 Miranda Palumbo1,2
Miranda Palumbo1,2 Ezequiel Sosa2,3
Ezequiel Sosa2,3 Florencia Castello1
Florencia Castello1 Gustavo Schottlender1
Gustavo Schottlender1 Federico Serral1Adrián Turjanski2,3
Federico Serral1Adrián Turjanski2,3 María Mercedes Palomino2,3*Darío Fernández Do Porto1,2*
María Mercedes Palomino2,3*Darío Fernández Do Porto1,2*- 1Instituto de Cálculo, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
- 2Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
- 3Instituto de Química Biológica de la Facultad de Ciencias Exactas y Naturales (IQUIBICEN) CONICET, Ciudad Universitaria, Buenos Aires, Argentina
Listeria monocytogenes (Lm) is a Gram-positive bacillus responsible for listeriosis in humans. Listeriosis has become a major foodborne illness in recent years. This illness is mainly associated with the consumption of contaminated food and ready-to-eat products. Recently, Lm has developed resistances to a broad range of antimicrobials, including those used as the first choice of therapy. Moreover, multidrug-resistant strains have been detected in clinical isolates and settings associated with food processing. This scenario punctuates the need for novel antimicrobials against Lm. On the other hand, increasingly available omics data for diverse pathogens has created new opportunities for rational drug discovery. Identification of an appropriate molecular target is currently accepted as a critical step of this process. In this work, we generated multiple layers of omics data related to Lm, aiming to prioritize proteins that could serve as attractive targets for antimicrobials against L. monocytogenes. We generated genomic, transcriptomic, metabolic, and protein structural information, and this data compendium was integrated onto a freely available web server (Target Pathogen). Thirty targets with desirable features from a drug development point of view were shortlisted. This set of target proteins participates in key metabolic processes such as fatty acid, pentose, rhamnose, and amino acids metabolism. Collectively, our results point towards novel targets for the control of Lm and related bacteria. We invite researchers working in the field of drug discovery to follow up experimentally on our revealed targets.
Introduction
Listeria monocytogenes (Lm) is a short, motile, non-sporulated, and Gram-positive bacillus responsible for listeriosis in humans. Lm infections have become a major foodborne illness in recent years. They are mainly associated with the consumption of contaminated food such as meat, fish and vegetables and the so-called ready-to-eat products (Olaimat et al., 2018). This disease has two different clinical presentations: an invasive and a non-invasive form. The non-invasive form includes symptoms such as fever, gastroenteritis, headache, and muscle pain. The invasive form is more severe and may cause bacteremia, meningitis, meningoencephalitis, abortion, and prenatal infection. Patients who are more susceptible to these clinical manifestations are immunosuppressed, older adults, infants, and pregnant women with an alarming fatality rate that can reach up to 30%, even with the current antibiotic therapy administration (Scallan et al., 2011; Authority, 2015; Garner and Kathariou, 2016).
The therapy of choice to treat listeriosis is based on β-lactams, penicillin G, or ampicillin combined with or without a classical aminoglycoside as gentamicin. An alternative treatment based on trimethoprim and sulfamethoxazole is considered for penicillin-allergic patients. Additionally, tetracycline, erythromycin, and vancomycin have also been used to fight human listeriosis (Temple and Nahata, 2000; Pagliano et al., 2017). In the last decades, Lm has developed resistances to a wide range of antimicrobial agents, even those used in reference treatments. The first multidrug-resistant Lm strain was discovered in France in 1988 (Poyart-Salmeron et al., 1990). Since then, multidrug-resistant strains have been detected in clinical isolates and food processing environment (Morvan et al., 2010; Pesavento et al., 2010; Gómez et al., 2014; Dahshan et al., 2016; Şanlıbaba et al., 2018; Basha et al., 2019; Kayode and Okoh, 2022).
Pesavento et al. (2010) studied the presence of Listeria spp. In raw meat and retail products, reporting that 20% of Lm strains showed resistance to three or more antibiotics. Şanlıbaba et al. (2018) evaluated the antibiotic resistance of 17 L. monocytogenes strains isolated from ready-to-eat food. All strains were resistant to nalidixic acid, ampicillin, penicillin G, linezolid, and clindamycin. In recent reports, Basha et al. (2019) showed that Lm isolated from fish and fishery environments harbored resistance to the most commonly β-lactams (ampicillin and penicillin), macrolides (erythromycin), tetracycline and clindamycin. Kayode and Okoh (2022) observed that 71.43% of Lm isolates obtained from milk and derivatives harbored one or more resistance genes.
In face of this critical situation, the design of novel antimicrobial agents is pressingly required. Currently, it is accepted that the identification of appropriate targets is a critical step in designing new drugs. In the postgenomic era, integrative computational approaches facilitate the identification and prioritization of candidate targets. In this sense, Target-Pathogen (http://target.sbg.qb.fcen.uba.ar) (Sosa E. et al., 2018) is a unique resource that combines structural druggability datasets, essentiality analysis, metabolic context, genomic and expression data to rank proteins according to their potential to be used as novel targets. Previous reports from our and other groups have selected and prioritized molecular targets of several relevant pathogens such as Mycobacterium tuberculosis (Defelipe et al., 2016), Klebsiella pneumoniae (Ramos et al., 2018; Serral et al., 2021, Serral et al., 2022), Bartonella bacilliformis (Farfán-López et al., 2020), Trypanosoma cruzi (Osorio-Méndez and Cevallos, 2018; Coutinho et al., 2021) and Schistosoma mansoni (Lobo-Silva et al., 2020) using Target-Pathogen.
Here, we report the application of a multidimensional data integration strategy to prioritize drug targets in Listeria monocytogenes EGD-e. Combining multilayers of genomic-scale information of Lm, which included genomic, transcriptomic, metabolic, and protein structural data sources, we were able to delineate candidate proteins with features that are relevant to target selection in Lm and related pathogens. We expect that our results will be particularly useful to accelerate the initial steps of drug discovery through the identification of attractive targets.
Materials and methods
Generation of structural homology-based models
L. monocytogenes EGD-e genome was obtained from NCBI GenBank (NC_003210.1). 184 experimental structures for Lm were obtained from the Protein Data Bank (PDB). For all remaining proteins, we attempted to predict their structure by homology modeling. Protein sequences were used as a query for PSI-BLAST searches (Iterations: three; E-value: 1e-05) against UniRef90 (Suzek et al., 2015). After generating each position-specific scoring matrix (PSSM), they were used as input to search against the PDB in order to find templates. When an adequate template was found, homology-based models were built using MODELLER (https://salilab.org/modeller/) (Webb and Sali, 2016) following a pipeline previously described (Defelipe et al., 2016; Sosa E. J. et al., 2018; Ramos et al., 2018). With this approach, we generated 1,741 homology-based models. Out of 2,867 predicted proteins that form the Lm proteome, we obtained 1,925 structures.
Structure-based druggability assessment
The druggability concept refers to the capacity of a protein to bind drug-like compounds, modulating its activity in the desired way. From the structural point of view, a protein is considered druggable if it has a well-defined pocket with suitable physicochemical properties to allow drug binding-sites prediction.
Druggability prediction of each target was performed following a pipeline developed by our group (Radusky et al., 2014). This methodology is based on fpocket (https://github.com/Discngine/fpocket) (Le Guilloux et al., 2009), an open-source pocket detection algorithm that integrates physicochemical descriptors in order to estimate the pocket druggability and can be used in a genomic scale.
Protein pockets were grouped in four categories according to their Druggability Score (DS): non-druggable (0.0 ≤ DS < 0.2), poorly druggable (0.2 ≤ DS < 0.5), druggable (0.5 ≤ DS < 0.7) and highly druggable (0.7 ≤ DS ≤ 1.0). This classification is based on a previous analysis of DS distribution of all pockets found in PDB structures that were crystallized binding a drug-like compound (Radusky et al., 2014; Sosa E. et al., 2018).
L. monocytogenes metabolic network construction and analysis
The metabolic network of Lm EGD-e was built using the PathoLogic module from Pathway Tools v. 23.0 (http://bioinformatics.ai.sri.com/ptools/) (Karp et al., 2011) that generates a Pathway/Genome Database (PGDB) containing the predicted reactions and metabolic pathways. This database was built using as input a Genbank file obtained from NCBI (NC_003210.1). The reconstruction consists in making gene-protein-reaction associations principally based on each gene product annotation or its enzyme commission (EC) number.
Once finished the automatic reconstruction process, the metabolic network was refined through manual curation. The Pathway Hole Filler algorithm was used to identify candidates that can fill the enzymatic holes present in incomplete pathways. Additionally, missing pathways with biological evidence were added and false-positive predicted pathways were removed.
The curated metabolic network was exported in systems biology markup language (SBML) format, considering only the reactions included in the small-molecules metabolism (Ramos et al., 2018). An in-house Python script was used to calculate the frequency at which each compound was present in the network reactions. The metabolites involved in a high number of reactions were filtered out (such as ATP, NAD, water, and protons) given that ubiquitous compounds can generate artificial associations in the network. After manual inspection, a total of 27 compounds were filtered to avoid the creation of artificial links on the reaction graph, and the resulting SBML file was transformed into a reaction graph in a simple interaction file (sif) format.
Cytoscape_v3.8.1 (https://cytoscape.org/) (Shannon et al., 2003) was used for network visualization and subsequent analysis. In this representation, reactions are represented as nodes and there is an edge connecting two nodes if the product of a reaction is consumed as a substrate for the reaction that follows. Choke-points (reactions that uniquely produce or consume a given metabolite) identification was also conducted. The Betweenness centrality of every node was also calculated as described previously (Sosa E. J. et al., 2018). This topological metric indicates the participation of a reaction as an intermediary in the network.
Essentiality analysis
The proteome of Lm was used as a query in BLASTp against the Database of Essential Genes (DEG) (http://origin.tubic.org/deg/public/index.php) (Luo et al., 2021). E-value = 1e−05, identity ≥50% were used as cut-off values. DEG is a database that stores the essential genes identified by high-throughput experiments. As essential genes are broadly conserved in microorganisms, homologous genes found in this analysis are likely to be also essential.
Off-target criteria
All Lm EGD-e proteins were subjected to NCBI BLASTp against the human proteome (version GRCh38. p10) to identify non-host homologous targets.
Hits with an E-value smaller than 10–5 and identity ≥50% were filtered out, as they can share a high structural similarity with a human protein. This minimizes the possibility of adverse effects produced by cross-interference. In this respect, protein inhibition in organisms that inhabit the human intestine could generate an impact on the host’s normal flora. To reduce this possibility, the proteome of Lm EGD-e was also compared to the proteins of the 226 representative microorganisms of the gut flora sequenced by the Human Microbiome Project (NIH HMP Working Group et al., 2009), obtaining the number of organisms that present at least one significant hit (E-value ≤10–5; identity ≥50%).
Analysis of conservation among pathogenic L. monocytogenes
Mauve (https://darlinglab.org/mauve/mauve.html) (Darling et al., 2004)was used to find groups of orthologs (identity ≥60% and coverage ≥70%) among 25 clinically relevant pathogenic strains of L. monocytogenes genomes available in NCBI (Bergholz et al., 2016; Chen et al., 2016; Kwong et al., 2016). Proteins conserved in several genomes are attractive targets because this implies that a drug could be used to control multiple strains of this microorganism including from different serotypes and serogroups.
Expression data of L. monocytogenes
Very important shifts in the expression profile of genes are involved during the passage of Lm from a saprophytism to virulence lifestyle. To capture the transcriptome landscape of L. monocytogenes we analyzed some previously published work that used microarrays analysis for gene expression from relevant physiological conditions that mimic the pathogenic environment during infection: intracellular replication in macrophages (Lobel et al., 2012) and intestinal lumen and blood (Toledo-Arana et al., 2009). The knowledge of protein targets with a critical role during infection might serve as new targets for the development of antilisterial compounds. Information about overexpressed proteins (436 in intestine, 541 in blood and 809 in macrophages) was added to Target Pathogen.
Target prioritization
All previously calculated data was integrated into Target-Pathogen (Sosa E. J. et al., 2018). Target-Pathogen has been designed and developed by our group as an online resource to allow genome-wide based data consolidation from diverse sources focusing on structural druggability, essentiality, and metabolic role of proteins. By allowing the integration and weighting of this information, this bioinformatic tool aims to facilitate the identification and prioritization of candidate drug targets for pathogens. Using Target Pathogen, non-essential, non-druggable proteins (DS < 0.5) and close human homologs were filtered out. Afterward, a score was assigned to each protein following the equation:
where
Eq. 2 incorporates expression data, where
Results and discussion
All results are organized with the following scheme. We will begin analyzing the structural druggability of the Lm proteome performing a broad classification of the proteins. Secondly, we will present a highly-cured metabolic network of this pathogen and at last, we combined this data with diverse layers of omic data in Target Pathogen in order to identify and prioritize novel drug targets with the potential to develop new antimicrobials against Lm. A general scheme of our prioritization pipeline is shown in Figure 1.
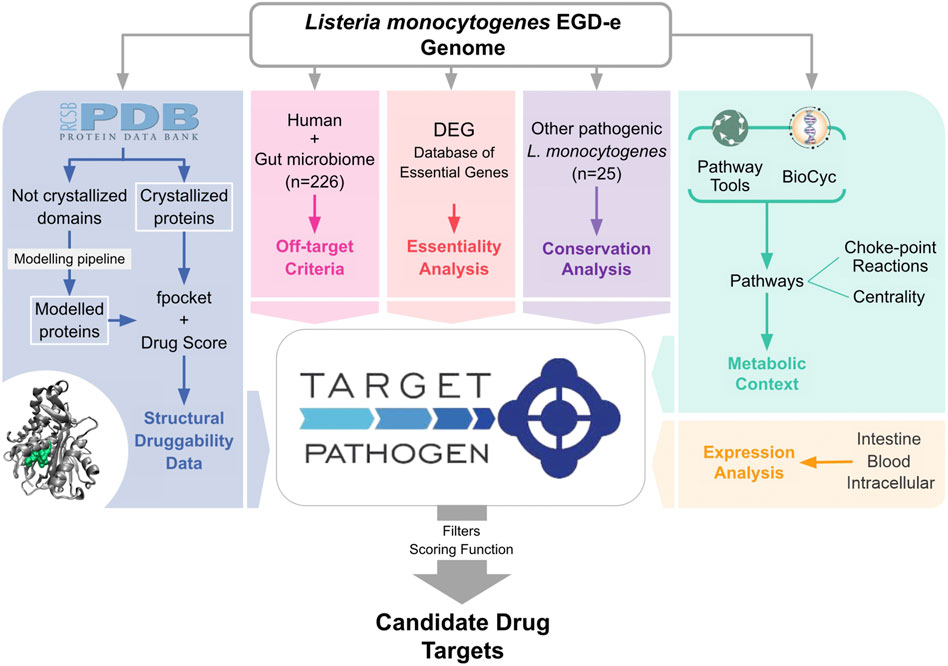
FIGURE 1. Schematic diagram of the prioritization pipeline.
Listeria monocytogenes protein structures are enriched in druggable pockets.
Our analysis began by classifying all the domain structures of Listeria monocytogenes, including our own generated homology-based models and those directly retrieved from PDB, according to their structural druggability. For comparison, we also calculated the DS for all ligand-bound structures in a non-redundant subset of PDB (PDB95), revealing an important enrichment of druggable pockets in the Listeria models (Figure 2) in line with previous results in other pathogenic bacteria (Defelipe et al., 2016; Ramos et al., 2018; Farfán-López et al., 2020). Afterward, we grouped the proteins, according to how their structure was retrieved and their DS, into four categories. The two first categories include proteins that were directly retrieved from the PDB. One corresponds to those which have been experimentally obtained bound to an inhibitor or drug-like compound (ED + group). The second one includes proteins also obtained from the PDB but without binding a drug (ED-). The remaining two categories include homology-based models.
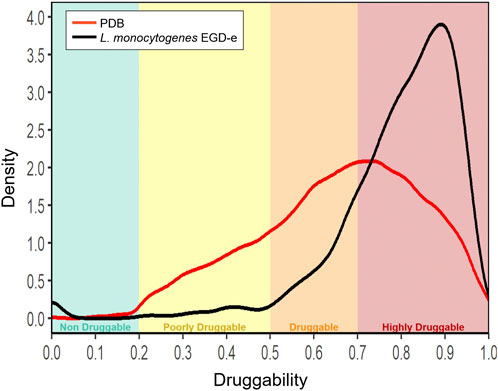
FIGURE 2. Histogram of Druggability Score (DS). Representation of all Protein Data Bank (PDB) structures that host a drug-like compound (red line) and all modeled structures of L. monocytogenes (black line). Protein pockets were grouped in four categories according to their DS: non-druggable (0.0 ≤ DS < 0.2), poorly druggable (0.2 ≤ DS < 0.5), druggable (0.5 ≤ DS < 0.7) and highly druggable (0.7 ≤ DS ≤ 1.0).
The Modeled With Drug group (MD+) includes protein structures modeled from a template that was co-crystallized with a drug-like compound, thus the proteins of this group are likely to be druggable. The last category groups all modeled proteins whose structure was resolved from proteins deposited in the PDB with no drugs or associated inhibitors present (MD-).
We then computed all the possible pockets and their corresponding Druggability Score (DS) for each protein using fpocket. According to their DS, we classified all the structures into four druggability groups (Table 1), according to the criteria set in materials and methods. As expected, most of the Listeria monocytogenes structures crystallized in the presence of a drug presented high DS. As expected, the results showed that most of the proteins in the MD + group (98.5%) have DS Scores higher than 0.5 (Table 1) validating our methodology, according to previous works (Defelipe et al., 2016; Ramos et al., 2018). The first group of interest from the target selection point of view concerns the MD + -D group (druggable proteins in the MD + group). The fact that both, an association (assignation to MD+) and a structural criteria (DS > 0.5) match is a strong argument for the selection of these 468 proteins as drug targets (Supplementary Table S1).
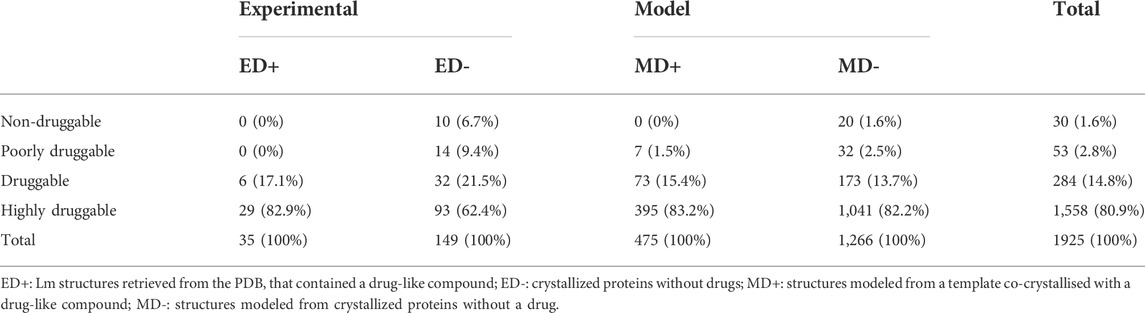
TABLE 1. List of the 1925 protein structures obtained for L. monocytogenes classified according to their Druggability Score (DS).
Listeria monocytogenes metabolic network reconstruction and refinement
We performed a whole-genome-based reconstruction of the Listeria monocytogenes metabolic network (Lm-MN) using Pathway Tools followed by manual curation. The automatic reconstruction of Lm-MN with Pathway Tools resulted in a draft metabolic model composed of 172 metabolic pathways and 1,272 reactions (1,165 with enzymatic activities and 11 with transport function). We identified 120 pathway holes (reactions without associated genes) in this draft model, which were distributed across 70 metabolic pathways with different degree of completeness. Refinement of Lm-MN began by manually inspecting the draft MN using the Pathway Hole Filler (PHF) algorithm. Based on this tool, 22 holes could be filled. During this process, reactions corresponding to 17 metabolic pathways were evaluated, and nine could be completed (i.e., all their reactions had a gene assigned). In the last refinement step, using bibliographic evidence, eight pathways were added and six were removed after being identified as false positives during manual curation. Correct and complete associations between reactions and genes are paramount to downstream uses of any metabolic network. Such is the case of target prioritization, our focus in the following sections. Once constructed, this network was analyzed using graph theory, allowing the calculation of topological metrics related to node importance as the betweenness centrality. High centrality nodes (reactions) are attractive from the target discovery point of view because their inhibition would lead to an imbalance in many different pathways. We also identified choke-points (CPs) in the Lm-MN, i.e. reactions that uniquely consume or produce a given compound. Choke-point reactions are attractive for drug targeting because their blockage can lead to the depletion of an essential metabolite or the accumulation of a toxic compound. A total of 89 proteins were associated with reactions strictly classified as producing CPs, while 297 proteins with strictly consuming CPs. On the other hand, 26 proteins were mapped with reactions classified as CPs on both producing and consuming sides. Since many CPs involve the transformation of indispensable compounds, they have been proposed as attractive drug targets (Yeh et al., 2004). We identified that while 21% of the Lm proteome is composed of predicted essential proteins, 40% of identified CPs are associated with essential proteins, reinforcing the usefulness of this parameter in our target prioritization strategy. The projection of Lm-MN onto a reaction graph also allowed the calculation of relevant topological metrics. Figure 3 depicts the resultant Lm-MN graph, with node sizes proportional to betweenness centrality. The presence of a few high-centrality nodes indicates that these hubs may be of special importance to the cohesiveness of the network. We did not limit our analysis to solely filtering choke-point nodes or hubs identification. Rather, this information was incorporated into the scoring function that allowed ranking of the potential Lm targets within the proteome of this organism. The complete list of choke-points and centrality measures is also available within Target-Pathogen, as well as the complete metabolic annotation of Lm.

FIGURE 3. Reaction graph of the L. monocytogenes metabolic network. Predicted Lm reactions are depicted as nodes and there is an edge connecting two nodes if the product of a reaction is consumed as a substrate for the reaction that follows. In this representation, node size is proportional to betweenness centrality and choke-point reactions are colored red. MetaCyc IDs (http://metacyc.org) are also shown.
Integration of structural and druggability information, conservation and metabolic analysis and off-target criteria allows prioritization of potential molecular targets for drug development against Listeria monocytogenes.
After integration of the generated multi-omic datasets, we score individual proteins according to their potential to be used as targets of novel drugs to combat L. monocytogenes. At first, we rule out all non-essential, non-druggable, and close human homologs proteins. A total of 434 essential, druggable proteins with no close homologs in the human genome were kept (Supplementary Figure S1, Supplementary Table S2). Afterward, we take into account different features that a protein should exhibit to serve as an attractive target. These include the contextualization of its function into the metabolic context and presence/absence in related pathogens, which degree of conservation ultimately defines the spectrum activity (Supplementary Figure S2). Based on the gathered data, we define a scoring function (see Eq. 1 in materials and methods) that allowed us to rank all proteins in the Lm proteome. The 15 highest-ranking proteins are presented in Table 2. This analysis can also be replicated in the Target-Pathogen interface.
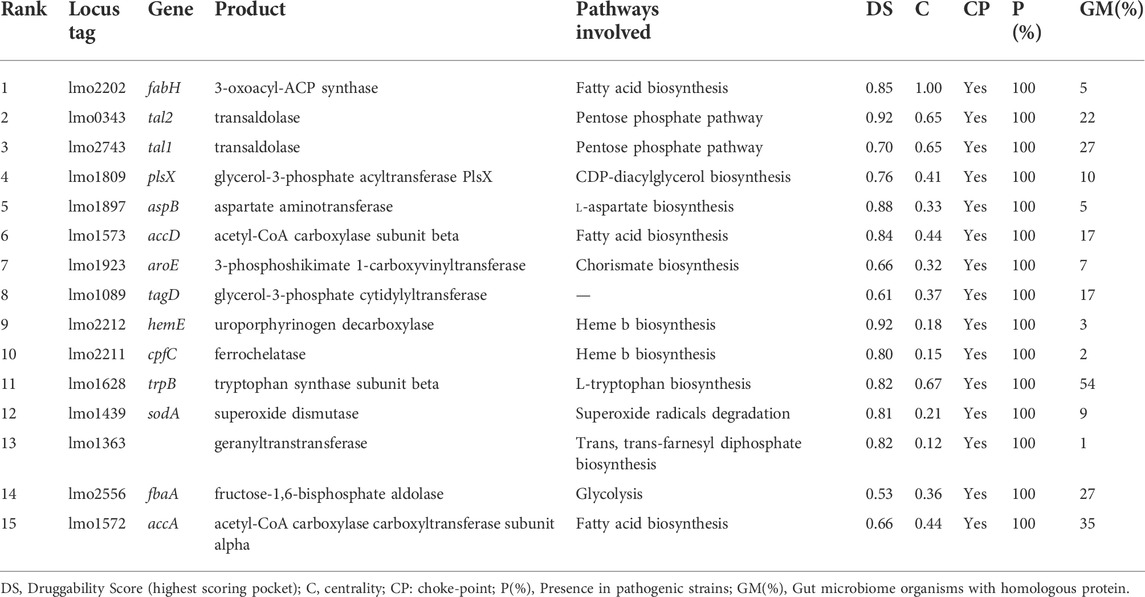
TABLE 2. List of L. monocytogenes highest-ranking proteins considering metabolic network metrics, conservation in pathogenic Lm and presence in microbiome (Eq. 1).
As can be appreciated in Table 2, most of the best-ranked proteins participate in pathways with great relevance in bacteria metabolism such as fatty acids and amino acid biosynthesis. Fatty acid pathway is essential for bacteria viability and growth, and is required for the production of phospholipids, lipoproteins, and lipopolysaccharides. There are significant differences between human and bacterial fatty acid biosynthesis systems that include the organization and structure of enzymes and the specific roles played by fatty acids (Wang et al., 2007). In Lm, β-ketoacyl-ACP synthase III (FabH, the best-ranked protein) catalyzes the first condensation reaction of short-chain fatty acid thioesters, i. e the conversion of acetyl-CoA and malonyl-ACP to β-ketoacyl-ACP products (Costa et al., 2016). This protein harbors a highly druggable pocket (SD: 0.85), is conserved in all the analyzed pathogenic strains, and does not present close homologs in humans and their microbiota. Moreover, from a metabolic point of view, it catalyzes a choke-point, highly central, reaction. Platensimycin, platencin, cerulenin, thiolactomycin, and thiotetromycin are known selective inhibitors of FabH. These natural compounds were exhaustively analyzed elsewhere (Wang et al., 2006; Jayasuriya et al., 2007; Shang et al., 2015). Two other genes, implicated in fatty acid biosynthesis, were also prioritized in our analysis: accA and accD encoding alpha and beta subunits of Acetyl-CoA Carboxylase Carboxyltransferase (ACC) respectively. Both subunits are part of the essential enzyme complex that catalyzes the acetyl-CoA carboxylation to form malonyl-CoA in the initiation of the fatty acid pathway (Freiberg et al., 2004). These proteins not only comply with our employed structural druggability criterion (presenting a high druggable score), but they are also essential and conserved in all Lm strains considered (Figure 4).
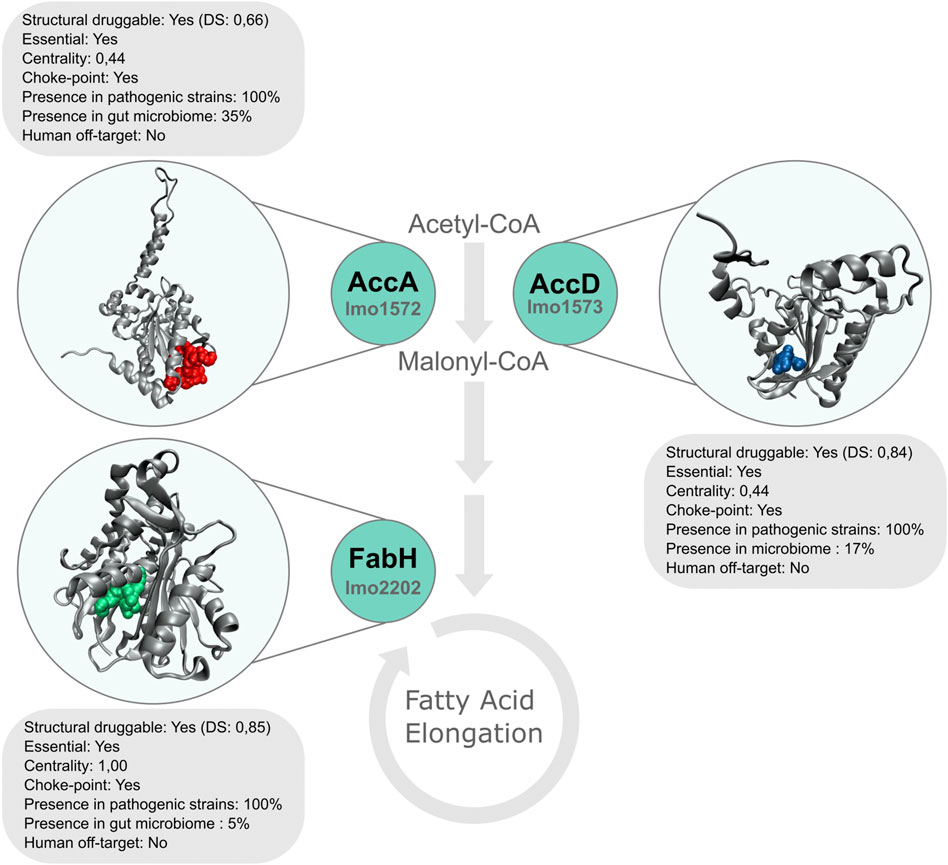
FIGURE 4. Representation of top-ranked proteins AccA, AccD, and FabH, involved in fatty acid biosynthesis. The structures of these targets are shown, with the most druggable pocket in colored spheres.
Nevertheless, not many antibiotics have been described to possess selective ACC inhibition until now. The first natural compounds analyzed were andrimid and their derivatives and, pseudopeptidic pyrrolidinedione moiramide B. These compounds present a broad-spectrum antibacterial activity (Fredenhagen et al., 1987; Freiberg et al., 2004). The effect of different herbicides was also evaluated as ACC inhibitors in M. tuberculosis. Haloxyfop was shown to inhibit the AccA3-AccD6 complex activity (Daniel et al., 2007) meanwhile Diclofop was described to inhibit AccA3-AccD5 acetyl-CoA carboxylation (Oh et al., 2006). Thus, enzymes of the fatty-acid biosynthesis pathway are attractive targets for the development of new drugs against pathogenic microorganisms.
Other molecular targets prioritized in Table 2 are those involved in central metabolism, particularly in the pentose phosphate pathway (PPP). Transaldolases are enzymes involved in the non-oxidative phase of PPP. PPP is considered the predominant pathway of sugar metabolism during infection (Fuchs et al., 2012). In line with this fact, genes of the oxidative and non-oxidative branches of PPP were found overexpressed in a macrophage cells infection model (Chatterjee et al., 2006). It was also shown that Proteus mirabilis talB knockout affected bacterial fitness during urinary tract infections in a murine model, showing that transaldolase has an important role in vivo metabolism (Alteri et al., 2015).
TrpB catalyzes the second-highest centrality reaction listed in Table 2. This protein is implicated in the l-tryptophan biosynthesis. The TrpB polypeptide functions as the β subunit of the tetrameric (α2-β2) tryptophan synthase complex, and catalyzes the final step of the synthesis of l-tryptophan from indole and l-serine. TrpB is highly drugable (SD: 0.82) and essential, it is conserved in all the strains analyzed and does not present homology with the human proteome. It was also associated with a choke-point reaction. The trp operon is highly conserved in bacteria and is finely regulated. In Lm the trp operon is composed of trpE, trpG, trpD, trpC, trpF, trpB and trpA genes (Gutierrez-Preciado et al., 2005). These enzymes are considered valuable therapeutic targets to combat pathogenic microorganisms and currently, the continuing search for effective inhibitors is of interest. Wellington et al. (2017) have identified a novel azetidine, BRD4592, effective against Mycobacterium tuberculosis through allosteric inhibition of the tryptophan synthase.
Additionally, Table 2 presents the 3-phosphoshikimate 1-carboxyvinyltransferase enzyme (aroE) which catalyzes the sixth step in the biosynthesis of the aromatic amino acids (shikimate pathway). This protein harbors a druggable pocket (DS: 0.66) and, like the candidates mentioned above, it is also essential. It is highly conserved among Listeria species and does not present homology with the human and microbiota proteome. Also, it has attractive features from a metabolic point of view (i.e catalyzes a choke-point and highly central reaction) (Table 2). Since the shikimate pathway is not present in vertebrates but is essential for bacteria, it is commonly considered a valuable target for antimicrobial discovery. Listeria monocytogenesaroE mutants showed a strong reduction of growth rates in epithelial cells and attenuated virulence in a murine model. Further, the aroE mutant was unable to grow in a minimal medium in presence of shikimate. Partial growth of the aroE mutant was shown when all aromatic amino acids were supplemented, describing an essential role in the aromatic amino acid pathway (Stritzker et al., 2004).
Prioritization of L. monocytogenes proteins according to their expression in infection-mimicking conditions
Once targets that complied with rules associated with gene essentiality, protein druggability, metabolic importance, and broad Lm conservation were identified, we further incorporated in our analysis a term related to overexpression in conditions that resemble infection in vivo.
It is known that upon ingestion with Lm contaminated food, the pathogen can proliferate in the intestinal lumen environment and, after passing the intestinal barrier, it can disseminate through the lymph and blood to reach the brain, spleen, liver or even cross the blood-placenta barrier. In our analysis, expression data (Toledo-Arana et al., 2009; Lobel et al., 2012) were taken into account to prioritize those genes that were upregulated during the infection-mimicking conditions. The selected conditions, which group different reports, comprise survival and replication in intestinal, blood, and macrophage environments. As shown in Table 3, the best-ranked protein, transaldolase Tal2, participates in the non-oxidative branch of the Pentose Phosphate Pathway. This protein is transcriptionally upregulated in the intestine, blood, and macrophage contexts and meets the requirements of an attractive target. This protein is essential, druggable (DS = 0.92), metabolically relevant, and widely conserved within Lm pathogens. Another upregulated gene involved in this pathway is lmo2661, which encodes a ribulose-5-phosphate 3-epimerase. Although we could not predict epimerase as an essential gene (and it was ruled out in the filtering step), our results revealed its association with a highly central and choke-point reaction. Moreover, lmo2661 is highly druggable (SD: 0.82), conserved in all the analyzed strains, and does not present close homologs in humans. The importance of this pathway lies in the production of xylulose and ribose-5-phosphate as precursors of nucleotides biosynthesis during intracellular growth as was shown in another macrophage infection model (Chatterjee et al., 2006).
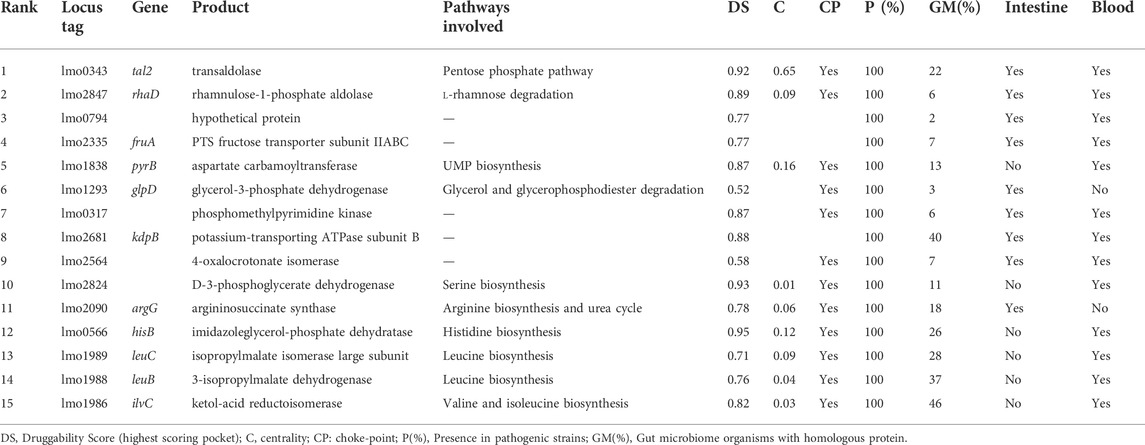
TABLE 3. List of L. monocytogenes highest-ranking proteins by incorporating protein overexpression in intestine, blood and intracellular environments (Eq. 2).
The second best-ranked protein is implicated in the rhamnose utilization pathway: rhamnulose-1-phosphate aldolase protein RhaD (Table 3). RhaD would be an excellent candidate to further study since we found it is essential and highly druggable, is conserved in all the analyzed pathogenic strains and does not present close homologs in humans, and their microbiota. Particularly, it appears overexpressed under different contexts that mimic infection. Although only RhaD is an essential target based on our bioinformatics pipeline, other proteins in the rhamnose utilization pathway, such as rhamnulokinase (RhaB) and rhamnose mutarotase (RhaM) also harbor many features that make them attractive targets (Figure 5). Interestingly, experimental data have shown that Listeria monocytogenes ΔrhaB mutants grow less efficiently in macrophage cells (Lobel et al., 2012). Is interesting to note, that regardless of whether intestine, blood, or macrophages were infected, the gene cluster involved in rhamnose catabolism is transcriptionally upregulated. Moreover, it has been established that the most abundant glycopolymer associated with the cell wall of Lm are wall teichoic acids (WTA) that can be decorated with rhamnose (Shen et al., 2017). WTA were postulated as antigenic determinants (Kamisango et al., 1983). Rhamnose metabolic pathway induction during infection could directly impact on the cell surface architecture allowing the pathogen to escape from immunity. Moreover, it was shown that rhamnosylation of L monocytogenes WTA promotes resistance to antimicrobial peptides by delaying interaction with the membrane (Carvalho et al., 2015; Meireles et al., 2020).
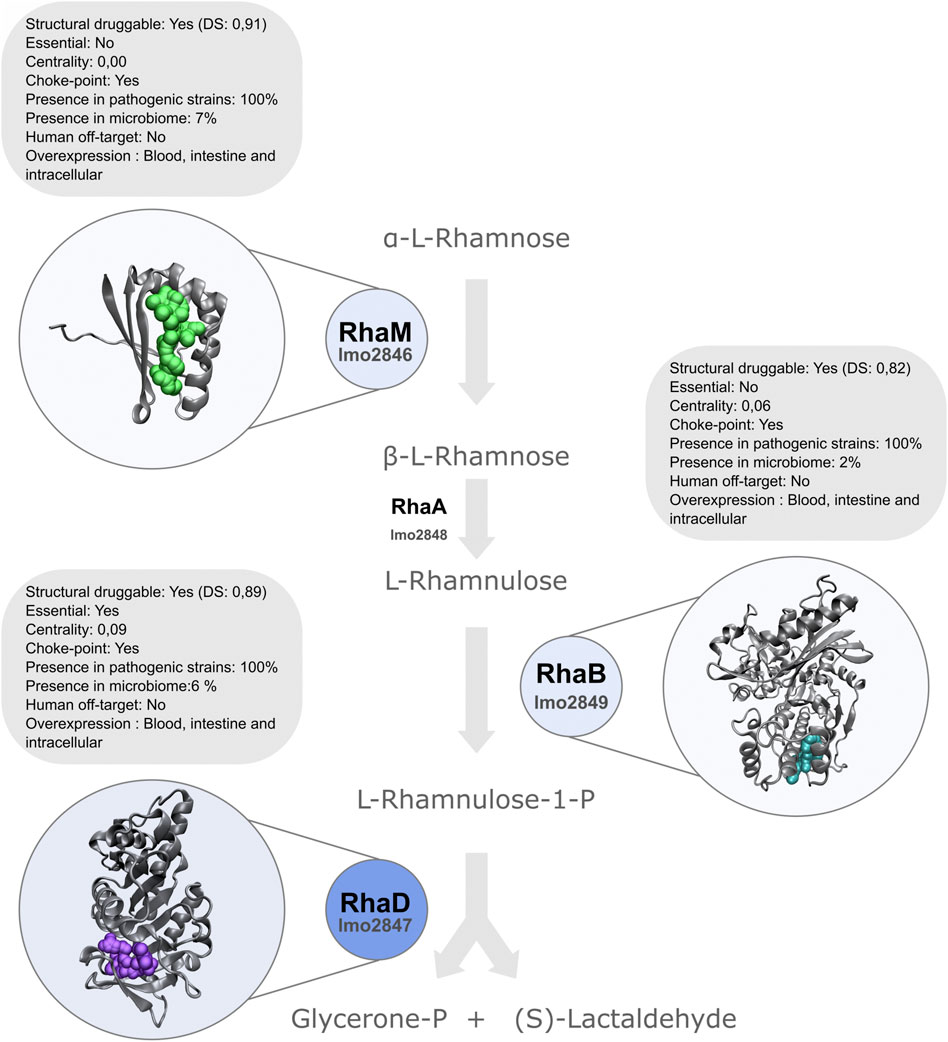
FIGURE 5. Schematic representation of l-rhamnose degradation pathway. RhaD is highlighted, as a top-ranked protein in our analyses.
Other top-ranking targets are those responsible for serine, arginine, histidine and, brand chain amino acids (BCAA) biosynthesis. It was shown that the novo biosynthesis of amino acids is a key process of intracellular replication of L. monocytogenes (Joseph et al., 2006; Schauer et al., 2010). Overexpression of enzymes involved in amino acids biosynthesis during intracellular replication (blood and macrophages) might reflect the limited availability of these important metabolites in the host cytosol. Moreover, experimental data have shown that L. monocytogenes amino acid metabolic mutants (ΔargD, ΔhisC, and ΔilvC) grow less efficiently in macrophage cells (Lobel et al., 2012). In this respect, our analysis has provided other interesting targets to be further investigated that include expression, essentiality, and druggability data (Table 3).
Conclusion
In this work, we developed and applied an integrative analysis framework for the prioritization of protein targets in L. monocytogenes. Various layers of information were combined, including genomic structural and metabolic data that allowed shortlisting of targets with desirable characteristics from a druggability standpoint. Our integrative approach, where multiple layers of omics information were overlaid, disclosed a series of potential targets for antibiotic drug discovery, with fatty-acid, pentose, rhamnose and amino acids metabolism emerging as interesting dominant biological themes warranting further consideration in the molecular target space of L. monocytogenes. Further studies are warranted to follow-up experimentally on our elicited targets, and we invite the scientific community dedicated to this subject to help pursue these goals, thus strengthening the ongoing fight against pathogenic bacteria.
Lm EGD-e 2,867 proteins were compared against DEG, human and gut flora proteome. Its conservation was evaluated among 25 clinically relevant pathogenic strains. The information obtained from these analyses was integrated with protein structures (184 experimental crystals and 1741 homology-based models), metabolic annotations and previously published expression data. Finally, a set of filters and a scoring function were applied.
The structures of these targets are shown, with the most druggable pocket in colored spheres.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
MaP and DD conceived the study design, MiP, ES, FC, GS, and FS contributed tools and performed data analysis. AT, MaP, and DD drafted the manuscript with input from the other authors. All authors read and approved the final version of the manuscript.
Funding
Fellowship supported from CONICET to GS, MiP, FS, and FC. DD, ES, AT, and MaP are members of CONICET research career. This work was supported by Agencia Nacional de Promoción Científica y Tecnológica [ANPCyT, PICT-2018-04663 to DD]. D.F. D; .J. M.; E. J. S.; N.Y.; M. M.; B. L.; A. T. and V. R.] and Universidad de Buenos Aires (20020190200275BA to DD).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.969415/full#supplementary-material
References
Alteri, C. J., Himpsl, S. D., and Mobley, H. L. T. (2015). Preferential use of central metabolism in vivo reveals a nutritional basis for polymicrobial infection. PLoS Pathog. 11, e1004601. doi:10.1371/journal.ppat.1004601
Authority, E. F. S. (2015). European Food Safety Authority, and European Centre for Disease Prevention and ControlThe European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2013. EFSA J. 13, 3991. doi:10.2903/j.efsa.2015.3991
Basha, K. A., Kumar, N. R., Das, V., Reshmi, K., Rao, B. M., Lalitha, K. V., et al. (2019). Prevalence, molecular characterization, genetic heterogeneity and antimicrobial resistance of Listeria monocytogenes associated with fish and fishery environment in Kerala, India. Lett. Appl. Microbiol. 69, 286–293. doi:10.1111/lam.13205
Bergholz, T. M., den Bakker, H. C., Katz, L. S., Silk, B. J., Jackson, K. A., Kucerova, Z., et al. (2016). Determination of evolutionary relationships of outbreak-associated Listeria monocytogenes strains of serotypes 1/2a and 1/2b by whole-genome sequencing. Appl. Environ. Microbiol. 82, 928–938. doi:10.1128/AEM.02440-15
Carvalho, F., Atilano, M. L., Pombinho, R., Covas, G., Gallo, R. L., Filipe, S. R., et al. (2015). L-rhamnosylation of Listeria monocytogenes wall teichoic acids promotes resistance to antimicrobial peptides by delaying interaction with the membrane. PLoS Pathog. 11, e1004919. doi:10.1371/journal.ppat.1004919
Chatterjee, S. S., Hossain, H., Otten, S., Kuenne, C., Kuchmina, K., Machata, S., et al. (2006). Intracellular gene expression profile of Listeria monocytogenes. Infect. Immun. 74, 1323–1338. doi:10.1128/iai.74.2.1323-1338.2006
Chen, Y., Gonzalez-Escalona, N., Hammack, T. S., Allard, M. W., Strain, E. A., and Brown, E. W. (2016). Core genome multilocus sequence typing for identification of globally distributed clonal groups and differentiation of outbreak strains of Listeria monocytogenes. Appl. Environ. Microbiol. 82, 6258–6272. doi:10.1128/AEM.01532-16
Costa, T. P. S., da Costa, T. P. S., Nanson, J. D., and Forwood, J. K. (2016). 1.35 angstrom resolution crystal structure of beta-ketoacyl-ACP synthase II (FabF) from Listeria monocytogenes. Scientific Reports 7 (January), 39277. doi:10.2210/pdb5sxo/pdb
Coutinho, J. V. P., Rosa-Fernandes, L., Mule, S. N., de Oliveira, G. S., Manchola, N. C., Santiago, V. F., et al. (2021). The thermal proteome stability profile of Trypanosoma cruzi in epimastigote and trypomastigote life stages. J. Proteomics 248, 104339. doi:10.1016/j.jprot.2021.104339
Dahshan, H., Merwad, A. M. A., and Mohamed, T. S. (2016). Listeria species in broiler poultry farms: Potential public health hazards. J. Microbiol. Biotechnol. 26, 1551–1556. doi:10.4014/jmb.1603.03075
Daniel, J., Oh, T.-J., Lee, C.-M., and Kolattukudy, P. E. (2007). AccD6, a member of the fas II locus, is a functional Carboxyltransferase subunit of the acyl-coenzyme A carboxylase in Mycobacterium tuberculosis. J. Bacteriol. 189, 911–917. doi:10.1128/jb.01019-06
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi:10.1101/gr.2289704
Defelipe, L. A., Do Porto, D. F., Pereira Ramos, P. I., Nicolás, M. F., Sosa, E., Radusky, L., et al. (2016). A whole genome bioinformatic approach to determine potential latent phase specific targets in Mycobacterium tuberculosis. Tuberculosis 97, 181–192. doi:10.1016/j.tube.2015.11.009
Farfán-López, M., Espinoza-Culupú, A., García-de-la-Guarda, R., Serral, F., Sosa, E., Palomino, M. M., et al. (2020). Prioritisation of potential drug targets against Bartonella bacilliformis by an integrative in-silico approach. Mem. Inst. Oswaldo Cruz 115, e200184. doi:10.1590/0074-02760200184
Fredenhagen, A., Tamura, S. Y., Kenny, P. T. M., Komura, H., Naya, Y., Nakanishi, K., et al. (1987). ChemInform abstract: Andrimid, a new peptide antibiotic produced by an intracellular bacterial symbiont isolated from a Brown planthopper. ChemInform 18. doi:10.1002/chin.198745305
Freiberg, C., Brunner, N. A., Schiffer, G., Lampe, T., Pohlmann, J., Brands, M., et al. (2004). Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity. J. Biol. Chem. 279, 26066–26073. doi:10.1074/jbc.M402989200
Fuchs, T. M., Eisenreich, W., Kern, T., and Dandekar, T. (2012). Toward a systemic understanding of Listeria monocytogenes metabolism during infection. Front. Microbiol. 3, 23. doi:10.3389/fmicb.2012.00023
Garner, D., and Kathariou, S. (2016). Fresh produce–associated listeriosis outbreaks, sources of concern, teachable moments, and insights. J. Food Prot. 79, 337–344. doi:10.4315/0362-028x.jfp-15-387
Gómez, D., Azón, E., Marco, N., Carramiñana, J. J., Rota, C., Ariño, A., et al. (2014). Antimicrobial resistance of Listeria monocytogenes and Listeria innocua from meat products and meat-processing environment. Food Microbiol. 42, 61–65. doi:10.1016/j.fm.2014.02.017
Gutierrez-Preciado, A., Jensen, R. A., Yanofsky, C., and Merino, E. (2005). New insights into regulation of the tryptophan biosynthetic operon in Gram-positive bacteria. Trends Genet. 21, 432–436. doi:10.1016/j.tig.2005.06.001
Jayasuriya, H., Herath, K. B., Zhang, C., Zink, D. L., Basilio, A., Genilloud, O., et al. (2007). Isolation and structure of platencin: A FabH and FabF dual inhibitor with potent broad-spectrum antibiotic activity. Angew. Chem. Int. Ed. Engl. 46, 4684–4688. doi:10.1002/anie.200701058
Joseph, B., Przybilla, K., Stühler, C., Schauer, K., Slaghuis, J., Fuchs, T. M., et al. (2006). Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J. Bacteriol. 188, 556–568. doi:10.1128/jb.188.2.556-568.2006
Kamisango, K., Fujii, H., Okumura, H., Saiki, I., Araki, Y., Yamamura, Y., et al. (1983). Structural and immunochemical studies of teichoic acid of Listeria monocytogenes. J. Biochem. 93, 1401–1409. doi:10.1093/oxfordjournals.jbchem.a134275
Karp, P. D., Latendresse, M., and Caspi, R. (2011). The pathway tools pathway prediction algorithm. Stand. Genomic Sci. 5, 424–429. doi:10.4056/sigs.1794338
Kayode, A. J., and Okoh, A. I. (2022). Assessment of multidrug-resistant Listeria monocytogenes in milk and milk product and One Health perspective. PloS One 17 (7), e0270993. doi:10.1371/journal.pone.0270993
Kwong, J. C., Mercoulia, K., Tomita, T., Easton, M., Li, H. Y., Bulach, D. M., et al. (2016). Prospective whole-genome sequencing enhances national surveillance of Listeria monocytogenes. J. Clin. Microbiol. 54, 333–342. doi:10.1128/JCM.02344-15
Le Guilloux, V., Schmidtke, P., and Tuffery, P. (2009). Fpocket: An open source platform for ligand pocket detection. BMC Bioinforma. 10, 168. doi:10.1186/1471-2105-10-168
Lobel, L., Sigal, N., Borovok, I., Ruppin, E., and Herskovits, A. A. (2012). Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet. 8, e1002887. doi:10.1371/journal.pgen.1002887
Lobo-Silva, J., Cabral, F. J., Amaral, M. S., Miyasato, P. A., de Freitas, R. P., Pereira, A. S. A., et al. (2020). The antischistosomal potential of GSK-J4, an H3K27 demethylase inhibitor: Insights from molecular modeling, transcriptomics and in vitro assays. Parasit. Vectors 13, 140. doi:10.1186/s13071-020-4000-z
Luo, H., Lin, Y., Liu, T., Lai, F.-L., Zhang, C.-T., Gao, F., et al. (2021). DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res. 49, D677–D686. doi:10.1093/nar/gkaa917
Meireles, D., Pombinho, R., Carvalho, F., Sousa, S., and Cabanes, D. (2020). Listeria monocytogenes wall teichoic acid glycosylation promotes surface anchoring of virulence factors, resistance to antimicrobial peptides, and decreased susceptibility to antibiotics. Pathogens 9, 290. doi:10.3390/pathogens9040290
Morvan, A., Moubareck, C., Leclercq, A., Hervé-Bazin, M., Bremont, S., Lecuit, M., et al. (2010). Antimicrobial resistance of Listeria monocytogenes strains isolated from humans in France. Antimicrob. Agents Chemother. 54, 2728–2731. doi:10.1128/AAC.01557-09
Oh, T.-J., Daniel, J., Kim, H.-J., Sirakova, T. D., and Kolattukudy, P. E. (2006). Identification and characterization of Rv3281 as a novel subunit of a biotin-dependent acyl-CoA Carboxylase in Mycobacterium tuberculosis H37Rv. J. Biol. Chem. 281, 3899–3908. doi:10.1074/jbc.M511761200
Olaimat, A. N., Al-Holy, M. A., Shahbaz, H. M., Al-Nabulsi, A. A., Abu Ghoush, M. H., Osaili, T. M., et al. (2018). Emergence of antibiotic resistance in Listeria monocytogenes isolated from food products: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 17, 1277–1292. doi:10.1111/1541-4337.12387
Osorio-Méndez, J. F., and Cevallos, A. M. (2018). Discovery and genetic validation of chemotherapeutic targets for chagas’ disease. Front. Cell. Infect. Microbiol. 8, 439. doi:10.3389/fcimb.2018.00439
Pagliano, P., Arslan, F., and Ascione, T. (2017). Epidemiology and treatment of the commonest form of listeriosis: Meningitis and bacteraemia. Infez. Med. 25, 210–216.
Pesavento, G., Ducci, B., Nieri, D., Comodo, N., and Lo Nostro, A. (2010). Prevalence and antibiotic susceptibility of Listeria spp. isolated from raw meat and retail foods. Food control. 21, 708–713. doi:10.1016/j.foodcont.2009.10.012
NIH HMP Working Group Peterson, J., Garges, S., Giovanni, M., McInnes, P., Wang, L., et al. (2009). The NIH human microbiome Project. Genome Res. 19 (12), 2317–2323. doi:10.1101/gr.096651.109
Poyart-Salmeron, C., Carlier, C., Trieu-Cuot, P., Courtieu, A. L., and Courvalin, P. (1990). Transferable plasmid-mediated antibiotic resistance in Listeria monocytogenes. Lancet 335, 1422–1426. doi:10.1016/0140-6736(90)91447-i
Radusky, L., Defelipe, L. A., Lanzarotti, E., Luque, J., Barril, X., Marti, M. A., et al. (2014). TuberQ: A Mycobacterium tuberculosis protein druggability database. Database 2014, bau035. doi:10.1093/database/bau035
Ramos, P. I. P., Fernández Do Porto, D., Lanzarotti, E., Sosa, E. J., Burguener, G., Pardo, A. M., et al. (2018). An integrative, multi-omics approach towards the prioritization of Klebsiella pneumoniae drug targets. Sci. Rep. 8, 10755. doi:10.1038/s41598-018-28916-7
Şanlıbaba, P., Tezel, B. U., and Çakmak, G. A. (2018). Prevalence and antibiotic resistance of Listeria monocytogenes isolated from ready-to-eat foods in Turkey. J. Food Qual. 2018, 1–9. doi:10.1155/2018/7693782
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M.-A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17, 7–15. doi:10.3201/eid1701.p11101
Schauer, K., Geginat, G., Liang, C., Goebel, W., Dandekar, T., and Fuchs, T. M. (2010). Deciphering the intracellular metabolism of Listeria monocytogenes by mutant screening and modelling. BMC Genomics 11, 573. doi:10.1186/1471-2164-11-573
Serral, F., Castello, F. A., Sosa, E. J., Pardo, A. M., Palumbo, M. C., Modenutti, C., et al. (2021). From genome to drugs: New approaches in antimicrobial discovery. Front. Pharmacol. 12, 647060. doi:10.3389/fphar.2021.647060
Serral, F., Pardo, A. M., Sosa, E., Palomino, M. M., Nicolás, M. F., Turjanski, A. G., et al. (2022). Pathway driven target selection in Klebsiella pneumoniae: Insights into carbapenem exposure. Front. Cell. Infect. Microbiol. 12, 773405. doi:10.3389/fcimb.2022.773405
Shang, R., Liang, J., Yi, Y., Liu, Y., and Wang, J. (2015). Review of platensimycin and platencin: Inhibitors of β-Ketoacyl-acyl carrier protein (ACP) synthase III (FabH). Molecules 20, 16127–16141. doi:10.3390/molecules200916127
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi:10.1101/gr.1239303
Shen, Y., Boulos, S., Sumrall, E., Gerber, B., Julian-Rodero, A., Eugster, M. R., et al. (2017). Structural and functional diversity in Listeria cell wall teichoic acids. J. Biol. Chem. 292, 17832–17844. doi:10.1074/jbc.m117.813964
Sosa, E., Burguener, G., Pardo, A., Marti, M., Turjanski, A., and Do Porto, D. A. F. (2018). Target-pathogen: A structural bioinformatic approach to prioritize drug targets in pathogens. Int. J. Infect. Dis. 73, 84. doi:10.1016/j.ijid.2018.04.3616
Sosa, E. J., Burguener, G., Lanzarotti, E., Defelipe, L., Radusky, L., Pardo, A. M., et al. (2018). Target-pathogen: A structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic Acids Res. 46, D413–D418. doi:10.1093/nar/gkx1015
Stritzker, J., Janda, J., Schoen, C., Taupp, M., Pilgrim, S., Gentschev, I., et al. (2004). Growth, virulence, and immunogenicity of Listeria monocytogenes aro mutants. Infect. Immun. 72, 5622–5629. doi:10.1128/iai.72.10.5622-5629.2004
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B., and Wu, C. H.UniProt Consortium (2015). UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. doi:10.1093/bioinformatics/btu739
Temple, M. E., and Nahata, M. C. (2000). Treatment of listeriosis. Ann. Pharmacother. 34, 656–661. doi:10.1345/aph.19315
Toledo-Arana, A., Dussurget, O., Nikitas, G., Sesto, N., Guet-Revillet, H., Balestrino, D., et al. (2009). The Listeria transcriptional landscape from saprophytism to virulence. Nature 459, 950–956. doi:10.1038/nature08080
Wang, J., Kodali, S., Lee, S. H., Galgoci, A., Painter, R., Dorso, K., et al. (2007). Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. U. S. A. 104, 7612–7616. doi:10.1073/pnas.0700746104
Wang, J., Soisson, S. M., Young, K., Shoop, W., Kodali, S., Galgoci, A., et al. (2006). Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441, 358–361. doi:10.1038/nature04784
Webb, B., and Sali, A. (2016). Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 86, 1–2. doi:10.1002/cpps.20
Wellington, S., Nag, P. P., Michalska, K., Johnston, S. E., Jedrzejczak, R. P., Kaushik, V. K., et al. (2017). A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 13, 943–950. doi:10.1038/nchembio.2420
Keywords: drug discovery, drug target, metabolic reconstructions, target prioritization, Listeria monocytogenes
Citation: Palumbo M, Sosa E, Castello F, Schottlender G, Serral F, Turjanski A, Palomino MM and Do Porto DF (2022) Integrating diverse layers of omic data to identify novel drug targets in Listeria monocytogenes. Front. Drug. Discov. 2:969415. doi: 10.3389/fddsv.2022.969415
Received: 15 June 2022; Accepted: 19 August 2022;
Published: 23 September 2022.
Edited by:
Didier Cabanes, University of Porto, PortugalReviewed by:
Carla Sabia, University of Modena and Reggio Emilia, ItalyMonika Kurpas, Medical University of Gdansk, Poland
Copyright © 2022 Palumbo, Sosa, Castello, Schottlender, Serral, Turjanski, Palomino and Do Porto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Mercedes Palomino, bXBhbG9taW5vQHFiLmZjZW4udWJhLmFy; Darío Fernández Do Porto, ZGFyaW9mZEBnbWFpbC5jb20=