Abstract
Cardiovascular disease (CVD) remains a predominant cause of morbidity and mortality globally, characterized by pathological mechanisms that encompass inflammation, oxidative stress, metabolic disturbances and immune dysregulation. Recently, the influence of gut microbiota and its metabolites on the onset and progression of CVD has garnered significant attention. Short-chain fatty acids (SCFAs), particularly butyrate, are the primary products of gut microbial fermentation of dietary fiber. Butyrate is instrumental in maintaining intestinal barrier function and immune homeostasis and exhibits notable anti-inflammatory, antioxidant, and metabolic regulatory potentials in cardiovascular diseases. Nonetheless, the precise molecular mechanisms of butyrate in various cardiovascular diseases and its clinical translational value necessitate a systematic review of the literature. This study conducted a comprehensive search of databases, such as PubMed and Web of Science, to synthesize recent basic and clinical research on butyrate and cardiovascular diseases, focusing on its role in hypertension, atherosclerosis, coronary artery disease, atrial fibrillation, diabetic cardiomyopathy, and heart failure. The findings indicate that butyrate can influence cardiovascular health through multiple pathways, including the modulation of G protein-coupled receptors (GPCRs), histone deacetylases (HDACs), and peroxisome proliferator-activated receptors (PPARs). Although numerous experimental studies have corroborated the protective effects of butyrate in cardiovascular diseases, its clinical translation remains challenging owing to factors such as optimal administration route, dose optimization, and individualized treatment strategies. Future research should integrate large-scale population cohort analyses and randomized controlled trials (RCTs) to ascertain the precise efficacy of butyrate in the prevention and treatment of cardiovascular diseases and explore its potential as a novel therapeutic target.
1 Introduction
CVD is the leading cause of mortality and morbidity worldwide. It encompasses conditions such as atherosclerosis, hypertension, and myocardial infarction, with heart failure constituting the terminal stage of various cardiovascular diseases (1, 2). Dyslipidemia, obesity, and insulin resistance are globally acknowledged as significant risk factors for CVD and are frequently associated with alterations in intestinal barrier integrity and gut microbiota composition. With the rapid advancement of the global economy and technology, individuals' lifestyles and dietary habits have undergone significant changes. Notably, the traditional “Western diet” pattern—characterized by high fat, high processed sugar, and low fiber—exerts a substantial impact on metabolic health. Empirical studies have demonstrated that such dietary patterns can markedly alter the composition and abundance of the gut microbiota, reduce the production of short-chain fatty acids (SCFAs), and concurrently increase the secretion of lipopolysaccharide (LPS), trimethylamine N-oxide (TMAO), and proinflammatory cytokines. This, in turn, exacerbates intestinal barrier dysfunction and systemic low-grade inflammation, thereby further promoting the onset and progression of obesity, type 2 diabetes mellitus, and cardiovascular diseases. Epidemiological data corroborate this trend: between 2017 and 2018, the prevalence of obesity increased significantly to 42.4%, compared to 30.5% in 2000 (3). Concurrently, the International Diabetes Federation 2021 Global Diabetes Atlas report indicated that among individuals aged 20–79, there are already 536.6 million patients with diabetes worldwide. Predictive models suggest that in the absence of effective interventions, this number will increase to 783.2 million by 2,045 (4). The intricate interplay among obesity, diabetes, and gut microbiota not only directly exacerbates the global disease burden through systemic chronic inflammation but also significantly elevates cardiovascular disease risk via multiple pathophysiological mechanisms, emerging as a critical public health challenge that demands urgent resolution (5–7). Currently, a range of strategies and policies have been formulated to mitigate the incidence of CVD and prevent its onset. In clinical practice, pharmacological interventions are pivotal in the management and treatment of cardiovascular disease and its associated complications. Furthermore, the public has increasingly adopted the approach of reducing the risk of CVD through the intake of functional foods and dietary supplements. These functional foods and dietary supplements are frequently associated with alterations in the gut microbiota (8, 9). Therefore, maintaining gut microbiota homeostasis and augmenting beneficial microbial metabolites may be effective strategies for the primary prevention of CVD. The metabolites produced by the gut microbiota include TMAO, SCFAs, and bile acid metabolites. Notably, SCFAs, primarily derived from the fermentation of dietary fibers in the colon, such as acetic, propionic, and butyric acids, play crucial protective roles in CVD (2).
Butyric acid, a four-carbon short-chain fatty acid, is predominantly synthesized by various intestinal microbiota through glycolysis via two distinct pathways: butyryl-CoA: acetate CoA-transferase, phosphotransbutyrylase, and butyric acid kinase pathways (10, 11). Within the intestinal tract, it exists in an ionic form and is primarily absorbed via the monocarboxylic acid transporter protein 1 (MCT 1), which is expressed in the parietal and basolateral membranes of colonic epithelial cells. This absorption not only provides an energy source for intestinal epithelial cells but also modulates intestinal immune function (12, 13). Upon entering the portal vein and reaching systemic circulation, butyric acid interacts with multiple host organs, playing a crucial role in anti-inflammatory and antioxidant processes, glucose and lipid metabolism regulation, immune modulation, and enhancement of neural signaling. In comparison to other dietary fibers, butyrate, recognized as the "final effector molecule" of their function, offers several advantages, including direct absorption, rapid onset of action, safety, and a range of effects.This study aimed to provide a comprehensive review of the efficacy of butyrate treatment for CVD and its mechanism of action on CVD risk factors in humans.
2 Main mechanism
The mechanism of action of butyric acid is complex and involves multiple host pathways. Primarily, it functions as a significant epigenetic regulator by inhibiting HDACs and thereby modulating gene expression (14). In the context of epigenetic regulation, histone acetyltransferases (HATs) and HDACs establish HATs that catalyze the acetylation of lysine residues in histone tails, facilitating chromatin relaxation and gene transcription. In contrast, HDACs remove acetyl groups, resulting in chromatin condensation and gene silencing. HDACs are categorized into four classes based on their structural characteristics and cofactor dependency. Classes I (HDAC1–3, 8), II (HDAC4-7, 9–10), and IV (HDAC11) are part of the zinc-dependent Rpd3/Hda1 superfamily, whereas Class III HDACs are NAD+-dependent deacetylases. Sirtuins, belonging to Class III HDACs, can catalyze various histone (e.g., H3K9, H3K18, and H3K56) and non-histone substrates and play a pivotal role in cardiovascular diseases (15, 16). Under steady-state conditions, nuclear factor erythroid 2-related factor 2 (NRF2), a core transcription factor that regulates cellular antioxidant defense and detoxification mechanisms, predominantly resides in the cytoplasm and forms a complex with Kelch-like ECH-associated protein 1 (Keap1). Upon stimulation by NRF2 agonists, NRF2 dissociates from Keap1, undergoes nuclear translocation, and binds to the antioxidant response element (ARE). This process activates the transcription and expression of the downstream target genes. Recent studies have demonstrated that histone acetylation exerts dual regulatory effects on NRF2. On the one hand, they enhance the binding affinity between NRF2 and transcription factors in the promoter regions of target genes through epigenetic modifications. However, they inhibit the ubiquitination of NRF2, significantly reducing its proteasomal degradation rate and effectively enhancing NRF2 protein stability. This increased stability further promotes the interaction of NRF2 with members of the basic region leucine zipper family at ARE sites, ultimately strengthening the cell's defense against oxidative stress (17). Butyric acid plays a crucial role in modulating cellular responses by activating multiple receptors on the surface of the target cells. The primary receptors targeted by butyric acid are GPR43, GPR41, and Olfr78 olfactory receptors. These receptors initiate downstream signaling pathways by coupling with various G protein subtypes, such as Gq and Gi/o, thereby influencing the secretion of gastrointestinal hormones, lipolysis, inflammation, the renin-angiotensin-aldosterone system (RAAS), and the sympathetic nervous system (18). Furthermore, butyric acid functions as an endogenous PPAR agonist, exerting extensive metabolic regulatory effects through the specific activation of PPAR subtypes. Three PPAR isoforms have been identified as integral members of the nuclear receptor superfamily: PPARα, PPARγ, and PPARδ, each exhibiting distinct tissue distribution patterns and physiological functions. PPARα is predominantly expressed in tissues with high fatty acid β-oxidation activity, such as the liver, heart, and skeletal muscle, and is involved in fatty acid uptake and oxidative metabolism by regulating the expression of target genes. In contrast, PPARγ is primarily expressed in white and brown adipose tissues and acts as a key transcription factor that regulates adipocyte differentiation and lipid storage. In contrast, PPARδ is widely distributed in various metabolically active tissues. Collectively, these receptor subtypes regulate various physiological processes, including fatty acid metabolism, glucose homeostasis, discrimination, and vascular function. Activation of PPARα has been shown to significantly reduce plasma triglyceride and very-low-density lipoprotein (VLDL) levels, increase high-density lipoprotein cholesterol (HDL-C) concentration, and inhibit the expression of pro-inflammatory factors. Additionally, PPARγ agonists have been demonstrated to enhance insulin sensitivity and exert anti-atherosclerotic effects by regulating the cholesterol reverse transport pathway (19–21). In summary, butyrate is pivotal in numerous physiological processes, including metabolic, immune, inflammatory, and neuroendocrine regulation via various pathways (Figure 1).
Figure 1
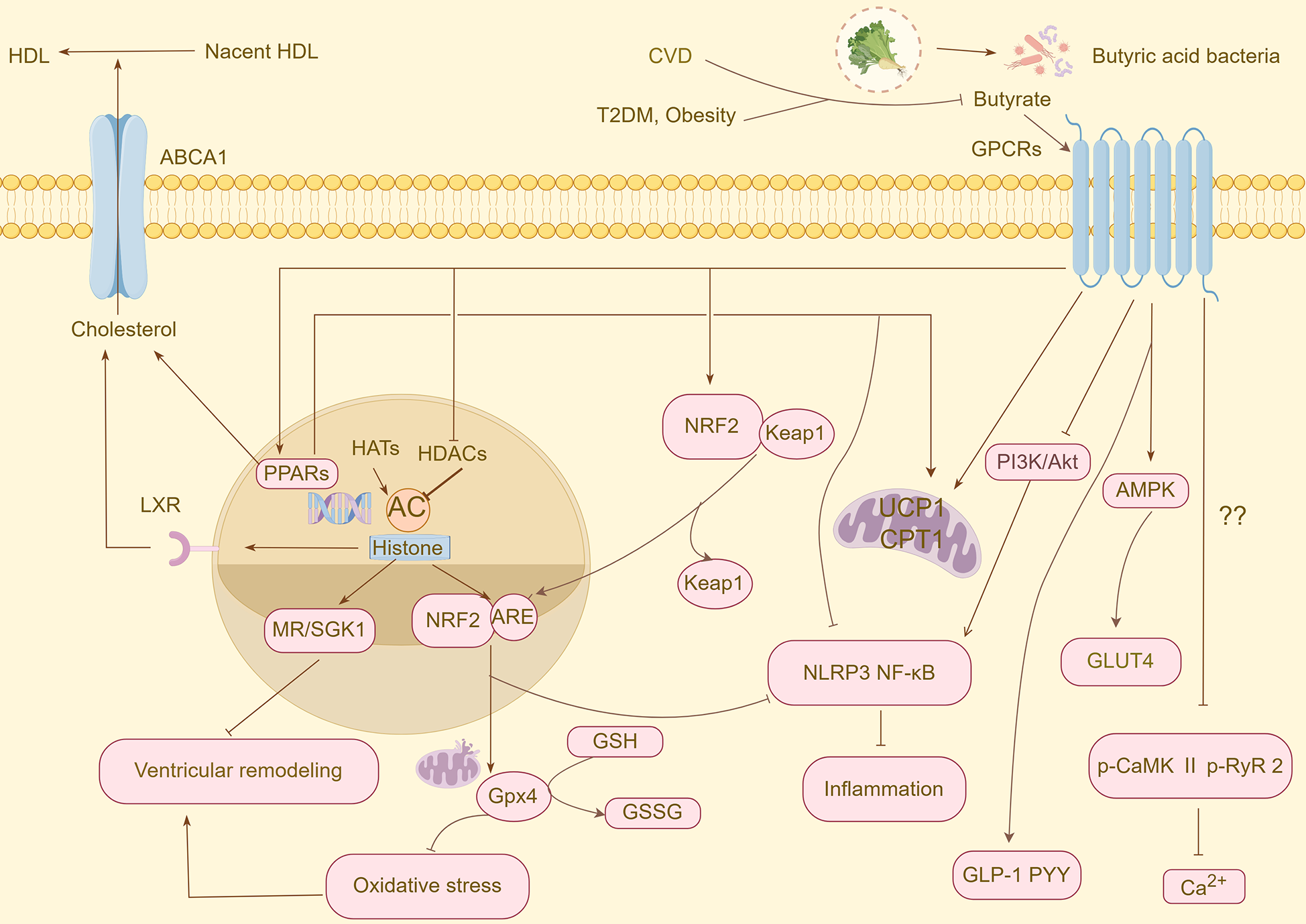
An overview of the protective mechanisms of butyrate against CVD and CVD risk factors. CVD, cardiovascular disease; T2DM, type 2 diabetes mellitus; HDL, high-density lipoprotein cholesterol; ABCA1, ATP-binding cassette transporter protein A1; GPCRs, G protein-coupled receptors; PPARs, peroxisome proliferator-activated receptors; HATs, histone acetyltransferases; HDACs, histone deacetylases; NRF2, nuclear factor erythroid 2-related factor 2; Keap 1, Kelch-like ECH-associated protein 1; ARE, antioxidant response element; GLP-1, glucagon-like peptide-1; PYY, peptide YY; MR, mineralocorticoid receptor; SGK1, glucocorticoid-dependent protein kinase 1; GSH, glutathione; GPX, glutathione peroxidase; GSSG, oxidised glutathione; AMPK, AMP-activated protein kinase; GLUT4, glucose transporter 4; PI3K, phosphatidylinositol, 3-kinase; Akt, protein kinase B; UCP1, uncoupling protein 1; CPT1, carnitine palmitoyltransferase 1; p-CaMK II, phospho-Ca2+/calmodulin-dependent protein kinase-II; p-RyR2, phospho-ryanodine receptor 2.
3 Butyric acid and hypertension
Hypertension is a major public health concern worldwide. Recent research has indicated that metabolites derived from the gut microbiota can influence blood pressure regulation through various mechanisms, including vasodilation, renal sodium/potassium regulation, immunomodulation, and neurotransmitter regulation (22). Consistent findings have been reported in both animal studies and clinical trials. Notably, the abundance of butyric acid-producing bacteria in the gut microbiota is markedly reduced in animal models of simple hypertension and in hypertensive patients. Furthermore, there is a negative correlation between the abundance of butyric acid-producing flora, serum butyric acid levels, and blood pressure (23–27). In a rat model of hypertension induced by obstructive sleep apnea (OSA), Durgan et al. reported a significant reduction in the abundance of butyric acid-producing bacteria in a rat model of hypertension induced by obstructive sleep apnea and established a causal link between intestinal flora dysbiosis and the synergistic effect of OSA on hypertension through cecum transplantation (28). Gomez-Arango et al. identified a significant negative correlation between the abundance of intestinal butyrate-producing bacteria, butyrate production, and blood pressure in obese pregnant women, with similar findings reported in pregnant women with late-onset preeclampsia (29, 30). Interestingly, fecal butyrate levels were elevated in hypertensive patients compared to normotensive individuals and showed a significant correlation with 24-hour mean blood pressure, a phenomenon potentially attributable to impaired sodium-coupled monocarboxylate transporter protein 1 (SMCT 1) in the colon (29, 31). The findings of these studies are presented in the table. To elucidate the causal relationship between butyric acid and hypertension, Kim et al. administered butyric acid to C57BL6 mice infused with angiotensin II (Ang II), resulting in a significant reduction in mean arterial pressure (24).
Extensive research has been conducted on the mechanisms by which butyric acid regulates blood pressure. Current understanding suggests that GPR41 and Olfr78 play roles in blood pressure regulation, with both being expressed in smooth muscle cells of small resistance vessels. Olfr78 activation is associated with renin-dependent antihypertensive effects (32). Wang et al. demonstrated that sodium butyrate (NaB) inhibits renal (pro)renin receptors and the intrarenal renin-angiotensin system, thereby suppressing Ang II-induced hypertension (33). GPR41 knockout mice exhibit systolic hypertension, and Onyszkiewicz et al. observed that the modulation of GPR41/GPR43 by butyric acid reduced the mean arterial pressure in Wistar rats (34, 35). Furthermore, a high-salt diet is a significant contributor to hypertension, with mineralocorticoid receptor (MR) and glucocorticoid-dependent protein kinase 1 (SGK1) being crucial regulators of the salt homeostasis. The transcriptional activities of MR and SGK1 are regulated by histone acetylation. NaB alleviates high salt-induced glomerular injury and regulates water-sodium balance to improve hypertension by affecting MR transcription levels (36). In addition, endothelial nitric oxide synthase (eNOS), which is specifically expressed in the vascular endothelium, is influenced by histone deacetylation. Researchers have shown that Histone deacetylase inhibitors can induce eNOS expression in vascular smooth muscle cells and play a vital role in reducing smooth muscle cell proliferation when administered with NaB (37). The expression of inflammatory factors such as IL17, INFγ, and IL1β leads to vascular endothelial dysfunction and phenotypic transformation of vascular smooth muscle cells (VSMC). Butyrate is implicated in blood pressure regulation by repairing the intestinal barrier and reversing Th17 cell polarization (24, 38, 39). NOD-like receptor thermal protein domain-associated protein 3 (NLRP3) is a critical inflammatory vesicle involved in the pathogenesis of various diseases, including atherosclerosis, heart failure, and metabolic syndrome. Sun et al. found that NLRP3 activation promotes phenotypic transformation and proliferation of hypertensive VSMCs, and subsequent studies confirmed its role in hypertension through pharmacological inhibition of NLRP3 (40, 41). Several studies have shown that butyrate, a significant short-chain fatty acid, inhibits NLRP3 and exhibits potent anti-inflammatory effects (42, 43). It is widely acknowledged that blood pressure regulation is mediated by the hypothalamus-adrenal-sympathetic neural axis, with neuroinflammation being a key factor in sympathetic nerve overexcitation. Microglia release various proinflammatory cytokines, including TNF-α, IL-1β, and IL-6, which contribute to hypertension development (44). Neurological studies have demonstrated that butyrate inhibits microglial-mediated neuroinflammation (45). Reduced expression levels of the butyrate-sensitive receptors FFAR2 and FFAR3 have been observed in the paraventricular nucleus of the hypothalamus in spontaneously hypertensive rats, suggesting that the gut-brain axis may represent a novel pathway for butyrate-mediated blood pressure regulation (29). Additionally, butyrate has been shown to modulates cardiac sympathetic and vagal excitability (24, 34) (Table 1).
Table 1
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Kim et al., (24) | in vivo | 1 g/kg | C57BL6 mice infused with angiotensin II | Improved spontaneous cardiac baroreceptor reflexes and cardiac sympathetic tone |
| Reduced mean arterial pressure | ||||
| Reversed the increase in Th17 cells | ||||
| Improved myocardial hypertrophy | ||||
| Wang et al., (33) | in vivo | 1 μg/kg min | SD rats with uninephrectomized and angiotensin II injection | Inhibited renal the PRR and renal angiotensin expression levels |
| Reduced mean arterial pressure | ||||
| ex vivo | 2 μmol/L | The innermedullary collecting duct cells | Regulated PRR expression and renin release | |
| Onyszkiewicz et al., (34) | in vivo | 0.14,1.4,2.8,5.6 mmol/kg | Wistar rats | Regulated GPR41/43 expression |
| ex vivo | 5 μM–1 mM | Mesentery and gracilis arteries | Improved arterial diastolic function | |
| Wu et al., (36) | in vivo | 1 g/kg | Deoxycorticosterone acetate/salt-induced hypertensive rats | Decreased renal TNF-α and IL-6 mRNA levels, and increased IL-10 mRNA levels |
| Inhibited MR and SGK1 expression on renal tubules | ||||
| ex vivo | 0.5 µM | HK2 cell line | Inhibited aldosterone-induced MR/SGK1 expression |
The effect of butyrate in hypertension.
Th17, T helper 17; PRR, (pro)renin receptor; GPR, G-protein-coupled receptor; TNF-α, tumor necrosis factor-alpha; IL-6, interleukin-6; IL-10, interleukin-10; MR, mineralocorticoid receptor; SGK1, glucocorticoid-dependent protein kinase 1; HK, human kidney.
4 Butyric acid and atherosclerosis
Atherosclerosis (AS), a predominant cause of cardiovascular disease, is characterized by a progressive inflammatory condition of the arterial vessel wall, facilitated by pro-inflammatory cytokines, inflammatory signaling pathways, bioactive lipids, and adhesion molecules (46, 47). An imbalance in gut microecology is a potential contributing factor to the onset of atherosclerosis, with butyric acid, a metabolite of intestinal flora, serving as a crucial short-chain fatty acid in mitigating atherosclerosis (48–51). Aguilar et al. demonstrated that dietary supplementation with butyrate diminishes atherosclerotic lesions and enhances plaque stability by inhibiting NF-κB activation (52). Butyrate decreases the levels of IL-1β, IL-6, and TNF-α in human aortic endothelial cells by upregulating intestinal tight junction proteins and restoring intestinal immunity, thereby ameliorating inflammatory damage in the arterial wall (53). Under pathological conditions, NLRP3 inflammasomes induce autocatalysis and activation of caspase-1, leading to the release of proinflammatory cytokines, which are pivotal in the progression of atherosclerosis. In a partially ligated carotid artery mouse model, butyrate significantly reduced the formation and activation of NLRP3 inflammasomes in the carotid artery wall of Asc+/+ mice, which was comparable to that observed in Asc-/- mice. In cultured EOMA cells, butyrate inhibited the formation and activation of NLRP3 inflammasomes induced by 7-ketocholesterol and cholesterol crystals (54). Previous studies have suggested that butyrate also influences the regulation of macrophage M1/M2 phenotypic transition. Butyrate stimulates hepatic ketogenesis, and the ketone body β-hydroxybutyrate may promote macrophage M2 polarization by inhibiting the NLRP3 inflammasome. However, this may be associated with the regulation of GPR43/HDAC-miRNAs (55–58). Adhesion molecules, which mediate cell-cell and cell-extracellular matrix recognition and adhesion, contribute to the development of atherosclerosis by mediating tissue inflammation, immune response, and other mechanisms (46). Butyrate significantly inhibits the expression of intercellular adhesion molecule-1, intercellular adhesion molecule, and E-selectin, thereby reducing the adhesion of human monocytes (THP-1 cells) to human aortic endothelial cells (HAECs) (53, 54, 59).
Oxidative stress induces inflammation, facilitating the development of arterial lipid plaques in atherosclerosis. Reactive oxygen species (ROS) influence the formation of oxidized low-density lipoproteins and promote various forms of cell death, including pyroptosis, apoptosis, and ferroptosis (60). Nicotinamide adenine dinucleotide oxidase (NOX) is an important source of ROS. Aguilar et al. demonstrated that oral administration of butyrate inhibited NOX activity in endothelial cells and reduced macrophage migration and activation at the lesion site, thereby mitigating oxidative stress and inflammation in the brain. This effect may be associated with the regulation of the PPARδ/miR-181 pathway (61, 62). NRF2 is a crucial transcription factor in the antioxidative stress response, primarily acting on the cystine/glutamate antiporter (System Xc-), GPX4, GSH, and heme oxygenase 1, among others. Its potent agonists have been shown to ameliorate nearly all disease-like phenotypes in cardiovascular disease models, including heart failure, myocardial infarction, cardiomyopathy, and atherosclerosis (63–65). Butyrate is a potent NRF2 agonist that modulates glutathione/glutathione S-transferase activity to scavenge ROS in VSMC (66, 67). Mathew et al. found that butyrate treatment of VSMC not only upregulated GPX4 expression but also enhanced the overall catalytic activity of GPX and inhibited the expression of inflammatory genes targeted by NF-κB (68). GPX4 is pivotal in regulating ferroptosis, and its unique function of inhibiting lipid peroxidation by converting lipid hydroperoxides into nontoxic alcohols. However, the mechanism by which butyric acid inhibits ferroptosis in atherosclerosis remains unclear (69, 70). Phagocytosis of oxidized low-density lipoproteins (LDL) by macrophages and smooth muscle cells to form foam cells is a critical step in the development of atherosclerosis. Butyrate has been shown to reduces cholesterol uptake by inhibiting Niemann-Pick C1-like protein 1 (NPC1L1), an intestinal phytosterol and cholesterol transporter protein, through the upregulation of hepatic X receptor transcriptional activity (71). ATP-binding cassette transporter protein A1 (ABCA1) is an essential cholesterol transporter protein regulated by liver X receptor alpha (LXRα) and PPARs that facilitates cholesterol efflux for HDL synthesis and inhibits foam cell formation (72, 73). Du et al. found that butyrate protects against high-fat diet-induced atherosclerosis by mediating the activation of ABCA1 in macrophages of ApoE-deficient mice through specificity protein 1 (Sp1) (74). Additionally, histone deacetylase 3 (HDAC3) reduces ABCA1 expression by decreasing LXRα promoter levels, suggesting a potential role for butyrate in inhibiting LDL and foam cell formation (50) (Table 2).
Table 2
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Aguilar et al., (52) | in vivo | Feed containing 1% sodium butyrate | ApoE-/- mice | Reduced macrophage migration at lesion sites and increased collagen deposition and plaque stability |
| Reduced aortic atherosclerotic lesions | ||||
| ex vivo | 0, 0.1, 0.5, 1.0 mM | EA.Hy926 cells | Reduced oxLDL uptake | |
| Reduced nuclear translocation of the p65 subunit | ||||
| ex vivo | 0.5 mM | Macrophages | Reduced oxLDL uptake and intracellular accumulation | |
| Yuan et al., (54) | in vivo | 500 mg/kg | Mice with partially ligated carotid arteries | Blocked the formation of NLRP3 inflammatory vesicles |
| ex vivo | 1 mM | EOMA cells | Inhibited the formation of NlRP3 inflammatory vesicles | |
| Ma et al., (55) | in vivo | 200 mg/kg | ApoE-/- mice | Promoted M2 polarization |
| Inhibited the expression of IL-1β, IL-6, IL-17A, IFN-γ | ||||
| Up-regulated GPR43 expression and down-regulated HDAC 3 | ||||
| Shen et al., (53) | ex vivo | 50, 100 μM | HAECs, THP-1 cells | Reduced ICAM-1 and VCAM-1 expression levels |
| Reduced levels of IL-1β, IL-6 and TNF-α | ||||
| Wang et al., (59) | ex vivo | 100, 200 μM | HUVECs, THP-1 cells | Reduced IL-8 and MCP-1 secretion |
| Inhibited VCAM-1 and E-selectin expression | ||||
| Aguilar et al., (61) | ex vivo | 0.5 mM | EA.Hy926 cells | Reduced expression of NOX subunit p22phox and CCL2 production |
| Tian et al., (62) | in vivo | 0.5 mg/g | ApoE-/- mice and PPARδ-/-mice | Reduced production of ROS |
| ex vivo | 100 μmol/L | mBMECs | Reduced NOX2 expression | |
| Increased miR-181b expression | ||||
| Ranganna et al., (66) | ex vivo | 5 mM | VSMCs | Improved the overall catalytic activity of GSH⁄GST |
| Reduced levels of ROS | ||||
| Mathew et al., (68) | ex vivo | 0, 1, 2, 3, 4, 5, 6, 7, 8 mM | VSMCs | Inhibited the synthesis of NF-κBp65, IKKα, IKKβ and IkBα |
| improved overall GPX activity | ||||
| Increased GPX3 and GPX4 expression | ||||
| Chen et al., (71) | ex vivo | 0, 0.01, 0.1, 1 mmol/L | Caco-2 | Reduced NPC1L1 and inhibited cholesterol uptake |
| Enhanced LXRE-Luc transcriptional activity | ||||
| Du et al., (74) | in vivo | 200 mg/kg·day,400 mg/kg·day | ApoE-/- mice | Reduced the formation of atherosclerotic lesions |
| Du et al., (74) | ex vivo | 0.5, 1, 2, 5 mM | Murine RAW 264.7 macrophages | Increased ABCA1 protein levels and cholesterol efflux |
| Decreased Sp1 levels |
The effect of butyrate in atherosclerosis.
ApoE, apolipoprotein E; oxLDL, oxidized low-density lipoprotein; NlRP3, NOD-like receptor thermal protein domain-associated protein 3; IL-1β, Interleukin-1β; IL-17A, interleukin-17A; IL-8, interleukin-8; IFN-γ, interferon-gamma; TNF-α, tumor necrosis factor-alpha; MCP-1, monocyte chemoattractant protein-1; CCL2, C-C motif chemokine ligand 2; PPARδ, peroxisome proliferator-activated receptor delta; mBMECs, mouse brain microvascular endothelial cells; VSMCs, vascular smooth muscle cells; NF-κB, nuclear factor-κB; IKK, IκB kinase; Caco, human colorectal adenocarcinoma cells; HDAC 3, histone deacetylase 3; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; NOX, NADPH oxidase; GSH, glutathione; GST, glutathione S-transferase; ROS, reactive oxygen species; GPX, glutathione peroxidase; NPC1L1, niemann-pick C1-like protein 1; LXRE-Luc, liver X receptor response element-driven luciferase; ABCA1, ATP-binding cassette transporter protein A1; Sp1, specificity protein 1.
5 Butyric acid and coronary artery disease
Coronary artery disease (CAD) is a major cause of mortality worldwide. One study identified associations between the gut microbiome and metabolic alterations with the severity of CAD, noting a significant reduction in butyric acid-producing Trichoniculidae and Rumen cocci in patients with CAD compared to healthy controls. It has been posited that butyric acid-producing Faecalibacterium and Bayeriella rosans may sustain normal coronary artery physiology through their interactions with various serum metabolites (75). Liu et al. analyzed the microbial community using 16S rRNA gene sequencing and shotgun metagenomic sequencing, revealing a low molar proportion of butyrate in stool samples from patients with acute myocardial infarction (AMI) and a negative correlation between the abundance of Roseberry and AMI severity (76). Additionally, patients with acute coronary syndrome (ACS) exhibit a relative reduction in butyrate-producing bacteria, including Clostridium spp., Hadrus anaerobic rods, Streptococcus thermophilus, and Blauderia (77). Similarly, research involving young patients with AMI indicated that the gut metabolite butyric acid may exert a protective effect against coronary artery disease (78).
Butyric acid, a gut microbiota-derived metabolite, has been implicated in the repair process after myocardial infarction (MI) (79, 80). Cheng et al. developed NaB-loaded poly(lactic acid-hydroxyacetic acid)-poly(N-isopropylacrylamide) microspheres (PP-N) to extend the release of NaB, which were then injected into the myocardial ischemic region of an AMI rat model. These findings indicate that NaB released from the PPN, in conjunction with Sirt3, activates various biological functions, reduces ROS production in the infarct area, and significantly enhances the systolic and diastolic functions of the rat heart, surpassing the effects of direct NaB injection (81). Furthermore, β-hydroxybutyric acid, a butyric acid metabolite, exhibits anti-inflammatory properties in patients with AS. Through clinical studies and animal experiments, Chen et al. suggested that the cardioprotective effect of butyric acid in myocardial infarction may be associated with increased plasma β-hydroxybutyric acid levels and improved intestinal function in a dose-dependent manner (80). The high mortality rate associated with acute myocardial infarction is attributed to malignant arrhythmias and heart failure. Malignant arrhythmias in infarcted hearts are associated with excessive sympathetic innervation and inflammatory responses. Jiang et al. demonstrated that butyrate administration could reduce myocardial infarction size and prevent post-infarction ventricular arrhythmia by inhibiting cardiac sympathetic remodeling and inflammatory responses (82).
Early ischemia-reperfusion therapy can reduce the mortality risk in patients with acute myocardial infarction. However, secondary damage to cardiomyocytes, termed Myocardial Ischemia-Reperfusion Injury (MIRI), occurs during this therapy. This process involves elevated ROS levels within cardiomyocytes, disrupting the balance of myocardial oxidative and antioxidant systems, accompanied by M1 macrophage polarization, calcium overload, and other factors, ultimately leading to cellular necrosis, apoptosis, and ferroptosis (83–86). Hu et al. demonstrated that an intraperitoneal injection of 300 mg/kg NaB significantly mitigated the increase in malondialdehyde levels and decrease in superoxide dismutase levels induced by myocardial ischemia-reperfusion, thereby alleviating oxidative stress and the inflammatory response (87). Conversely, Lim et al. explored whether intraperitoneal injection of 10 mg/kg sodium acetate, sodium propionate, and NaB administered one hour prior to coronary artery ligation could positively affect myocardial ischemia-reperfusion injury. Their results indicated that NaB and sodium propionate reduced cardiac infarct size by inhibiting apoptosis, with the anti-apoptotic effects of NaB being evident in both the infarct and border zones (88). A subsequent study further elucidated the role of butyric acid in reversing MIRI-induced autonomic dysfunction by inhibiting the paraventricular nucleus and superior cervical ganglia (89) (Table 3).
Table 3
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Cheng et al., (81) | in vivo | 5 μg | Rats with acute myocardial infarction | Improved systolic and diastolic function of the heart |
| Inhibited the expression of the NOX subunit gp91phox | ||||
| Activated Sirt3 | ||||
| Jiang et al., (82) | in vivo | 1 mol/L (butyric acid,7.5 ml/kg) | Rats with acute myocardial infarction | Reduced myocardial infarct size and improved susceptibility to malignant ventricular tachyarrhythmias |
| Inhibited TNF-α and IL-1β expression and promoted IL-10 production | ||||
| Improved cardiac sympathetic remodeling | ||||
| Hu et al., (87) | in vivo | 300 mg/kg | Rats with myocardial ischemia-reperfusion injury | Inhibited TNF-α and IL-6 expression |
| Reduced MDA levels and increased SOD levels | ||||
| Reduced myocardial I/R-induced infarct size | ||||
| Lim, (88) | in vivo | 10 mg/kg | Rats with myocardial ischemia-reperfusion injury | Reduced the proportion of apoptotic cells in the area of myocardial infarction and the border zone |
| Yu et al., (89) | in vivo | 200 mmol/L | Rats with myocardial ischemia-reperfusion injury | Improved oxidative stress response and apoptosis |
| Inhibited cFos expression in PVN and SCG |
The effect of butyrate in coronary artery disease.
NOX, NADPH oxidase; Sirt3, sirtuin 3; TNF-α, tumor necrosis factor-alpha; IL-1β, interleukin-1β; IL-6, interleukin-6; MDA, malondialdehyde; SOD, superoxide dismutase; PVN, paraventricular; SCG, nucleus superior cervical ganglion.
6 Butyric acid and atrial fibrillation
Atrial fibrillation (AF) is one of the most prevalent cardiac arrhythmias and is associated with an increased risk of vascular embolic disease, mortality, and disability. Current research suggests a connection between gut dysbiosis, inflammation, and the development of AF. Zhang et al. investigated the hypothesis that gut dysbiosis promotes age-related AF through activation of the NLRP3 inflammasome induced by LPS and glucose (90). In examining the potential effects of gut microbiota-derived SCFAs on AF, some researchers have identified disruptions in genes related to SCFA synthesis and observed reduced levels of fecal SCFAs, including acetic, propionic, and butyric acids, in patients with AF. Ex vivo experiments have demonstrated that SCFAs may prevent AF by improving cardiac electrical remodeling and fibrosis via the GPR43/NLRP3 signaling pathway. The primary mechanism involves SCFAs preventing the overexpression of calcium/calmodulin-dependent protein kinase II(CaMKII) phosphorylation and its associated ryanodine receptor 2 (RyR2) phosphorylation in the atria, thereby alleviating calcium disruption (91). Given its potent anti-inflammatory properties, butyric acid may offer a protective effect against AF; however, further investigation is required.
7 Butyric acid and diabetic cardiomyopathy
In 1972, Rubler et al. identified a novel cardiomyopathy in patients with diabetes, termed diabetic cardiomyopathy (DCM). This condition is characterized by hyperglycemia-induced diastolic contractile dysfunction of the heart in the absence of coronary artery disease, hypertension, or valvular heart disease (92). DCM is characterized by cardiac fibrosis, cardiomyocyte hypertrophy, and microvascular pathology, with primary mechanisms involving impaired metabolic pathways, oxidative stress, inflammation, and cell death. Intestinal flora and its metabolites are significantly associated with diabetic cardiomyopathy (93, 94). Guo et al. conducted a study examining the diagnostic value of plasma SCFAs levels and hypoxia-inducible factor 3A (HIF 3A) intron 1 methylation in patients with DCM. The study revealed that patients with DCM exhibited lower plasma butyric acid levels and increased overall mean methylation in HIF 3A intron 1 compared with patients with diabetes mellitus (DM). The receiver operating characteristic curve indicated that the area under the curve for the combination of plasma butyric acid levels and HIF 3A intron 1 CpG 6 methylation was 0.737, with a sensitivity of 75% and specificity of 79% at a cutoff value of 0.69. This suggests that the association between plasma butyric acid and HIF 3A intron 1 CpG 6 methylation may represent a potential mechanism for DCM pathogenesis and that the combined detection of these levels may serve as a diagnostic tool for DCM (95). The addition of 1% butyrate to the drinking water of streptozotocin-induced diabetic mice demonstrated that butyrate ameliorated myocardial hypertrophy and fibrosis by inhibiting histone deacetylase activity and apoptosis, while inducing angiogenesis in the myocardium (96). Furthermore, butyrate plays a role in regulating impaired insulin metabolism signaling, which is discussed below (Table 4).
Table 4
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Chen et al., (96) | in vivo | 1% NaB drinking water | Diabetic mice | Reduced cardiomyocyte diameter and myocardial interstitial collagen deposition |
| Reduced HDAC activity | ||||
| Improved apoptotic signaling |
The effect of butyrate in diabetic cardiomyopathy.
NaB, sodium butyrate; HDAC, histone deacetylase.
8 Butyric acid and heart failure
Heart failure (HF) is characterized by impaired systolic and diastolic functions of the heart and represents the terminal stage of nearly all cardiovascular diseases, imposing an economic burden and significantly diminishing the quality of life of patients with HF. Disruptions in cardiac energy metabolism are critical in the pathogenesis of HF, leading to an inadequate cardiac energy supply, which ultimately results in cardiac pump failure and systemic energy metabolism. Fatty acids serve as the primary carbon source for cardiomyocytes, and the fatty acid oxidation pathway depends on the carnitine palmitoyltransferase system to transport fatty acids to the mitochondria for energy production (97). The reduced activity of carnitine palmitoyltransferase 1 (CPT 1) on the outer mitochondrial membrane in failing hearts indicates impaired fatty acid oxidation in cardiomyocytes, prompting ketone bodies to become an alternative carbon source for ATP production to meet the demands of myocardial energy metabolism. Carley et al. induced cardiac hypertrophy and dysfunction in rats through transverse aortic narrowing and subsequently perfused the isolated hearts with 13C-labeled β-hydroxybutyric acid and butyrate. They observed that the oxidative utilization of butyrate exceeded that of β-hydroxybutyric acid, suggesting that butyrate may serve as a superior alternative fuel for failing heart (98). Another study confirmed that butyrate upregulates CPT1A expression to regulate fatty acid oxidative energy supply (99). In addition to regulating cardiac metabolism, butyric acid modulates cardiac hypertrophy and exerts antifibrotic effects (100). Seefeldt et al. demonstrated that butyric acid functions as a positive inotropic agent with vasodilatory effects (101). Collectively, these findings, along with the previously established antihypertensive and antiatherosclerotic effects of butyric acid, highlight its potential anti-HF properties (Table 5).
Table 5
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Carley et al., (98) | in vivo | 1 mmol/L | Rats with coarctation of the transverse aorta | Entered the TCA produces more acetyl-CoA |
| Increased coupling between β-oxidation and TCA cycling activities | ||||
| Zhang et al., (100) | in vivo | 1 g/kg/d | Male SD rats for chronic infusion of Ang II | Reduced cardiomyocyte hypertrophy |
| Reduced collagen fiber deposition | ||||
| Inhibited the expression of NLRP3 and IL-1β | ||||
| Seefeldt et al., (101) | in vivo | 0.033,0.165, 0.661 g/kg | Male SD rats | Increased cardiac output |
| Increased left ventricular ejection fraction | ||||
| 0.1,0.5,5 mM | Isolated heart | Increased left ventricular systolic blood pressure | ||
| 0.1–30 mM | Isolated coronary arteries | Reduced vascular resistance | ||
| Dilated blood vessels |
The effect of butyrate in heart failure.
TCA, tricarboxylic acid cycle; Ang II, angiotensin II; NLRP3, NOD-like receptor thermal protein domain-associated protein 3; IL-1β, interleukin-1β.
9 Butyric acid and obesity, diabetes
Obesity and diabetes, which are interconnected metabolic disorders, substantially increase the risk of cardiovascular disease through their combined effects. The fundamental pathological mechanisms involve a self-perpetuating cycle of chronic, low-grade inflammation and metabolic disturbances. Excessive accumulation of visceral adipose tissue serves as an independent risk factor for cardiovascular lesions via various mechanisms. At the tissue level, caloric surplus induces abnormal hypertrophy and proliferation of adipocytes, leading to pathological expansion of white adipose tissue and a concomitant reduction in the amount of brown fat. This remodeling of adipose tissue drives changes in adipokine secretion profiles, marked by a significant increase in pro-inflammatory factors (TNF-α, IL-6, and MCP-1) and a decrease in adiponectin, which exerts protective effects while simultaneously activating the NLRP3 inflammasome to promote IL-1β secretion. At the microenvironment level, hypoxia in adipose tissue results in a reduction in regulatory T cells and polarization of macrophages towards the pro-inflammatory M1 phenotype, establishing a chronic low-grade inflammatory state. This inflammatory milieu interacts with the characteristic metabolic abnormalities of diabetes: persistent hyperglycemia activates the polyol pathway and promotes the formation of advanced glycation end products (AGEs), which activate the NF-κB signaling pathway through RAGE receptors, leading to excessive ROS production and endothelial dysfunction. At the myocardial level, metabolic remodeling occurs, including increased fatty acid oxidation, impaired glucose utilization, mitochondrial dysfunction, and increased lipotoxicity. Overactivation of the RAAS and endothelin-1 systems further promotes myocardial fibrosis and vascular remodeling, ultimately culminating in various cardiovascular complications, including atherosclerosis, acute coronary syndrome, and heart failure. The synergistic mechanism involves an inflammatory microenvironment associated with obesity, which exacerbates insulin resistance, whereas the hyperglycemic state linked to diabetes further intensifies the inflammatory response, creating a positive feedback loop that exacerbates the condition. This interaction results in an exponential increase in cardiovascular risk among patients with metabolic disorders, underscoring the need for multitargeted interventions to address the pathological network (94, 102–107). A multicenter randomized controlled trial involving overweight or obese adults with type 2 diabetes mellitus demonstrated that individuals who achieved a weight loss of at least 10% within a year experienced a 21% reduction in the risk of major outcomes, such as acute myocardial infarction and stroke, and a 24% reduction in the risk of secondary outcomes, such as hospitalization for congestive heart failure (108). Effective blood glucose control can enhance the prognosis and quality of life of patients with diabetes mellitus. Research suggests that dapagliflozin can reduce the hospitalization rate for heart failure (109). Therefore, effective management of weight and blood glucose levels is crucial for improving cardiovascular outcomes.
Although numerous strategies exist for lowering blood glucose levels and reducing body weight, the outcomes of coexisting cardiovascular conditions remain poor. To enhance the understanding of the diagnosis and treatment of metabolic disorders, researchers have identified the gut microbiota as a crucial "hidden organ" within the human body that plays a significant role in regulating energy metabolism and maintaining immune homeostasis (110, 111). While exploring therapeutic interventions for diabetes, researchers have identified that butyric acid-producing microbiota can enhance glucose metabolism, although this is not universally applicable to all bacteria (112, 113). The interplay between glucotoxicity, oxidative stress, inflammation, and other mechanisms results in the progressive decline of pancreatic B-cell function, transitioning from insulin resistance to impaired insulin secretion. NaB has been shown to effectively enhance pancreatic B cell function and insulin sensitivity. Powers et al. demonstrated that NaB modulates the expression of glucagon and insulin genes (114). Conversely, another study indicated that butyric acid might influence the expression of lysine at position 18 of histone H3. Butyrylation of lysine at this position in the promoter region induces the expression of insulin genes and promotes insulin secretion in rats. However, this process adversely affects the expression of genes associated with pancreatic β-cell identity, including Pdx 1, MafA, NeuroD1, Gck, and Slc 2a2 (115).Furthermore, NaB ameliorated endoplasmic reticulum stress by inhibiting the PERK-CHOP pathway, thereby preventing histological changes and functional impairment of pancreatic islets in diabetic rats (116). Yan et al. demonstrated that NaB inhibits autophagy and NLRP3 activation by blocking the PI3K/Akt/NF-κB pathway in THP-1 cells, which in turn reduces oxidative stress, inflammation, and metabolic disorders induced by glycosylation end products (117). Intraperitoneal administration of NaB (500 mg/kg/d) in juvenile DM rats indicated that NaB facilitated the proliferation of pancreatic islet B cells by modulating the p38/ERK MAPK and apoptosis pathways (118). In investigating the anti-inflammatory properties of butyric acid in diabetes, NaB was found to suppress NF-κB activation and the release of inflammatory cytokines in high-glucose-treated THP-1 cells. Conversely, in lipopolysaccharide (LPS)-treated THP-1 cells, 1 mM NaB reduced TNF-α and IFN-γ release. However, the difference was not statistically significant in patients with type 2 diabetes with poor glycemic control compared to normal subjects, and it is important to note that the study's sample size was small, resulting in a relatively large margin of error (119). Muscle tissue, which is primarily responsible for glucose uptake, comprises type I and II skeletal muscle fibers. Type I fibers exhibit a higher oxidative capacity and greater number of mitochondria than type II fibers. Evidence indicates that NaB significantly increases the number of type I skeletal muscle fibers and ameliorates insulin resistance in mice fed a high-fat diet (HFD). This effect may be associated with epigenetic regulation of nucleosome localization. Additionally, oral administration of NaB to mice alters mitochondrial function, enhances fatty acid oxidation, and increases thermogenesis (120, 121). Intestinal gluconeogenesis (IGN) positively influences glucose and energy homeostasis by signaling glucose release from the IGN to the brain via the portal glucose sensor through the peripheral nervous system, thereby affecting glucose metabolism. Butyric acid may activate IGN gene expression to promote intestinal gluconeogenesis (122). Glucagon-like peptide-1 (GLP-1), peptide YY (PYY), and glucose-dependent insulinotropic polypeptide (GIP), which are expressed in the intestinal mucosal epithelium, are crucial for insulin secretion and maintaining glucose homeostasis. Following butyrate administration, the pattern of insulin release in high-fat diet-fed mice coincided with the release of GLP-1, PYY, and GIP, whereas other short-chain fatty acids (SCFA) did not induce intestinal hormone secretion, except for propionate-induced GIP secretion. This suggests an indirect regulation of insulin production (123, 124). Clinical data indicate that oral butyric acid administration in patients with type 2 diabetes mellitus (T2DM) also elevates serum GLP-1 levels (125). Regarding the hypoglycemic effects of butyric acid compared with those of clinically used drugs, some studies have demonstrated that NaB improves blood glucose, glycated hemoglobin (HbA1c), insulin resistance, and lipid profiles compared with metformin. Furthermore, butyric acid may act as a co-agent with metformin to enhance insulin sensitization (126, 127). In a study by de Groot et al., targeting patients with type 1 diabetes mellitus, oral butyric acid supplementation did not significantly affect innate or adaptive immunity in patients with long-term diabetes, affecting only peripheral blood immune cells. This finding does not fully reflect the overall immune response of the human body or pancreatic tissues (128). Notably, differences exist between rat, mouse, and human tissues, necessitating further clinical studies to elucidate the efficacy of NaB in reducing glucose levels in humans (Table 6).
Table 6
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Wang et al., (115) | ex vivo | 5 mM | Rat islet cells | Inhibited the expression of Pdx 1, MafA, NeuroD 1, Gck and Slc2a2 |
| Increased H3K18bu levels in the Ins1 and Ins2 promoter regions | ||||
| ex vivo | 5 mM | INS-1 cells | Promoted insulin secretion | |
| Powers et al., (114) | ex vivo | 0.2,1, 2, 5, and 20 mM | RIN1056A and RIN1027-B2 cell line | Increased glucagon and insulin mRNA levels |
| Hu et al., (116) | in vivo | 500 mg/kg | Type 2 diabetes rats | Regulated the pancreatic endoplasmic reticulum stress PERK-CHOP pathway |
| Yan et al., (117) | ex vivo | 400 μmol/L | THP-1 cells | Increased the expression of SOD and decreased the expression of ROS and MDA. |
| Regulated the PI3K/Akt/NF-κB pathway to inhibit autophagy and NLRP3 | ||||
| Khan & Jena, (118) | in vivo | 500 mg/kg | Juvenile diabetic rats | Inhibited pancreatic B cell apoptosis Modulated the p38/ERK MAPK pathway |
| Maintained glucose tolerance and homeostasis | ||||
| Moon & Yun, (141) | ex vivo | 0–30 μM | THP-1 cells | Inhibited mRNA levels of IL-6 and TNF-α |
| Reduced the interaction of p300 in acetylated NF-κB and TNF-α | ||||
| Wibowo et al., (119) | ex vivo | 1 mM | peripheral blood mononuclear cells | TNF-α and IFN-γ expression were not significantly reduced |
| Gao et al., (120) | in vivo | High-fat diet with 5% WT/WT NaB | C57BL/6 mice | Increased energy expenditure and fatty acid oxidation |
| Improved insulin sensitivity | ||||
| Increased type I oxide fiber | ||||
| Henagan et al., (121) | in vivo | High-fat diet with 5% WT/WT NaB | C57BL/6 mice | Increased the proportion of type 1 oxide fibers |
| Improved mitochondrial dysfunction. | ||||
| Modulated nuclear-encoding mitochondrial genes associated with the PPAR signaling pathway | ||||
| De Vadder et al., (122) | in vivo | Diet with 5% WT/WT NaB | Male SD rats | Increases IGN gene expression, including G6PC and PCK1 |
| Lin et al., (124) | in vivo | High-fat diet with 5% WT/WT NaB | C57BL/6 mice | Increased levels of GLP-1, GIP, PYY in plasma |
| Roshanravan et al., (125) | Clinical trial | 600 mg/day | T2DM Patients | Increased postprandial GLP-1 concentration |
| Khan & Jena, (127) | in vivo | 400 mg/kg, 800 mg/kg | T2DM rats | Reduced plasma triglyceride, cholesterol, LDL, VLDL,glucose and HbA1c levels |
| de Groot et al., (128) | Clinical trial | 4 g/day | Patients with T1DM | Does not improve innate or adaptive immunity |
The effect of butyrate in diabetes.
INS-1, Insulinoma cell line 1; Pdx 1, pancreatic and duodenal homeobox 1; MafA, V-maf musculoaponeurotic fibrosarcoma oncogene homolog A; NeuroD 1, neuronal differentiation 1; Gck, glucokinase; Slc2a2, solute carrie; family 2 member 2; H3K18bu, histone H3 lysine 18 butyrylation; Ins1, insulin 1; Ins1, insulin 2; RIN1056A, rat insulinoma1056A; RIN1027-B2, rat insulinoma 1027-B2; SOD, superoxide dismutase; ROS, reactive oxygen species; MDA, malondialdehyde; NLRP3, NOD-like receptor thermal protein domain-associated protein 3; IL-6, interleukin-6; TNF-α, tumor necrosis factor-alpha; NaB, sodium butyrate; PPAR, peroxisome proliferator-activated receptor; IGN, intestinal gluconeogenesis; G6PC, glucose-6-phosphatase catalytic subunit; PCK1, phosphoenolpyruvate carboxykinase 1; GLP-1, glucagon-like peptide-1; GIP, glucose-dependent insulinotropic polypeptide; PYY, peptide YY; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein; HbA1c, hemoglobin A1c; T2DM, type 2 diabetes mellitus; T1DM, type 1 diabetes.
The effects of butyric acid on weight loss have been demonstrated in animal studies. Aguilar et al. found that NaB inhibited adipocyte hypertrophy by upregulating PPAR-γ in obese ApoE- mice, while reducing leptin secretion in adipose tissue and enhancing lipocalin production, thereby decreasing adipose tissue NF-κB expression and mitigating inflammatory responses in obese mice (129). Compared with metformin, NaB supplementation in diabetic mice resulted in smaller adipocytes and reduced fat accumulation, significantly inhibiting NLRP3 activation and diminishing the release of downstream inflammatory factors in the testicular and subcutaneous adipose tissues (43). Zhu et al. demonstrated that NaB modulates the expression of beige-specific genes, such as Hoxa9 and Tmem 26, and enhances the expression of mitochondrial uncoupling protein 1 (UCP1) and peroxisome proliferator-activated receptor γ. NaB also augments the thermogenic properties of brown adipose tissue, a phenomenon partially attributed to increased sympathetic innervation of adipose tissue (130). Furthermore, NaB enhances adipose tissue metabolic function by promoting the phosphorylation of adenosine monophosphate-activated protein kinase (AMPK) and facilitating glucose transporter 4 (GLUT4) activity in adipose tissue (131). GPR43 has been identified as a potential therapeutic target for metabolic disorders, and GPR43 deficiency leads to obesity in mice. Conversely, mice overexpressing GPR43 specifically in adipose tissue exhibit a significant reduction in body weight, suggesting that GPR43 activation in adipose tissue is a potential mechanism for butyrate-induced weight loss (132). Li et al. explored the effect of butyrate on appetite and found that oral administration of butyrate suppressed appetite by inhibiting neuropeptide Y-expressing orexigenic neurons in the hypothalamus and reducing the number of FOS-positive neurons in the nucleus tractus solitarius of the brainstem and the dorsal complex of the vagus nerve (133). In contrast, Whitt et al. reported that intestinal epithelial expression of HDAC3 promotes diet-induced obesity, whereas butyrate may mitigate HDAC3 activity to prevent obesity (134). The liver plays a crucial role in coordinating metabolic changes to ensure continuous energy production and delivery to the body. Numerous studies have investigated whether butyrate enhances liver functions. A rapid influx of energy through repeated fasting and refeeding stimulates fatty acid synthesis, resulting in triglyceride accumulation and ROS production. NaB reduces the risk of non-alcoholic fatty liver disease (NAFLD) by inhibiting the mRNA levels of fatty acid synthase and increasing the mRNA levels of fatty acid oxidation-associated Cpt1a expression. However, another study found that NaB did not elevate the expression of genes involved in fatty acid β-oxidation, which might be related to the dosage of NaB used (135, 136). Additionally, insulin-inducible genes (Insig), potent inhibitors of the cleavage and maturation of sterol regulatory element-binding proteins (SREBP), which are key transcription factors in adipogenesis, were found to reduce hepatic steatosis and improve lipid profiles in mice fed a high-fat diet by activating hepatic kinase B1 and increasing Insig-1 protein levels (137). However, relatively few studies have examined the effects of butyrate on weight control in humans with obesity. Data from a cross-sectional study indicated a negative correlation between plasma butyrate levels and body mass index (BMI) (138). Fecal microbiota transplantation can increase butyrate-producing bacteria in the gut of obese patients with T2DM, and when combined with lifestyle interventions, it can lead to more favorable changes in the recipients' microbiota and improved lipid profiles (139). In obese children, butyrate supplementation as an adjunct to standard treatment resulted in reductions in BMI, waist circumference, and insulin levels after six months (140). However, there is a paucity of research on obese populations, necessitating further investigation (Table 7).
Table 7
| Article | Type of study | Butyrate dose | Research target | Result |
|---|---|---|---|---|
| Aguilar et al., (129) | in vivo | Diet with 1% NaB | Diet-induced obese ApoE-/- mice | Inhibited fat cell hypertrophy |
| Reduced leptin and increased adiponectin | ||||
| Increased PPAR-γ expression and inhibited NF-κB expression | ||||
| Wang et al., (43) | in vivo | 1.0 g/kg | db/db mice | Reduced the expression of IL-1, IL-6 and TNF-α |
| Reduced protein levels of NLRP3 and IL-1β | ||||
| Zhu et al., (130) | in vivo | High-fat diet with 0.4% WT/WT NaB | C57BL/6 mice | Increased mRNA expression of beige-selective genes such as Hoxa9 and Tmem26 |
| Increased UCP-1 and PGC-1α protein levels | ||||
| Enhanced the activity of the adipose sympathetic nerve | ||||
| Gao et al., (131) | in vivo | 400 mg/kg | High-fat diet C57BL/6 mice | Increased the levels of p-AMPK, GLUT4, and HDAC in liver and adipose tissue |
| Li et al., (133) | in vivo | High-fat diet with 5% WT/WT NaB | APOE*3-Leiden.CETP mice | Reduced the number of FOS-positive neurons and neuronal ratios of neuropeptide Y and c-FOS within the arcuate nucleus of the hypothalamus |
| Reduced the number of FOS-positive neurons in the brainstem nucleus solitary tract and dorsal vagus complex | ||||
| Whitt et al., (134) | in vivo | 80 mg/d | HDAC3ΔIEC, HDAC3ΔIEC-IND mice | Reduced HDAC enzyme activity in IECs |
| Reduced body weight | ||||
| Honma et al., (135) | in vivo | A diet high in sucrose with 5% NaB | SD male rats | Inhibited mRNA levels of Fas |
| Increased the mRNA level of Cpt1a | ||||
| Hattori et al., (136) | in vivo | A high-sucrose diet supplemented with 1% and 3% NaB | Wistar rats | Does not improve CPT1 gene expression |
| Zhao et al., (137) | in vivo | 200 mg/kg | High-fat diet mice | Increased hepatic protein levels of Insig-1 |
| Activated LKB1 | ||||
| Coppola et al., (140) | Randomized, quadruple-blind, parallel-group, placebo-controlled trial | 20 mg/kg body weight per day, up to a maximum of 800 mg/d | Obese children | Reduced BMI, waist circumference, and plasma insulin levels in obese children |
The effect of butyrate in obesity.
NaB, sodium butyrate; ApoE, apolipoprotein E; PPAR-γ, peroxisome proliferator-activated receptor gamma; NF-κB, nuclear factor-κB; IL-1, interleukin-1; IL-6, interleukin-6; TNF-α, tumor necrosis factor-alpha; NLRP3, NOD-like receptor thermal protein domain-associated protein 3; Hoxa9, homeobox A9; Tmem26, transmembrane protein 26; UCP-1, uncoupling protein 1; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; AMPK, AMP-activated protein kinase; HDAC, histone acetylation.
10 Conclusions and prospects
Butyrate plays a significant role in the prevention and treatment of CVD through various pathways, including anti-inflammatory, antioxidant, and metabolic regulation (Figure 2). However, current research remains predominantly in the basic stage, necessitating advancements in clinical translational research. Future research should investigate the effects of butyrate on CVD in depth from the perspectives of clinical validation, intervention optimization, mechanistic exploration, and safety assessment. Initially, large-scale RCTs are required to evaluate the differences in the efficacy of butyrate in diverse CVD populations, such as patients with hypertension and atherosclerosis. Moreover, intervention strategies should be optimized by comparing different supplementation methods (butyrate salts, probiotics, or dietary fibers) and determining optimal dosage and safety. Additionally, it is essential to assess the interactions between butyrate and cardiovascular drugs such as statins and antihypertensive medications to avoid adverse reactions. Ultimately, integrating multi-omics technologies to elucidate the regulatory mechanisms of the "butyrate-gut microbiota-cardiovascular" axis will provide a foundation for precise interventions. This framework will facilitate the translation of butyrate from basic research to clinical application, supporting the precise prevention and treatment of CVD.
Figure 2
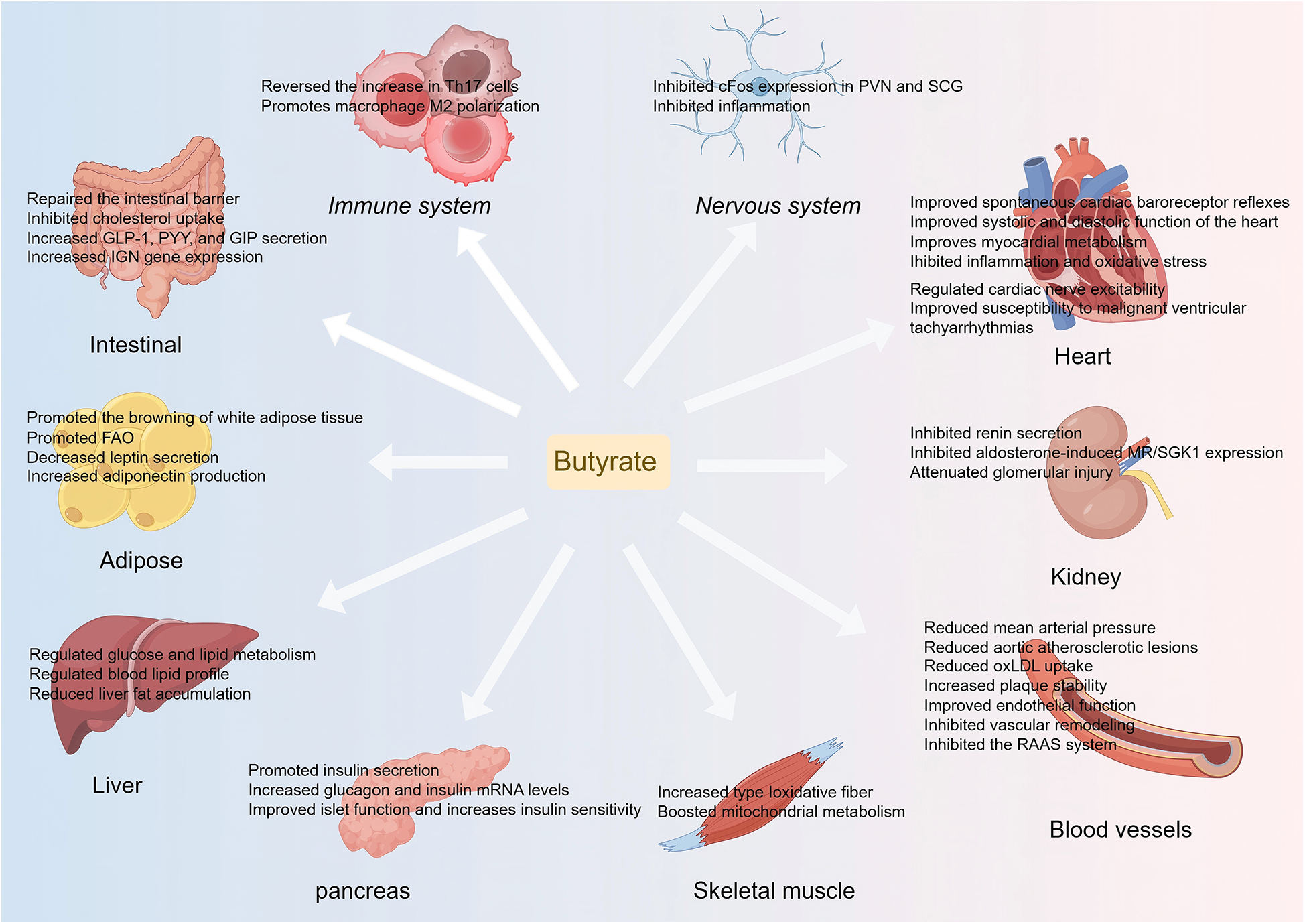
An overview of butyrate's protective effects in CVD and CVD risk factors. Th 17, T helper 17; PVN, paraventricular; SCG, nucleus superior cervical ganglion; GLP-1, glucagon-like peptide-1; GIP, glucose-dependent insulinotropic polypeptide; PYY, peptide YY; IGN, intestinal gluconeogenesis; oxLDL, oxidized low-density lipoprotein; FAO, fatty acid oxidation; MR, mineralocorticoid receptor; SGK1, glucocorticoid-dependent protein kinase 1; RAAS, renin-angiotensin-aldosterone system.
Statements
Author contributions
QX: Data curation, Conceptualization, Methodology, Writing – original draft. XL: Data curation, Methodology, Conceptualization, Writing – original draft. ZW: Writing – review & editing, Visualization, Formal analysis. XL: Visualization, Formal analysis, Writing – review & editing. QJ: Writing – review & editing, Conceptualization, Validation, Supervision, Funding acquisition. MX: Validation, Supervision, Writing – review & editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Collaborative Research Fund of the Zunyi Science and Technology Program (Grant No.: Zunshikehe HZ [2024] No. 110) and the Science and Technology Fund of the Guizhou Provincial Health Commission (Grant No.: gzwkj2024-341).
Acknowledgments
We sincerely appreciate the support and assistance provided by the Science and Technology Fund of Guizhou Provincial Health Commission and the Collaborative Research Fund of the Zunyi Science and Technology Program for this study. Their valuable insights and resources were instrumental in the successful completion of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
KwanGFMayosiBMMocumbiAOMirandaJJEzzatiMJainYet alEndemic cardiovascular diseases of the poorest billion. Circulation. (2016) 133:2561–75. 10.1161/circulationaha.116.008731
2.
DuttaroyAK. Role of gut microbiota and their metabolites on atherosclerosis, hypertension and human blood platelet function: a review. Nutrients. (2021) 13:144. 10.3390/nu13010144
3.
BeamAClingerEHaoL. Effect of diet and dietary components on the composition of the gut microbiota. Nutrients. (2021) 13:2795. 10.3390/nu13082795
4.
SunHSaeediPKarurangaSPinkepankMOgurtsovaKDuncanBBet alIDF Diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. 10.1016/j.diabres.2021.109119
5.
ChartrandDJMurphy-DesprésAAlmérasNLemieuxILaroseEDesprésJP. Overweight, obesity, and CVD risk: a focus on visceral/ectopic fat. Curr Atheroscler Rep. (2022) 24:185–95. 10.1007/s11883-022-00996-x
6.
La SalaLPontiroliAE. Prevention of diabetes and cardiovascular disease in obesity. Int J Mol Sci. (2020) 21:8178. 10.3390/ijms21218178
7.
LeeCJSearsCLMaruthurN. Gut microbiome and its role in obesity and insulin resistance. Ann N Y Acad Sci. (2020) 1461:37–52. 10.1111/nyas.14107
8.
CheangWSChenX. Protective effects of piceatannol on cardiovascular diseases. PHYTONutrients. (2024) 3:36–45. 10.62368/pn.v3i.30
9.
WanCWWongCNPinWKWongMHKwokCYChanRYet alChlorogenic acid exhibits cholesterol lowering and fatty liver attenuating properties by up-regulating the gene expression of PPAR-α in hypercholesterolemic rats induced with a high-cholesterol diet. Phytother Res. (2013) 27:545–51. 10.1002/ptr.4751
10.
KohADe VadderFKovatcheva-DatcharyPBäckhedF. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. (2016) 165:1332–45. 10.1016/j.cell.2016.05.041
11.
LouisPHoldGLFlintHJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. (2014) 12:661–72. 10.1038/nrmicro3344
12.
CummingsJHPomareEWBranchWJNaylorCPMacfarlaneGT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. (1987) 28:1221–7. 10.1136/gut.28.10.1221
13.
SivaprakasamSBhutiaYDYangSGanapathyV. Short-chain fatty acid transporters: role in colonic homeostasis. Compr Physiol. (2017) 8:299–314. 10.1002/cphy.c170014
14.
HazimeHDucasaGMSantanderAMBritoNGonzalez-HortaEEQuinteroMAet alDUOX2 activation drives bacterial translocation and subclinical inflammation in IBD-associated dysbiosis. Gut. (2025):1–13. 10.1136/gutjnl-2024-334346
15.
EvansLWAthukoralaMMartinez-GurynKFergusonBS. The role of histone acetylation and the microbiome in phytochemical efficacy for cardiovascular diseases. Int J Mol Sci. (2020) 21:4006. 10.3390/ijms21114006
16.
XuSKamatoDLittlePJNakagawaSPelisekJJinZG. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther. (2019) 196:15–43. 10.1016/j.pharmthera.2018.11.003
17.
YangXLiuYCaoJWuCTangLBianWet alTargeting epigenetic and post-translational modifications of NRF2: key regulatory factors in disease treatment. Cell Death Discov. (2025) 11:189. 10.1038/s41420-025-02491-z
18.
LymperopoulosASusterMSBorgesJI. Short-chain fatty acid receptors and cardiovascular function. Int J Mol Sci. (2022) 23:3303. 10.3390/ijms23063303
19.
AjithTAJayakumarTG. Peroxisome proliferator-activated receptors in cardiac energy metabolism and cardiovascular disease. Clin Exp Pharmacol Physiol. (2016) 43:649–58. 10.1111/1440-1681.12579
20.
LiuHJLiaoHHYangZTangQZ. Peroxisome proliferator-activated receptor-γ is critical to cardiac fibrosis. PPAR Res. (2016) 2016:2198645. 10.1155/2016/2198645
21.
StaelsBFruchartJC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. (2005) 54:2460–70. 10.2337/diabetes.54.8.2460
22.
MuralitharanRRJamaHAXieLPehASnelsonMMarquesFZ. Microbial peer pressure: the role of the gut microbiota in hypertension and its complications. Hypertension. (2020) 76:1674–87. 10.1161/HYPERTENSIONAHA.120.14473
23.
YangTSantistebanMMRodriguezVLiEAhmariNCarvajalJMet alGut dysbiosis is linked to hypertension. Hypertension. (2015) 65:1331–40. 10.1161/HYPERTENSIONAHA.115.05315
24.
KimSGoelRKumarAQiYLobatonGHosakaKet alImbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci. (2018) 132:701–18. 10.1042/CS20180087
25.
ChenLHeFJDongYHuangYWangCHarshfieldGAet alModest sodium reduction increases circulating short-chain fatty acids in untreated hypertensives: a randomized, double-blind, placebo-controlled trial. Hypertension. (2020) 76:73–9. 10.1161/HYPERTENSIONAHA.120.14800
26.
TilvesCYehHCMaruthurNJuraschekSPMillerEWhiteKet alIncreases in circulating and fecal butyrate are associated with reduced blood pressure and hypertension: results from the SPIRIT trial. J Am Heart Assoc. (2022) 11:e024763. 10.1161/JAHA.121.024763
27.
LuoXHanZKongQWangYMouHDuanX. Clostridium butyricum prevents dysbiosis and the rise in blood pressure in spontaneously hypertensive rats. Int J Mol Sci. (2023) 24:4955. 10.3390/ijms24054955
28.
DurganDJGaneshBPCopeJLAjamiNJPhillipsSCPetrosinoJFet alRole of the gut microbiome in obstructive sleep apnea-induced hypertension. Hypertension. (2016) 67:469–74. 10.1161/HYPERTENSIONAHA.115.06672
29.
YangTMageeKLColon-PerezLMLarkinRLiaoYSBalazicEet alImpaired butyrate absorption in the proximal colon, low serum butyrate and diminished central effects of butyrate on blood pressure in spontaneously hypertensive rats. Acta Physiol. (2019) 226:e13256. 10.1111/apha.13256
30.
Gomez-ArangoLFBarrettHLMcIntyreHDCallawayLKMorrisonMDekker NitertMet alIncreased systolic and diastolic blood pressure is associated with altered gut microbiota composition and butyrate production in early pregnancy. Hypertension. (2016) 68:974–81. 10.1161/HYPERTENSIONAHA.116.07910
31.
HuartJLeendersJTaminiauBDescyJSaint-RemyADaubeGet alGut microbiota and fecal levels of short-chain fatty acids differ upon 24-hour blood pressure levels in men. Hypertension. (2019) 74:1005–13. 10.1161/HYPERTENSIONAHA.118.12588
32.
PluznickJLProtzkoRJGevorgyanHPeterlinZSiposAHanJet alOlfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. (2013) 110:4410–5. 10.1073/pnas.1215927110
33.
WangLZhuQLuALiuXZhangLXuCet alSodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin–angiotensin system. J Hypertens. (2017) 35:1899–908. 10.1097/hjh.0000000000001378
34.
OnyszkiewiczMGawrys-KopczynskaMKonopelskiPAleksandrowiczMSawickaAKozniewskaEet alButyric acid, a gut bacteria metabolite, lowers arterial blood pressure via colon-vagus nerve signaling and GPR41/43 receptors. Pflugers Arch. (2019) 471:1441–53. 10.1007/s00424-019-02322-y
35.
NatarajanNHoriDFlavahanSSteppanJFlavahanNABerkowitzDEet alMicrobial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. (2016) 48:826–34. 10.1152/physiolgenomics.00089.2016
36.
WuCChenZZhangLZhuYDengMHuangCet alSodium butyrate ameliorates deoxycorticosterone acetate/salt-induced hypertension and renal damage by inhibiting the MR/SGK1 pathway. Hypertens Res. (2021) 44:168–78. 10.1038/s41440-020-00548-3
37.
GanYShenYHWangJWangXUtamaBWangJet alRole of histone deacetylation in cell-specific expression of endothelial nitric-oxide synthase. J Biol Chem. (2005) 280:16467–75. 10.1074/jbc.M412960200
38.
GuzikTJHochNEBrownKAMcCannLARahmanADikalovSet alRole of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. (2007) 204:2449–60. 10.1084/jem.20070657
39.
DeussenAKopalianiI. Targeting inflammation in hypertension. Curr Opin Nephrol Hypertens. (2023) 32:111–7. 10.1097/MNH.0000000000000862
40.
SunHJRenXSXiongXQChenYZZhaoMXWangJJet alNLRP3 Inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. (2017) 8:e3074. 10.1038/cddis.2017.470
41.
KrishnanSMLingYHHuuskesBMFerensDMSainiNChanCTet alPharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. (2019) 115:776–87. 10.1093/cvr/cvy252
42.
BayazidABKimJGAzamSJeongSAKimDHParkCWet alSodium butyrate ameliorates neurotoxicity and exerts anti-inflammatory effects in high fat diet-fed mice. Food Chem Toxicol. (2022) 159:112743. 10.1016/j.fct.2021.112743
43.
WangXHeGPengYZhongWWangYZhangB. Sodium butyrate alleviates adipocyte inflammation by inhibiting NLRP3 pathway. Sci Rep. (2015) 5:12676. 10.1038/srep12676
44.
LiYWeiBLiuXShenXZShiP. Microglia, autonomic nervous system, immunity and hypertension: is there a link?Pharmacol Res. (2020) 155:104451. 10.1016/j.phrs.2019.104451
45.
WeiHYuCZhangCRenYGuoLWangTet alButyrate ameliorates chronic alcoholic central nervous damage by suppressing microglia-mediated neuroinflammation and modulating the microbiome-gut-brain axis. Biomed Pharmacother. (2023) 160:114308. 10.1016/j.biopha.2023.114308
46.
ZhuYXianXWangZBiYChenQHanXet alResearch progress on the relationship between atherosclerosis and inflammation. Biomolecules. (2018) 8:80. 10.3390/biom8030080
47.
De MeyerGRYZurekMPuylaertPMartinetW. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat Rev Cardiol. (2024) 21:312–25. 10.1038/s41569-023-00957-0
48.
GregoryJCBuffaJAOrgEWangZLevisonBSZhuWet alTransmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. (2015) 290:5647–60. 10.1074/jbc.M114.618249
49.
JieZXiaHZhongSLFengQLiSLiangSet alThe gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. (2017) 8:845. 10.1038/s41467-017-00900-1
50.
ChenWZhangSWuJYeTWangSWangPet alButyrate-producing bacteria and the gut-heart axis in atherosclerosis. Clin Chim Acta. (2020) 507:236–41. 10.1016/j.cca.2020.04.037
51.
KasaharaKKrautkramerKAOrgERomanoKAKerbyRLVivasEIet alInteractions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol. (2018) 3:1461–71. 10.1038/s41564-018-0272-x
52.
AguilarECLeonelAJTeixeiraLGSilvaARSilvaJFPelaezJMet alButyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr Metab Cardiovasc Dis. (2014) 24:606–13. 10.1016/j.numecd.2014.01.002
53.
ShenHRWangZYShenZLiuTTGuoYDGaoTLet alBacterial butyrate mediates the anti-atherosclerotic effect of silybin. Biomed Pharmacother. (2023) 169:115916. 10.1016/j.biopha.2023.115916
54.
YuanXWangLBhatOMLohnerHLiPL. Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: antioxidant action of butyrate. Redox Biol. (2018) 16:21–31. 10.1016/j.redox.2018.02.007
55.
MaHYangLLiuYYanRWangRZhangPet alButyrate suppresses atherosclerotic inflammation by regulating macrophages and polarization via GPR43/HDAC-miRNAs axis in ApoE-/- mice. PLoS One. (2023) 18:e0282685. 10.1371/journal.pone.0282685
56.
LiXLiRYouNZhaoXLiJJiangW. Butyric acid ameliorates myocardial fibrosis by regulating M1/M2 polarization of macrophages and promoting recovery of mitochondrial function. Front Nutr. (2022) 9:875473. 10.3389/fnut.2022.875473
57.
LiHGaoZZhangJYeXXuAYeJet alSodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes. (2012) 61:797–806. 10.2337/db11-0846
58.
YoumYHNguyenKYGrantRWGoldbergELBodogaiMKimDet alThe ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–9. 10.1038/nm.3804
59.
WangYXuYYangMZhangMXiaoMLiX. Butyrate mitigates TNF-alpha-induced attachment of monocytes to endothelial cells. J Bioenerg Biomembr. (2020) 52:247–56. 10.1007/s10863-020-09841-9
60.
ZhengDLiuJPiaoHZhuZWeiRLiuK. ROS-triggered endothelial cell death mechanisms: focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol. (2022) 13:1039241. 10.3389/fimmu.2022.1039241
61.
AguilarECSantosLCLeonelAJde OliveiraJSSantosEANavia-PelaezJMet alOral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down-regulation in endothelial cells. J Nutr Biochem. (2016) 34:99–105. 10.1016/j.jnutbio.2016.05.002
62.
TianQLeungFPChenFMTianXYChenZTseGet alButyrate protects endothelial function through PPARdelta/miR-181b signaling. Pharmacol Res. (2021) 169:105681. 10.1016/j.phrs.2021.105681
63.
LobodaADamulewiczMPyzaEJozkowiczADulakJ. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. (2016) 73:3221–47. 10.1007/s00018-016-2223-0
64.
HeFRuXWenT. NRF2, A transcription factor for stress response and beyond. Int J Mol Sci. (2020) 21:4777. 10.3390/ijms21134777
65.
RobertsJARainbowRDSharmaP. Mitigation of cardiovascular disease and toxicity through NRF2 signalling. Int J Mol Sci. (2023) 24:6723. 10.3390/ijms24076723
66.
RangannaKMathewOPYatsuFMYousefipourZHayesBEMiltonSG. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. (2007) 274:5962–78. 10.1111/j.1742-4658.2007.06119.x
67.
ChenHQianYJiangCTangLYuJZhangLet alButyrate ameliorated ferroptosis in ulcerative colitis through modulating Nrf2/GPX4 signal pathway and improving intestinal barrier. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:166984. 10.1016/j.bbadis.2023.166984
68.
MathewOPRangannaKMiltonSG. Involvement of the antioxidant effect and anti-inflammatory response in butyrate-inhibited vascular smooth muscle cell proliferation. Pharmaceuticals. (2014) 7:1008–27. 10.3390/ph7111008
69.
LiuYWanYJiangYZhangLChengW. GPX4: the hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. (2023) 1878:188890. 10.1016/j.bbcan.2023.188890
70.
ZhangWLiuYLiaoYZhuCZouZ. GPX4, Ferroptosis, and diseases. Biomed Pharmacother. (2024) 174:116512. 10.1016/j.biopha.2024.116512
71.
ChenYXuCHuangRSongJLiDXiaM. Butyrate from pectin fermentation inhibits intestinal cholesterol absorption and attenuates atherosclerosis in apolipoprotein E-deficient mice. J Nutr Biochem. (2018) 56:175–82. 10.1016/j.jnutbio.2018.02.011
72.
BabaevVRIshiguroHDingLYanceyPGDoveDEKovacsWJet alMacrophage expression of peroxisome proliferator-activated receptor-alpha reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2007) 116:1404–12. 10.1161/CIRCULATIONAHA.106.684704
73.
GaidarovIChenXAnthonyTMaciejewski-LenoirDLiawCUnettDJ. Differential tissue and ligand-dependent signaling of GPR109A receptor: implications for anti-atherosclerotic therapeutic potential. Cell Signal. (2013) 25:2003–16. 10.1016/j.cellsig.2013.06.008
74.
DuYLiXSuCXiMZhangXJiangZet alButyrate protects against high-fat diet-induced atherosclerosis via up-regulating ABCA1 expression in apolipoprotein E-deficiency mice. Br J Pharmacol. (2020) 177:1754–72. 10.1111/bph.14933
75.
LiuHChenXHuXNiuHTianRWangHet alAlterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome. (2019) 7:68. 10.1186/s40168-019-0683-9
76.
LiuCSunZShaliSMeiZChangSMoHet alThe gut microbiome and microbial metabolites in acute myocardial infarction. J Genet Genomics. (2022) 49:569–78. 10.1016/j.jgg.2021.12.007
77.
Talmor-BarkanYBarNShaulAAShahafNGodnevaABussiYet alMetabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat Med. (2022) 28:295–302. 10.1038/s41591-022-01686-6
78.
GuoMFanXTuerhongjiangGWangCWuHLouBet alTargeted metabolomic analysis of plasma fatty acids in acute myocardial infarction in young adults. Nutr Metab Cardiovasc Dis. (2021) 31:3131–41. 10.1016/j.numecd.2021.06.024
79.
TangTWHChenHCChenCYYenCYTLinCJPrajnamitraRPet alLoss of gut microbiota alters immune system composition and cripples postinfarction cardiac repair. Circulation. (2019) 139:647–59. 10.1161/circulationaha.118.035235
80.
ChenHCLiuYWChangKCWuYWChenYMChaoYKet alGut butyrate-producers confer post-infarction cardiac protection. Nat Commun. (2023) 14:7249. 10.1038/s41467-023-43167-5
81.
ChengPZengWLiLHuoDZengLTanJet alPLGA-PNIPAM microspheres loaded with the gastrointestinal nutrient NaB ameliorate cardiac dysfunction by activating Sirt3 in acute myocardial infarction. Adv Sci. (2016) 3:1600254. 10.1002/advs.201600254
82.
JiangXHuangXTongYGaoH. Butyrate improves cardiac function and sympathetic neural remodeling following myocardial infarction in rats. Can J Physiol Pharmacol. (2020) 98:391–9. 10.1139/cjpp-2019-0531
83.
HeJLiuDZhaoLZhouDRongJZhangLet alMyocardial ischemia/reperfusion injury: mechanisms of injury and implications for management (review). Exp Ther Med. (2022) 23:430. 10.3892/etm.2022.11357
84.
WuQXuRZhangKSunRYangMLiKet alCharacterization of early myocardial inflammation in ischemia-reperfusion injury. Front Immunol. (2022) 13:1081719. 10.3389/fimmu.2022.1081719
85.
MurphyESteenbergenC. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. (2008) 88:581–609. 10.1152/physrev.00024.2007
86.
XuMLiXSongL. Baicalin regulates macrophages polarization and alleviates myocardial ischaemia/reperfusion injury via inhibiting JAK/STAT pathway. Pharm Biol. (2020) 58:655–63. 10.1080/13880209.2020.1779318
87.
HuXZhangKXuCChenZJiangH. Anti-inflammatory effect of sodium butyrate preconditioning during myocardial ischemia/reperfusion. Exp Ther Med. (2014) 8:229–32. 10.3892/etm.2014.1726
88.
LimSH. Tyrate and propionate, short chain fatty acids, attenuate myocardial damages by inhibition of apoptosis in a rat model of ischemia-reperfusion. J Korean Soc Appl Biol Chem. (2010) 53:570–7. 10.3839/jksabc.2010.088
89.
YuZHanJChenHWangYZhouLWangMet alOral supplementation with butyrate improves myocardial ischemia/reperfusion injury via a gut-brain neural circuit. Front Cardiovasc Med. (2021) 8:718674. 10.3389/fcvm.2021.718674
90.
ZhangYZhangSLiBLuoYGongYJinXet alGut microbiota dysbiosis promotes age-related atrial fibrillation by lipopolysaccharide and glucose-induced activation of NLRP3-inflammasome. Cardiovasc Res. (2022) 118:785–97. 10.1093/cvr/cvab114
91.
ZuoKFangCLiuZFuYLiuYLiuLet alCommensal microbe-derived SCFA alleviates atrial fibrillation via GPR43/NLRP3 signaling. Int J Biol Sci. (2022) 18:4219–32. 10.7150/ijbs.70644
92.
ZhaoXLiuSWangXChenYPangPYangQet alDiabetic cardiomyopathy: clinical phenotype and practice. Front Endocrinol. (2022) 13:1032268. 10.3389/fendo.2022.1032268
93.
JiaGHillMASowersJR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. 10.1161/circresaha.117.311586
94.
RitchieRHAbelED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. 10.1161/circresaha.120.315913
95.
GuoYZouJXuXZhouHSunXWuLet alShort-chain fatty acids combined with intronic DNA methylation of HIF3A: potential predictors for diabetic cardiomyopathy. Clin Nutr. (2021) 40:3708–17. 10.1016/j.clnu.2021.04.007
96.
ChenYDuJZhaoYTZhangLLvGZhuangSet alHistone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. (2015) 14:99. 10.1186/s12933-015-0262-8
97.
ChenCWangJZhuXHuJLiuCLiuL. Energy metabolism and redox balance: how phytochemicals influence heart failure treatment. Biomed Pharmacother. (2024) 171:116136. 10.1016/j.biopha.2024.116136
98.
CarleyANMauryaSKFasanoMWangYSelzmanCHDrakosSGet alShort-chain fatty acids outpace ketone oxidation in the failing heart. Circulation. (2021) 143:1797–808. 10.1161/CIRCULATIONAHA.120.052671
99.
HaoFTianMZhangXJinXJiangYSunXet alButyrate enhances CPT1A activity to promote fatty acid oxidation and iTreg differentiation. Proc Natl Acad Sci U S A. (2021) 118:e2014681118. 10.1073/pnas.2014681118
100.
ZhangLDengMLuAChenYChenYWuCet alSodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J Cell Mol Med. (2019) 23:8139–50. 10.1111/jcmm.14684
101.
SeefeldtJMHomiliusCHansenJLassenTRJespersenNRJensenRVet alShort-chain fatty acid butyrate is an inotropic agent with vasorelaxant and cardioprotective properties. J Am Heart Assoc. (2024) 13:e033744. 10.1161/JAHA.123.033744
102.
RosengrenADikaiouP. Cardiovascular outcomes in type 1 and type 2 diabetes. Diabetologia. (2023) 66:425–37. 10.1007/s00125-022-05857-5
103.
StrainWDPaldaniusPM. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc Diabetol. (2018) 17:57. 10.1186/s12933-018-0703-2
104.
KhanAWJandeleit-DahmKAM. Atherosclerosis in diabetes mellitus: novel mechanisms and mechanism-based therapeutic approaches. Nat Rev Cardiol. (2025) 22:482–96. 10.1038/s41569-024-01115-w
105.
Powell-WileyTMPoirierPBurkeLEDespresJPGordon-LarsenPLavieCJet alObesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. (2021) 143:e984–e1010. 10.1161/CIR.0000000000000973
106.
MarcelinGGautierELClementK. Adipose tissue fibrosis in obesity: etiology and challenges. Annu Rev Physiol. (2022) 84:135–55. 10.1146/annurev-physiol-060721-092930
107.
JungUJChoiMS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. (2014) 15:6184–223. 10.3390/ijms15046184
108.
GreggEWJakicicJMBlackburnGBloomquistPBrayGAClarkJMet alAssociation of the magnitude of weight loss and changes in physical fitness with long-term cardiovascular disease outcomes in overweight or obese people with type 2 diabetes: a post-hoc analysis of the Look AHEAD randomised clinical trial. Lancet Diabetes Endocrinol. (2016) 4:913–21. 10.1016/s2213-8587(16)30162-0
109.
WiviottSDRazIBonacaMPMosenzonOKatoETCahnAet alDapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. 10.1056/NEJMoa1812389
110.
GomesACHoffmannCMotaJF. The human gut microbiota: metabolism and perspective in obesity. Gut Microbes. (2018) 9:308–25. 10.1080/19490976.2018.1465157
111.
ScheithauerTPMRampanelliENieuwdorpMVallanceBAVerchereCBvan RaalteDHet alGut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol. (2020) 11:571731. 10.3389/fimmu.2020.571731
112.
ChenZRadjabzadehDChenLKurilshikovAKavousiMAhmadizarFet alAssociation of insulin resistance and type 2 diabetes with gut microbial diversity: a microbiome-wide analysis from population studies. JAMA Netw Open. (2021) 4:e2118811. 10.1001/jamanetworkopen.2021.18811
113.
CuiJRameshGWuMJensenETCragoOBertoniAGet alButyrate-producing bacteria and insulin homeostasis: the microbiome and insulin longitudinal evaluation study (MILES). Diabetes. (2022) 71:2438–46. 10.2337/db22-0168
114.
PowersACPhilippeJHermannHHabenerJF. Sodium butyrate increases glucagon and insulin gene expression by recruiting immunocytochemically negative cells to produce hormone. Diabetes. (1988) 37:1405–10. 10.2337/diab.37.10.1405
115.
WangSYuanMZhangLZhuKShengCZhouFet alSodium butyrate potentiates insulin secretion from rat islets at the expense of compromised expression of β cell identity genes. Cell Death Dis. (2022) 13:67. 10.1038/s41419-022-04517-1
116.
HuYLiuJYuanYChenJChengSWangHet alSodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ Toxicol Pharmacol. (2018) 64:112–21. 10.1016/j.etap.2018.09.002
117.
YanMLiXSunCTanJLiuYLiMet alSodium butyrate attenuates AGEs-induced oxidative stress and inflammation by inhibiting autophagy and affecting cellular metabolism in THP-1 cells. Molecules. (2022) 27:8715. 10.3390/molecules27248715
118.
KhanSJenaGB. Protective role of sodium butyrate, a HDAC inhibitor on beta-cell proliferation, function and glucose homeostasis through modulation of p38/ERK MAPK and apoptotic pathways: study in juvenile diabetic rat. Chem Biol Interact. (2014) 213:1–12. 10.1016/j.cbi.2014.02.001
119.
WibowoHHarbuwonoDSTahaparyDLKartikaRPradiptaSLarasatiRA. Impact of sodium butyrate treatment in LPS-stimulated peripheral blood mononuclear cells of poorly controlled type 2 DM. Front Endocrinol. (2021) 12:652942. 10.3389/fendo.2021.652942
120.
GaoZYinJZhangJWardREMartinRJLefevreMet alButyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58:1509–17. 10.2337/db08-1637
121.
HenaganTMStefanskaBFangZNavardAMYeJLenardNRet alSodium butyrate epigenetically modulates high-fat diet-induced skeletal muscle mitochondrial adaptation, obesity and insulin resistance through nucleosome positioning. Br J Pharmacol. (2015) 172:2782–98. 10.1111/bph.13058
122.
De VadderFKovatcheva-DatcharyPGoncalvesDVineraJZitounCDuchamptAet alMicrobiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. (2014) 156:84–96. 10.1016/j.cell.2013.12.016
123.
van DeurenTBlaakEECanforaEE. Butyrate to combat obesity and obesity-associated metabolic disorders: current status and future implications for therapeutic use. Obes Rev. (2022) 23:e13498. 10.1111/obr.13498
124.
LinHVFrassettoAKowalikEJJrNawrockiARLuMMKosinskiJRet alButyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS One. (2012) 7:e35240. 10.1371/journal.pone.0035240
125.
RoshanravanNMahdaviRAlizadehEJafarabadiMAHedayatiMGhavamiAet alEffect of butyrate and inulin supplementation on glycemic status, lipid profile and glucagon-like peptide 1 level in patients with type 2 diabetes: a randomized double-blind, placebo-controlled trial. Horm Metab Res. (2017) 49:886–91. 10.1055/s-0043-119089
126.
PalaciosTVitettaLCoulsonSMadiganCDLamYYManuelRet alTargeting the intestinal microbiota to prevent type 2 diabetes and enhance the effect of metformin on glycaemia: a randomised controlled pilot study. Nutrients. (2020) 12:2041. 10.3390/nu12072041
127.
KhanSJenaG. Sodium butyrate reduces insulin-resistance, fat accumulation and dyslipidemia in type-2 diabetic rat: a comparative study with metformin. Chem Biol Interact. (2016) 254:124–34. 10.1016/j.cbi.2016.06.007
128.
De GrootPFNikolicTImangaliyevSBekkeringSDuinkerkenGKeijFMet alOral butyrate does not affect innate immunity and islet autoimmunity in individuals with longstanding type 1 diabetes: a randomised controlled trial. Diabetologia. (2020) 63:597–610. 10.1007/s00125-019-05073-8
129.
AguilarECda SilvaJFNavia-PelaezJMLeonelAJLopesLGMenezes-GarciaZet alSodium butyrate modulates adipocyte expansion, adipogenesis, and insulin receptor signaling by upregulation of PPAR-gamma in obese apo E knockout mice. Nutrition. (2018) 47:75–82. 10.1016/j.nut.2017.10.007
130.
ZhuWPengKZhaoYXuCTaoXLiuYet alSodium butyrate attenuated diet-induced obesity, insulin resistance and inflammation partly by promoting fat thermogenesis via intro-adipose sympathetic innervation. Front Pharmacol. (2022) 13:938760. 10.3389/fphar.2022.938760
131.
GaoFLvYWLongJChenJMHeJMRuanXZet alButyrate improves the metabolic disorder and gut microbiome dysbiosis in mice induced by a high-fat diet. Front Pharmacol. (2019) 10:1040. 10.3389/fphar.2019.01040
132.
KimuraIOzawaKInoueDImamuraTKimuraKMaedaTet alThe gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. (2013) 4:1829. 10.1038/ncomms2852
133.
LiZYiCXKatiraeiSKooijmanSZhouEChungCKet alButyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut. (2018) 67:1269–79. 10.1136/gutjnl-2017-314050
134.
WhittJWooVLeePMoncivaizJHabermanYDensonLet alDisruption of epithelial HDAC3 in intestine prevents diet-induced obesity in mice. Gastroenterology. (2018) 155:501–13. 10.1053/j.gastro.2018.04.017
135.
HonmaKOshimaKTakamiSGodaT. Regulation of hepatic genes related to lipid metabolism and antioxidant enzymes by sodium butyrate supplementation. Metabol Open. (2020) 7:100043. 10.1016/j.metop.2020.100043
136.
HattoriYTsutsuiSYamadaCKobayashiYNakagawaTShimadaM. Dietary supplementation with sodium butyrate reduces high-sucrose diet-induced hepatic accumulation of triacylglycerols and expression of fatty acid synthesis enzymes in rats. J Oleo Sci. (2022) 71:1189–93. 10.5650/jos.ess22112
137.
ZhaoZHWangZXZhouDHanYMaFHuZet alSodium butyrate supplementation inhibits hepatic steatosis by stimulating liver kinase B1 and insulin-induced gene. Cell Mol Gastroenterol Hepatol. (2021) 12:857–71. 10.1016/j.jcmgh.2021.05.006
138.
MullerMHernandezMAGGoossensGHReijndersDHolstJJJockenJWEet alCirculating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and GLP-1 concentrations in humans. Sci Rep. (2019) 9:12515. 10.1038/s41598-019-48775-0
139.
NgSCXuZMakJWYYangKLiuQZuoTet alMicrobiota engraftment after faecal microbiota transplantation in obese subjects with type 2 diabetes: a 24-week, double-blind, randomised controlled trial. Gut. (2022) 71:716–23. 10.1136/gutjnl-2020-323617
140.
CoppolaSNocerinoRPaparoLBedogniGCalignanoADi ScalaCet alTherapeutic effects of butyrate on pediatric obesity: a randomized clinical trial. JAMA Netw Open. (2022) 5:e2244912. 10.1001/jamanetworkopen.2022.44912
141.
MoonH-RYunJ-M. Sodium butyrate inhibits high glucose-induced inflammation by controlling the acetylation of NF-κB p65 in human monocytes. Nutr Res Pract. (2023) 17(1):164–73. 10.4162/nrp.2023.17.1.164
Summary
Keywords
butyric acid, cardiovascular disease, hypertension, atherosclerosis, diabetes, obesity
Citation
Xu Q, Liu X, Wang Z, Li X, Jiang Q and Xu M (2025) Recent advancements and comprehensive analyses of butyric acid in cardiovascular diseases. Front. Cardiovasc. Med. 12:1608658. doi: 10.3389/fcvm.2025.1608658
Received
09 April 2025
Accepted
14 July 2025
Published
28 July 2025
Volume
12 - 2025
Edited by
Antoni Sureda, University of the Balearic Islands, Spain
Reviewed by
Haroon Khan, Abdul Wali Khan University Mardan, Pakistan
Giuseppe Annunziata, Pegaso University, Italy
Updates
Copyright
© 2025 Xu, Liu, Wang, Li, Jiang and Xu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianfeng Jiang jiangqianfeng2005@126.com Min Xu 540618316@qq.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.