 Vanessa Machado
Vanessa Machado Rui Carvalho1
Rui Carvalho1 João Botelho
João Botelho- 1Evidence-Based Hub, Clinical Research Unit, Centro de Investigação Interdisciplinar Egas Moniz, Instituto Universitário Egas Moniz, Caparica, Portugal
- 2Periodontology Department, Clinical Research Unit, Centro de Investigação Interdisciplinar Egas Moniz (CiiEM), Egas Moniz, CRL, Caparica, Portugal
The turnover of intracellular proteins is a highly selective and regulated process. This process is responsible for avoiding injury and irreparable breakdown of cellular constituents. Its impairment disrupts cellular stability, integrity, and homeostasis. The ubiquitin-proteasome system (UPS) is responsible for this programmed degradation of most intracellular proteins. This process involves a cascade of enzymes that involves the ubiquitin conjugation to a target substrate protein, its recognition and degradation by the proteasome. The turn-over of intracellular proteins is a non-stop ubiquitous process that regulates a series of mechanisms, for instance transcription, translation, endocytosis. In addition, proteasome act by releasing peptides that may serve to other purposes, such as antigen presentation in immune actions and enzymatic flagging toward biosynthesis and gluconeogenesis. The role of the UPS impairment in periodontal diseases is gaining growing. This acquaintance might contribute to the development of novel therapeutic applications. Thus, this review focuses on the latest progresses on the role of the UPS and its signaling pathways in Periodontal Medicine. Furthermore, we discuss the potential of UPS-based drugs development to be used in periodontal disease therapy.
Introduction
The ubiquitin-proteasome system (UPS) is a vital intracellular protein degradation network in eukaryotes (1–4) (Figure 1). This highly regulated, selective and programmed protein degradation system is involved in proteolytic and non-proteolytic functions (such as proteasomal degradation of proteins, receptor internalization and downregulation, multiprotein complexes assembly, intracellular transportation, inflammatory signaling, autophagy, DNA repair and regulation of enzymatic activity) (5–8). Therefore, this recycling process prevents damage or surplus proteins from building up and becoming toxic to the cell, and it is estimated to degrade more than 80% of normal and abnormal intracellular proteins (8).
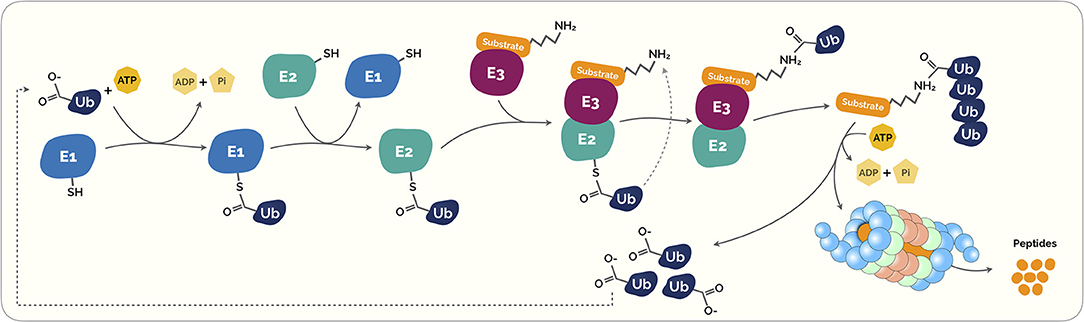
Figure 1. The Ubiquitin-Proteasome System (UPS). Ubiquitination is a highly regulated control system that post-translationally modifies marked proteins for degradation. Initially, the ubiquitin (Ub) is covalently coupled to lysine residues on target protein carried out by activating (E1). This process requires energy in the form of adenosine triphosphate (ATP). The activated Ub is then transferred through transfer ubiquitination reaction from E1 to a second enzyme called E2 ubiquitin-conjugating enzyme. This enzyme acts as an escort for Ub to its next destination, the E3 ubiquitin ligase enzyme. This last acts as a platform in which the target protein substrate and the active E2 Ub complex can meet and interact. Subsequently, both proteins and the Ub are loaded onto the E3 ligase enzyme, the Ub is transferred to the target protein substrate. This process can be repeated several times to create a polyubiquitin chain on the protein. This polyubiquitin creates a clear signal to the proteasome. Proteasomes are shaped like barrels that remove polyubiquitin and open at one end to allow target protein to enter and at the other end to allow broken down pieces of protein to exit (5).
The consequences of UPS impairment have been of great interest. Currently, UPS dysfunction has been implicated in several diseases, for instance Alzheimer's disease, Amyotrophic Lateral Sclerosis, cancer, diabetes mellitus, renal disease, inflammatory bowel disease or rheumatic conditions (9–15). Comprehensively, the impact of UPS in these illnesses relies on a clearance deficit toward misfolded proteins (16, 17), and as a consequence, these result in intracellular protein aggregation, cytotoxicity, and cell death.
The involvement of the UPS disability in the Periodontal Medicine field has been a topic of growing interest for their role in the periodontal diseases and their prospective application as therapeutic (18). This review aimed to focus on the latest advances on the role of UPS in the field of Periodontal Medicine. Furthermore, we discuss the potential of UPS-based drugs development to be used in periodontal disease therapy.
The Ubiquitin-Proteasome System
Ubiquitins
At a cellular level, the crucial plethora of physiological processes, including cell survival and differentiation and innate and adaptive immunity, is carried out by the ubiquitin conjugation system (5, 19–21). As described in Figure 1, ubiquitins are small peptides that “flag” a target protein toward its destruction by the proteasome through polyubiquitination, a process that results from a cascade of enzymes. These small peptides are composed by a 76-amino acid protein that can covalently link to lysine or N-terminal residues, through a process known as ubiquitylation, of the target proteins (22).
This intricate process starts with the formation of a thioester bond of a ubiquitin-activating enzyme (E1) with ubiquitin via an ATP-dependent formation process. Then, ubiquitin is transferred to ubiquitin conjugases (E2) and, then, it is conjugated with a ubiquitin ligase (E3), that provides substrate recognition and catalyze the covalent attachment of ubiquitin to the target substrate via an isopeptide bond. Subsequently, both proteins and the ubiquitin are loaded onto the E3 ligase enzyme, the ubiquitin is transferred to the target protein substrate. This process takes place repeatedly causing the polyubiquitination of the substrate leading to its signal for degradation by the proteasome complex (Figure 1) (23, 24).
Recent studies unraveled the human genome contains two E1 enzymes, more than 40 E2 enzymes, and more than 600 E3 ligase, thus the potential of a mutation is quite high in one of those proteins (21, 25). This structural hierarchy, that is, a limited number of E2s and a higher number of E3s foment specific interactions.
Proteasome
Following polyubiquitination, signaled proteins are degraded by a multiprotein complex called the 26S proteasome (24). The 26S proteasome is a multicatalytic enzim complex expressed in the nucleus and cytoplasm of all eukariotic cells (24), and its structure consists of a 20S core particle that is capped by one or two 19S regulatory particles. The 20S core is a cylinder with four rings, composed with 14 different but related subunits, with low proteolytic activity in its cellular milieu (24). The two external rings of the 20S core, each containing seven different α subunits (α rings), form a narrow channel where denatured proteins may pass only, while the two internal heptameric rings each containing seven different β subunits (β rings), constitute the catalytic chamber that degrades proteins (26). The α-subunits bind to the 19S regulatory proteins, regulating the selectivity and entry of specific substrates into the polymeric chain (13). Target proteins are then degraded by the core particle in a progressive fashion, generating peptides with about 3–25 amino acids in length (27). Each 19S unit can bind polyubiquitin chains that are signalizing proteins, forwarding the protein into the proteolytic core where protein degradation takes place (28). The 19S regulatory proteins is a large complex composed by two heteromeric subcomplexes term “base” and “lid”, the “base” recognizes ubiquitin-tagged proteins, a step that is followed by the unfolding and translocation of the substrates through the proteolytic chamber of the 20S proteasome, the “lid” main function is the deubiquitylation of the entering substrates (29).
Dysregulation of the Ubiquitin-Proteasome System
To ensure homeostasis, cells evolved to adapt to new environmental conditions and to avoid unnecessary damage to survival (30). The dysregulation of the UPS is consistently interplaying with several conditions. From a pathological point-of-view, dysfunctional UPS is associated with Alzheimer's disease, Parkinson's disease, Amyotrophic Lateral Sclerosis, neuronal degeneration, remodeling and regeneration after spinal cord injury, Transmissible Spongiform Encephalopathies, and Huntington's disease (31), Renal Diseases (12), and others. The mechanism through which the UPS impact neurological or renal disorders is related to the impairment in the clearance of misfolded proteins that cause an abnormal aggregation of intracellular protein and, as a consequence, cytotoxicity and cell death (16, 17).
The Ubiquitin-Proteasome System in Periodontal Disease
Periodontal Diseases
Periodontal diseases are a group of inflammatory non-communicable conditions triggered by a dysbiotic plaque that damages the periodontium (32–34). Periodontitis is an endemic disease and a worldwide public health problem (35, 36), and its symptoms start with gum inflammation (gingivitis) to an exacerbated and unrestrained inflammatory reaction from the host immune system (37–39). The destruction of the periodontium permits bacterial penetration through the ulcerated epithelium and, then, they spread through the circulatory system.
The plausible mechanism through which periodontitis impacts systemic homeostasis might be the result of an inflammatory cascade triggered by subgingival periodontopathogenic bacteria. Initially a local acute inflammatory response, subsequently evolves to a systemic inflammation reaction, consistently associated with vascular dysfunction (40, 41) and systemic inflammation (42). Consequently, the infectious nature of periodontitis promote bacterial invasion trough the periodontal tissues via the ulcerated epithelium, and immune and inflammatory pathways are triggered toward this periodontal injury (43–45).
For the past decades, periodontitis has been consistently linked with several systemic diseases, such as diabetes mellitus (46), cardiovascular diseases (47, 48), adverse pregnancy outcomes (49), rheumatic diseases (50), stress (51), male and female infertility-related conditions (52, 53), Alzheimer's Disease (54), inflammatory bowel disease (55), metabolic syndrome or even some types of cancers (56). In a patient-centered view, periodontitis has a negative impact on oral health-related quality of life (57), though periodontal treatment through non-surgical procedures showed to be effective to its recovery (58).
Several of these pathologies that have already been associated with periodontitis have also been thoroughly studied for the role of the UPS on its pathogenesis, for instance Alzheimer's Disease or Renal Disease (12, 31).
Mindful of the strong link of periodontitis with systemic health, the role of genetics and epigenetics in Periodontal Medicine is still in its early stages. The involvement of inflammasome components in periodontal health and subtypes of disease as well the recent genome-wide association studies unraveled conceivable routes of association (59). Recent researches in transcriptomic, proteomic, and metabolomic have deepened the molecular pathogenesis of periodontitis highlighting new host-microbe interactions, processes, and pathways that might improve diagnostic and treatment strategies for periodontitis (60, 61). Further, the role of UPS in periodontitis has undergone important advances increasing our knowledge of molecular pathways and potential new drugs. These lines of evidence are contributing to move forward a tailored and personalized treatment for each patient according to its particular characteristics (62). However, the notion of the role of UPS is slightly different from expected since in periodontitis the UPS is exacerbated and promotes destruction of proteins possibly resulting in the destruction of the periodontium. Therefore, the rationale for an action has been emphasized in blocking these pathways with blocking therapeutics.
Periodontopathogenic Bacteria
The current postulated scheme for periodontitis onset and progression relies as symbiosis between a dysbiotic plaque and host immune system. This concept is based on microbial succession followed by reciprocal host–bacterial interaction (63), where this microbial progression triggers gingival inflammation, and the increase of inflammation results in increased growth of colonizing species (64, 65). Then, a comprehensive and thorough DNA analysis by Socransky et al. (66) found associations between all pairs of species resulted in similarity matrices subjected to cluster analysis. These analyses supported the existence of distinct complexes of microorganisms in subgingival bio-films, now named complexes (63, 67–70). These works were fundamental and valuable in that they confirmed the complexes described by the pairwise cluster analyses and proposed the nature of the relationships of the complexes (communities) to each other (63). The establishment of such complexes provided a framework for describing and understanding the subgingival ecosystem, where a red complex stands out (Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola), since this group categorization is based on their association with severe forms of periodontal disease.
Remarkably, several lines of evidence have emerged novel considerations about subgingival microbiota of the aging mouth. The complex organization of the oral microbiota, poses enduring difficulties due to a huge environment diversity and a unique ability to promote rapid phenotypic alterations that occur within the environment where they stand (63, 67, 68). Following this rationale, a multicentre research was carried out using data from two centers, the Center for Clinical Research at Guarulhos University (São Paulo, Brazil) and The Forsyth Institute (Cambridge, United States of America [USA]) (71). This data comprised data from patient within periods longer than 10 years, and this study aimed to investigate the evolution of the periodontal microbiota throughout life using DNA counts of about forty bacterial species. Hence, subgingival microbiota composition might not be age-related and, consequently, age is not a determinant of the oral microbiome (71). Individuals with severe periodontitis had higher levels of Tannerella forsythia, Porphyromonas gingivalis and Treponema denticola, pathogens that compose the red complex (71). Besides, a previous research also evidenced this apparent microbial community stability across lifespan, however human microbiota can rapidly and markedly be changed due to host lifestyle, for instance traveling to developing countries or enteric infections (72). Interestingly, patients with persistent aggressive periodontitis have more dysbiotic subgingival biofilm than patients that have been treated with clinical success (73). Nevertheless, even well-maintained periodontitis cases present more dysbiotic microbial community than healthy individuals, with described higher diversity, disordered structure, pathogenic microbiota, and more host-destructive metabolism pathways (74). For this reason, the bacterial load is theorized to happen throughout life and its associations and biological interactions may perpetuate.
Studies on UPS in Human Periodontal and Oral Cell Lineages
In 2002, Krisanaprakornkit et al. (75) investigated the role of NF-kB in human β-defensins (hBDs) mRNA of human gingival epithelial cells (hGECs) after activation through Fusobacterium nucleatum cell wall extract (FnCW). As a result, the authors hypothesized that MG132 likely promotes the accumulation of ubiquitinated proteins, resulting in superinduction of hBD-2 mRNA. Subsequently, Yin et al. (76) in 2007 investigated how hGECs gene expression was altered on the presence of Fusobacterium nucleatum (through F. nucleatum Cell Wall (FnCW) extracts) or hBDs (mimicking the secretion of hBDs inducted by commensal flora). Both FnCW and hBD2 were found to induce significant changes in the expression of genes associated with immune and defense responses (76). One particular finding was that FnCW extracts suppressed the UPS. Such suppression was seen as likely inhibiting inflammatory markers, similarly to previous reports in intestinal epithelial cells in response to non-pathogenic Salmonella spp. (77) and Lactobacillus casei (78).
In 2010, Ghosh et al. (79) investigated the extracellular matrix in signaling for tissue remodeling and cell survival in inflammatory diseases such as periodontitis. In human primary periodontal ligament cell culture isolated from extracted tooth they found that proapoptotic fibronectin matrix induces ubiquitination and degradation of p53 in the proteasome. Further, pre-treatment using proteasomal inhibitors MG132 and lactacystin rescued the cells from apoptosis (79).
The role of nuclear factor (NF)-kB gene expression in the initiation of inflammation signaling and subsequent release of proinflammatory cytokines has led to the development of inhibitors of this pathway for the treatment of chronic inflammation (80). The zinc finger protein A20 is encoded by an immediate early response gene and inhibits activation of NF-kB via inflammatory cytokines receptors (80). The induction of A20 occur by different stimulus such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and bacterial lipopolysaccharides (LPS) in a variety of cell types (81). The NF-kB signaling pathway is regulated by ubiquitination, and although it was studied as a regulator of numerous chronic inflammatory conditions, literature is still scarce on its function in the oral mucosa (82). In 2015, Hong et al. assessed the A20 expression in patients with periodontitis, the effects of A20 overexpression on inflammatory response and on osteoblastic differentiation in LPS- and nicotine-stimulated human periodontal ligament cells (hPDLCs) (83). They found that pre-treatment with A20 overexpression markedly had anti-inflammatory effects and blocked osteoclastic differentiation in LPS- and nicotine-stimulated hPDLCs models. More specifically, A20 overexpression reduced LPS- and nicotine-induced production of prostaglandin E2, cyclooxygenase-2 and proinflammatory cytokines, inhibited the number and size of tartrate-resistant acid phosphatase-stained osteoclasts, and downregulated osteoclast-specific gene expression (83). Later, Li et al. (84) reported alike results regarding the A20 inflammatory response regulation through its effect on NF-kB signaling and cytokine production using a combination of in vivo and ex vivo disease models. Further, A20 depletion via gene editing in human macrophage-like cells (THP-1) significantly increased cytokine secretion (84). Still, in mice infected with Porphyromonas gingivalis, bone marrow–derived macrophages from A20-deficient showed increased NF-kB activity and cytokine production compared with cells isolated from A20-competent mice (84). Recently, Li et al. (85) in 2020 reported a specific role of an ubiquitin editing molecule in gingival keratinocyte inflammatory response, indicated that sufficient A20 levels is a major determinant to control apoptosis in response to microbial and inflammatory injury. Also, the A20 expression in response to oral bacterial depends among different cells and, therefore, it is essential studying the cell-type specific A20 responses and their effect on disease phenotype to develop targeted therapies (85). Another mechanism that could explain the ability of transforming growth factor β (TGFβ) to inhibit Toll-like receptor (TLR)-NFκB signaling is protein arginine methyltransferase 1 (PRMT1)–induced Smad6 methylation. This Smad6 is expressed in the gingival epithelium and the methylation signaling promotes tissue homeostasis by limiting inflammation (86).
Others, the importance of hypoxic entourage during the chronic periodontal inflammation showed that oxygen concentration is a crucial regulating factor of physiological processes and pathogenesis in periodontal disease (87, 88). Regarding the proteomic profile, 220 differentially expressed proteins under hypoxia and three main processes were identified in hypoxia-treated hPDLCs, namely energy metabolism, autophagy and adaptive response to stimuli such as adhesion and inflammation (87). Further, hPDLCs obtained from premolars extracted under 2% O2 exhibited decreased A20 expression and increased receptor activator of nuclear factor-kappa B (RANKL)/osteoprotegerin (OPG) ratio. These findings suggest that A20 may be a key gene target during bone loss in periodontitis (88).
A study investigating Apolipoprotein mice deficient in nucleotide binding oligomerization domain-containing protein 2 (NOD2) and subjected to an oral gavage of P. gingivalis reported elevated serum inflammatory cytokines, cholesterol, alveolar bone loss, and atherosclerosis (89). This research proposed a pivotal role of a Muramyl DiPeptide (MDP)/NOD2 signaling pathways in P. gingivalis-induced periodontitis/atherosclerosis. Later, Maekawa et al. (90) in 2014 unveiled a possible model of P. gingivalis subversion of neutrophils resulting in dysbiotic inflammation. In this proposed model in mice, P. gingivalis coactivates Toll-like receptor-2 (TLR2) and C5a receptor (C5aR) in neutrophils, causing ubiquitination and proteasomal degradation of MyD88, thereby inhibiting a host-protective antimicrobial response (90). Accordingly, this mechanism could contribute to dysbiotic diseases through microbial communities persistence. Additionally, Cai et al. (91) in 2016 have further explored a new signaling pathway for TNF-α induction activated after exposing macrophages to both P. gingivalis and muramyl dipeptide (MDP)-high, where they found that MDP-high affected the ubiquitin editing-enzyme A20 and activator protein 1. On the other hand, Bengtsson et al. (92) in 2017 showed that low concentrations of bacteriocin PLNC8 αβ have a potent antimicrobial action on P. gingivalis and stimulate cell proliferation. Thus, PLNC8 αβ antagonizes the pathogenic activity of P. gingivalis in cell culture model, prevents cytotoxicity mediated by this bacteria, increases the expression of proteins associated with the cytoskeleton rearrangement, increases the vesicle transport and restores the levels of inflammatory mediators.
Recently, the outcome of hPDLCs exposure to hydrogen peroxide (H2O2) was studied (93). The authors demonstrated that 400 and 500 μM concentration of H2O2 decreases Periodontal Ligament Stem Cells (PDLSCs) viability in 40% to 50% with the, respectively, with a downregulation of Pellino-1 mRNA. The Pellino proteins family are a defining feature of the RING class of E3 ubiquitin ligase. Further, in this study the author found that overexpression of Pellino-1 inhibited H2O2-induced cellular apoptosis through the activation of the NF-κB signaling pathway. Therefore, Pellino-1 might be relevant for cell survival in the presence of oxidative elements (93).
Proteasome Inhibitors in the Treatment of Periodontal Disease
A number of drugs that target the proteasome have been developed in the past decades. Presently, only Bortezomib (BTZ) has been investigated within a potential application in the diseased periodontium. BTZ was the first proteasome inhibitor approved by the US Food and Drugs Administration for anti-multiple myeloma (94). Ever since proteasome inhibitors have been mainly used in the management of patients with multiple myeloma and mantle-cell lymphoma (95), and emerged a potential therapeutic in the treatment of periodontitis (Figure 2).
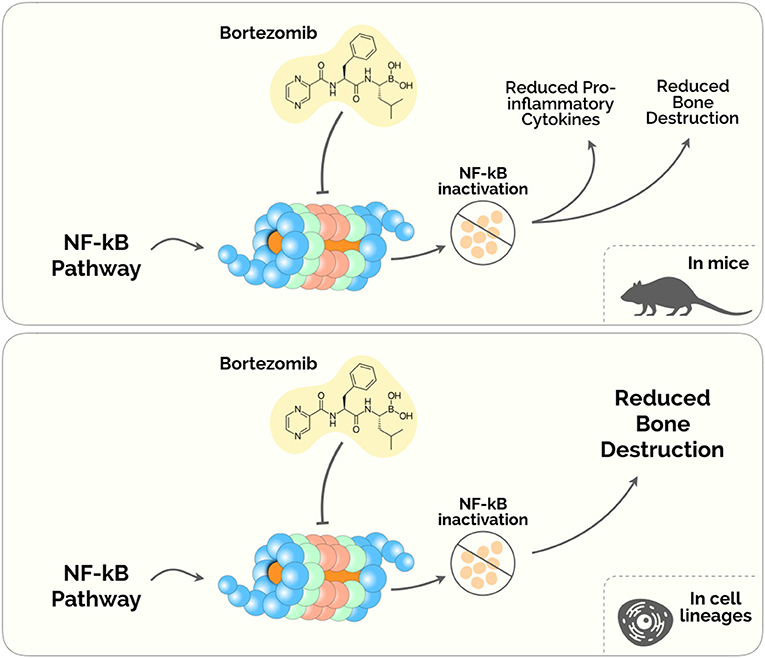
Figure 2. Proteasome Inhibitors in the treatment of Periodontal disease. The diagram depicts the plausible biological mechanisms unveiled in mice (upper) and human cell lineages (lower).
Despite its anti-tumoral effects, BTZ has been shown to ameliorate the inflammatory milieu and to regulate bone remodeling pathways (96, 97). Under this rationale, Kitagaki et al. (98) tested whether BTZ could induce the differentiation of hPDLCs into hard-tissue-forming cells. This study was the first to demonstrate a cytodifferentiation and mineralization roles of BTZ on hPDLCs, by enhancing the accumulation of β-catenin (inside the cytosol and the nucleus) and increasing the expression of Bmp-2,−4 and−6 mRNAs (98). A subsequent study from the same group found that safe concentrations of BTZ (between 0.25 and 1 nM) inhibited proinflammatory cytokines releasing in LPS-stimulated hPDLCs via NF-κB, p38/extracellular- signal-regulated kinase (ERK5), and mitogen-activated protein kinase (MAPK)/activator protein-1 (AP-1) pathways (99). Also, this experiment showed that BTZ was able to suppress the expression of TNF-α, IL-1β, IL-6, and IL-8, reduced the ratio of RANKL/OPG, and prevented alveolar bone absorption in a lipopolysaccharide and ligature-induced periodontitis rat model (99).
Also, Kim et al. (100) showed that BTZ directly inhibited the receptor activator NF-κB ligand (RANKL)– and LPS-dependent osteoclast differentiation in a mouse model of periodontitis. BTZ showed to prevent alveolar bone erosion induced by P. gingivalis-LPS (100). These results corroborated the primary results on the novel applications of BTZ beyond its use as an antimyeloma agent. Nevertheless, BTZ presents noteworthy adverse effects, namely fatigue, nausea, diarrhea, thrombocytopenia, peripheral edema and dyspnea (101, 102). Therefore, although some evidence has been describing BTZ as a possible therapy, further studies are imperative to weigh the advantage and disadvantage of prescribing this drug in the Periodontal Medicine setting.
Very recently, the potential of nanosized Alumina (Al) particles and low doses of BTZ were investigated for treating titanium particle-induced inflammatory reaction in human osteosarcoma cells and in mouse calvarial osteolysis model (103). This study concluded that Al particles and BTZ attenuated the expression of inflammatory cytokines (such as IL-1β, IL-6, TNF-α).
Discussion and Future Trends in Periodontal Research
The understanding of the UPS has escalated for the past decades (9–15). Interestingly, periodontal diseases have been consistently linked to these conditions both in associative and mechanistic relationships (46–48, 50, 54, 104, 105). Ergo, we anticipate that the number of pre-clinical and clinical studies reporting the association of periodontal disease with these diseases through UPS will increase. It is never too much to stress the importance of bioinformatics in the study of these associations since strategies like protein-protein analysis, arrays studies, and other methodologies might strength possible biological routes of communication.
The way in which periodontal disease can interfere with systemic diseases has been based on the existence of chronic subclinical inflammation, bacterial dispersion in the bloodstream and metabolic influence. However, the confirmation of a systemic interference path through the UPS can open an unprecedented area of investigation in Periodontal Medicine.
Henceforth, it will not be surprising to see the first randomized clinical trials taking place in the coming years, namely the use of BTZ as an adjunct in non-surgical and periodontal surgical treatment. Additionally, it is expected the discovery of novel ubiquitin-regulatory enzymes, factors and pathways that will support the development of new selective therapeutic compounds.
Conclusions
The UPS might be involved in the regulation of physiological and pathophysiological processes in periodontium. Despite increasing knowledge regarding the process of the highly complex UPS in periodontal disease, the unknown by far exceeds the currently known. Nevertheless, the recent advancements on the UPS in periodontal disease have unveiled potentially new therapeutics to prevent bone destruction. Future research will elucidate deeper the mechanisms of ubiquitination-associated periodontal disease and increase the likelihood of new treatment in Periodontal Medicine.
Author Contributions
VM and JB conceived and wrote the manuscript. All Authors contributed to the article and approved the submitted version.
Funding
This work was financed by national funds through the FCT–Foundation for Science and Technology, I.P., under the project UIDB/04585/2020.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. (2005) 12:1191–7. doi: 10.1038/sj.cdd4401702
2. Ciechanover A, and Iwai K. The ubiquitin system: from basic mechanisms to the patient bed. IUBMB Life. (2004) 56:193–201. doi: 10.1080/1521654042000223616
3. Rose I. Ubiquitin at fox chase. Cell Death Differ. (2005) 12:1198–201. doi: 10.1038/sj.cdd4401710
4. Ciechanover A. Intracellular protein degradation: from a Vague Idea, through the lysosome and the ubiquitin-proteasome system, and onto human diseases and drug targeting (Nobel Lecture). Angew Chemie-Int Ed. (2005) 44:5944–67. doi: 10.1002/anie200501428
5. Hershko A, and Ciechanover A. The ubiquitin system. Annu Rev Biochem. (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1425
6. Grabbe C, Husnjak K, and Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. (2011) 12:295–307. doi: 10.1038/nrm3099
7. Reiter KH, and Klevit RE. Characterization of RING-between-RING E3 ubiquitin transfer mechanisms. Methods Mol Biol. (2018) 1844:3–17. doi: 10.1007/978-1-4939-8706-1_1
8. Wang J, and Maldonado MA. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. (2006) 3:255–61.
9. Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE, et al. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis: morphology, distribution, and specificity. Brain. (1991) 114:775–88. doi: 10.1093/brain/114.2775
10. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. (2006) 314:130–3. doi: 10.1126/science1134108
11. Versari D, Herrmann J, Gössl M, Mannheim D, Sattler K, Meyer FB, et al. Dysregulation of the ubiquitin-proteasome system in human carotid atherosclerosis. Arterioscler Thromb Vasc Biol. (2006) 26:2132–9. doi: 10.1161/01.ATV.0000232501.0857673
12. Meyer-Schwesinger C. The ubiquitin–proteasome system in kidney physiology and disease. Nat Rev Nephrol. (2019) 15:393–411. doi: 10.1038/s41581-019-0148-1
13. Bustamante HA, González AE, Cerda-Troncoso C, Shaughnessy R, Otth C, Soza A, et al. Interplay between the autophagy-lysosomal pathway and the ubiquitin-proteasome system: a target for therapeutic development in alzheimer's disease. Front Cell Neurosci. (2018) 12:1–18. doi: 10.3389/fncel.201800126
14. Cao J, Zhong MB, Toro CA, Zhang L, and Cai D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer's disease pathogenesis. Neurosci Lett. (2019) 703:68–78. doi: 10.1016/j.neulet.2019.03016
15. Hong L, Huang HC, and Jiang ZF. Relationship between amyloid-beta and the ubiquitin-proteasome system in alzheimer's disease. Neurol Res. (2014) 36:276–82. doi: 10.1179/1743132813Y0000000288
16. Demuro A, Mina E, Kayed R, Milton SC, Parker I, and Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. (2005) 280:17294–300. doi: 10.1074/jbcM500997200
17. Schwartz AL, and Ciechanover A. Targeting Proteins for Destruction by the Ubiquitin System: Implications for Human Pathobiology. Annu Rev Pharmacol Toxicol. (2009) 49:73–96. doi: 10.1146/annurev.pharmtox.051208165340
18. Tsuchida S, Satoh M, Takiwaki M, and Nomura F. Ubiquitination in periodontal disease: a review. Int J Mol Sci. (2017) 18:1–15. doi: 10.3390/ijms18071476
19. Özkaynak E, Finley D, and Varshavsky A. The yeast ubiquitin gene: head-to-tail repeats encoding a polyubiquitin precursor protein. Nature. (1984) 312:663–6. doi: 10.1038/312663a0
20. Rosenstein ED, Greenwald RA, Kushner LJ, and Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. (2004) 28:311–8. doi: 10.1007/s10753-004-6641-z
21. Popovic D, Vucic D, and Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. (2014) 20:1242–53. doi: 10.1038/nm3739
22. Komander D, and Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
23. Bence NF, Sampat RM, and Kpito RR. Impairment of the ubiquitin-proeasome system by protein aggregation. Science. (2001) 292:1552–5. doi: 10.1126/science.292.55211552
24. Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev. (2003) 29:3–9. doi: 10.1016/S0305-7372(03)00081-1
25. Swatek KN, and Komander D. Ubiquitin modifications. Cell Res. (2016) 26:399–422. doi: 10.1038/cr.201639
26. Groll M, Koguchi Y, Huber R, and Kohno J. Crystal structure of the 20 S proteasome:TMC-95A complex: a non-covalent proteasome inhibitor. J Mol Biol. (2001) 311:543–8. doi: 10.1006/jmbi.20014869
27. Nussbaum AK, Dick TP, Keilholz W, Schirle M, Stevanović S, Dietz K, et al. Cleavage motifs of the yeast 20S proteasome β subunits deduced from digests of enolase. Proc Natl Acad Sci USA. (1998) 95:12504–9. doi: 10.1073/pnas.95.2112504
28. Zwickl P, Seemüller E, Kapelari B, and Baumeister W. The proteasome: a supramolecular assembly designed for controlled proteolysis. Adv Protein Chem. (2001) 59:187–222. doi: 10.1016/S0065-3233(01)59006-3
29. Meyer U, Weingart D, Deng MC, Scheld HH, and Joos U. Heart transplants–assessment of dental procedures. Clin Oral Investig. (1999) 3:79–83. doi: 10.1007/s007840050082
30. Pohl C, and Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. (2019) 366:818–22. doi: 10.1126/scienceaax3769
31. Gong B, Radulovic M, Figueiredo-Pereira ME, and Cardozo C. The ubiquitin-proteasome system: potential therapeutic targets for alzheimer's disease and spinal cord injury. Front Mol Neurosci. (2016) 9:1–16. doi: 10.3389/fnmol.201600004
32. Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri - implant diseases and conditions. J Clin Periodontol. (2018) 45:162–70. doi: 10.1111/jcpe12946
33. Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. (2010) 8:481–90. doi: 10.1038/nrmicro2337
34. Curtis MA, Diaz PI, and Van Dyke TE. The role of the microbiota in periodontal disease. Periodontol 2000. (2020) 83:14–25. doi: 10.1111/prd12296
35. Kassebaum NJ., Bernabé E, Dahiya M, Bhandari B, Murray CJL, Marcenes W. Global burden of severe periodontitis in 1990-2010: a systematic review and meta-regression. J Dent Res. (2014) 93:1045–53. doi: 10.1177/0022034514552491
36. Peres MA, Macpherson LMD, Weyant RJ, Daly B, Venturelli R, Mathur MR, et al. Oral diseases: a global public health challenge. Lancet. (2019) 394:249–60. doi: 10.1016/S0140-6736(19)31146-8
37. Ebersole JL, Dawson D, Emecen-Huja P, Nagarajan R, Howard K, Grady ME, et al. The periodontal war: microbes and immunity. Periodontol 2000. (2017) 75:52–115. doi: 10.1111/prd12222
38. Ebersole JL, Schuster JL, Stevens J, Dawson D, Kryscio RJ, Lin Y, et al. Patterns of salivary analytes provide diagnostic capacity for distinguishing chronic adult periodontitis from health. J Clin Immunol. (2013) 33:271–9. doi: 10.1007/s10875-012-9771-3
39. Zhang S, Yu N, and Arce RM. Periodontal inflammation: integrating genes and dysbiosis. Periodontol 2000. (2020) 82:129–42. doi: 10.1111/prd12267
40. Tonetti MS, D'Aiuto F, and Nibali L. Treatment of periodontitis and endothelial function. N Engl J Med. (2007) 356:911–20. doi: 10.1056/NEJMoa063186
41. Orlandi M, Suvan J, Petrie A, Donos N, Masi S, Hingorani A, et al. Association between periodontal disease and its treatment, flow-mediated dilatation and carotid intima-media thickness: a systematic review and meta-analysis. Atherosclerosis. (2014) 236:39–46. doi: 10.1016/j.atherosclerosis.2014.06002
42. D'Aiuto F, Nibali L, Parkar M, Patel K, Suvan J, and Donos N. Oxidative stress, systemic inflammation, and severe periodontitis. J Dent Res. (2010) 89:1241–6. doi: 10.1177/0022034510375830
43. Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000. (2010) 53:124–37. doi: 10.1111/j.1600-0757.2009.00327x
44. Aboodi GM, Goldberg MB, and Glogauer M. Refractory periodontitis population characterized by a hyperactive oral neutrophil phenotype. J Periodontol. (2011) 82:726–33. doi: 10.1902/jop.2010100508
45. Hirschfeld J. Dynamic interactions of neutrophils and biofilms. J Oral Microbiol. (2014) 6:1–10. doi: 10.3402/jom.v626102
46. Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, et al. Periodontitis and diabetes: a two-way relationship. Diabetologia. (2012) 55:21–31. doi: 10.1007/s00125-011-2342-y
47. Muñoz Aguilera E, Suvan J, Buti J, Czesnikiewicz-Guzik M, Barbosa Ribeiro A, Orlandi M, et al. Periodontitis is associated with hypertension: a systematic review and meta-analysis. Cardiovasc Res. (2020) 116:28–39. doi: 10.1093/cvr/cvz201
48. Sanz M, Marco del Castillo A, Jepsen S, Gonzalez-Juanatey JR, D'Aiuto F, Bouchard P, et al. Periodontitis and cardiovascular diseases: consensus report. J Clin Periodontol. (2020) 47:268–88. doi: 10.1111/jcpe13189
49. Manrique-Corredor EJ, Orozco-Beltran D, Lopez-Pineda A, Quesada JA, Gil-Guillen VF, and Carratala-Munuera C. Maternal periodontitis and preterm birth: systematic review and meta-analysis. Community Dent Oral Epidemiol. (2019) 47:243–51. doi: 10.1111/cdoe12450
50. Hussain SB, Botelho J, Machado V, Zehra SA, Mendes JJ, Ciurtin C, et al. Is there a bidirectional association between rheumatoid arthritis and periodontitis? A systematic review and meta-analysis. Semin Arthritis Rheum. (2020) 50:414–22. doi: 10.1016/j.semarthrit.2020.01009
51. Botelho J, Machado V, Mascarenhas P, Rua J, Alves R, Cavacas MA, et al. Stress, salivary cortisol and periodontitis: a systematic review and meta-analysis of observational studies. Arch Oral Biol. (2018) 96:58–65. doi: 10.1016/j.archoralbio.2018.08016
52. Machado V, Lopes J, Patrão M, Botelho J, Proença L, and Mendes JJ. Validity of the association between periodontitis and female infertility conditions: a concise review. Reproduction. (2020) 160:R41–54. doi: 10.1530/REP-20-0176
53. Kellesarian SV, Kellesarian TV, Ros Malignaggi V, Al-Askar M, Ghanem A, Malmstrom H, et al. Association between periodontal disease and erectile dysfunction: a systematic review. Am J Mens Health. (2018) 12:338–46. doi: 10.1177/1557988316639050
54. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:eaau3333. doi: 10.1126/sciadvaau3333
55. Papageorgiou SN, Hagner M, Nogueira AVB, Franke A, Jäger A, and Deschner J. Inflammatory bowel disease and oral health: systematic review and a meta-analysis. J Clin Periodontol. (2017) 44:382–93. doi: 10.1111/jcpe12698
56. Xiao L, Zhang Q, Peng Y, Wang D, and Liu Y. The effect of periodontal bacteria infection on incidence and prognosis of cancer. Medicine. (2020) 99:e19698. doi: 10.1097/MD0000000000019698
57. Buset SL, Walter C, Friedmann A, Weiger R, Borgnakke WS, and Zitzmann NU. Are periodontal diseases really silent? A systematic review of their effect on quality of life. J Clin Periodontol. (2016) 43:333–44. doi: 10.1111/jcpe12517
58. Botelho J, Machado V, Proença L, Bellini D, Chambrone L, Alcoforado G, et al. The impact of nonsurgical periodontal treatment on oral health-related quality of life: a systematic review and meta-analysis. Clin Oral Investig. (2020) 24:585–96. doi: 10.1007/s00784-019-03188-1
59. Marchesan JT, Girnary MS, Moss K, Monaghan ET, Egnatz GJ, Jiao Y, et al. Role of inflammasomes in the pathogenesis of periodontal disease and therapeutics. Periodontol 2000. (2020) 82:93–114. doi: 10.1111/prd12269
60. Golub LM, and Lee HM. Periodontal therapeutics: current host-modulation agents and future directions. Periodontol 2000. (2020) 82:186–204. doi: 10.1111/prd12315
61. Nguyen T, Sedghi L, Ganther S, Malone E, Kamarajan P, and Kapila YL. Host-microbe interactions: profiles in the transcriptome, the proteome, and the metabolome. Periodontol 2000. (2020) 82:115–28. doi: 10.1111/prd12316
62. Beck JD, Philips K, Moss K, Divaris K, Morelli T, and Offenbacher S. Advances in precision oral health. Periodontol 2000. (2020) 82:268–85. doi: 10.1111/prd12314
63. Socransky SS, and Haffajee A. Periodontal microbial ecology. Periodontol 2000. (2005) 38:135–87. doi: 10.1111/j.1600-0757.2005.00107x
64. Ramberg P, Furuichi Y, Volpe AR, Gaffar A, and Lindhe J. The effects of antimicrobial mouthrinses on de novo plaque formation at sites with healthy and inflamed gingivae. J Clin Periodontol. (1996) 23:7–11. doi: 10.1111/j.1600-051X.1996.tb00497x
65. Daly CG, and Highfield JE. Effect of localized experimental gingivitis on early supragingival plaque accumulation. J Clin Periodontol. (1996) 23:160–4. doi: 10.1111/j.1600-051X.1996.tb02071x
66. Socransky SS, Haffajee AD, Cugini MA, Smith C, and Kent RL. Microbial complexes in subgingival plaque. J Clin Periodontol. (1998) 25:134–44. doi: 10.1111/j.1600-051X.1998.tb02419x
67. Goodson JM, Tanner ACR, Haffajee AD, Sornberger GC, and Socransky SS. Patterns of progression and regression of advanced destructive periodontal disease. J Clin Periodontol. (1982) 9:472–81. doi: 10.1111/j.1600-051X.1982.tb02108x
68. Socransky SS, Haffajee AD, Goodson JM, and Lindhe J. New concepts of destructive periodontal disease. J Clin Periodontol. (1984) 11:21–32. doi: 10.1111/j.1600-051X.1984.tb01305x
69. Craig RG, Yip JK, So MK, Boylan RJ, Socransky SS, and Haffajee AD. Relationship of destructive periodontal disease to the acute-phase response. J Periodontol. (2003) 74:1007–16. doi: 10.1902/jop.2003.74.71007
70. Mdala I, Haffajee AD, Socransky SS, de Blasio BF, Thoresen M, Olsen I, et al. Multilevel analysis of clinical parameters in chronic periodontitis after root planing/scaling, surgery, and systemic and local antibiotics: 2-year results. J Oral Microbiol. (2012) 4:17535. doi: 10.3402/jom.v4i017535
71. Feres M, Teles F, Teles R, Figueiredo LC, and Faveri M. The subgingival periodontal microbiota of the aging mouth. Periodontol 2000. (2016) 72:30–53. doi: 10.1111/prd12136
72. David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. (2014) 15:1–15. doi: 10.1186/gb-2014-15-7-r89
73. Nibali L, Sousa V, Davrandi M, Spratt D, Alyahya Q, Dopico J, et al. Differences in the periodontal microbiome of successfully treated and persistent aggressive periodontitis. J Clin Periodontol. (2020) 47:980–990. doi: 10.1111/jcpe13330
74. Lu H, He L, Xu J, Song W, Feng X, Zhao Y, et al. Well-maintained patients with a history of periodontitis still harbor a more disbiotic microbiome than health. J Periodontol. (2020) 26:1–11. doi: 10.1002/JPER.19-0498
75. Krisanaprakornkit S, Kimball JR, and Dale BA. Regulation of human β-defensin-2 in gingival epithelial cells: the involvement of mitogen-activated protein kinase pathways, but not the NF-κB transcription factor family. J Immunol. (2002) 168:316–24. doi: 10.4049/jimmunol.168.1316
76. Yin L, and Dale BA. Activation of protective responses in oral epithelial cells by Fusobacterium nucleatum and human β-defensin-2. J Med Microbiol. (2007) 56:976–87. doi: 10.1099/jmm.047198-0
77. Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, et al. Prokaryotic regulation of epithelial responses by inhibition of IκB-α ubiquitination. Science. (2000) 289:1560–3. doi: 10.1126/science.289.54841560
78. Tien M-T, Girardin SE, Regnault B, Le Bourhis L, Dillies M-A, Coppée J-Y, et al. Anti-inflammatory effect of lactobacillus casei on shigella -infected human intestinal epithelial cells. J Immunol. (2006) 176:1228–37. doi: 10.4049/jimmunol.176.21228
79. Ghosh A, Joo NE, Chen TC, and Kapila YL. Proapoptotic fibronectin fragment induces the degradation of ubiquitinated p53 via proteasomes in periodontal ligament cells. J Periodontal Res. (2010) 45:481–7. doi: 10.1111/j.1600-0765.2009.01261x
80. Karin M, Yamamoto Y, and Wang QM. The IKK NF-κB system: a treasure trove for drug development. Nat Rev Drug Discov. (2004) 3:17–26. doi: 10.1038/nrd1279
81. Heyninck K, and Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-κB activation at the level of TRAF6. FEBS Lett. (1999) 442:147–50. doi: 10.1016/S0014-5793(98)01645-7
82. Malynn BA, and Ma A. Ubiquitin makes its mark on immune regulation. Immunity. (2010) 33:843–52. doi: 10.1016/j.immuni.2010.12007
83. Hong JY, Bae WJ, JK Yi, Kim GT, and Kim EC. Anti-inflammatory and anti-osteoclastogenic effects of zinc finger protein A20 overexpression in human periodontal ligament cells. J Periodontal Res. (2016) 51:529–39. doi: 10.1111/jre12332
84. Li Y, Mooney EC, Holden SE, Xia X-J, Cohen DJ, Walsh SW, et al. A20 orchestrates inflammatory response in the oral mucosa through restraining NF-κB activity. J Immunol. (2019) 202:2044–56. doi: 10.4049/jimmunol1801286
85. Li Y, Mooney EC, Xia XJ, Gupta N, and Sahingur SE. A20 restricts inflammatory response and desensitizes gingival keratinocytes to apoptosis. Front Immunol. (2020) 11:365. doi: 10.3389/fimmu.202000365
86. Zhang T, Wu J, Ungvijanpunya N, Jackson-Weaver O, Gou Y, Feng J, et al. Smad6 methylation represses NFκB activation and periodontal inflammation. J Dent Res. (2018) 97:810–9. doi: 10.1177/0022034518755688
87. Li Q, Luo T, Lu W, Yi X, Zhao Z, and Liu J. Proteomic analysis of human periodontal ligament cells under hypoxia. Proteome Sci. (2019) 17:1–11. doi: 10.1186/s12953-019-0151-2
88. Yan K, Wu C, Ye Y, Li L, Wang X, He W, et al. A20 inhibits osteoclastogenesis via TRAF6-dependent autophagy in human periodontal ligament cells under hypoxia. Cell Prolif . (2020) 53:1–10. doi: 10.1111/cpr12778
89. Yuan H, Zelka S, Burkatovskaya M, Gupte R, Leeman SE, and Amar S. Pivotal role of NOD2 in inflammatory processes affecting atherosclerosis and periodontal bone loss. Proc Natl Acad Sci USA. (2013) 110:E5059–68. doi: 10.1073/pnas1320862110
90. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. (2014) 15:768–78. doi: 10.1016/j.chom.2014.05012
91. Cai B, Panek JS, and Amar S. Convergent synthesis of novel muramyl dipeptide analogues: inhibition of porphyromonas gingivalis-induced pro-inflammatory effects by high doses of muramyl dipeptide. J Med Chem. (2016) 59:6878–90. doi: 10.1021/acs.jmedchem6b00681
92. Bengtsson T, Zhang B, Selegård R, Wiman E, Aili D, and Khalaf H. Dual action of bacteriocin PLNC8 α β through inhibition of porphyromonas gingivalis infection and promotion of cell proliferation. Pathog Dis. (2017) 75:1–10. doi: 10.1093/femspd/ftx064
93. Tian J, Gu L, Adams A, Wang X, and Huang R. Pellino-1 protects periodontal ligament stem cells against H2O2-induced apoptosis via activation of NF-κB signaling. Mol Biotechnol. (2018) 60:533–8. doi: 10.1007/s12033-018-0067-6
94. Chen D, Frezza M, Schmitt S, Kanwar J, and Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. (2011) 11:239–53. doi: 10.2174/156800911794519752
95. Terpos E, and Christoulas D. Effects of proteasome inhibitors on bone cancer. Bonekey Rep. (2013) 2:1–5. doi: 10.1038/bonekey.2013129
96. Pellom ST, Dudimah DF, Thounaojam MC, Sayers TJ, and Shanker A. Modulatory effects of bortezomib on host immune cell functions. Immunotherapy. (2015) 7:1011–22. doi: 10.2217/imt.1566
97. Toscani D, Palumbo C, Dalla Palma B, Ferretti M, Bolzoni M, Marchica V, et al. The proteasome inhibitor bortezomib maintains osteocyte viability in multiple myeloma patients by reducing both apoptosis and autophagy: a new function for proteasome inhibitors. J Bone Miner Res. (2016) 31:815–27. doi: 10.1002/jbmr2741
98. Kitagaki J, Miyauchi S, Xie CJ, Yamashita M, Yamada S, Kitamura M, et al. Effects of the proteasome inhibitor, bortezomib, on cytodifferentiation and mineralization of periodontal ligament cells. J Periodontal Res. (2015) 50:248–55. doi: 10.1111/jre12202
99. Jiang L, Song J, Hu X, Zhang H, Huang E, Zhang Y, et al. The proteasome inhibitor bortezomib inhibits inflammatory response of periodontal ligament cells and ameliorates experimental periodontitis in rats. J Periodontol. (2017) 88:473–83. doi: 10.1902/jop.2016160396
100. Kim YG, Kang JH, Kim HJ, Kim HJ, Kim HH, Kim JY, et al. Bortezomib inhibits osteoclastogenesis and porphyromonas gingivalis lipopolysaccharide-induced alveolar bone resorption. J Dent Res. (2015) 94:1243–50. doi: 10.1177/0022034515592592
101. Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. (2003) 348:2609–17. doi: 10.1056/NEJMoa030288
102. Jagannath S, Barlogie B, Berenson JR, Siegel DS, Irwin D, Richardson PG, et al. Updated survival analyses after prolonged follow-up of the phase 2, multicenter CREST study of bortezomib in relapsed or refractory multiple myeloma. Br J Haematol. (2008) 143:537–40. doi: 10.1111/j.1365-2141.2008.07359x
103. Zhang Z, Fu X, Xu L, Hu X, Deng F, Yang Z, et al. Nanosized alumina particle and proteasome inhibitor bortezomib prevented inflammation and osteolysis induced by titanium particle via autophagy and NF-κB signaling. Sci Rep. (2020) 10:1–12. doi: 10.1038/s41598-020-62254-x
104. Komine-Aizawa S, Aizawa S, and Hayakawa S. Periodontal diseases and adverse pregnancy outcomes. J Obstet Gynaecol Res. (2019) 45:5–12. doi: 10.1111/jog13782
Keywords: ubiquitin-proteasome system, ubiquitin, proteasome, ubiquitination, periodontal disease, periodontitis, oral health
Citation: Machado V, Carvalho R, Mendes JJ and Botelho J (2020) The Ubiquitin Proteasome System in Periodontal Disease: A Comprehensive Review. Front. Dent. Med. 1:613080. doi: 10.3389/fdmed.2020.613080
Received: 01 October 2020; Accepted: 30 November 2020;
Published: 17 December 2020.
Edited by:
Jamil Awad Shibli, Guarulhos University, BrazilReviewed by:
Carina Maciel Silva-Boghossian, Federal University of Rio de Janeiro, BrazilOleh Andrukhov, University Dental Clinic Vienna, Austria
Copyright © 2020 Machado, Carvalho, Mendes and Botelho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanessa Machado, dm1hY2hhZG9AZWdhc21vbml6LmVkdS5wdA==