 Jun Yasuhara
Jun Yasuhara Karlee Schultz
Karlee Schultz Amee M. Bigelow2,4
Amee M. Bigelow2,4 Vidu Garg
Vidu Garg- 1Center for Cardiovascular Research, Abigail Wexner Research Institute, Nationwide Children’s Hospital, Columbus, OH, United States
- 2Heart Center, Nationwide Children’s Hospital, Columbus, OH, United States
- 3Medical Student Research Program, The Ohio State University College of Medicine, Columbus, OH, United States
- 4Department of Pediatrics, The Ohio State University, Columbus, OH, United States
- 5Department of Molecular Genetics, The Ohio State University, Columbus, OH, United States
Congenital aortic valve stenosis (AVS) is one of the most common valve anomalies and accounts for 3%–6% of cardiac malformations. As congenital AVS is often progressive, many patients, both children and adults, require transcatheter or surgical intervention throughout their lives. While the mechanisms of degenerative aortic valve disease in the adult population are partially described, the pathophysiology of adult AVS is different from congenital AVS in children as epigenetic and environmental risk factors play a significant role in manifestations of aortic valve disease in adults. Despite increased understanding of genetic basis of congenital aortic valve disease such as bicuspid aortic valve, the etiology and underlying mechanisms of congenital AVS in infants and children remain unknown. Herein, we review the pathophysiology of congenitally stenotic aortic valves and their natural history and disease course along with current management strategies. With the rapid expansion of knowledge of genetic origins of congenital heart defects, we also summarize the literature on the genetic contributors to congenital AVS. Further, this increased molecular understanding has led to the expansion of animal models with congenital aortic valve anomalies. Finally, we discuss the potential to develop novel therapeutics for congenital AVS that expand on integration of these molecular and genetic advances.
Introduction
Congenital heart disease (CHD) is the most frequently occurring birth defect with an incidence of 0.8%–1.2% among live births (1, 2). Congenital aortic valve stenosis (AVS) has an incidence of 3.8–4.9 per 10,000 live births, representing ∼3%–6% of all CHD (1–3). Congenital AVS occurs more commonly in males as compared to females, with a reported ratio ranging from 3 to 5:1 (4, 5). It is defined as an obstruction of the aortic valve orifice due to a congenital valve malformation, which could be in the form of a bicuspid aortic valve (BAV), unicuspid aortic valve, or fused or malformed aortic valve cusps (6, 7) Associated CHD is found in approximately 20% of patients with congenital AVS, including ventricular septal defect (VSD), coarctation of the aorta (CoA), and patent ductus arteriosus (8). In this review, we aim to describe our understanding of aortic valve development with a particular focus on the embryology, anatomy and pathology relevant to congenital AVS. Further, we discuss the clinical characteristics of congenital AVS, including the natural history and current treatment options. Lastly, we highlight the molecular genetics of congenital AVS and discuss the prospects for the development of future potential therapeutics.
Embryology
Heart development occurs during the first trimester of pregnancy in humans from the embryonic gestational ages of 6 to 9 weeks. Here, we focus on semilunar valve development and refer the reader to comprehensive reviews on cardiac morphogenesis for details on this process (9, 10). Development of semilunar valves, which include the aortic valve and pulmonary valve, initiates between 7 and 9 weeks of gestation in the human embryo, and development of mature valve cusps continues after birth (11). This process has been well studied in mouse embryogenesis and is detailed here (12, 13). The primitive linear heart tube consists of an outer layer of myocardium and an inner layer of endocardium at embryonic day (E) 8.0. By E9.5, the heart has undergone rightward looping and is composed of the following 4 segments: the atrium, atrioventricular canal (AVC), ventricle, and outflow tract (OFT). During this time, a subset of endocardial cells forms swellings known as endocardial cushions within the AVC and embryonic cardiac OFT. In response to molecular signals from the adjacent myocardium, endocardial cells covering the cushions undergo endothelial-to-mesenchymal transformation (EMT) between E9.5 and E11.5 (Figure 1A). These mesenchymal cells migrate into the cardiac jelly and proliferate to occupy the endocardial cushions in the proximal OFT, while in the distal OFT the cardiac neural crest cells (CNC) are the major contributor. By E11.5, the AVC and OFT cushions have completed cellularization. After E11.5, the AVC and OFT cushions rapidly grow and remodel. This involves both apoptosis of valvular interstitial cells (VICs) as well as dynamic ECM arrangement (Figure 1B). Complex molecular networks tightly regulate each step of EMT, including the transforming growth factor-β (TGF-β) (14, 15), bone morphogenetic protein (BMP) (15, 16), WNT (17, 18), Notch (19, 20) and vascular endothelial growth factor A (VEGF) signaling pathways (21, 22). Although EMT is required for pooling mesenchyme valve precursors within the endocardial cushions, other cell lineages also play an essential role, including the CNC and secondary heart field (SHF) cells (23–27). CNC cells occupy the distal outflow tract cushions after migration from the aortic sac. By E12.5, the distal outflow tract is divided into the into the aorta and pulmonary artery by the aortopulmonary septum. On the other hand, the proximal outflow tract cushion contains mostly EMT-derived mesenchymal cells. During cushion development, the two cushions fuse at the distal-proximal boundary, where neural crest- and endothelium-derived mesenchymal cells meet, and the proximal outflow tract is separated into two ventricular outlets. The non-fused cushions then undergo extensive ECM remodeling and morphological sculpting and extend to become the coronary apex of the aortic valve. Mature left and right coronary cusps are derived primarily from endothelium-derived mesenchymal progenitor cells, with little contribution from CNC cells, while non-coronary cusp contains cells of the SHF lineage (28, 29).
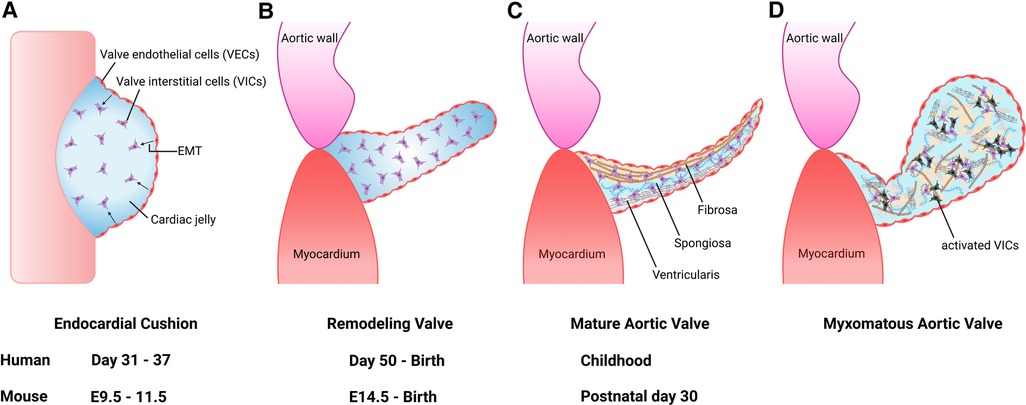
Figure 1. Aortic valve development and progression to myxomatous aortic valve. (A) Valve development begins with endocardial cushion formation. Valve endothelial cells (VECs) lining the cushion undergo endothelial-to-mesenchymal transformation (EMT) to differentiate into valve interstitial cells (VICs) between 7 and 9 weeks gestation in humans, and embryonic day (E) 9.5 and E11.5 in the mouse. (B) The outflow tract endocardial cushions undergo remodeling into mature valve leaflet. (C) Mature valve layers consist of fibrosa, spongiosa, and ventricularis. (D) Myxomatous alterations associated with leaflet thickening, activated VICs (black cells), disorganized extracellular matrix, diffuse accumulation of proteoglycans and fragmentation of collagen and elastin fibers.
Aortic valve anatomy
The aortic valve is located between the left ventricle (LV) and ascending thoracic aorta (6). Like all valves in the heart, it serves to maintain unidirectional flow. The aortic valve is avascular and is connected to the aortic root by the fibrous annulus (30). Each leaflet is named according to the location corresponding to the coronary artery ostia and are accordingly referred to as the right coronary cusp, left coronary cusp and noncoronary cusp (31). Each cusp is composed of three layers of extracellular matrix (ECM) components that are oriented relative to blood flow, including the fibrosa comprised of collagen, the proteoglycan-rich spongiosa, and the elastin fiber-containing ventricularis (32) (Figure 1C). In addition, fibronectin and lamin are included in the aortic valve cusps as minor ECM components (33). The cellular composition of the aortic valve is comprised of VICs and valve endothelial cells (VECs). VICs are found within the interior of each cusp and important for ECM synthesis and homeostasis, while VECs form a protective cellular layer that surrounds each cusp. The communication between VICs and VECs via paracrine signaling is necessary to preserve ECM homeostasis and prevent disease (34–36).
Aortic valve pathology
The pathologic features of diseased heart valves include myxomatous degeneration, which is characterized histologically by leaflet thickening, diffuse accumulation of proteoglycans and fragmentation of collagen and elastin fibers (Figure 1D) (37). Myxomatous valve disease is more commonly found in the mitral valve. It can be caused by congenital valve malformations, genetic abnormalities such as pathogenic variants in ECM genes, or can be acquired due to autoimmune diseases, progressive cardiovascular dysfunction/heart failure and infective endocarditis (38–40). Myxomatous valves also demonstrate an increased recruitment of pro-inflammatory macrophages and immunogenic ECM remodeling consistent with an inflammatory micro-environment (41). These findings suggest that macrophages play a role in the initiation and progression of myxomatous valve disease.
Aortic valves in infants with severe congenital AVS were reported to be described as “greatly thickened” and “nodular” and often had bicuspid morphology (42). Pathological and histological findings of congenital AVS demonstrated the primitive mesenchymal tissue, located between the valve endothelial layers, to be increased as well as containing a loose myxomatous ground substance. Further, the cells within the ground substance were described as spindle-shaped, similar to that seen in myxomatous valve tissue (Figure 1D). Interestingly, the valvular tissue resembled that of the endocardial cushions in the developing fetal heart. In normal heart development, loose connective tissue within the endocardial cushions is supposed to thin out and transition into dense, mature tissue. As a result, it is suggested this persistence of this loose embryonic connective tissue and likely its continued growth is a defining feature of congenital AVS.
Inflammation remains unclear as a contributor to congenital AVS. However, inflammation plays a significant role in many of the chronic diseases of adulthood, including cardiovascular disorders (43). Valvular inflammation can result in fibrosis, thickening, and calcification. Calcific aortic valve disease (CAVD) is the most common valvular condition in the developed world and increases in prevalence with age (44). Evidence of chronic inflammatory infiltrates in tissue exhibiting CAVD has been demonstrated along with a positive correlation between rate of progression and density of leukocytes (45). In the case of BAV, chronic inflammation may explain the earlier onset of disease, given that stenosis of a BAV is associated with increased inflammatory cells and vascularity in comparison to AVS in a tricuspid aortic valve (46). Together, these findings demonstrate that congenital AVS and adult-onset AVS may differ in regard to inflammation. There are multiple glycoproteins which regulate the aortic valve structure during development and are associated with progression (47, 48). N-glycosylation was found to be spatially regulated within the normal aortic valve and sialylated N-glycans were increased in pediatric end-stage congenital AVS (49). Collagen deregulation is a distinctive feature of congenital AVS, while the regulation of the collagen fibers in the aortic valve remains largely elusive. A recent study identified the collagen types and hydroxylated prolines (HYP) modifications, which are critical to stabilizing the triple helix of collagen, that are seen during human aortic valve development and at pediatric end-stage congenital AVS (50). Histological and proteomic analysis identified a unique region of high-density collagen present in pediatric end-stage congenital AVS and reported that specific collagen peptides were modified by HYP. In addition, network analysis identified BAMBI (BMP and Activin Membrane Bound Inhibitor) as a prospective regulator of the collagen interactome.
Clinical characteristics, natural history and management of congenital AVS
Clinical presentations of congenital AVS vary widely, ranging from mild to critical, depending on the aortic valve morphology and the severity of AVS, but progress over time. During the fetal period, mild or moderate AVS leads to increased LV pressures and LV hypertrophy. Severe AVS results in severe LV hypertrophy and decreased flow through LV, which may ultimately lead to hypoplastic left heart syndrome (HLHS) (51–53). The fetus usually tolerates severe AVS, but symptoms can develop rapidly after birth. Critical AVS in neonates often presents with heart failure, cardiogenic shock, and other end organ dysfunction and can lead to death within the first weeks of life (54). Older children and adolescents with AVS tend to be asymptomatic with approximately 10% experiencing symptoms and signs of congestive heart failure, including dyspnea, angina, or syncope especially upon exercise (55). A combination of maximum aortic velocity (Vmax), mean pressure gradient (MPG), and aortic valve area (AVA) are used to assess the severity of stenosis as published in the most recent American College of Cardiology (ACC) and American Heart Association (AHA) guidelines (56).
The natural history of patients with congenital AVS shows that progressive obstruction is likely to occur by late adulthood. In childhood, significant progression was seen in one third of all medically managed patients in the Natural History Study (57). A follow-up study for 30 years found that the diagnosis of mild AVS before 6 months of age was associated with a significantly increased risk of requiring aortic valvotomy and balloon valvuloplasty with age (58). The likelihood that the stenosis remains mild was reported to be less than 20%, thus supporting the need for long-term follow-up of mild AS into adulthood. Recently, the probability of requiring balloon valvuloplasty is shown to be 20% in patients with catheter-measured peak pressure gradients less than 25 mmHg, and 40% and 70% in patients with gradients 25–49 mmHg and >50 mmHg, respectively (59). Notably, congenital AVS is a progressive disorder as the risk of morbid events such as heart failure, sudden death, and ventricular arrhythmia increase at a rate of 1%–1.5% per year, if left untreated (6, 59). Similarly, the risk of developing AVS in children with isolated BAV increases along with age (6). As with all CHD, bacterial endocarditis remains a potential complication of AVS, with an incidence of 27.1 per 10,000 person years (60). As congenital AVS is a progressive condition, guidelines have been developed which outline the indications for intervention. These interventions are limited to balloon valvuloplasty in the cardiac catheterization laboratory and transcatheter aortic valve replacement or surgical aortic valve replacement. We refer readers to recent clinical management guidelines from the ACC/AHA or European Society of Cardiology (ESC) for timing of intervention and different valve replacement options as these details are beyond the scope of this review (56, 61, 62).
Molecular genetics of congenital aortic valve disease
Recent advances in genetic sequencing technologies, such as massively parallel sequencing, as well as interpretation of the clinical presentation and genetic variants, have made it easier for establishing a diagnosis and discovering new genetic etiologies for congenital aortic valve disease, including ADAMTS19, SMAD6 and ROBO gene family members (63, 64). Multiple human genetic abnormalities associated with syndromic and non-syndromic congenital aortic valve disease have been identified, including AVS and BAV (Tables 1, 2). However, compared to BAV, well established genetic contributors to congenital AVS are scarce. Common syndromes associated with congenital AVS are Turner syndrome and Jacobson syndrome. Turner syndrome is caused by chromosomal aneuploidies (Monosomy X) and associated CHD is observed in 30% of cases, including AVS, BAV, CoA and HLHS (65). Jacobsen syndrome is caused by terminal deletion of chromosome 11q and associated CHD is found in 56% of cases, including AVS, HLHS, CoA and VSD (66). In addition to Turner syndrome and Jacobsen syndrome, several common syndromes associated with BAV are known. Congenital heart valve anomalies associated with Trisomy 18 (Edwards syndrome) include BAV, bicuspid pulmonary valve and polyvalvular nodular dysplasia (67, 68). BAV is present in 1p36 deletion syndrome as well as Kabuki syndrome caused primarily by KMT2D and KDM6A variants (69–71). BAV and thoracic aortic aneurysm (TAA) have been also described in Marfan syndrome associated with FBN1 variants (72) or Loeys-Dietz syndrome associated with TGFBR1 and TGFBR2 variants (73).
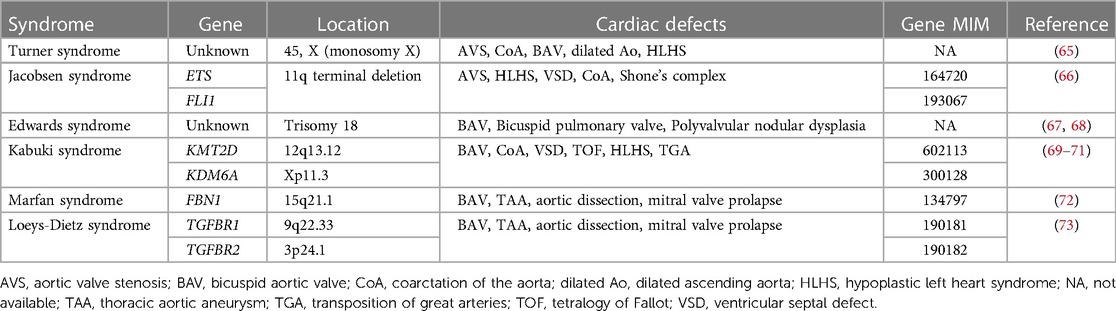
Table 1. Genetic syndromes associated with congenital aortic valve disease.
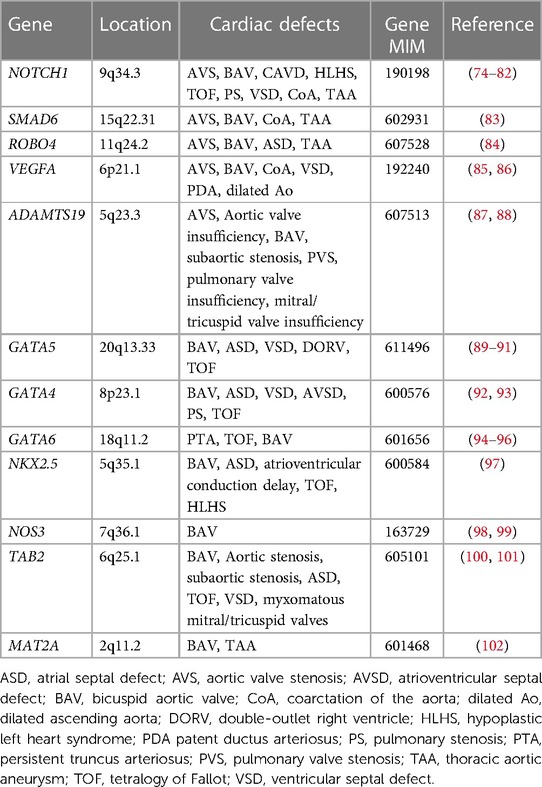
Table 2. Human genes associated with congenital aortic valve disease.
For non-syndromic congenital AVS, only a few genes have been implicated. NOTCH1 pathogenic variants were identified in individuals with left ventricular outflow tract (LVOT) malformations, including congenital AVS, CoA and HLHS (74, 75). SMAD6 variants were observed in patients with AVS and BAV (83). ROBO4 variants were also identified in individuals with AVS and atrial septal defect (ASD) (84). Furthermore, variants in vascular endothelial growth factor-A (VEGFA) were found in a patient with congenital tricuspid AVS and LVOT obstruction (85, 86). Moreover, ADAMTS19 variants were found to cause a spectrum of congenital heart valve diseases, including AVS, aortic valve insufficiency, subaortic stenosis, pulmonary valve stenosis, pulmonary valve insufficiency and atrioventricular valve insufficiency (87, 88). However, no other genes have been reported as monogenic causes of congenital AVS. Previous studies reported that common variants in genes linked to cardiac development such as ERBB4, BMP4, and ISL1, may bestow risk for LVOT defects, including congenital AVS (103–105).
Taking another approach, genome-wide DNA methylation analysis identified significant alterations in CpG methylation at 59 sites in 52 genes for congenital AVS (106). A significant epigenetic change in the APOA5 and PCSK9 genes, which are known to be important in lipid metabolism, was also observed associated with AVS. It remains to be determined if pathogenic variation in the other 50 genes will be implicated in congenital AVS.
As BAV is often found in the setting of congenital AVS, there is likely overlap in the genetic etiologies of BAV and congenital AVS. BAV is widely acknowledged to have genetic contributors with a reported heritability of 89% (107). Insights into the genetic contributors of BAV were first provided from studies of familial BAV, where NOTCH1 variants were discovered to segregate in familial aortic valve disease through linkage analysis, including BAV, AVS and CAVD (74). Since then, pathogenic variants in NOTCH1 have been found to cause not only left -sided CHD, including BAV, CoA and HLHS (75, 76–79), but also other types of CHD such as tetralogy of Fallot (TOF) and VSD (80–82). The GATA family of zinc-finger transcription factors, particularly GATA4, GATA5, and GATA6, play essential roles in cardiac development. Pathogenic variation in GATA5 is well characterized in human BAV (89–91), but GATA5 variants have also been associated with a spectrum of CHD, including TOF, VSD, ASD and double outlet right ventricle (DORV) (108–110). GATA4 pathogenic variants were identified in BAV cases (92) and in addition, the burden of rare variants in GATA4 were shown to be significantly enriched in early-onset BAV (93). Although GATA6 variants have been mainly implicated in conotruncal heart defects (94, 95), GATA6 loss-of-function variants were identified in a family with BAV (96). Furthermore, a deleterious variant in NKX2.5 was identified in a family with BAV (97), while pathogenic variants in NKX2.5 have been reported in ASD along with atrioventricular conduction abnormalities, VSD, TOF and HLHS (111–113). The contribution of other candidate genes, such as NOS3, TAB2, and MAT2A has been also suggested in BAV (98–102).
Mouse models of congenital aortic valve disease
Since congenitally stenotic aortic valves have often been manipulated due to current management strategies, there is limited access to diseased human tissues to investigate mechanisms of disease initiation and progression. Accordingly, animal models serve an important role in understanding the genetic etiologies and progression of congenital AVS (114). A summary of reported genetic mouse models found to exhibit congenital aortic valve disease can be found in Table 3, including congenital AVS and BAV. Morphological phenotypes in BAV are summarized in Figure 2 (133). Although NOTCH1 has been implicated in human aortic valve disease, Notch1 haploinsufficiency causes CAVD and ascending aortic aneurysms in mice, but not congenital aortic valve abnormalities (134, 135). Interestingly, while mice which are homozygous for a null mutation in endothelial NOS (Nos3−/−) display a partially penetrant BAV at an incidence of ∼25% (117, 118), Notch1; Nos3 compound mutant mice (Notch1+/−; Nos3−/−) display congenital aortic valve disease such as BAV and AVS at a penetrance of 64%. These aortic valve abnormalities are accompanied by additional cardiac outflow tract defects resulting in ≈65% lethality by postnatal day 10 (35, 115). Telomere shortening in Notch1 haploinsufficient mice (Notch1+/− mTRG1–3) elicit age-dependent tricuspid AVS and aortic valve calcification, however, early lethality is observed (116). In addition, cell-specific deletion or inactivation of mediators of Notch signaling pathway, such as JAG1 or RBPJ, in mice demonstrate BAV at a penetrance of 47%–54% with high perinatal lethality (121, 122). Interestingly, no endocardial cushion defects are observed in these murine models of BAV targeting Notch ligands, suggesting that congenital aortic valve disease may result from a disruption in the process that occurs after EMT, potentially during the valve remodeling. Genetic deletion of Gata5 in mice (Gata5−/−) lead to R/NC subtype BAV at a partially penetrance of 25% (119). Gata6 haploinsufficient mice (Gata6+/−) develop R/L subtype BAV with incidence of 56% in males and 27% in females (120). Furthermore, SHF-specific deletion of Gata6 within the Isl1-lineage recapitulated the BAV phenotype, suggesting the role of Gata6 in SHF during valve development. Heterozygous Nkx2.5 knockout mice display a variety of cardiac phenotypes, depending on the genetic background, including BAV with AVS at a low penetrance of 8.2% (123). In addition to these mice, other genetic mouse models of congenital aortic valve disease have been described, whereas the majority have limitations with regards to a low penetrance or high lethality (84, 87, 124–132). Further studies are needed to generate murine models with a high penetrance that survive to adulthood, allowing us to validate the role of cardiac developmental genes in the etiology of aortic valve disease as well as investigate their role in disease progression and to serve as models to test novel therapies.
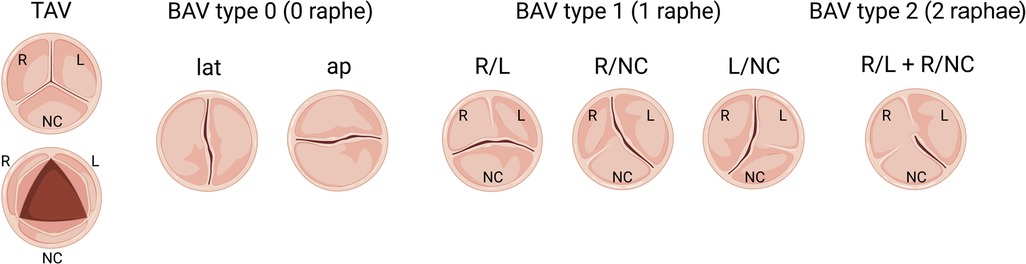
Figure 2. Schematic of morphologic phenotypes in bicuspid aortic valve (BAV). Schematic depiction of a normal tricuspid aortic valve (TAV) and three subtypes of BAV based on the raphe position relative to coronary artery origins: Type 0 (no raphe), Type 1 (one fibrous raphe) and Type 2 (two raphae). Type 1 is the most common, including BAV R/L (right-left fusion), R/NC (right-noncoronary fusion) and L/NC (left-noncoronary fusion), followed by Type 0, including lat (lateral arrangement of the free edge of the cusps) and ap (anterior-posterior arrangement of the free edge of the cusps), and the most infrequent is Type 2.
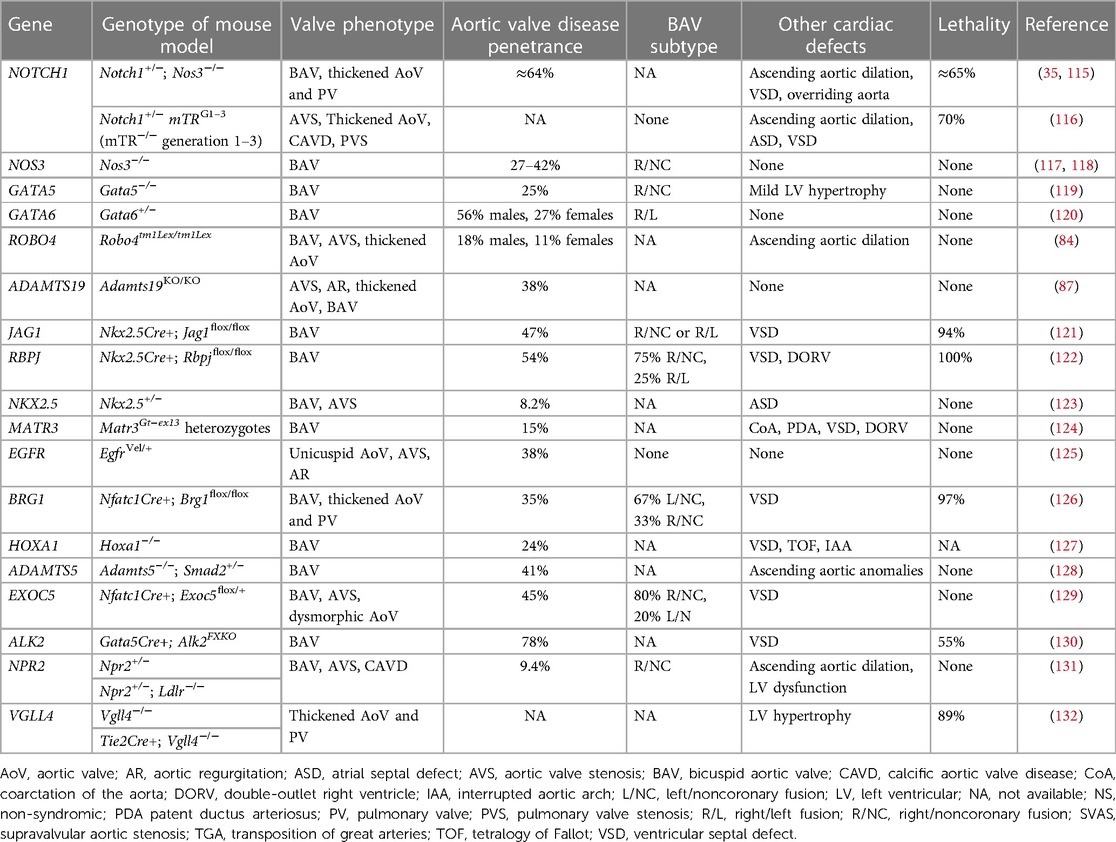
Table 3. Mouse models of congenital aortic valve disease.
Clinical implications and future directions
Given the limited translation of molecular mechanisms found in animal models to human patients, effective pharmacologic therapies for congenital AVS remain elusive. The current standard treatment is transcatheter/surgical repair or replacement of the diseased valve. Recently, fetal aortic valvuloplasty has been performed in fetuses with AVS and evolving HLHS. However, outcomes for achieving biventricular circulation remain controversial and this has not been applied to isolated congenital AVS (136, 137). Thus, development of novel medical treatment for congenital AVS is essential, while pharmacological therapies are limited. One limited treatment option is statin therapy to treat elevated cholesterol levels. Statins have been demonstrated to reduce cardiovascular risk and prevent cardiovascular disease such as coronary artery disease (138, 139). Although some attempts have been made to utilize pharmacologic treatments such as statins to treat CAVD with associated AVS in adults, these studies did not show consistent beneficial effects for progression of AVS (140, 141). The pathophysiology of degenerative AVS in adults is similar to coronary artery disease, whereas atherosclerosis pathway may not play a significant role in the development of congenital aortic valve disease, including BAV. Therefore, statins have not been tested in children with aortic valve disease and the clinical impact on congenital AVS in children remains unknown. Increased understanding the molecular pathways regulating aortic valve development along with advances in genetic sequencing technologies have allowed for the discovery of new candidate genes for congenital aortic valve disease. In addition, genetic murine models of aortic valve disease have been generated and uncovered molecular pathways as potential therapeutic targets. Specifically, TGF-β signaling is activated in degenerative valves with ECM abnormalities and may be a potential therapeutic target, as TGF-β antagonists such as the angiotensin II type 1 receptor blocker, losartan, could prevent abnormal aortic root growth in a mouse model of Marfan syndrome (142). In addition, targeting monocyte-derived macrophages has emerged as a potential therapeutic approach to prevent myxomatous valve disease in Marfan syndrome mice (41). Recent studies by using single-cell RNA-sequencing, human induced pluripotent stem cell (iPSC) technology and machine learning, identified new pathogenic pathways and a new therapeutic candidate to prevent aortic valve disease in mouse models of CAVD, although the implications for congenital AVS are unknown (36, 143). Further, the molecular and genetic links between congenital aortic valve disease and adult CAVD need to be defined. Improved identification of human aortic valve disease genes, generation of murine models for clinically relevant congenital AVS, and new technologies such as single cell genomics, iPSC modeling and machine learning may reveal novel therapeutic targets to develop effective treatments for early intervention.
Conclusions
Congenital AVS is a complex and progressive disease that affects children and adults throughout their lives. Although advances in transcatheter aortic valvuloplasty and transcatheter or surgical aortic valve implantation have improved morbidity and mortality in this patient population, pharmacologic therapies for congenital AVS remain elusive. Several potential therapeutic targets have been proposed in animal models to prevent the myxomatous valve disease by using next-generation sequencing, single-cell genomics, machine learning, cardiac organoid and bioengineering technologies. A continued escalation of our understanding in molecular genetics of congenital AVS has clinical implications as it will facilitate the development of new treatment options to prevent the progression or treat congenital AVS.
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. All authors contributed to the article and approved the submitted version.
Funding
JY is supported by Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad and NIH/NHLBI (R01-HL144009). K.S. is supported by Ohio State University College of Medicine Roessler Research Scholarship. V.G. is supported by funding from the National Institutes of Health NIH/NHLBI (R01HL144009; R21HL161823).
Acknowledgments
The authors acknowledge the use of Biorender.com for generation of figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. (2002) 39(12):1890–900. doi: 10.1016/s0735-1097(02)01886-7
2. Hoffman JI, Kaplan S, Liberthson RR. Prevalence of congenital heart disease. Am Heart J. (2004) 147(3):425–39. doi: 10.1016/j.ahj.2003.05.003
3. Kitchiner DJ, Jackson M, Walsh K, Peart I, Arnold R. Incidence and prognosis of congenital aortic valve stenosis in liverpool (1960-1990). Br Heart J. (1993) 69(1):71–9. doi: 10.1136/hrt.69.1.71
4. Campbell M. The natural history of congenital aortic stenosis. Br Heart J. (1968) 30(4):514%–26. doi: 10.1136/hrt.30.4.514
5. Frank S, Johnson A, Ross J Jr. Natural history of valvular aortic stenosis. Br Heart J. (1973) 35(1):41–6. doi: 10.1136/hrt.35.1.41
6. Singh GK. Congenital aortic valve stenosis. Children. (2019) 6(5):69. doi: 10.3390/children6050069
7. Pujari SH, Agasthi P. Aortic stenosis. StatPearls. Treasure Island, FL: StatPearls Publishing. Copyright © 2022, StatPearls Publishing LLC, (2022).
8. Rosenquist GC. Congenital mitral valve disease associated with coarctation of the aorta: a spectrum that includes parachute deformity of the mitral valve. Circulation. (1974) 49(5):985–93. doi: 10.1161/01.cir.49.5.985
9. Meilhac SM, Buckingham ME. The deployment of cell lineages that form the mammalian heart. Nat Rev Cardiol. (2018) 15(11):705–24. doi: 10.1038/s41569-018-0086-9
10. Choudhury TZ, Garg V. Molecular genetic mechanisms of congenital heart disease. Curr Opin Genet Dev. (2022) 75:101949. doi: 10.1016/j.gde.2022.101949
11. Dhanantwari P, Lee E, Krishnan A, Samtani R, Yamada S, Anderson S, et al. Human cardiac development in the first trimester: a high-resolution magnetic resonance imaging and episcopic fluorescence image capture atlas. Circulation. (2009) 120(4):343–51. doi: 10.1161/circulationaha.108.796698
12. Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. (2009) 105(5):408–21. doi: 10.1161/circresaha.109.201566
13. Lincoln J, Garg V. Etiology of valvular heart disease-genetic and developmental origins. Circ J. (2014) 78(8):1801–7. doi: 10.1253/circj.cj-14-0510
14. Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. (2004) 166(3):359–67. doi: 10.1083/jcb.200403050
15. Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat Rec. (2000) 258(2):119–27. doi: 10.1002/(sici)1097-0185(20000201)258:2%3C119::Aid-ar1%3E3.0.Co;2-u
16. Délot EC, Bahamonde ME, Zhao M, Lyons KM. BMP Signaling is required for septation of the outflow tract of the mammalian heart. Development. (2003) 130(1):209–20. doi: 10.1242/dev.00181
17. Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. (2010) 338(2):127–35. doi: 10.1016/j.ydbio.2009.11.030
18. Gessert S, Kühl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ Res. (2010) 107(2):186–99. doi: 10.1161/circresaha.110.221531
19. Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. (2004) 18(1):99–115. doi: 10.1101/gad.276304
20. Luxán G, D'Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. (2016) 118(1):e1–e18. doi: 10.1161/circresaha.115.305350
21. Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P, et al. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. (2001) 128(9):1531–8. doi: 10.1242/dev.128.9.1531
22. von Gise A, Pu WT. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ Res. (2012) 110(12):1628–45. doi: 10.1161/circresaha.111.259960
23. Hinton RB J, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. (2006) 98(11):1431–8. doi: 10.1161/01.RES.0000224114.65109.4e
24. Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. (2000) 127(8):1607–16. doi: 10.1242/dev.127.8.1607
25. George RM, Maldonado-Velez G, Firulli AB. The heart of the neural crest: cardiac neural crest cells in development and regeneration. Development. (2020) 147(20):dev188706. doi: 10.1242/dev.188706
26. Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. (2005) 287(1):134–45. doi: 10.1016/j.ydbio.2005.08.041
27. Dyer LA, Kirby ML. The role of secondary heart field in cardiac development. Dev Biol. (2009) 336(2):137–44. doi: 10.1016/j.ydbio.2009.10.009
28. Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental mechanisms of aortic valve malformation and disease. Annu Rev Physiol. (2017) 79:21–41. doi: 10.1146/annurev-physiol-022516-034001
29. Eley L, Alqahtani AM, MacGrogan D, Richardson RV, Murphy L, Salguero-Jimenez A, et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. eLife. (2018) 7:e34110. doi: 10.7554/eLife.34110
30. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the national heart and lung and blood institute aortic stenosis working group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. (2011) 124(16):1783–91. doi: 10.1161/circulationaha.110.006767
31. Chen JH, Chen WL, Sider KL, Yip CY, Simmons CA. β-catenin mediates mechanically regulated, transforming growth factor-β1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler, Thromb, Vasc Biol. (2011) 31(3):590–7. doi: 10.1161/atvbaha.110.220061
32. Koenig SN, Lincoln J, Garg V. Genetic basis of aortic valvular disease. Curr Opin Cardiol. (2017) 32(3):239–45. doi: 10.1097/hco.0000000000000384
33. Latif N, Sarathchandra P, Taylor PM, Antoniw J, Brand N, Yacoub MH. Characterization of molecules mediating cell-cell communication in human cardiac valve interstitial cells. Cell Biochem Biophys. (2006) 45(3):255–64. doi: 10.1385/cbb:45:3:255
34. Anstine LJ, Bobba C, Ghadiali S, Lincoln J. Growth and maturation of heart valves leads to changes in endothelial cell distribution, impaired function, decreased metabolism and reduced cell proliferation. J Mol Cell Cardiol. (2016) 100:72–82. doi: 10.1016/j.yjmcc.2016.10.006
35. Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. (2013) 60:27–35. doi: 10.1016/j.yjmcc.2013.04.001
36. Majumdar U, Manivannan S, Basu M, Ueyama Y, Blaser MC, Cameron E, et al. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci Adv. (2021) 7(6):eabe3706. doi: 10.1126/sciadv.abe3706
37. Kim AJ, Xu N, Yutzey KE. Macrophage lineages in heart valve development and disease. Cardiovasc Res. (2021) 117(3):663–73. doi: 10.1093/cvr/cvaa062
38. Levine RA, Hagége AA, Judge DP, Padala M, Dal-Bianco JP, Aikawa E, et al. Mitral valve disease–morphology and mechanisms. Nat Rev Cardiol. (2015) 12(12):689–710. doi: 10.1038/nrcardio.2015.161
39. Geirsson A, Singh M, Ali R, Abbas H, Li W, Sanchez JA, et al. Modulation of transforming growth factor-β signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation. (2012) 126(11 Suppl 1):S189–97. doi: 10.1161/circulationaha.111.082610
40. Meier LA, Auger JL, Engelson BJ, Cowan HM, Breed ER, Gonzalez-Torres MI, et al. CD301b/MGL2(+) Mononuclear phagocytes orchestrate autoimmune cardiac valve inflammation and fibrosis. Circulation. (2018) 137(23):2478–93. doi: 10.1161/circulationaha.117.033144
41. Kim AJ, Xu N, Umeyama K, Hulin A, Ponny SR, Vagnozzi RJ, et al. Deficiency of circulating monocytes ameliorates the progression of myxomatous valve degeneration in marfan syndrome. Circulation. (2020) 141(2):132–46. doi: 10.1161/circulationaha.119.042391
42. Serck-Hanssen A. Congenital valvular aortic stenosis. Histological changes in the valves and myocardium in 3 cases. Acta Pathol Microbiol Scand. (1968) 72(4):465–77. doi: 10.1111/j.1699-0463.1968.tb00462.x
43. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25(12):1822–32. doi: 10.1038/s41591-019-0675-0
44. Rajamannan NM, Gersh B, Bonow RO. Calcific aortic stenosis: from bench to the bedside–emerging clinical and cellular concepts. Heart. (2003) 89(7):801–5. doi: 10.1136/heart.89.7.801
45. Coté N, Mahmut A, Bosse Y, Couture C, Pagé S, Trahan S, et al. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation. (2013) 36(3):573–81. doi: 10.1007/s10753-012-9579-6
46. Manno G, Bentivegna R, Morreale P, Nobile D, Santangelo A, Novo S, et al. Chronic inflammation: a key role in degeneration of bicuspid aortic valve. J Mol Cell Cardiol. (2019) 130:59–64. doi: 10.1016/j.yjmcc.2019.03.013
47. Satta J, Melkko J, Pöllänen R, Tuukkanen J, Pääkkö P, Ohtonen P, et al. Progression of human aortic valve stenosis is associated with tenascin-C expression. J Am Coll Cardiol. (2002) 39(1):96–101. doi: 10.1016/s0735-1097(01)01705-3
48. Della Corte A, Quarto C, Bancone C, Castaldo C, Di Meglio F, Nurzynska D, et al. Spatiotemporal patterns of smooth muscle cell changes in ascending aortic dilatation with bicuspid and tricuspid aortic valve stenosis: focus on cell-matrix signaling. J Thorac Cardiovasc Surg. (2008) 135(1):8–18. 18.e1-2. doi: 10.1016/j.jtcvs.2007.09.009
49. Angel PM, Drake RR, Park Y, Clift CL, West C, Berkhiser S, et al. Spatial N-glycomics of the human aortic valve in development and pediatric endstage congenital aortic valve stenosis. J Mol Cell Cardiol. (2021) 154:6–20. doi: 10.1016/j.yjmcc.2021.01.001
50. Clift CL, Su YR, Bichell D, Jensen Smith HC, Bethard JR, Norris-Caneda K, et al. Collagen fiber regulation in human pediatric aortic valve development and disease. Sci Rep. (2021) 11(1):9751. doi: 10.1038/s41598-021-89164-w
51. Hornberger LK, Sanders SP, Rein AJ, Spevak PJ, Parness IA, Colan SD. Left heart obstructive lesions and left ventricular growth in the midtrimester fetus. A longitudinal study. Circulation. (1995) 92(6):1531–8. doi: 10.1161/01.cir.92.6.1531
52. Simpson JM, Sharland GK. Natural history and outcome of aortic stenosis diagnosed prenatally. Heart. (1997) 77(3):205–10. doi: 10.1136/hrt.77.3.205
53. Mäkikallio K, McElhinney DB, Levine JC, Marx GR, Colan SD, Marshall AC, et al. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: patient selection for fetal intervention. Circulation. (2006) 113(11):1401–5. doi: 10.1161/circulationaha.105.588194
54. Zeevi B, Keane JF, Castaneda AR, Perry SB, Lock JE. Neonatal critical valvar aortic stenosis. A comparison of surgical and balloon dilation therapy. Circulation. (1989) 80(4):831–9. doi: 10.1161/01.cir.80.4.831
55. Hastreiter AR, Oshima M, Miller RA, Lev M, Paul MH. Congenital aortic stenosis syndrome in infancy. Circulation. (1963) 28:1084–95. doi: 10.1161/01.cir.28.6.1084
56. Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin JP 3rd, Gentile F, et al. 2020 ACC/AHA guideline for the management of patients with valvular heart disease: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2021) 143(5):e72–e227. doi: 10.1161/cir.0000000000000923
57. Wagner HR, Ellison RC, Keane JF, Humphries OJ, Nadas AS. Clinical course in aortic stenosis. Circulation. (1977) 56(1 Suppl):I47–56. PMID: 872345.872345
58. Kitchiner D, Jackson M, Walsh K, Peart I, Arnold R. The progression of mild congenital aortic valve stenosis from childhood into adult life. Int J Cardiol. (1993) 42(3):217–23. doi: 10.1016/0167-5273(93)90051-h
59. Keane JF, Driscoll DJ, Gersony WM, Hayes CJ, Kidd L, O'Fallon WM, et al. Second natural history study of congenital heart defects. Results of treatment of patients with aortic valvar stenosis. Circulation. (1993) 87(2 Suppl):I16–27. PMID: 8425319.8425319
60. Gersony WM, Hayes CJ, Driscoll DJ, Keane JF, Kidd L, O'Fallon WM, et al. Bacterial endocarditis in patients with aortic stenosis, pulmonary stenosis, or ventricular septal defect. Circulation. (1993) 87(2 Suppl):I121–6. PMID: 8425318.8425318
61. Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin JP, Gentile F, et al. 2020 ACC/AHA guideline for the management of patients with valvular heart disease: executive summary: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2021) 143(5):e35–71. doi: 10.1161/cir.0000000000000932
62. Vahanian A, Beyersdorf F, Praz F, Milojevic M, Baldus S, Bauersachs J, et al. 2021 ESC/EACTS guidelines for the management of valvular heart disease. Eur Heart J. (2022) 43(7):561–632. doi: 10.1093/eurheartj/ehab395
63. Yasuhara J, Garg V. Genetics of congenital heart disease: a narrative review of recent advances and clinical implications. Transl Pediatr. (2021) 10(9):2366–86. doi: 10.21037/tp-21-297
64. Ackah RL, Yasuhara J, Garg V. Genetics of aortic valve disease. Curr Opin Cardiol. (2023) 38(3):169–78. doi: 10.1097/hco.0000000000001028
65. Sybert VP, McCauley E. Turner's syndrome. N Engl J Med. (2004) 351(12):1227–38. doi: 10.1056/NEJMra030360
66. Jacobsen P, Hauge M, Henningsen K, Hobolth N, Mikkelsen M, Philip J. An (11;21) translocation in four generations with chromosome 11 abnormalities in the offspring. A clinical, cytogenetical, and gene marker study. Hum Hered. (1973) 23(6):568–85. doi: 10.1159/000152624
67. Balderston SM, Shaffer EM, Washington RL, Sondheimer HM. Congenital polyvalvular disease in trisomy 18: echocardiographic diagnosis. Pediatr Cardiol. (1990) 11(3):138–42. doi: 10.1007/bf02238843
68. Springett A, Wellesley D, Greenlees R, Loane M, Addor MC, Arriola L, et al. Congenital anomalies associated with trisomy 18 or trisomy 13: a registry-based study in 16 European countries, 2000-2011. Am J Med Genet Part A. (2015) 167a(12):3062–9. doi: 10.1002/ajmg.a.37355
69. Battaglia A, Hoyme HE, Dallapiccola B, Zackai E, Hudgins L, McDonald-McGinn D, et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. (2008) 121(2):404–10. doi: 10.1542/peds.2007-0929
70. Yuan SM. Congenital heart defects in Kabuki syndrome. Cardiol J. (2013) 20(2):121–4. doi: 10.5603/cj.2013.0023
71. Digilio MC, Gnazzo M, Lepri F, Dentici ML, Pisaneschi E, Baban A, et al. Congenital heart defects in molecularly proven kabuki syndrome patients. Am J Med Genet Part A. (2017) 173(11):2912–22. doi: 10.1002/ajmg.a.38417
72. Nistri S, Porciani MC, Attanasio M, Abbate R, Gensini GF, Pepe G. Association of marfan syndrome and bicuspid aortic valve: frequency and outcome. Int J Cardiol. (2012) 155(2):324–5. doi: 10.1016/j.ijcard.2011.12.009
73. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. (2005) 37(3):275–81. doi: 10.1038/ng1511
74. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. (2005) 437(7056):270–4. doi: 10.1038/nature03940
75. McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, Belmont JW, et al. NOTCH1 Mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet. (2008) 17(18):2886–93. doi: 10.1093/hmg/ddn187
76. Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, Wrobel S, et al. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun. (2006) 345(4):1460–5. doi: 10.1016/j.bbrc.2006.05.046
77. Foffa I, Ait Alì L, Panesi P, Mariani M, Festa P, Botto N, et al. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet. (2013) 14:44. doi: 10.1186/1471-2350-14-44
78. Theis JL, Hrstka SC, Evans JM, O'Byrne MM, de Andrade M, O'Leary PW, et al. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum Genet. (2015 Sep) 134(9):1003–11. doi: 10.1007/s00439-015-1582-1
79. Kerstjens-Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med. (2016) 18(9):914–23. doi: 10.1038/gim.2015.193
80. Preuss C, Capredon M, Wünnemann F, Chetaille P, Prince A, Godard B, et al. Family based whole exome sequencing reveals the multifaceted role of notch signaling in congenital heart disease. PLoS Genet. (2016) 12(10):e1006335. doi: 10.1371/journal.pgen.1006335
81. Debiec RM, Hamby SE, Jones PD, Safwan K, Sosin M, Hetherington SL, et al. Contribution of NOTCH1 genetic variants to bicuspid aortic valve and other congenital lesions. Heart. (2022) 108(14):1114–20. doi: 10.1136/heartjnl-2021-320428
82. Debiec R, Hamby SE, Jones PD, Coolman S, Asiani M, Kharodia S, et al. Novel loss of function mutation in NOTCH1 in a family with bicuspid aortic valve, ventricular septal defect, thoracic aortic aneurysm, and aortic valve stenosis. Mol Genet Genomic Med. (2020) 8(10):e1437. doi: 10.1002/mgg3.1437
83. Tan HL, Glen E, Töpf A, Hall D, O'Sullivan JJ, Sneddon L, et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat. (2012) 33(4):720–7. doi: 10.1002/humu.22030
84. Gould RA, Aziz H, Woods CE, Seman-Senderos MA, Sparks E, Preuss C, et al. ROBO4 Variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat Genet. (2019) 51(1):42–50. doi: 10.1038/s41588-018-0265-y
85. Zhao W, Wang J, Shen J, Sun K, Zhu J, Yu T, et al. Mutations in VEGFA are associated with congenital left ventricular outflow tract obstruction. Biochem Biophys Res Commun. (2010) 396(2):483–8. doi: 10.1016/j.bbrc.2010.04.124
86. Zhao W, Wang J, Shen J, Sun K, Chen Y, Ji W, et al. A nonsense variation p.Arg325X in the vascular endothelial growth factor-A gene may be associated with congenital tricuspid aortic valve stenosis. Cardiol Young. (2012) 22(3):316–22. doi: 10.1017/s104795111100151x
87. Wünnemann F, Ta-Shma A, Preuss C, Leclerc S, van Vliet PP, Oneglia A, et al. Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat Genet. (2020) 52(1):40–7. doi: 10.1038/s41588-019-0536-2
88. Massadeh S, Alhashem A, van de Laar I, Alhabshan F, Ordonez N, Alawbathani S, et al. ADAMTS19-associated Heart valve defects: novel genetic variants consolidating a recognizable cardiac phenotype. Clin Genet. (2020) 98(1):56–63. doi: 10.1111/cge.13760
89. Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C. Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol. (2012) 53(2):277–81. doi: 10.1016/j.yjmcc.2012.05.009
90. Bonachea EM, Chang SW, Zender G, LaHaye S, Fitzgerald-Butt S, McBride KL, et al. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr Res. (2014) 76(2):211–6. doi: 10.1038/pr.2014.67
91. Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med. (2014) 33(5):1219–26. doi: 10.3892/ijmm.2014.1700
92. Li RG, Xu YJ, Wang J, Liu XY, Yuan F, Huang RT, et al. GATA4 Loss-of-Function mutation and the congenitally bicuspid aortic valve. Am J Cardiol. (2018) 121(4):469–74. doi: 10.1016/j.amjcard.2017.11.012
93. Musfee FI, Guo D, Pinard AC, Hostetler EM, Blue EE, Nickerson DA, et al. Rare deleterious variants of NOTCH1, GATA4, SMAD6, and ROBO4 are enriched in BAV with early onset complications but not in BAV with heritable thoracic aortic disease. Mol Genet Genomic Med. (2020) 8(10):e1406. doi: 10.1002/mgg3.1406
94. Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, et al. GATA6 Mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci U S A. (2009) 106(33):13933–8. doi: 10.1073/pnas.0904744106
95. Maitra M, Koenig SN, Srivastava D, Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res. (2010) 68(4):281–5. doi: 10.1203/PDR.0b013e3181ed17e4
96. Xu YJ, Di RM, Qiao Q, Li XM, Huang RT, Xue S, et al. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene. (2018) 663:115–20. doi: 10.1016/j.gene.2018.04.018
97. Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM, Liu XY, et al. A novel NKX2.5 loss-of-function mutation associated with congenital bicuspid aortic valve. Am J Cardiol. (2014) 114(12):1891–5. doi: 10.1016/j.amjcard.2014.09.028
98. Dargis N, Lamontagne M, Gaudreault N, Sbarra L, Henry C, Pibarot P, et al. Identification of gender-specific genetic variants in patients with bicuspid aortic valve. Am J Cardiol. (2016) 117(3):420–6. doi: 10.1016/j.amjcard.2015.10.058
99. Girdauskas E, Geist L, Disha K, Kazakbaev I, Groß T, Schulz S, et al. Genetic abnormalities in bicuspid aortic valve root phenotype: preliminary results. Eur J Cardiothorac Surg. (2017) 52(1):156–62. doi: 10.1093/ejcts/ezx065
100. Weiss K, Applegate C, Wang T, Batista DA. Familial TAB2 microdeletion and congenital heart defects including unusual valve dysplasia and tetralogy of fallot. Am J Med Genet Part A. (2015) 167a(11):2702–6. doi: 10.1002/ajmg.a.37210
101. Thienpont B, Zhang L, Postma AV, Breckpot J, Tranchevent LC, Van Loo P, et al. Haploinsufficiency of TAB2 causes congenital heart defects in humans. Am J Hum Genet. (2010) 86(6):839–49. doi: 10.1016/j.ajhg.2010.04.011
102. Guo DC, Gong L, Regalado ES, Santos-Cortez RL, Zhao R, Cai B, et al. MAT2A Mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet. (2015) 96(1):170–7. doi: 10.1016/j.ajhg.2014.11.015
103. McBride KL, Zender GA, Fitzgerald-Butt SM, Seagraves NJ, Fernbach SD, Zapata G, et al. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth defects research part A. Clin Mol Teratol. (2011) 91(3):162–8. doi: 10.1002/bdra.20764
104. Qian B, Mo R, Da M, Peng W, Hu Y, Mo X. Common variations in BMP4 confer genetic susceptibility to sporadic congenital heart disease in a han Chinese population. Pediatr Cardiol. (2014) 35(8):1442–7. doi: 10.1007/s00246-014-0951-1
105. Stevens KN, Hakonarson H, Kim CE, Doevendans PA, Koeleman BP, Mital S, et al. Common variation in ISL1 confers genetic susceptibility for human congenital heart disease. PloS one. (2010) 5(5):e10855. doi: 10.1371/journal.pone.0010855
106. Radhakrishna U, Albayrak S, Alpay-Savasan Z, Zeb A, Turkoglu O, Sobolewski P, et al. Genome-Wide DNA methylation analysis and epigenetic variations associated with congenital aortic valve stenosis (AVS). PloS one. (2016) 11(5):e0154010. doi: 10.1371/journal.pone.0154010
107. Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. (2004) 44(1):138–43. doi: 10.1016/j.jacc.2004.03.050
108. Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ, Fang WY, et al. Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int J Cardiol. (2013) 165(3):570–3. doi: 10.1016/j.ijcard.2012.09.039
109. Kassab K, Hariri H, Gharibeh L, Fahed AC, Zein M, El-Rassy I, et al. GATA5 Mutation homozygosity linked to a double outlet right ventricle phenotype in a Lebanese patient. Mol Genet Genomic Med. (2016) 4(2):160–71. doi: 10.1002/mgg3.190
110. Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li RG, et al. GATA5 loss-of-function mutations underlie tetralogy of fallot. Int J Med Sci. (2013) 10(1):34–42. doi: 10.7150/ijms.5270
111. Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science (New York, NY). (1998) 281(5373):108–11. doi: 10.1126/science.281.5373.108
112. Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. (1999) 104(11):1567–73. doi: 10.1172/jci8154
113. McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 Mutations in patients with congenital heart disease. J Am Coll Cardiol. (2003) 42(9):1650–5. doi: 10.1016/j.jacc.2003.05.004
114. Majumdar U, Yasuhara J, Garg V. In vivo and in vitro genetic models of congenital heart disease. Cold Spring Harb Perspect Biol. (2021) 13(4):a036764. doi: 10.1101/cshperspect.a036764
115. Koenig SN, Bosse K, Majumdar U, Bonachea EM, Radtke F, Garg V. Endothelial Notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J Am Heart Assoc. (2016) 5(4):e003075. doi: 10.1161/jaha.115.003075
116. Theodoris CV, Mourkioti F, Huang Y, Ranade SS, Liu L, Blau HM, et al. Long telomeres protect against age-dependent cardiac disease caused by NOTCH1 haploinsufficiency. J Clin Invest. (2017) 127(5):1683–8. doi: 10.1172/jci90338
117. Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. (2000) 101(20):2345–8. doi: 10.1161/01.cir.101.20.2345
118. Peterson JC, Chughtai M, Wisse LJ, Gittenberger-de Groot AC, Feng Q, Goumans MTH, et al. Bicuspid aortic valve formation: nos3 mutation leads to abnormal lineage patterning of neural crest cells and the second heart field. Dis Model Mech. (2018) 11(10):dmm034637. doi: 10.1242/dmm.034637
119. Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest. (2011) 121(7):2876–87. doi: 10.1172/jci44555
120. Gharibeh L, Komati H, Bossé Y, Boodhwani M, Heydarpour M, Fortier M, et al. GATA6 Regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation. (2018) 138(10):1025–38. doi: 10.1161/circulationaha.117.029506
121. MacGrogan D, D'Amato G, Travisano S, Martinez-Poveda B, Luxán G, Del Monte-Nieto G, et al. Sequential ligand-dependent notch signaling activation regulates valve primordium formation and morphogenesis. Circ Res. (2016) 118(10):1480–97. doi: 10.1161/circresaha.115.308077
122. Salguero-Jiménez A, Grego-Bessa J, D'Amato G, Jiménez-Borreguero LJ, de la Pompa JL. Myocardial Notch1-rbpj deletion does not affect NOTCH signaling, heart development or function. PloS one. (2018) 13(12):e0203100. doi: 10.1371/journal.pone.0203100
123. Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res. (2000) 87(10):888–95. doi: 10.1161/01.res.87.10.888
124. Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, Lee JH, Higgins AW, Anchan RM, et al. MATR3 Disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet. (2015) 24(8):2375–89. doi: 10.1093/hmg/ddv004
125. Weiss RM, Chu Y, Brooks RM, Lund DD, Cheng J, Zimmerman KA, et al. Discovery of an experimental model of unicuspid aortic valve. J Am Heart Assoc. (2018) 7(13):e006908. doi: 10.1161/jaha.117.006908
126. Akerberg BN, Sarangam ML, Stankunas K. Endocardial Brg1 disruption illustrates the developmental origins of semilunar valve disease. Dev Biol. (2015) 407(1):158–72. doi: 10.1016/j.ydbio.2015.06.015
127. Makki N, Capecchi MR. Cardiovascular defects in a mouse model of HOXA1 syndrome. Hum Mol Genet. (2012) 21(1):26–31. doi: 10.1093/hmg/ddr434
128. Dupuis LE, Nelson EL, Hozik B, Porto SC, Rogers-DeCotes A, Fosang A, et al. Adamts5(-/-) mice exhibit altered aggrecan proteolytic profiles that correlate with ascending aortic anomalies. Arterioscler, Thromb, Vasc Biol. (2019) 39(10):2067–81. doi: 10.1161/atvbaha.119.313077
129. Fulmer D, Toomer K, Guo L, Moore K, Glover J, Moore R, et al. Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation. (2019) 140(16):1331–41. doi: 10.1161/circulationaha.119.038376
130. Thomas PS, Sridurongrit S, Ruiz-Lozano P, Kaartinen V. Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PloS one. (2012) 7(4):e35539. doi: 10.1371/journal.pone.0035539
131. Blaser MC, Wei K, Adams RLE, Zhou YQ, Caruso LL, Mirzaei Z, et al. Deficiency of natriuretic peptide receptor 2 promotes bicuspid aortic valves, aortic valve disease, left ventricular dysfunction, and ascending aortic dilatations in mice. Circ Res. (2018) 122(3):405–16. doi: 10.1161/circresaha.117.311194
132. Yu W, Ma X, Xu J, Heumüller AW, Fei Z, Feng X, et al. VGLL4 Plays a critical role in heart valve development and homeostasis. PLoS Genet. (2019) 15(2):e1007977. doi: 10.1371/journal.pgen.1007977
133. Sievers HH, Schmidtke C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J Thorac Cardiovasc Surg. (2007) 133(5):1226–33. doi: 10.1016/j.jtcvs.2007.01.039
134. Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. (2009) 47(6):828–34. doi: 10.1016/j.yjmcc.2009.08.008
135. Koenig SN, LaHaye S, Feller JD, Rowland P, Hor KN, Trask AJ, et al. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight. (2017) 2(21):e91353. doi: 10.1172/jci.insight.91353
136. Tulzer A, Arzt W, Gitter R, Sames-Dolzer E, Kreuzer M, Mair R, et al. Valvuloplasty in 103 fetuses with critical aortic stenosis: outcome and new predictors for postnatal circulation. Ultrasound Obstet Gynecol. (2022) 59(5):633–41. doi: 10.1002/uog.24792
137. Vorisek CN, Zurakowski D, Tamayo A, Axt-Fliedner R, Siepmann T, Friehs I. Postnatal circulation in patients with aortic stenosis undergoing fetal aortic valvuloplasty: systematic review and meta-analysis. Ultrasound Obstet Gynecol. (2022) 59(5):576–84. doi: 10.1002/uog.24807
138. Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults: evidence report and systematic review for the US preventive services task force. Jama. (2016) 316(19):2008–24. doi: 10.1001/jama.2015.15629
139. Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. (2011) 365(22):2078–87. doi: 10.1056/NEJMoa1110874
140. Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ, Enriquez-Sarano M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. J Am Coll Cardiol. (2002) 40(10):1723–30. doi: 10.1016/s0735-1097(02)02496-8
141. Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. (2005) 352(23):2389–97. doi: 10.1056/NEJMoa043876
142. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of marfan syndrome. Science. (2006) 312(5770):117–21. doi: 10.1126/science.1124287
Keywords: congenital aortic valve stenosis, bicuspid aortic valve, human genetics, cardiac development, animal models
Citation: Yasuhara J, Schultz K, Bigelow AM and Garg V (2023) Congenital aortic valve stenosis: from pathophysiology to molecular genetics and the need for novel therapeutics. Front. Cardiovasc. Med. 10:1142707. doi: 10.3389/fcvm.2023.1142707
Received: 12 January 2023; Accepted: 14 April 2023;
Published: 28 April 2023.
Edited by:
Roney Orismar Sampaio, University of São Paulo, BrazilReviewed by:
Layara Fernanda Vicente Pereira Lipari, Instituto do Coração, BrazilKatherine Yutzey, Cincinnati Children's Hospital Medical Center, United States
© 2023 Yasuhara, Schultz, Bigelow and Garg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yasuhara anVueWFzdWhhcmExMDE2QGdtYWlsLmNvbQ== Vidu Garg dmlkdS5nYXJnQG5hdGlvbndpZGVjaGlsZHJlbnMub3Jn