-type, and P/Q-type voltage gated calcium channels in cardiomyocytes and neurons (7, 101, 102, 104). Specifically, two clinically relevant ANKB variants, p.E1458G and p.S646F, differentially modulate levels of CaV2.1, the pore forming subunit of P/Q-type voltage gated calcium channels, in HEK293T cells (102). The p.E1458G variant was found to decrease surface CaV2.1 levels while the p.S646F variant increased intracellular CaV2.1 levels. Another variant, p.Q879R, which to our knowledge has not yet been associated with disease, is located at the linker region required for proper ANKB localization. Expression of p.Q879R increased the surface level expression of CaV2.1 in the presence of the CaV accessory subunits (102). Additionally, ANKB may also regulate the key intracellular calcium release channel, ryanodine receptor 2 (RYR2). RYR2 hyperphosphorylation in the mouse Ank2 knock-in model harboring the p.Q1283H variant suggests ANKB's interaction with the regulatory subunit, B56α, of the protein phosphatase PP2A, of the protein phosphatase PP2A (PPP2R5A) is necessary for PP2A activity on RYR2 (Figure 2) (95). Abnormal calcium handling associated with ANKB variant expression is a plausible pathophysiological mechanism underlying the increase in frequency of delayed afterdepolarizations and susceptibility for cardiac arrhythmias observed with ANK2 cardiac variants (104, 106), as well as a possible mechanism for the increased susceptibility to epileptic seizures associated with some ANK2 variants.
PP2A is a key regulator in most signal transduction pathways and cellular processes (107, 108). Other targets of PP2A and the resulting impact of ANKB dysfunction has not yet been investigated and should be an area of research in the future. Of the many PP2A targets some include other ANKB interactors such as the inositol trisphosphate receptor (109) and the sodium potassium ATPase (110) of whose phosphorylation and therefore function may also be altered as a result of ANKB dysfunction. In neuronal cells PP2A is one of the major enzymes associated with regulating microtubules, neurofilaments, and the actin cytoskeleton (111–113). While ANKB's interaction with PP2A in neurons has not yet been confirmed, this likely regulation of signaling events has key implications to the functioning and development of neuronal cells as well.
ANKB functions in glucose and fat metabolism regulation
ANKB has also been linked to regulating glucose and fat metabolism. An earlier study by Healy et al. (114) described that mice with partial global loss of Ank2 (Ank2+/−) exhibit impaired oral glucose tolerance likely secondary to decreased expression of the inositol trisphosphate receptor in pancreatic islets, which mediates the signaling for augmented glucose-induced insulin secretion after parasympathetic stimulation (115, 116). Knock-in mice harboring the type-2 diabetes-associated ANK2 p.R1788W variant exhibit decreased insulin secretion following parasympathetic stimulation and increased peripheral glucose uptake (coupled with increased plasma membrane density of the glucose co-transporter 4 in skeletal muscle and adipose tissue) (19). Notably, older ANK2 R1788W mice had increased adiposity and showed insulin resistance (19). The increase in adiposity is also observed in adipose tissue-specific Ank2 knockout mice, which develop progressive pancreatic islet dysfunction, accumulation of fat with age or high fat diet, and insulin resistance associated with impaired glucose co-transporter clathrin-mediated endocytosis (117). Importantly, a subset of ANK2 variants associated with cardiac arrhythmias failed to rescue the metabolic defects in Ank2−/− adipocytes (117), calling to attention additional cardiovascular risk considerations for individuals with known ANK2 cardiac arrhythmia variants. A knock in ANK2 p.L1622I model exhibited a measurable and distinct cardiac phenotype, reduced ANKB expression, and even developed insulin resistance and age-dependent increases in adiposity (19).
Discussion
Variants in ANK2 are associated primarily to complex cardiac phenotypes; however, some functional ANK2 variants also have neurological or metabolic manifestations. Cardiac phenotypes associated with ANK2 functional variants are characterized by a predisposition to arrhythmias, conduction anomalies, and congenital and adult-onset structural heart disease, and in some cases, seizure (Table 1). Other ANK2 variants may contribute to risk for ASD and epilepsy (Table 2). With type 2 diabetes also linked to specific ANK2 antecedents, the putative compounding effects of metabolic perturbation on cardiac and neurological phenotypes may pose additional risk to individuals carrying ANK2 variants. The diversity of manifestations associated with ANK2 variants could result, in part, from complex ANKB protein interaction networks involving critical proteins that regulate cellular structure and function (Table 3). Overall, improved knowledge of ANKB cellular roles and regulation is now needed to advance understanding of clinical phenotypes associated with ANK2 variants and, ultimately to develop improved, targeted therapeutic approaches.
As there is such diversity in features reported across ANK2 variants with cardiac phenotypes, further studies are required to better understand which features are truly linked to Ankyrin-B syndrome. For example, congenital heart defects have been described in association with only one variant to date, p.S464F (7), and a structural chromosomal re-arrangement involving breakpoints in ANK2 (39). Whether congenital heart defects are part of the ANKB spectrum of manifestations or just isolated events remains to be determined. In favor of the notion that ANK2 functional variants can also contribute to structural heart disease, a British study on hypertrophic cardiomyopathy reported that the proportion of patients with a maximum left ventricular wall thickness >30 mm (i.e., extreme wall thickness) was higher in carriers of ANK2 variants (43). This effect was still present when restraining the analysis to patients carrying sarcomeric protein variants (43), suggesting that ANK2 might play a role of a disease modifier in cases of hypertrophic cardiomyopathy (43, 118, 119). Further population and laboratory studies are required to fully elucidate the connection between ANK2 variants and hypertrophic cardiomyopathy, which could involve ANKB interactions with structural/cytoskeletal elements within cardiac cells.
Given the cases of structural malformations it is also important to investigate the potential role of ANKB in cardiac cell development. Of note, the p.S6464F variant is less stable and experiences reduced expression only in undifferentiated H9c2 cells suggesting this variant's impact to cells occurs during their development (94) and provides some additional rationale behind investigating ANKB's roles during cardiac development. As seen in patients with both the p.S646F (7) and p.E1458G (4) variants there appears to be an age-related effect. This implies that not only is ANKB function important in early development but also over a lifespan. Some possible mechanisms behind ANKB's role in cardiac development include its interactions with beta-catenin, PP2A, and ion channels. Understanding the developmental expression of ANKB and the impact of variants may provide insight into the cardiac dysfunction observed in patients over their lifetime.
While ANKB's link to neuronal development has been better pieced together through studies with model organisms, we have highlighted key knowledge gaps and areas of future investigations. Early observations revealed neuroanatomical defects in the global Ank2 knockout mouse (57) and model organisms have highlighted homologous ankyrin roles in neuronal polarization (79). Other mainly in vitro studies point to a role for ANKB in GABAergic synaptic development (120), axonal branching (51, 58, 98), and voltage-gated calcium channel trafficking (101, 102) (Figure 3). These studies suggest ANKB regulates neurodevelopmental processes and could help explain its putative role in risk for ASD, as well as its association with epilepsy and seizure. Given the important roles of giant ANKB in the development of the nervous system, future studies aiming to elucidate the roles of giant ANKB in the development of the heart and conduction system are warranted. Moreover, recent single cell transcriptomics surveys identifying several non-myocyte cells that contribute to heart development, such as cardiac neural crest cells, neuronal cells, and glial-like cells (all with detectable ANK2 transcripts levels) (26, 121–123) open the door to novel lines of research investigating ANK2's functions within these cells and their impact in shaping heart development. Notably, many studies in the brain have focused primarily on the giant isoform of ANKB, the putative central nervous system-specific, neonatal isoform (51, 58, 98); however, the roles of the smaller, more prominent 220 kDa isoform are vastly understudied. ANKB's roles at the mammalian synapse are yet to be studied even though ANKB is not only enriched at synapses, but also seems to associate with multiple postsynaptic scaffolding proteins (124, 125). Given ANKB's interactions with ion channels, βII-spectrin, and components of the cytoskeleton (126), it is possible that ANKB plays important roles in regulating the shape of postsynaptic structures and protein sorting therein contributing to maturation of synapses and establishment of neuronal circuits.
Recently, disease associations of ANK2 variants and LQTS and CPVT have been debated in part due to the population frequency of certain previously reported variants (34–36, 127). Although the minor allele frequency is certainly a useful predictive tool (128), an elevated minor allele frequency may not completely eliminate a role for the variants in disease. For example, while the ANK2 p.L1622I variant is associated with prolonged QTc and ventricular tachyarrhythmias, which is modeled in a knock-in homozygous mouse, the study was limited by the use of juvenile homozygous mice. This cardiac phenotype likely exceeds that of the carriers in the general population, who are most likely heterozygous for the ANK2 p.L1622I variant (37). It is possible that ANK2 variants are part of an oligogenic/polygenic disease (129). Such a possibility is seen with the p.E1813K variant which has been shown to aggravate the cardiac phenotype of an individual carrying KCNH2 p.H562R variant (130). In isolation, the p.E1813K variant was associated with age-related conduction disease, and the individual carrying only the KCNH2 p.H562R variant was asymptomatic. ANK2 is a gene that appears to tolerate mutations well as seen by the allele frequencies of many variants. This variant toleration may be a result of a compensatory mechanism to protect the overall function of the protein given its apparent importance in cellular biology. Overall, this evidence highlights the importance of integrating allele frequency, genetic ancestry, and environmental and genetic factors in the analysis and determination of cardiovascular gene-disease associations of ANK2 variants.
Insights from model organism studies have highlighted the significance of ANKB's many roles within cells. ANK2 variants are linked with cardiac, neurological, or metabolomic phenotypes consisting of electrical, structural, and signaling impacts. The mechanisms behind ANK2 variant dysfunction can be explained in part due to ANKB's protein interactions and cellular partners outlined within the review. With many interactions in both signaling and cytoskeletal components, ANKB can easily be implicated in a variety of cellular events and basic functions. Furthermore, interactions identified and studied within one cell type could hold relevance across multiple cell types in which ANKB is expressed. With the large number of ANKB protein-protein interactions the phenotype associated with one particular variant could be anticipated to be vastly different from another depending on the amino acid location and the degree of conservation (chemical similarity). A variant located within the membrane binding domain is likely to have a different phenotype than a variant located within the spectrin binding domain as an ion channel disruption will result in altered signaling compared to losing a structural interaction.
Improved understanding of ANKB cellular roles and the effects of variant expression at a mechanistic level is needed to advance the identification possible therapeutic targets and biomarkers for individuals with ANK2 variants. Comprehensive characterization of ANKB's interacting and signaling partners would facilitate the design of small molecule modulators or repurposing of compounds to mitigate cellular pathology associated with ANKB variants. For example, inhibition of CamKII with KN-93 was able to mitigate RYR2 hyperphosphorylation and subsequent excessive calcium release in Ank2+/− pro-arrhythmogenic mouse hearts, resulting in a net reduction of RYR2 phosphorylation, calcium spark frequency, and delayed afterdepolarizations (95, 131, 132). More recently, inhibition of the GSK-3β pathway with SB-216763 (resulting in a net activation of the Wnt/beta-catenin signaling cascade) was effective in ameliorating cardiac remodeling in mice presenting with arrhythmogenic cardiomyopathy associated with cardiac specific loss of ANKB (44). However, given ANKB expression in other excitable tissues and the important roles linked to signaling pathways in which ANKB directly or indirectly participates, it is paramount to continue advancing the understanding of ANKB's role in cells and molecular pathways before defining and launching ANKB-targeting therapeutic programs. This is particularly important given the limited mechanistic appreciation of neurological phenotypes associated with ANK2 variants, such as seizure and white matter abnormalities. By exploiting the relatively conserved amino acid sequence and biological functions of ANKB and the availability of experimental model organisms, high-throughput cellular and molecular characterization of variants can bridge the gap to improved clinical understanding and development of targeted, specific therapeutic interventions.
The variability of clinical phenotypes associated with ANK2 variants poses challenges for treatment. At present, the understanding of the source of this variability is incomplete but could be partly due to the pleiotropic effects of ANKB, as well as surreptitious layering of variants in related pathways and/or environmental factors. The complexity and incomplete mechanistic understanding of ANKB cellular roles and regulation pose significant challenges for development of precise therapeutic interventions. As technological advances in personalized and precision medicine continue to expand, successful therapeutic strategies will arise from testing and modeling ANK2 variants directly on induced pluripotent stem cells derived from affected individuals themselves (133). A combination of experimental approaches, including personalized and precision medicine methods such as in vitro studies using patient-derived induced pluripotent stem cells, and model organism approaches will help to bridge the gaps to the identification of key pathways and therapeutics that target them safely and effectively. Current clinical efforts should therefore focus on monitoring carriers of ANK2 functional variants for arrhythmia and cardiomyopathy, along with symptomatic and treatment and control of co-morbidities (106).
Highlighted within this review are a variety of ANK2 variants and the different disease-linked phenotypes that arise as a result of their expression. Bringing together studies from model organisms and laboratory findings this review identifies potential mechanisms underlying ANKB dysfunction and possible contributions to disease. Investigating mechanisms underlying this link to disease will not only aid in our understanding of cellular pathways and ANKB's roles within them but will provide insight into disease risk and presentation. Understanding ANKB's roles in health and disease will advance the ability to translate this information into clinic and provide insights into developing treatments and therapies.
Funding
This work was supported by a Canadian Institutes for Health Research Project Grant (CIHR; PJT-169064) awarded to LTA and LAS. NY was supported by a CIHR Graduate Scholarship-Master's Program scholarship and University of Victoria Donor Awards. JCSA was supported by a Michael Smith Health Research BC Trainee Award (RT-2021-1735). LTA was supported by a BC Children's Hospital Research Institute Investigator award.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
Conceived by NY, LAS, and LTA. All authors wrote and revised the manuscript, contributed to the article, and approved the submitted version.
Acknowledgments
We acknowledge the Indigenous traditional territory of the W̱SÁNEĆ, Lekwungen, and Wyomilth peoples of the Coast Salish Nation and appreciate the continued partnership with the Gitxsan peoples and the Gitxsan Health Society as we address their research priorities together.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
RodenDM. Keep the QT interval: it is a reliable predictor of ventricular arrhythmias. Heart Rhythm. (2008) 5:1213–5. 10.1016/j.hrthm.2008.05.008
2.
O'NealWTSingletonMJRobertsJDTereshchenkoLGSotoodehniaNChenLYet al. The Association between QT interval components and sudden cardiac death: the atherosclerosis risk in communities study. Circ Arrhythm Electrophysiol. (2017) 10:e005485. 10.1161/CIRCEP.117.005485
3.
BohnenMSPengGRobeySHTerrenoireCIyerVSampsonKJet al. Molecular pathophysiology of congenital long QT syndrome. Physiol Rev. (2017) 97:89–134. 10.1152/physrev.00008.2016
4.
MohlerPJSchottJ-JGramoliniAODillyKWGuatimosimSduBellWHet al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. (2003) 421:634–9. 10.1038/nature01335
5.
MohlerPJSplawskiINapolitanoCBottelliGSharpeLTimothyKet al. cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U A. (2004) 101:9137–42. 10.1073/pnas.0402546101
6.
SmithSASturmACCurranJKlineCFLittleSCBonillaIMet al. Dysfunction in the βII Spectrin-dependent cytoskeleton underlies human arrhythmia. Circulation. (2015) 131:695–708. 10.1161/CIRCULATIONAHA.114.013708
7.
SwayneLAMurphyNPAsuriSChenLXuXMcIntoshSet al. A novel variant in the ANK2 membrane-binding domain is associated with Ankyrin-B syndrome and structural heart disease in a first nations population with a high rate of long QT syndrome. Circ Cardiovasc Genet. (2017) 10:e001537. 10.1161/CIRCGENETICS.116.001537
8.
MohlerPJLe ScouarnecSDenjoyILoweJSGuicheneyPCaronLet al. Defining the cellular phenotype of “Ankyrin-B Syndrome” variants. Circulation. (2007) 115:432–41. 10.1161/CIRCULATIONAHA.106.656512
9.
SatterstromFKKosmickiJAWangJBreenMSDe RubeisSAnJ-Yet al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. (2020) 180:568–84.e23. 10.1016/j.cell.2019.12.036
10.
WillseyAJSandersSJLiMDongSTebbenkampATMuhleRAet al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. (2013) 155:997–1007. 10.1016/j.cell.2013.10.020
11.
PengJZhouYWangK. Multiplex gene and phenotype network to characterize shared genetic pathways of epilepsy and autism. Sci Rep. (2021) 11:952. 10.1038/s41598-020-78654-y
12.
DavisJQBennettV. Brain ankyrin. a membrane-associated protein with binding sites for spectrin, tubulin, and the cytoplasmic domain of the erythrocyte anion channel. J Biol Chem. (1984) 259:13550–9. 10.1016/S0021-9258(18)90728-3
13.
BennettVStenbuckPJ. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membranes. Nature. (1979) 280:468–73. 10.1038/280468a0
14.
MohlerPJGramoliniAOBennettV. The Ankyrin-B C-terminal domain determines activity of Ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-Trisphosphate and ryanodine receptor distribution in Ankyrin-B (–/–) Neonatal Cardiomyocytes*. J Biol Chem. (2002) 277:10599–607. 10.1074/jbc.M110958200
15.
AbdiKMMohlerPJDavisJQBennettV. Isoform specificity of ankyrin-b: a site in the divergent C-TERMINAL domain is required for intramolecular association*. J Biol Chem. (2006) 281:5741–9. 10.1074/jbc.M506697200
16.
GalianoMRJhaSHoTSZhangCOgawaYChangK-Jet al. distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell. (2012) 149:1125–39. 10.1016/j.cell.2012.03.039
17.
ParkHHLoY-CLinS-CWangLYangJKWuH. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. (2007) 25:561–86. 10.1146/annurev.immunol.25.022106.141656
18.
TuviaSBuhusiMDavisLReedyMBennettV. Ankyrin-B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. J Cell Biol. (1999) 147:995–1008. 10.1083/jcb.147.5.995
19.
LorenzoDNHealyJAHostettlerJDavisJYangJWangCet al. Ankyrin-B metabolic syndrome combines age-dependent adiposity with pancreatic β cell insufficiency. J Clin Invest. (2015) 125:3087–102. 10.1172/JCI81317
20.
OttoEKunimotoMMcLaughlinTBennettV. Isolation and characterization of cDNAs encoding human brain ankyrins reveal a family of alternatively spliced genes. J Cell Biol. (1991) 114:241–53. 10.1083/jcb.114.2.241
21.
TseWTMenningerJCYang-FengTLFranckeUSahrKELuxSEet al. Isolation and chromosomal localization of a novel nonerythroid ankyrin gene. Genomics. (1991) 10:858–66. 10.1016/0888-7543(91)90173-C
22.
KunimotoMOttoEBennettV. A new 440-kD isoform is the major ankyrin in neonatal rat brain. J Cell. (1991) 115:1319–31. 10.1083/jcb.115.5.1319
23.
KunimotoMAdachiTIshidoM. Expression and localization of brain ankyrin isoforms and related proteins during early developmental stages of rat nervous system. J Neurochem. (1998) 71:2585–92. 10.1046/j.1471-4159.1998.71062585.x
24.
WuHCYamankurtGLuoJSubramaniamJHashmiSSHuHet al. Identification and characterization of two ankyrin-B isoforms in mammalian heart. Cardiovasc Res. (2015) 107:466–77. 10.1093/cvr/cvv184
25.
CunhaSRLe ScouarnecSSchottJ-JMohlerPJ. Exon organization and novel alternative splicing of the human ANK2 gene: implications for cardiac function and human cardiac disease. J Mol Cell Cardiol. (2008) 45:724–34. 10.1016/j.yjmcc.2008.08.005
26.
AspMGiacomelloSLarssonLWuCFürthDQianXet al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. (2019) 179:1647–60.e19. 10.1016/j.cell.2019.11.025
27.
KangHJKawasawaYIChengFZhuYXuXLiMet al. Spatiotemporal transcriptome of the human brain. Nature. (2011) 478:483–9. 10.1038/nature10523
28.
FertuzinhosSLiMKawasawaYIIvicVFranjicDSinghDet al. Laminar and temporal expression dynamics of coding and noncoding RNAs in the mouse neocortex. Cell Rep. (2014) 6:938–50. 10.1016/j.celrep.2014.01.036
29.
KawanoSBabaMFukushimaHMiuraDHashimotoHNakazawaT. Autism-associated ANK2 regulates embryonic neurodevelopment. Biochem Biophys Res Commun. (2022) 605:45–50. 10.1016/j.bbrc.2022.03.058
30.
ZhangYChenKSloanSABennettMLScholzeARO'KeeffeSet al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. 10.1523/JNEUROSCI.1860-14.2014
31.
BakkenTEJorstadNLHuQLakeBBTianWKalmbachBEet al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. (2021) 598:111–9. 10.1038/s41586-021-03465-8
32.
KoenigSNMohlerPJ. The evolving role of Ankyrin-B in cardiovascular disease. Heart Rhythm. (2017) 14:1884–9. 10.1016/j.hrthm.2017.07.032
33.
GiudicessiJRWildeAAMAckermanMJ. The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc Med. (2018) 28:453–64. 10.1016/j.tcm.2018.03.003
34.
GhouseJHaveCTWeekePBille NielsenJAhlbergGBalslev-HarderMet al. Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur Heart J. (2015) 36:2523–9. 10.1093/eurheartj/ehv297
35.
AdlerANovelliVAminASAbiusiECareMNannenbergEAet al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation. (2020) 141:418–28. 10.1161/CIRCULATIONAHA.119.043132
36.
GiudicessiJRAckermanMJ. Established loss-of-function variants in ANK2-Encoded Ankyrin-B rarely cause a concerning cardiac phenotype in humans. Circ Genomic Precis Med. (2020) 13:80–2. 10.1161/CIRCGEN.119.002851
37.
MusaHMurphyNPCurranJHigginsJDWebbTRMakaraMAet al. Common human ANK2 variant confers in vivo arrhythmia phenotypes. Heart Rhythm. (2016) 13:1932–40. 10.1016/j.hrthm.2016.06.012
38.
ArbourLRezazadehSEldstromJWeget-SimmsGRuppsRDyerZet al. A KCNQ1 V205M missense mutation causes a high rate of long QT syndrome in a first nations community of northern British Columbia: a community-based approach to understanding the impact. Genet Med Off J Am Coll Med Genet. (2008) 10:545–50. 10.1097/GIM.0b013e31817c6b19
39.
HuqAPertileMDavisALandonHJamesPKlineCet al. Novel mechanism for human cardiac Ankyrin-B syndrome due to reciprocal chromosomal translocation. Heart Lung Circ. (2017) 26:612–8. 10.1016/j.hlc.2016.09.013
40.
SongJSasmitaBRDengG. Ankyrin-2 genetic variants: a case of Ankyrin-B syndrome. Ann Noninvasive Electrocardiol. (2022). 10.1111/anec.12933
41.
SumitomoN. Current topics in catecholaminergic polymorphic ventricular tachycardia. J Arrhythmia. (2016) 32:344–51. 10.1016/j.joa.2015.09.008
42.
Coban-AkdemirZHCharngW-LAzamianMPaineISPunethaJGrochowskiCMet al. Wolff-parkinson-white syndrome: de novo variants and evidence for mutational burden in genes associated with atrial fibrillation. Am J Med Genet A. (2020) 182:1387–99. 10.1002/ajmg.a.61571
43.
LopesLRSyrrisPGuttmannOPO'MahonyCTangHCDalageorgouCet al. Novel genotype–phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. (2015) 101:294–301. 10.1136/heartjnl-2014-306387
44.
RobertsJDMurphyNPHamiltonRMLubbersERJamesCAKlineCFet al. Ankyrin-B dysfunction predisposes to arrhythmogenic cardiomyopathy and is amenable to therapy. J Clin Invest. (2019) 129:3171–84. 10.1172/JCI125538
45.
MohlerPJHealyJAXueHPucaAAKlineCFRand AllinghamRet al. Ankyrin-B syndrome: enhanced cardiac function balanced by risk of cardiac death and premature senescence. PLoS ONE. (2007) 2:e1051. 10.1371/journal.pone.0001051
46.
IchikawaMAibaTOhnoSShigemizuDOzawaJSonodaKet al. Phenotypic variability of ANK2 mutations in patients with inherited primary arrhythmia syndromes. Circ J. (2016) 80:2435–42. 10.1253/circj.CJ-16-0486
47.
LeppaVMKravitzSNMartinCLAndrieuxJLe CaignecCMartin-CoignardDet al. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am J Hum Genet. (2016) 99:540–54. 10.1016/j.ajhg.2016.06.036
48.
YuenRKThiruvahindrapuramBMericoDWalkerSTammimiesKHoangNet al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. (2015) 21:185–91. 10.1038/nm.3792
49.
IossifovIRonemusMLevyDWangZHakkerIRosenbaumJet al. De novo gene disruptions in children on the autistic spectrum. Neuron. (2012) 74:285–99. 10.1016/j.neuron.2012.04.009
50.
BowlingKMThompsonMLAmaralMDFinnilaCRHiattSMEngelKLet al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. (2017) 9:43. 10.1186/s13073-017-0433-1
51.
YangRWalder-ChristensenKKKimNWuDLorenzoDNBadeaAet al. ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc Natl Acad Sci U S A. (2019) 116:15262–71. 10.1073/pnas.1904348116
52.
GuoWShangD-MCaoJ-HFengKHeY-CJiangYet al. Identifying and analyzing novel epilepsy-related genes using random walk with restart algorithm. BioMed Res Int. (2017) 2017:1–13. 10.1155/2017/6132436
53.
HataYYoshidaKKinoshitaKNishidaN. Epilepsy-related sudden unexpected death: targeted molecular analysis of inherited heart disease genes using next-generation DNA sequencing. Brain Pathol. (2017) 27:292–304. 10.1111/bpa.12390
54.
CostagliolaGOrsiniACollMBrugadaRParisiPStrianoP. The brain–heart interaction in epilepsy: implications for diagnosis, therapy, and SUDEP prevention. Ann Clin Transl Neurol. (2021) 8:1557–68. 10.1002/acn3.51382
55.
ChengCYReichDHaimanCATandonAPattersonNElizabethSet al. African Ancestry and its correlation to type 2 diabetes in African Americans: a genetic admixture analysis in three US population cohorts. PLoS ONE. (2012) 7:e32840. 10.1371/journal.pone.0032840
56.
NgMC. Genetics of Type 2 diabetes in African Americans. Curr Diab Rep. (2015) 15:74. 10.1007/s11892-015-0651-0
57.
ScotlandPZhouDBenvenisteHBennettV. Nervous system defects of AnkyrinB (–/–) Mice suggest functional overlap between the cell adhesion molecule L1 and 440-kD AnkyrinB in premyelinated axons. J Cell Biol. (1998) 143:1305–15. 10.1083/jcb.143.5.1305
58.
CreightonBAAfriyieSAjitDCasingalCRVoosKMRegerJet al. Giant ankyrin-B mediates transduction of axon guidance and collateral branch pruning factor sema 3A. Elife. (2021) 10:e69815. 10.7554/eLife.69815
59.
LorenzoDNBadeaAZhouRMohlerPJZhuangXBennettV. βII-spectrin promotes mouse brain connectivity through stabilizing axonal plasma membranes and enabling axonal organelle transport. Proc Natl Acad Sci U S A. (2019) 116:15686–95. 10.1073/pnas.1820649116
60.
MohlerPJDavisJQBennettV. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. (2005) 3:e423. 10.1371/journal.pbio.0030423
61.
SmithRSWalshCA. Ion channel functions in early brain development. Trends Neurosci. (2020) 43:103–14. 10.1016/j.tins.2019.12.004
62.
Porter GAJrMakuckRFRivkeesSA. Intracellular calcium plays an essential role in cardiac development. Dev Dyn. (2003) 227:280–90. 10.1002/dvdy.10307
63.
RosenbergSSSpitzerNC. Calcium signaling in neuronal development. Cold Spring Harb Perspect Biol. (2011) 3:a004259. 10.1101/cshperspect.a004259
64.
CaiXZhangY. Molecular evolution of the ankyrin gene family. Mol Biol Evol. (2006) 23:550–8. 10.1093/molbev/msj056
65.
HopitzanAABainesAJKordeliE. Molecular evolution of Ankyrin: gain of function in vertebrates by acquisition of an Obscurin/Titin-Binding–related domain. Mol Biol Evol. (2006) 23:46–55. 10.1093/molbev/msj004
66.
BennettVBainesAJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. (2001) 81:1353–92. 10.1152/physrev.2001.81.3.1353
67.
HeMTsengW-CBennettV. A single divergent exon inhibits Ankyrin-B association with the plasma membrane. J Biol Chem. (2013) 288:14769–79. 10.1074/jbc.M113.465328
68.
KordeliELambertSBennettV. AnkyrinG: a new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier (*). J Biol Chem. (1995) 270:2352–9. 10.1074/jbc.270.5.2352
69.
MohlerPJHoffmanJADavisJQAbdiKMKimC-RJonesSKet al. Isoform specificity among ankyrins: an amphipathic α-helix in the divergent regulatory domain of ankyrin-b interacts with the molecular co-chaperone Hdj1/Hsp40*. J Biol Chem. (2004) 279:25798–804. 10.1074/jbc.M401296200
70.
ChangKJZollingerDRSusukiKShermanDLMakaraMABrophyPJet al. Glial ankyrins facilitate paranodal axoglial junction assembly. Nat Neurosci. (2014) 17:1673–81. 10.1038/nn.3858
71.
HoTSZollingerDRChangK-JXuMCooperECStankewichMCet al. Hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci. (2014) 17:1664–72. 10.1038/nn.3859
72.
RappelW-JEdelstein-KeshetL. Mechanisms of cell polarization. Curr Opin Syst Biol. (2017) 3:43–53. 10.1016/j.coisb.2017.03.005
73.
Vicente-ManzanaresMSánchez-MadridF. Cell polarization: a comparative cell biology and immunological view. Dev Immunol. (2000) 7:51–65. 10.1155/2000/70801
74.
HirschyASchatzmannFEhlerEPerriardJ-C. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. (2006) 289:430–41. 10.1016/j.ydbio.2005.10.046
75.
HendersonDJAndersonRH. The development and structure of the ventricles in the human heart. Pediatr Cardiol. (2009) 30:588–96. 10.1007/s00246-009-9390-9
76.
TakanoTXuCFunahashiYNambaTKaibuchiK. Neuronal polarization. Development. (2015) 142:2088–93. 10.1242/dev.114454
77.
BoontrakulpoontaweePOtsukaA. Mutational analysis of the Caenorhabditis elegans ankyrin gene unc-44 demonstrates that the large spliceoform is critical for neural development. Mol Genet Genomics. (2002) 267:291–302. 10.1007/s00438-002-0661-x
78.
OtsukaAJFrancoRYangBShimKHTangLZZhangYYet al. An ankyrin-related gene (unc-44) is necessary for proper axonal guidance in Caenorhabditis elegans. J Cell Biol. (1995) 129:1081–92. 10.1083/jcb.129.4.1081
79.
ManiarTAKaplanMWangGJShenKWeiLShawJEet al. UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nat Neurosci. (2011) 15:48–56. 10.1038/nn.2970
80.
HeLKooistraRDasROudejansEvan LeenEZieglerJet al. Cortical anchoring of the microtubule cytoskeleton is essential for neuron polarity. Elife. (2020) 9:e55111. 10.7554/eLife.55111
81.
HarterinkMEdwardsSLde HaanBYauKWvan den HeuvelSKapiteinLCet al. Local microtubule organization promotes cargo transport in C. elegans dendrites. J Cell Sci. (2018) 131:jcs223107. 10.1242/jcs.223107
82.
McIntireSLGarrigaGWhiteJJacobsonDRobert HorvitzH. Genes necessary for directed axonal elongation or fasciculation in C. elegans. Neuron. (1992) 8:307–22. 10.1016/0896-6273(92)90297-Q
83.
DesaiCGarrigaGMclntireSLHorvitzHR. A genetic pathway for the development of the Caenorhabditis elegans HSN motor neurons. Nature. (1988) 336:638–46. 10.1038/336638a0
84.
MengLChenCYanD. Regulation of gap junction dynamics by UNC-44/ankyrin and UNC-33/CRMP through VAB-8 in C. elegans neurons. PLoS Genet. (2016) 12:e1005948. 10.1371/journal.pgen.1005948
85.
MontoroRJYusteR. Gap junctions in developing neocortex: a review. Brain Res Rev. (2004) 47:216–26. 10.1016/j.brainresrev.2004.06.009
86.
BelousovABFontesJD. Neuronal gap junctions: making and breaking connections during development and injury. Trends Neurosci. (2013) 36:227–36. 10.1016/j.tins.2012.11.001
87.
HortschMPaisleyKLTianM-ZQianMBouleyMChandlerR. The axonal localization of large drosophila Ankyrin2 protein isoforms is essential for neuronal functionality. Mol Cell Neurosci. (2002) 20:43–55. 10.1006/mcne.2002.1113
88.
KochISchwarzHBeuchleDGoellnerBLangeggerMAberleH. Drosophila Ankyrin 2 is required for synaptic stability. Neuron. (2008) 58:210–22. 10.1016/j.neuron.2008.03.019
89.
PielageJChengLFetterRDCarltonPMSedatJWDavisGWet al. Presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion. Neuron. (2008) 58:195–209. 10.1016/j.neuron.2008.02.017
90.
FanYHoBXPangJKSPekNMQHorJHNgS-Yet al. Wnt/β-catenin-mediated signaling re-activates proliferation of matured cardiomyocytes. Stem Cell Res Ther. (2018) 9:338. 10.1186/s13287-018-1086-8
91.
KomiyaYHabasR. Wnt signal transduction pathways. Organogenesis. (2008) 4:68–75. 10.4161/org.4.2.5851
92.
PahnkeAConantGHuyerLDZhaoYFericNRadisicM. The role of Wnt regulation in heart development, cardiac repair and disease: a tissue engineering perspective. Biochem Biophys Res Commun. (2016) 473:698–703. 10.1016/j.bbrc.2015.11.060
93.
MénardCPupierSMornetDKitzmannMNargeotJLoryP. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J Biol Chem. (1999) 274:29063–70. 10.1074/jbc.274.41.29063
94.
ChenLChoiCSWSanchez-AriasJCArbourLTSwayneLA. Ankyrin-B pS646F undergoes increased proteasome degradation and reduces cell viability in the H9c2 rat ventricular cardiomyoblast cell line. Biochem Cell Biol. (2020) 98:299–306. 10.1139/bcb-2019-0082
95.
ZhuWWangCHuJWanRYuJXieJet al. Ankyrin-B Q1283H variant linked to arrhythmias via loss of local protein phosphatase 2A activity causes ryanodine receptor hyperphosphorylation. Circulation. (2018) 138:2682–97. 10.1161/CIRCULATIONAHA.118.034541
96.
DurakOde AndaFCSinghKKLeussisMPPetryshenTLSklarPet al. Ankyrin-G regulates neurogenesis and Wnt signaling by altering the subcellular localization of β-catenin. Mol Psychiatry. (2015) 20:388–97. 10.1038/mp.2014.42
97.
ThompsonJJWilliamsCS. Protein phosphatase 2A in the regulation of Wnt signaling, stem cells, and cancer. Genes. (2018) 9:121. 10.3390/genes9030121
98.
ChenKYangRLiYZhouJCZhangM. Giant ankyrin-B suppresses stochastic collateral axon branching through direct interaction with microtubules. J Cell Biol. (2020) 219:e201910053. 10.1083/jcb.201910053
99.
BenkarimOPaquolaCParkBHongS-JRoyerJVos de WaelRet al. Connectivity alterations in autism reflect functional idiosyncrasy. Commun Biol. (2021) 4:1–15. 10.1038/s42003-021-02572-6
100.
NomiJSUddinLQ. Developmental changes in large-scale network connectivity in autism. NeuroImage Clin. (2015) 7:732–41. 10.1016/j.nicl.2015.02.024
101.
KlineCFScottJCurranJHundTJMohlerPJ. Ankyrin-B regulates Cav21 and Cav22 channel expression and targeting*. J Biol Chem. (2014) 289:5285–95. 10.1074/jbc.M113.523639
102.
ChoiCSWSouzaIASanchez-AriasJCZamponiGWArbourLT. Swayne LA. Ankyrin B and Ankyrin B variants differentially modulate intracellular and surface Cav21 levels. Mol Brain. (2019) 12:75. 10.1186/s13041-019-0494-8
103.
LencesovaLO'NeillAResneckWGBlochRJBlausteinMP. Plasma membrane-cytoskeleton-endoplasmic reticulum complexes in neurons and astrocytes. J Biol Chem. (2004) 279:2885–93. 10.1074/jbc.M310365200
104.
CamorsEMohlerPJBersDMDespaS. Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J Mol Cell Cardiol. (2012) 52:1240–8. 10.1016/j.yjmcc.2012.02.010
105.
CunhaSRBhasinNMohlerPJ. Targeting and stability of Na/Ca Exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor Ankyrin-B*. J Biol Chem. (2007) 282:4875–83. 10.1074/jbc.M607096200
106.
SucharskiHCDudleyEKKeithCBREl RefaeyMKoenigSNMohlerPJ. Mechanisms and alterations of cardiac ion channels leading to disease: role of Ankyrin-B in cardiac function. Biomolecules. (2020) 10:211. 10.3390/biom10020211
107.
SchuhmacherDSontagJ-MSontagE. Protein phosphatase 2A: more than a passenger in the regulation of epithelial cell–cell junctions. Front Cell Dev Biol. (2019) 7:30. 10.3389/fcell.2019.00030
108.
WlodarchakNXingY. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol. (2016) 51:162–84. 10.3109/10409238.2016.1143913
109.
KirchheferUHeinickAKönigSKristensenTMüllerFUSeidlMDet al. Protein phosphatase 2A is regulated by protein kinase Cα (PKCα)-dependent phosphorylation of its targeting subunit B56α at Ser41*. J Biol Chem. (2014) 289:163–76. 10.1074/jbc.M113.507996
110.
LecuonaEDadaLASunHButtiMLZhouGChewT-Let al. Na,K-ATPase alpha1-subunit dephosphorylation by protein phosphatase 2A is necessary for its recruitment to the plasma membrane. FASEB J. (2006) 20:2618–20. 10.1096/fj.06-6503fje
111.
HoffmanATaleskiGSontagE. The protein serine/threonine phosphatases PP2A, PP1 and calcineurin: a triple threat in the regulation of the neuronal cytoskeleton. Mol Cell Neurosci. (2017) 84:119–31. 10.1016/j.mcn.2017.01.005
112.
YehP-AChangC-J. A novel function of twins, B subunit of protein phosphatase 2A, in regulating actin polymerization. PLoS ONE. (2017) 12:e0186037. 10.1371/journal.pone.0186037
113.
WestphalRSColbranRJ.EbnerFFWadzinskiBE. Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res. (1997) 49:15–28. 10.1016/s0169-328x(97)00117-4
114.
HealyJANilssonKRHohmeierHEBerglundJDavisJHoffmanJet al. Cholinergic augmentation of insulin release requires Ankyrin-B. Sci Signal. (2010) 3:ra19–ra19. 10.1126/scisignal.2000771
115.
KomatsuMTakeiMIshiiHSatoY. Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Investig. (2013) 4:511–6. 10.1111/jdi.12094
116.
AhrénB. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia. (2000) 43:393–410. 10.1007/s001250051322
117.
LorenzoDNBennettV. Cell-autonomous adiposity through increased cell surface GLUT4 due to ankyrin-B deficiency. Proc Natl Acad Sci U S A. (2017) 114:12743–8. 10.1073/pnas.1708865114
118.
LopesLRZekavatiASyrrisPHubankMGiambartolomeiCDalageorgouCet al. Uk10k Consortium, Plagnol V, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. (2013) 50:228–39. 10.1136/jmedgenet-2012-101270
119.
Sabater-MolinaMPérez-SánchezIHernández Del RincónJPGimenoJR. Genetics of hypertrophic cardiomyopathy: a review of current state. Clin Genet. (2018) 93:3–14. 10.1111/cge.13027
120.
GuanHManessPF. Perisomatic GABAergic innervation in prefrontal cortex is regulated by ankyrin interaction with the L1 cell adhesion molecule. Cereb Cortex N Y NY. (2010) 20:2684–93. 10.1093/cercor/bhq016
121.
LitvinukováMTalavera-LópezCMaatzHReichartDWorthCLLindbergELet al. Cells of the adult human heart. Nature. (2020) 588:466–72. 10.1038/s41586-020-2797-4
122.
TuckerNRChaffinMFlemingSJHallAWParsonsVABediKCet al. Transcriptional and cellular diversity of the human heart. Circulation. (2020) 142:466–82. 10.1161/CIRCULATIONAHA.119.045401
123.
Kikel-CouryNLBrandtJPCorreiaIAO'DeaMRDeSantisDFSterlingFet al. Identification of astroglia-like cardiac nexus glia that are critical regulators of cardiac development and function. PLoS Biol. (2021) 19:e3001444. 10.1371/journal.pbio.3001444
124.
SmithKRPenzesP. Ankyrins: roles in synaptic biology and pathology. Mol Cell Neurosci. (2018) 91:131–9. 10.1016/j.mcn.2018.04.010
125.
LiJZhangWYangHHowriganDPWilkinsonBSouaiaiaTet al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat Neurosci. (2017) 20:1150–61. 10.1038/nn.4594
126.
LorenzoDN. Cargo hold and delivery: ankyrins, spectrins, and their functional patterning of neurons. Cytoskeleton. (2020) 77:129–48. 10.1002/cm.21602
127.
WalshRAdlerAAminASAbiusiECareMBikkerHet al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur Heart J. (2022) 43:1500–10. 10.1093/eurheartj/ehab687
128.
KidoTSikora-WohlfeldWKawashimaMKikuchiSKamataniNPatwardhanAet al. Are minor alleles more likely to be risk alleles?BMC Med Genomics. (2018) 11:3. 10.1186/s12920-018-0322-5
129.
CerroneMRemmeCATadrosRBezzinaCRDelmarM. Beyond the one gene-one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. (2019) 140:595–610. 10.1161/CIRCULATIONAHA.118.035954
130.
GessnerGRungeSKoenenMHeinemannSHKoenenMHaasJet al. ANK2 functionally interacts with KCNH2 aggravating long QT syndrome in a double mutation carrier. Biochem Biophys Res Commun. (2019) 512:845–51. 10.1016/j.bbrc.2019.03.162
131.
DeGrandeSNixonDKovalOCurranJWWrightPWangQet al. CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm. (2012) 9:2034–41. 10.1016/j.hrthm.2012.08.026
132.
PopescuIGaliceSMohlerPJDespaS. Elevated local [Ca 2+] and CaMKII promote spontaneous Ca 2+ release in ankyrin-B-deficient hearts. Cardiovasc Res. (2016) 111:287–94. 10.1093/cvr/cvw093
133.
MatsaEAhrensJHWuJC. Human induced pluripotent stem cells as a platform for personalized and precision cardiovascular medicine. Physiol Rev. (2016) 96:1093–126. 10.1152/physrev.00036.2015
134.
KrummNTurnerTNBakerCVivesLMohajeriKWitherspoonKet al. Excess of rare, inherited truncating mutations in autism. Nat Genet. (2015) 47:582–8. 10.1038/ng.3303
135.
IossifovIO'RoakBJSandersSJRonemusMKrummNLevyDet al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. 10.1038/nature13908
136.
Le ScouarnecSBhasinNVieyresCHundTJCunhaSRKovalOet al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci U S A. (2008) 105:15617–22. 10.1073/pnas.0805500105
137.
CunhaSRHundTJHashemiSVoigtNLiNWrightPet al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. (2011) 124:1212–22. 10.1161/CIRCULATIONAHA.111.023986
138.
Garcia-CaballeroAZhangF-XHodgkinsonVHuangJChenLSouzaIAet al. T-type calcium channels functionally interact with spectrin (α/β) and ankyrin B. Mol Brain. (2018) 11:24. 10.1186/s13041-018-0368-5
139.
KlineCFKurataHTHundTJCunhaSRKovalOMWrightPJet al. Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc Natl Acad Sci U S A. (2009) 106:16669–74. 10.1073/pnas.0907138106
140.
LiJKlineCFHundTJAndersonMEMohlerPJ. Ankyrin-B regulates Kir62 membrane expression and function in heart*. J Biol Chem. (2010) 285:28723–30. 10.1074/jbc.M110.147868
141.
LiuXSpicarováZRydholmSLiJBrismarHAperiaA. Ankyrin B modulates the function of Na,K-ATPase/inositol 1,4,5-trisphosphate receptor signaling microdomain. J Biol Chem. (2008) 283:11461–8. 10.1074/jbc.M706942200
142.
GudmundssonHHundTJWrightPJKlineCFSnyderJSQianLet al. EH domain proteins regulate cardiac membrane protein targeting. Circ Res. (2010) 107:84–95. 10.1161/CIRCRESAHA.110.216713
143.
GilODSakuraiTBradleyAEFinkMYCassellaMRKuoJAet al. Ankyrin binding mediates L1CAM interactions with static components of the cytoskeleton and inhibits retrograde movement of L1CAM on the cell surface. J Cell Biol. (2003) 162:719–30. 10.1083/jcb.200211011
144.
AyalonGHostettlerJDHoffmanJKizhatilKDavisJQBennettV. Ankyrin-B Interactions with Spectrin and Dynactin-4 Are Required for Dystrophin-based protection of skeletal muscle from exercise injury. J Biol Chem. (2011) 286:7370–8. 10.1074/jbc.M110.187831
145.
AyalonGDavisJQScotlandPBBennettV. An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell. (2008) 135:1189–200. 10.1016/j.cell.2008.10.018
146.
LorenzoDNBadeaADavisJHostettlerJHeJZhongGet al. PIK3C3–Ankyrin-B–Dynactin pathway promotes axonal growth and multiorganelle transport. J Cell Biol. (2014) 207:735–52. 10.1083/jcb.201407063
147.
MohlerPJYoonWBennettV. Ankyrin-B Targets β2-Spectrin to an intracellular compartment in neonatal cardiomyocytes*. J Biol Chem. (2004) 279:40185–93. 10.1074/jbc.M406018200
148.
QuFLorenzoDNKingSJBrooksRBearJEBennettV. Ankyrin-B is a PI3P effector that promotes polarized α5β1-integrin recycling via recruiting RabGAP1L to early endosomes. Elife. (2016) 5:e20417. 10.7554/eLife.20417
149.
BhasinNCunhaSRMudannayakMGigenaMSRogersTBMohlerPJ. Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am J Heart Circ Physiol. (2007) 293:H109–19. 10.1152/ajpheart.00059.2007
150.
LittleSCCurranJMakaraMAKlineCFHoH-TXuZet al. Protein phosphatase 2A regulatory subunit B56α limits phosphatase activity in the heart. Sci Signal. (2015) 8:ra72. 10.1126/scisignal.aaa5876
151.
YeJLiJYeFZhangYZhangMWangC. Mechanistic insights into the interactions of dynein regulator Ndel1 with neuronal ankyrins and implications in polarity maintenance. Proc Natl Acad Sci U S A. (2020) 117:1207–15. 10.1073/pnas.1916987117
152.
CunhaSRMohlerPJ. Obscurin targets Ankyrin-B and protein phosphatase 2A to the Cardiac M-line. J Biol Chem. (2008) 283:31968–80. 10.1074/jbc.M806050200
153.
Di MeoDRavindranPSadhanasatishTDhumalePPüschelAW. The balance of mitochondrial fission and fusion in cortical axons depends on the kinases SadA and SadB. Cell Rep. (2021) 37:110141. 10.1016/j.celrep.2021.110141
Summary
Keywords
scaffolding protein, cellular morphology, calcium homeostasis, excitation-contraction coupling, arrhythmia, sudden cardiac death, seizure, autism spectrum disorders
Citation
York NS, Sanchez-Arias JC, McAdam ACH, Rivera JE, Arbour LT and Swayne LA (2022) Mechanisms underlying the role of ankyrin-B in cardiac and neurological health and disease. Front. Cardiovasc. Med. 9:964675. doi: 10.3389/fcvm.2022.964675
Received
08 June 2022
Accepted
11 July 2022
Published
04 August 2022
Volume
9 - 2022
Edited by
Joanne E. Curran, The University of Texas Rio Grande Valley, United States
Reviewed by
Chantal Josephina Maria Van Opbergen, Grossman School of Medicine, New York University, United States; Jinzhu Hu, Nanchang University, China
Updates
Copyright
© 2022 York, Sanchez-Arias, McAdam, Rivera, Arbour and Swayne.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura T. Arbour larbour@uvic.caLeigh Anne Swayne lswayne@uvic.ca
This article was submitted to Cardiovascular Genetics and Systems Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.