Abstract
Aging is a complex biological process characterized by a gradual decline in cellular and physiological function, increasing vulnerability to chronic diseases and mortality. It involves a set of interconnected mechanisms known as the hallmarks of aging, including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, and dysregulated nutrient sensing. These processes act at molecular, cellular, and systemic levels, contributing to age-related disorders such as neurodegeneration, cardiovascular disease, and metabolic syndromes. Emerging therapeutic strategies aim to delay or reverse aging by targeting specific hallmarks. These include senolytics to eliminate senescent cells, NAD+ boosters and mitophagy inducers to improve mitochondrial health, epigenetic reprogramming, and caloric restriction mimetics such as metformin and rapamycin to modulate nutrient-sensing pathways. Advances in regenerative medicine, gene editing, and organ cross-talk modulation are also contributing to the development of personalized, multi-targeted anti-aging therapies. Integration of omics technologies and biomarker research is expected to enhance our ability to monitor biological aging and optimize interventions for healthy longevity. This review highlights the current understanding of the hallmarks of aging and explores potential treatment strategies in light of our recent findings.
1 Introduction
Aging is not merely the passage of time—it reflects how well the body maintains its function. Chronological age counts the number of years lived, while biological age (or health-based aging) reflects the actual condition of cells, tissues, and organs, revealing how quickly or slowly the body is aging (Figure 1). Biological age is shaped by the body's ability to maintain dynamic equilibrium—a stable internal environment despite ongoing internal and external stressors. This balance supports essential processes such as cellular repair, metabolic regulation, and immune function. When disrupted, it can lead to physiological decline, thereby accelerating aging and increasing susceptibility to age-related diseases.
Figure 1
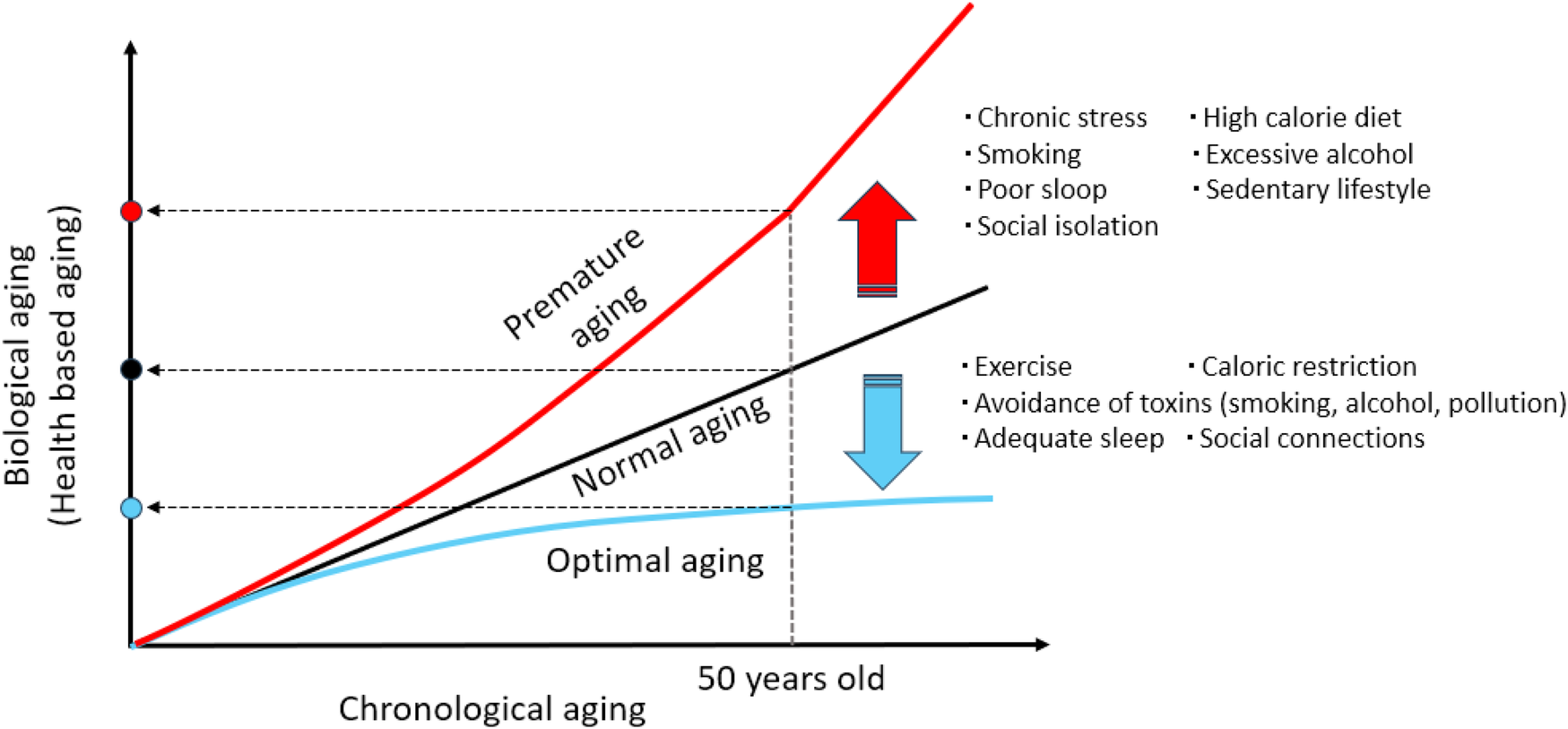
Chronological aging and biological aging. Three people are 50 years old chronologically, but one may be as healthy as a 40-year-old (optimal aging), another is age appropriate (normal aging), while another may have the biological characteristics of a 60-year-old (premature aging). Lifestyle has a profound influence on biological aging, either accelerating (premature aging) or decelerating (optimal aging) the process through its impact on cellular stress, inflammation, metabolism, and DNA stability, etc.
This equilibrium is governed by a network of interconnected biological processes known as the “Hallmarks of Aging”—the key drivers of aging. First introduced in 2013, the hallmarks framework consolidated emerging scientific insights into the mechanisms of aging and identified potential points of intervention (1). In 2023, the hallmarks were updated to incorporate a decade of advances in both basic and clinical aging research (2). Each hallmark corresponds to a specific molecular or cellular alteration, often measurable through established biomarkers. These hallmarks progress in distinct yet interrelated stages, collectively contributing to aging phenotypes and age-associated diseases.
Ongoing research seeks to determine the optimal stages for therapeutic intervention and to identify lifestyle factors that may delay or prevent the onset of premature aging. In this review, we summarize the current understanding of the Hallmarks of Aging and present our related findings.
2 Hallmarks of aging and possible interventions
The hallmarks of aging are key biological processes that drive the progressive decline in function and increase the risk of age-related diseases (Figure 2). With recent advances in aging research, biological aging can now be modulated by targeting fundamental cellular and molecular processes (Table 1).
Figure 2
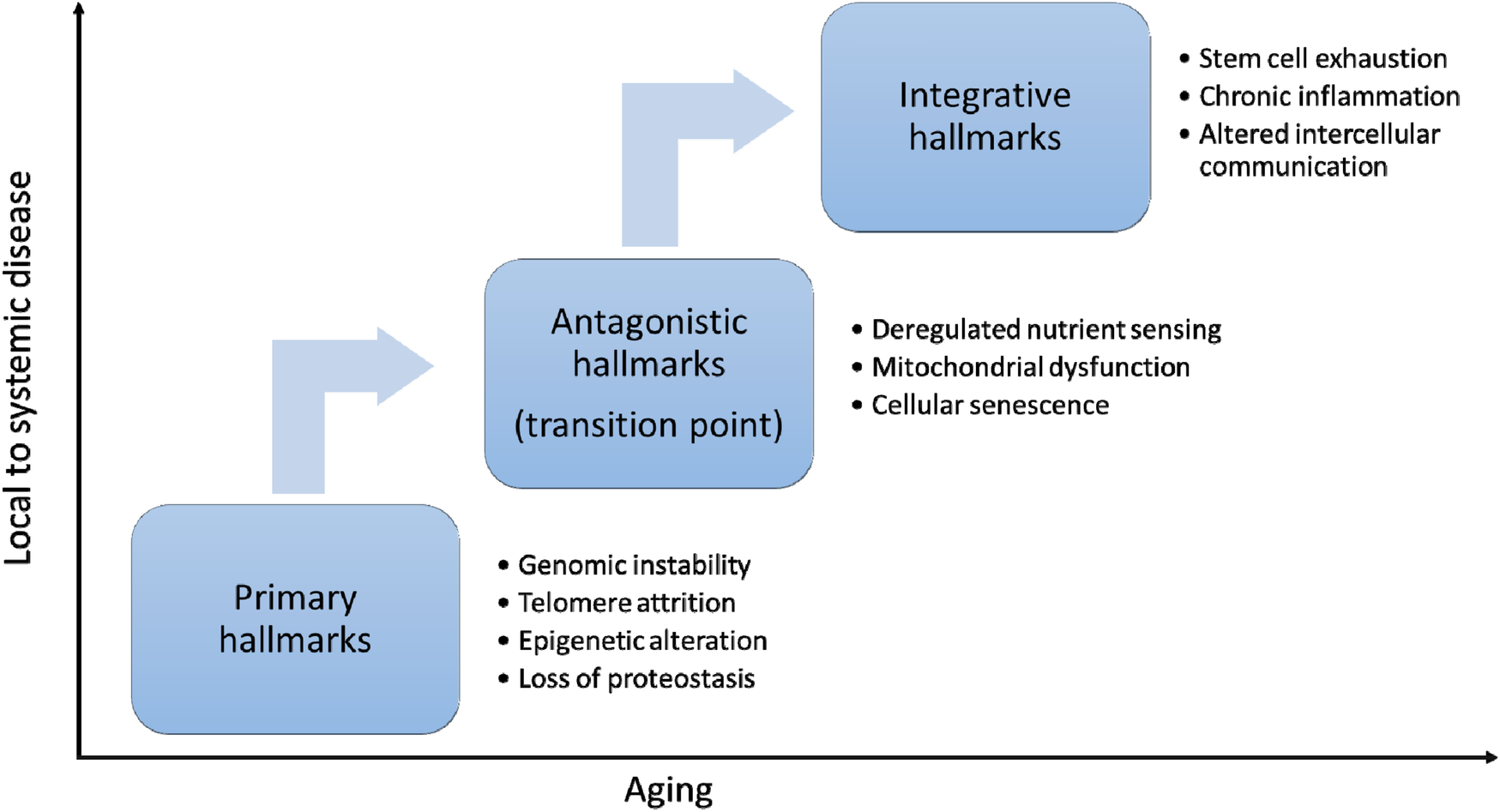
Hallmarks or aging. The hallmarks of aging can be categorized into three interconnected layers. Primary hallmarks—such as genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis—reflect the accumulation of molecular and cellular damage over time. In response, antagonistic hallmarks emerge as compensatory mechanisms, including deregulated nutrient sensing, mitochondrial dysfunction and cellular senescence. When these fail or become deleterious, they lead to integrative hallmarks, such as stem cell exhaustion, chronic inflammation, and altered intercellular communication, which drive systemic aging and functional decline. Antagonistic hallmarks represent a critical transition point in aging.
Table 1
| Hallmark | Category | Targeting strategy | Examples/Therapeutics |
|---|---|---|---|
| Genomic instability | Primary | Enhance DNA repair, reduce mutagenic stress | NAD+ boosters (NR/NMN), PARP activators, antioxidants |
| Telomere attrition | Primary | Promote telomerase activity, protect telomeres | TA-65, TERT gene therapy, lifestyle (stress reduction, exercise) |
| Epigenetic alterations | Primary | Modify chromatin state, reset epigenetic marks | Yamanaka factors, HDAC inhibitors, NAD+ precursors |
| Loss of proteostasis | Primary | Enhance protein folding and clearance | Rapamycin, spermidine, proteasome activators, HSP inducers |
| Deregulated nutrient sensing | Antagonistic | Restore metabolic balance via nutrient signaling pathways | Metformin, rapamycin, caloric restriction, intermittent fasting |
| Mitochondrial dysfunction | Antagonistic | Promote mitophagy, mitochondrial biogenesis | Urolithin A, MitoQ, exercise, NAD+ boosters |
| Cellular senescence | Antagonistic | Eliminate or suppress senescent cells and SASP | Senolytics (e.g., dasatinib, vaccine), metformin |
| Stem cell exhaustion | Integrative | Reactivate endogenous stem cells or replace with cell therapy | MSC therapy, GDF11, fasting, exercise |
| Altered intercellular communication | Integrative | Improve systemic signaling and tissue cross-talk | NAD+ boosters, probiotics, exercise, exosome therapy |
| Chronic inflammation (“Inflammaging”) | Integrative | Suppress inflammatory pathways and immune dysfunction | IL-1/IL-6 inhibitors, omega-3s, senolytics, anti-inflammatory diet |
Strategies for targeting the hallmarks of aging.
2.1 Primary hallmarks: the underlying causes of cellular decline
The primary hallmarks of aging—genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis—are considered the root causes of cellular aging. These hallmarks are associated with major age-related diseases: DNA damage accumulation contributes to cancer and Werner syndrome (3); telomere shortening is implicated in idiopathic pulmonary fibrosis and aplastic anemia (4); epigenetic dysregulation underlies Alzheimer's disease and Hutchinson-Gilford progeria syndrome (5); and impaired protein homeostasis is linked to neurodegenerative conditions such as Parkinson's and Huntington's diseases (6).
Interventions targeting these primary hallmarks—such as telomerase gene therapy to counter telomere attrition (7), NAD+ precursors like NMN to support DNA repair and genomic stability (8), partial epigenetic reprogramming with Yamanaka factors to reverse transcriptional aging (9), and autophagy enhancers like rapamycin to restore proteostasis (10)—have shown promise in preclinical models. However, translation to humans faces significant challenges, including cancer risk, off-target effects, delivery limitations, and insufficient long-term safety data. Therapies that stimulate cellular activity—such as those activating mTOR or telomerase—may paradoxically accelerate aging if not precisely regulated, leading to cellular senescence, stem cell exhaustion, or tumorigenesis (1, 7). Therefore, future strategies must emphasize precise, tissue-specific modulation, transient reprogramming, and combinatorial therapies that achieve rejuvenation without compromising long-term safety.
2.2 Antagonistic (secondary) hallmarks: protective responses that become detrimental
The antagonistic hallmarks of aging—deregulated nutrient sensing, mitochondrial dysfunction, and cellular senescence—initially serve protective or adaptive roles but become damaging when chronically activated. They are termed “antagonistic” because their effects vary depending on duration and context. Thus, these antagonistic hallmarks represent a critical transition point in aging.
Deregulated nutrient sensing, especially via the mTOR pathway, contributes to metabolic diseases such as type 2 diabetes and obesity through altered insulin/IGF-1 signaling (1). Mitochondrial dysfunction, characterized by impaired bioenergetics and oxidative stress, plays a central role in diseases like Alzheimer's, Parkinson's, and cardiomyopathy (11). Cellular senescence, marked by the secretion of inflammatory factors (SASP), contributes to conditions such as osteoporosis, osteoarthritis, pulmonary fibrosis, and cancer by promoting chronic inflammation and tissue dysfunction (12).
Modulating these hallmarks is a key goal of gerotherapeutics, with strategies including senolytics and caloric restriction mimetics. Recent research has explored vaccination as a novel approach. For example, a vaccine targeting CD153—a surface marker of senescent CD4+ T cells in visceral adipose tissue—was developed using a CD153 peptide conjugated with KLH and adjuvanted with CpG. It selectively reduced CD153+ senescent T cells via complement-dependent cytotoxicity, improving glucose tolerance and insulin sensitivity in obese mice (13). Another vaccination strategy targets GPNMB, a senescence-associated transmembrane protein enriched in vascular endothelial cells and leukocytes during atherosclerosis. Immunization against GPNMB reduced senescent cell burden, improved metabolic function, and alleviated atherosclerosis in high-fat diet-fed and ApoE-deficient mice. In progeroid mice, it ameliorated age-related phenotypes and extended lifespan, highlighting the therapeutic potential of targeting senescence-associated antigens (14).
While activating certain pathways may slow aging or treat diseases, chronic or unregulated stimulation can accelerate aging or promote cancer. For instance, we compared the effects of HGF and VEGF on endothelial progenitor cells under angiotensin II—a known atherosclerosis risk factor (15). HGF, but not VEGF, attenuated angiotensin II-induced senescence via suppression of the PI(3,4,5)P₃/Rac1 pathway and reduced oxidative stress. In vivo, HGF also enhanced neovascularization under angiotensin II stimulation, unlike VEGF. Although both factors promote angiogenesis in ischemic models, HGF has shown consistent clinical benefit in peripheral artery disease (PAD), whereas VEGF therapies have largely failed in Phase III trials. Notably, VEGF deficiency can induce aging-like features, but excessive VEGF contributes to pathological angiogenesis, inflammation, and cancer (16–19). Our data may explain these discrepancies (15). Therefore, temporal and tissue-specific control of modulators is essential to mitigate risks.
2.3 Integrative hallmarks: the downstream consequences of aging
As cellular damage accumulates and compensatory mechanisms fail, integrative hallmarks—stem cell exhaustion, chronic inflammation and altered intercellular communication—emerge. These reflect the systemic decline driven by earlier hallmarks and contribute to diseases such as sarcopenia and immunosenescence, where stem cell depletion impairs tissue regeneration (20), as well as chronic inflammatory conditions like atherosclerosis, Alzheimer's disease, and type 2 diabetes, exacerbated by SASP and systemic inflammaging (1, 2, 21).
We have reported that coagulation factor Xa induces cell senescence and activates inflammatory signaling through IGFBP-5, beyond its role in coagulation (22, 23). Similarly, Other groups also reported that low-grade activation of coagulation factor X contributes sterile chronic inflammation and atherogenesis via Protease-activated receptor-2 (24–28). Aging is associated with a hypercoagulable state involving increased procoagulant factors (e.g., fibrinogen, factors VIII and X, von Willebrand factor), endothelial dysfunction, reduced fibrinolysis, and increased platelet activity. This contributes to thrombotic events such as stroke and myocardial infarction, as well as microvascular damage, possibly linking coagulation and chronic inflammation in aging (29–31).
Therapies targeting integrative hallmarks include stem cell transplantation (32), heterochronic parabiosis and plasma exchange to rejuvenate systemic signaling (33), and senolytics like dasatinib and quercetin to reduce SASP (34). However, these approaches face limitations: immune rejection, short-lived effects, senescent cell heterogeneity, and delivery challenges.
Because integrative hallmarks involve multiple tissues and feedback loops, systemic targeting is inherently complex and prone to off-target effects. Early detection of localized aging (e.g., in the heart, kidney, or brain) is critical to prevent progression to systemic dysfunction. This can be achieved through biomarkers (e.g., NT-proBNP, creatinine, cystatin C), imaging (MRI, echocardiography), functional assessments (spirometry, gait speed), and emerging omics-based tools such as epigenetic clocks (35) and transcriptomic profiling (36).
3 Organs vulnerable to age-related damage and strategies for early detection
While aging affects all organs, those with high metabolic demands, limited regenerative capacity, and chronic exposure to stress—such as the brain, heart, kidneys, lungs, liver, bones, eyes, and skin—are particularly vulnerable. The brain is susceptible to neuronal loss, protein aggregation, and inflammation, increasing the risk of neurodegenerative diseases such as Alzheimer's (37). The heart undergoes arterial stiffening and myocardial hypertrophy, contributing to heart failure and arrhythmias (38). The kidneys experience nephron loss and vascular decline, elevating the risk of chronic kidney disease (39). Lung aging reduces tissue elasticity and impairs immune defense, making the lungs more prone to infections and chronic obstructive pulmonary disease (COPD) (40). The liver shows diminished detoxification capacity and accumulates fat, leading to non-alcoholic fatty liver disease (NAFLD) (41). Bone loss and cartilage degeneration result in osteoporosis and arthritis (42). The eyes are subject to oxidative stress, promoting cataracts and macular degeneration (43). Skin aging manifests as thinning, delayed wound healing, and UV-induced damage, which increases the risk of wrinkles and skin cancers.
A pivotal study by Dr. Tony Wyss-Coray's group at Stanford, published in Nature (44), demonstrated that organs can age at different rates—even in apparently healthy individuals. Using the SomaScan assay on blood samples from 5,676 participants, the team identified 856 organ-specific proteins across 11 organs and applied machine learning to estimate organ-specific biological age. The study revealed that: 18.4% of individuals over age 50 had at least one rapidly aging organ; 1.7% had multiple rapidly aging organs; Accelerated aging in 10 of 11 organs (excluding the intestine) was associated with a 15%–50% increased risk of mortality over 15 years. Notably, organ-specific accelerated aging was linked to: Heart: 250% higher risk of heart failure; Brain and vasculature: Strong predictor of Alzheimer's disease; Kidneys: Increased risk of hypertension and diabetes. These findings suggest that organ-specific biological aging could serve as an early warning system—potentially detectable through routine blood tests—even before clinical symptoms emerge. This implies that certain organs may undergo early biological aging (corresponding to primary and secondary hallmarks) and eventually engage in cross-organ communication that contributes to integrative hallmarks and systemic aging.
4 Conclusions and future directions
While aging is inevitable, its rate and impact can be modulated. A growing body of evidence suggests that specific behaviors and interventions can delay or attenuate the cellular and molecular hallmarks of aging, thereby enhancing both healthspan and lifespan (1). Lifestyle factors—including nutrition, physical activity, and stress management—play a critical role in regulating these hallmarks. On the other hand, several existing therapies—originally developed for other indications—are being repurposed or explored as anti-aging drug candidates. These drugs typically act by modulating key hallmarks of aging, such as genomic instability, cellular senescence, mitochondrial dysfunction, deregulated nutrient sensing, and stem cell exhaustion. Metformin, a widely used antidiabetic drug, has shown geroprotective potential by activating AMPK and inhibiting mTOR, thereby improving mitochondrial function and reducing inflammation and insulin resistance—key features of metabolic aging (45, 46). Rapamycin, an mTOR inhibitor originally used as an immunosuppressant, extends lifespan in multiple species by enhancing autophagy, improving proteostasis, and reducing stem cell exhaustion (10, 47). Senolytics such as dasatinib and quercetin selectively clear senescent cells, reducing the pro-inflammatory secretory phenotype (SASP) and restoring tissue homeostasis in aged organisms (34, 48). NAD+ precursors, including nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), replenish declining NAD+ levels in aging tissues, enhance sirtuin activity, and improve mitochondrial function and DNA repair (8, 49). Acarbose, an α-glucosidase inhibitor, improves glucose homeostasis and reduces postprandial insulin spikes, mimicking some effects of caloric restriction and extending lifespan in mice (50). Low-dose aspirin, through COX inhibition, exerts anti-inflammatory effects that may counteract chronic low-grade inflammation (“inflammaging”), although its net benefit in elderly populations remains controversial (51, 52). Lithium, used in bipolar disorder, inhibits GSK-3β and promotes autophagy and mitochondrial stability, with emerging evidence suggesting neuroprotective and potential lifespan-extending effects (53, 54).
SGLT2 inhibitors, such as empagliflozin, enhance metabolic flexibility, reduce oxidative stress, and may improve cardiovascular and renal aging phenotypes by mimicking fasting-like states (55, 56). The anti-aging effects of these existing drugs, the timing of administration, and long-term safety are expected to be studied. Looking ahead, it may become possible to detect organ-specific abnormalities before the emergence of integrative hallmarks and systemic dysfunction. Early identification of dysregulated nutrient sensing, mitochondrial dysfunction, or cellular senescence could enable timely, targeted interventions. Developing a simple, reliable method to monitor these early changes will be essential to identifying optimal therapeutic windows for age-delaying strategies.
Statements
Author contributions
FS: Writing – original draft, Writing – review & editing. SH: Writing – original draft, Writing – review & editing. RM: Funding acquisition, Project administration, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
RM received honoraria, consulting fees, and funds from Boehringer Ingelheim, AnGes, Inc., and FunPep Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
López-OtínCBlascoMAPartridgeLSerranoMKroemerG. The hallmarks of aging. Cell. (2013) 153(6):1194–217. 10.1016/j.cell.2013.05.039
2.
López-OtínCBlascoMAPartridgeLSerranoMKroemerG. Hallmarks of aging: an expanding universe. Cell. (2023) 186(2):243–78. 10.1016/j.cell.2022.11.001
3.
LombardDBChuaKFMostoslavskyRFrancoSGostissaMAltFW. DNA repair, genome stability, and aging. Cell. (2005) 120(4):497–512. 10.1016/j.cell.2005.01.028
4.
ArmaniosMBlackburnEH. The telomere syndromes. Nat Rev Genet. (2012) 13(10):693–704. 10.1038/nrg3246
5.
PalSTylerJK. Epigenetics and aging. Sci Adv. (2016) 2(7):e1600584. 10.1126/sciadv.1600584
6.
HippMSKasturiPHartlFU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. (2019) 20(7):421–35. 10.1038/s41580-019-0101-y
7.
Bernardes de JesusBVeraESchneebergerKTejeraAMAyusoEBoschFet alTelomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. (2012) 4(8):691–704. 10.1002/emmm.201200245
8.
GomesAPPriceNLLingAJMoslehiJJMontgomeryMKRajmanLet alDeclining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. (2013) 155(7):1624–38. 10.1016/j.cell.2013.11.037
9.
OcampoAReddyPMartinez-RedondoPPlatero-LuengoAHatanakaFHishidaTet alIn vivo amelioration of age-associated hallmarks by partial reprogramming. Cell. (2016) 167(7):1719–33.e12. 10.1016/j.cell.2016.11.052
10.
HarrisonDEStrongRSharpZDNelsonJFAstleCMFlurkeyKet alRapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. (2009) 460(7253):392–5. 10.1038/nature08221
11.
PietronigroEZenaroEConstantinG. Imaging of leukocyte trafficking in Alzheimer’s disease. Front Immunol. (2016) 7:33. 10.3389/fimmu.2016.00033
12.
van DeursenJM. The role of senescent cells in ageing. Nature. (2014) 509(7501):439–46. 10.1038/nature13193
13.
YoshidaSNakagamiHHayashiHIkedaYSunJTenmaAet alThe CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat Commun. (2020) 11(1):2482. 10.1038/s41467-020-16347-w
14.
SudaMShimizuIKatsuumiGYoshidaYHayashiYIkegamiRet alSenolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat Aging. (2021) 1(12):1117–26. 10.1038/s43587-021-00151-2
15.
SanadaFTaniyamaYAzumaJIekushiKDosakaNYokoiTet alHepatocyte growth factor, but not vascular endothelial growth factor, attenuates angiotensin II-induced endothelial progenitor cell senescence. Hypertension. (2009) 53(1):77–82. 10.1161/HYPERTENSIONAHA.108.120725
16.
GerberHPFerraraN. The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med. (2003) 81(1):20–31. 10.1007/s00109-002-0397-4
17.
OlssonAKDimbergAKreugerJClaesson-WelshL. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. (2006) 7(5):359–71. 10.1038/nrm1911
18.
CoppéJPDesprezPYKrtolicaACampisiJ. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. 10.1146/annurev-pathol-121808-102144
19.
UngvariZTarantiniSKissTWrenJDGilesCBGriffinCTet alEndothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. (2018) 15(9):555–65. 10.1038/s41569-018-0030-z
20.
RandoTAWyss-CorayT. Asynchronous, contagious and digital aging. Nat Aging. (2021) 1(1):29–35. 10.1038/s43587-020-00015-1
21.
FranceschiCGaragnaniPPariniPGiulianiCSantoroA. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. (2018) 14(10):576–90. 10.1038/s41574-018-0059-4
22.
SanadaFTaniyamaYMuratsuJOtsuRIwabayashiMCarracedoMet alActivated factor X induces endothelial cell senescence through IGFBP-5. Sci Rep. (2016) 6:35580. 10.1038/srep35580
23.
SanadaFTaniyamaYMuratsuJOtsuRShimizuHRakugiHet alIGF binding protein-5 induces cell senescence. Front Endocrinol. (2018) 9:53. 10.3389/fendo.2018.00053
24.
MatsuuraTSoekiTFukudaDUematsuETobiumeTHaraTet alActivated factor X signaling pathway via protease-activated receptor 2 is a novel therapeutic target for preventing atrial fibrillation. Circ J. (2021) 85(8):1383–91. 10.1253/circj.CJ-20-1006
25.
HaraTSataMFukudaD. Emerging roles of protease-activated receptors in cardiometabolic disorders. J Cardiol. (2023) 81(4):337–46. 10.1016/j.jjcc.2022.09.013
26.
HaraTPhuongPTFukudaDYamaguchiKMurataCNishimotoSet alProtease-activated receptor-2 plays a critical role in vascular inflammation and atherosclerosis in apolipoprotein E-deficient mice. Circulation. (2018) 138(16):1706–19. 10.1161/CIRCULATIONAHA.118.033544
27.
HaraTFukudaDTanakaKHigashikuniYHirataYYagiSet alInhibition of activated factor X by rivaroxaban attenuates neointima formation after wire-mediated vascular injury. Eur J Pharmacol. (2018) 820:222–8. 10.1016/j.ejphar.2017.12.037
28.
HaraTFukudaDTanakaKHigashikuniYHirataYNishimotoSet alRivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis. (2015) 242(2):639–46. 10.1016/j.atherosclerosis.2015.03.023
29.
MariDOgliariGCastaldiDVitaleGBolliniEMLioD. Hemostasis and ageing. Immun Ageing. (2008) 5:12. 10.1186/1742-4933-5-12
30.
WilkersonWRSaneDC. Aging and thrombosis. Semin Thromb Hemost. (2002) 28(6):555–68. 10.1055/s-2002-36700
31.
FranceschiCCapriMMontiDGiuntaSOlivieriFSeviniFet alInflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. (2007) 128(1):92–105. 10.1016/j.mad.2006.11.016
32.
KatsimpardiLLittermanNKScheinPAMillerCMLoffredoFSWojtkiewiczGRet alVascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. (2014) 344(6184):630–4. 10.1126/science.1251141
33.
ConboyIMConboyMJWagersAJGirmaERWeissmanILRandoTA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. (2005) 433(7027):760–4. 10.1038/nature03260
34.
ZhuYTchkoniaTPirtskhalavaTGowerACDingHGiorgadzeNet alThe Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. (2015) 14(4):644–58. 10.1111/acel.12344
35.
BuckleyMTSunEDGeorgeBMLiuLSchaumNXuLet alCell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain. Nat Aging. (2023) 3(1):121–37. 10.1038/s43587-022-00335-4
36.
RutledgeJOhHWyss-CorayT. Measuring biological age using omics data. Nat Rev Genet. (2022) 23(12):715–27. 10.1038/s41576-022-00511-7
37.
HouYDanXBabbarMWeiYHasselbalchSGCroteauDLet alAgeing as a risk factor for neurodegenerative disease. Nat Rev Neurol. (2019) 15(10):565–81. 10.1038/s41582-019-0244-7
38.
LakattaEGLevyD. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. (2003) 107(1):139–46. 10.1161/01.CIR.0000048892.83521.58
39.
DenicAGlassockRJRuleAD. Structural and functional changes with the aging kidney. Adv Chronic Kidney Dis. (2016) 23(1):19–28. 10.1053/j.ackd.2015.08.004
40.
LoweryEMBrubakerALKuhlmannEKovacsEJ. The aging lung. Clin Interv Aging. (2013) 8:1489–96. 10.2147/CIA.S51152
41.
BruntEM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2010) 7(4):195–203. 10.1038/nrgastro.2010.21
42.
KhoslaSShaneE. A crisis in the treatment of osteoporosis. J Bone Miner Res. (2016) 31(8):1485–7. 10.1002/jbmr.2888
43.
BeattySKohHPhilMHensonDBoultonM. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. (2000) 45(2):115–34. 10.1016/S0039-6257(00)00140-5
44.
OhHSRutledgeJNachunDPálovicsRAbioseOMoran-LosadaPet alOrgan aging signatures in the plasma proteome track health and disease. Nature. (2023) 624(7990):164–72. 10.1038/s41586-023-06802-1
45.
BarzilaiNCrandallJPKritchevskySBEspelandMA. Metformin as a tool to target aging. Cell Metab. (2016) 23(6):1060–5. 10.1016/j.cmet.2016.05.011
46.
KulkarniASGubbiSBarzilaiN. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. (2020) 32(1):15–30. 10.1016/j.cmet.2020.04.001
47.
MannickJBDel GiudiceGLattanziMValianteNMPraestgaardJHuangBet almTOR inhibition improves immune function in the elderly. Sci Transl Med. (2014) 6(268):268ra179. 10.1126/scitranslmed.3009892
48.
JusticeJNNambiarAMTchkoniaTLeBrasseurNKPascualRHashmiSKet alSenolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. 10.1016/j.ebiom.2018.12.052
49.
YoshinoMYoshinoJKayserBDPattiGJFranczykMPMillsKFet alNicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science. (2021) 372(6547):1224–9. 10.1126/science.abe9985
50.
HarrisonDEStrongRAllisonDBAmesBNAstleCMAtamnaHet alAcarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. (2014) 13(2):273–82. 10.1111/acel.12170
51.
RidkerPMCookNRLeeIMGordonDGazianoJMMansonJEet alA randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. (2005) 352(13):1293–304. 10.1056/NEJMoa050613
52.
McNeilJJNelsonMRWoodsRLLockeryJEWolfeRReidCMet alEffect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. (2018) 379(16):1519–28. 10.1056/NEJMoa1803955
53.
MartinssonLWeiYXuDMelasPAMathéAASchallingMet alLong-term lithium treatment in bipolar disorder is associated with longer leukocyte telomeres. Transl Psychiatry. (2013) 3(5):e261. 10.1038/tp.2013.37
54.
MutzJWongWLEPowellTRYoungAHDaweGSLewisCM. The duration of lithium use and biological ageing: telomere length, frailty, metabolomic age and all-cause mortality. Geroscience. (2024) 46(6):5981–94. 10.1007/s11357-024-01142-y
55.
VermaSMcMurrayJJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia. (2018) 61:2108–17. 10.1007/s00125-018-4670-7
56.
YuristaSRChongCRBadimonJJKellyDPde BoerRAWestenbrinkBD. Therapeutic potential of ketone bodies for patients with cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol. (2021) 77(13):1660–9. 10.1016/j.jacc.2020.12.065
Summary
Keywords
chronological aging, biological aging, aging related disease, senscence, chronic inflammation
Citation
Sanada F, Hayashi S and Morishita R (2025) Targeting the hallmarks of aging: mechanisms and therapeutic opportunities. Front. Cardiovasc. Med. 12:1631578. doi: 10.3389/fcvm.2025.1631578
Received
20 May 2025
Accepted
12 June 2025
Published
01 July 2025
Volume
12 - 2025
Edited by
Masanori Aikawa, Brigham and Women’s Hospital and Harvard Medical School, United States
Reviewed by
Masataka Sata, Tokushima University, Japan
Takehiro Funamizu, Juntendo University, Japan
Updates
Copyright
© 2025 Sanada, Hayashi and Morishita.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fumihiro Sanada sanada@cgt.med.osaka-u.ac.jp
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.