 Elaine O. C. Cardoso1,2
Elaine O. C. Cardoso1,2 Noah Fine1Michael Glogauer1,2,3
Noah Fine1Michael Glogauer1,2,3 Francis Johnson4Michael Goldberg1,2Lorne M. Golub5
Francis Johnson4Michael Goldberg1,2Lorne M. Golub5 Howard C. Tenenbaum1,2*
Howard C. Tenenbaum1,2*- 1Faculty of Dentistry, University of Toronto, Toronto, ON, Canada
- 2Department of Dentistry, Centre for Advanced Dental Research and Care, Mount Sinai Hospital, University of Toronto, Toronto, ON, Canada
- 3University Health Network (UHN), Toronto, ON, Canada
- 4Department of Pharmacological Sciences, School of Medicine, Stony Brook University, Stony Brook, NY, United States
- 5Department of Oral Biology and Pathology, School of Dental Medicine, Stony Brook University, Stony Brook, NY, United States
The pathophysiology of SARS-CoV-2 infection is characterized by rapid virus replication and aggressive inflammatory responses that can lead to acute respiratory distress syndrome (ARDS) only a few days after the onset of symptoms. It is suspected that a dysfunctional immune response is the main cause of SARS-CoV-2 infection-induced lung destruction and mortality due to massive infiltration of hyperfunctional neutrophils in these organs. Similarly, neutrophils are recruited constantly to the oral cavity to combat microorganisms in the dental biofilm and hyperfunctional neutrophil phenotypes cause destruction of periodontal tissues when periodontitis develops. Both disease models arise because of elevated host defenses against invading organisms, while concurrently causing host damage/disease when the immune cells become hyperfunctional. This represents a clear nexus between periodontal and medical research. As researchers begin to understand the link between oral and systemic diseases and their potential synergistic impact on general health, we argue that translational research from studies in periodontology must be recognized as an important source of information that might lead to different therapeutic options which can be effective for the management of both oral and non-oral diseases. In this article we connect concepts from periodontal research on oral inflammation while exploring host modulation therapy used for periodontitis as a potential strategy for the prevention of ARDS a deadly outcome of COVID-19. We suggest that host modulation therapy, although developed initially for management of periodontitis, and which inhibits proteases, cytokines, and the oxidative stress that underlie ARDS, will provide an effective and safe treatment for COVID-19.
Introduction
The outbreak of viral pneumonia cases from SARS-CoV-2 was first reported by the Chinese government in December 2019 (1). As with other viral diseases SARS-CoV-2 can cause various respiratory infections, including multifocal interstitial pneumonia which was leading to admission to intensive care and death in infected patients (2). This infection, named Coronavirus disease 2019 (COVID-19) (3), can cause complications including the development of acute respiratory distress syndrome (ARDS); an often fatal disorder (2, 4).
ARDS is caused by many pathogens including influenza and coronavirus. Although its precise pathophysiologic mechanisms are not completely clear, it could be the result of direct damage caused by the viral pathogen and then, more importantly, the triggering of a complex dysregulation of the inflammatory environment (5, 6). Indeed, it has been argued that the host-mediated lung and other tissue damage has more to do with the massive infiltration of polymorphonuclear neutrophils (PMNs) in the lungs rather than purely direct viral effects in relation to morbidity and mortality (4, 6, 7).
Immuno-Inflammatory Pathogenesis of COVID-19
Data from cohorts of critically ill patients with COVID-19-related pneumonia provide evidence of cytokine profiles like those of hyperinflammatory states seen in bacterial and viral pneumonias (4, 8). SARS-CoV-2 invades the host cell by binding of its viral spike glycoprotein to the host's cellular receptor for ACE2. Once in the cell, the virus may “deceive” the immune system through strategies that prevent pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) from recognizing pathogen-associated molecular patterns (PAMPs) and will start replicating freely within the infected cells using their own organelles and other cellular components (9). In addition, SARS- CoV-2 has also evolved strategies that interfere in the production of type I/III IFN which are essential for the development of effective immunity (9). As a result of this state of unchecked replication, SARS-CoV-2 can reach high titres shortly after initial infection that leads to an exponential production of PAMPS, cell damage and release of damage-associated molecular patterns (DAMPS), all of which triggering a hyperactive inflammatory responses (10).
The attachment of SARS-CoV-2 to ACE2 for host cell entry leads to down-regulation of ACE2 and a subsequent increase of angiotensin II (ANGII) (11–16) which dysregulate the renin-angiotensin system (RAS) (17). In elevated levels, ANGII acts as a pro-inflammatory mediator that ultimately activates NFκB, disintegrin, and metalloprotease 17 (ADAM17) (18). This activated pro-inflammatory environment triggers the production of reactive oxygen species (ROS), fibrosis, matrix metalloproteinases (MMPs), production of cytokines such as IL-6 and IL-8 by macrophages and recruitment of PMNs. The virus also activates NFκB (11, 15) that amplifies downstream signaling for cytokine production (14, 15). The release of cytokines activates pathogenic T helper type 1 (Th1) cells rapidly which then secrete additional pro-inflammatory cytokines (11, 12, 19). This is followed by additional infiltration of macrophages and PMNs into alveolar cavities where they begin to contribute to the hyper-inflammatory response (11, 14, 15). ANGII is also known to trigger the coagulation cascade by activating platelets through surface AngII receptors binding and inducing platelet shape change (20) both of which associated with thrombosis (21). In summary, SARS-CoV-2 binding to ACE2 for cell invasion is likely the first step for activation of the cytokine storm which releases uncontrolled levels of cytokines, including IL-1β, 1L-6, IL-8, and IL-10 (22), that prime the host for development of hyperactive inflammatory responses. Manifestation of the cytokine storm is extremely complex but in general in addition to virus-induced infiltration of inflammatory cells to the lungs causing oxidative stress and initial inflammation it relies on even more PMN infiltration into the lung whereby cytokines, MMPs, PMN elastase, ROS, and nitric oxide (NO) are released into the inflamed tissue (22, 23) causing diffuse alveolar damage, pulmonary edema, pulmonary fibrosis, acute lung tissue destruction, multiple organ failure and death. These developments essentially describe ARDS as seen in patients suffering from COVID-19 (11–16).
PMNs are the first and most numerous innate immune cells to reach the infection site and therefore play a central role in the resolution of inflammation through specific mechanisms of virus inactivation including the release of MMPs, cytokines, ROS, peroxidases and PMN extracellular traps (NETs) (24). This is of course protective. But PMNs can also become “hyperactive,” and when this happens, PMNs contribution to antiviral defense can cause harmful effects to the host including the development of pneumonia and ARDS (25–27). Paradoxically then, despite the critical roles played by PMN cells insofar as clearance of viral pathogens and other infectious disease is concerned it's recognized that excessively sensitized/activated PMN responses promote a vicious cycle of inflammatory damage to the very tissues to which they were dispatched as a consequence of a PMN-induced cytokine storm (24). Notably, MMP-2 and−9 destroy the extracellular matrix in the lungs by degrading collagen found in the basement membrane comprising their parenchymal architecture (22). The virucidal effects of ROS and the recruitment and activation of even more PMNs through the production of cytokines can perpetuate the hyperinflammatory response thereby leading to lung and other tissue injuries including the development of vasculitides and thrombotic conditions characteristic of ARDS (24, 27). In addition, ROS production further increases vascular and epithelial permeability, allowing for continuous infiltration of PMNs and serosanguinous exudates into the alveolar space (27). Finally, the formation of NETs aided by activated platelets in response to endothelial damage, ROS and IL-1β production and virus replication may increase the risk of thromboembolic events in COVID-19 patients by triggering complement activation and further fuelling the coagulation cascade (9) (Figure 1).
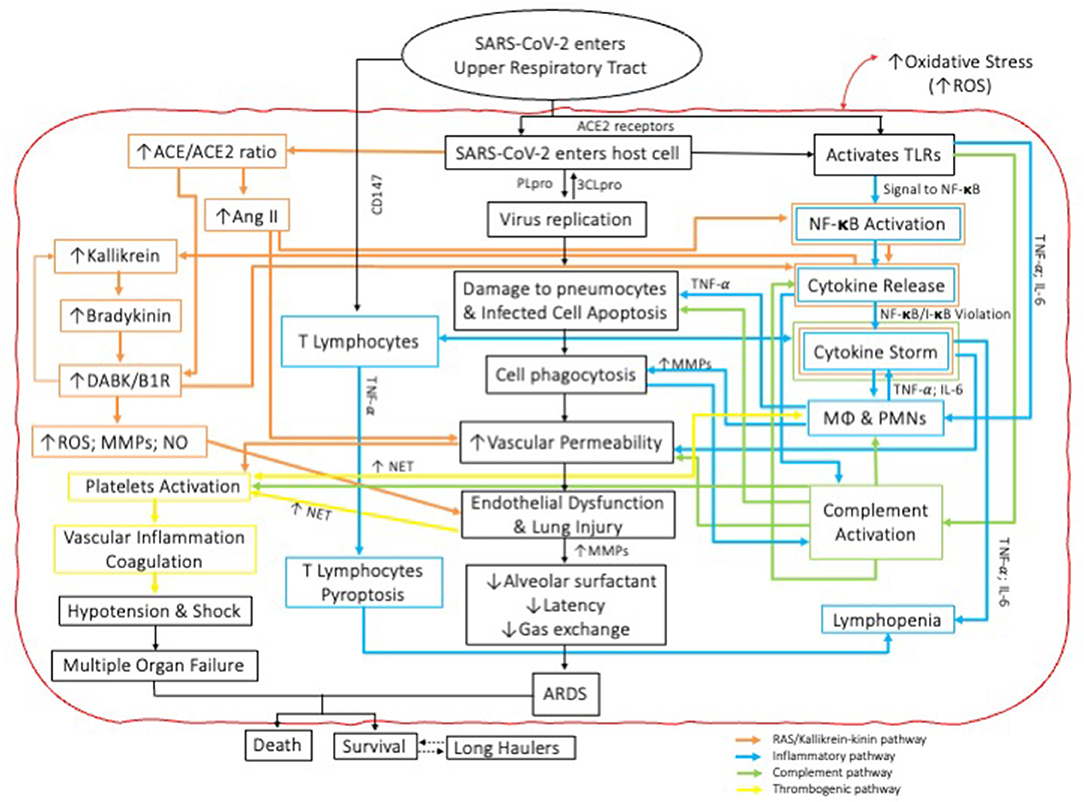
Figure 1. Schematic representation of the interactions amongst and between four independent pathophysiologic pathways involved in the cytokine storm cascade during COVID-19. Note mechanisms for COVID parallel those also described for periodontal diseases.
A summary of the role of PMNs on the severity of COVID-19 in recent studies is shown in Table 1.
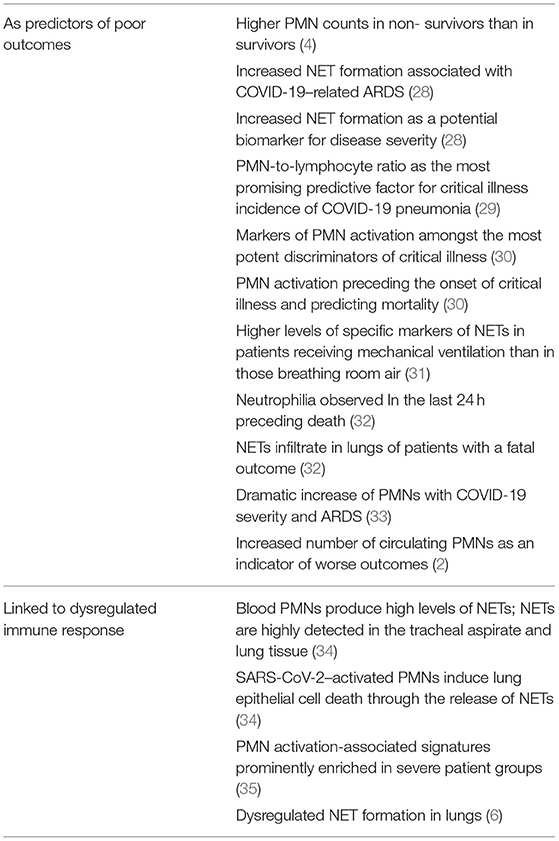
Table 1. Summary of the role of PMNs on the severity of COVID-19 in recent studies.
To prevent this, we hypothesize that any treatment which could prevent excess PMN infiltration and hyperactivation while also blocking excessive levels of MMP activity, elastase activities and simultaneously reducing excessive ROS levels or activity might represent a useful approach to the prevention and/or amelioration of the morbidity and mortality associated with the cytokine storm/ARDS in patients with COVID-19.
Links Between Oral Inflammation and Systemic Disease
As researchers begin to understand the link between oral and systemic diseases more clearly and their potential synergistic impact on general health, we argue that translational research from studies in periodontology must be recognized as an important source of information that might lead to new and different therapeutic options which can be effective for the management of both oral and non-oral diseases.
While evidence of associations between periodontal diseases and systemic conditions have long been noted (36), there has been increased interest in determining the underlying mechanisms that might explain the oral-systemic pathophysiology. We suggest that a causal and indeed bidirectional link may exist between periodontitis and systemic non-communicable diseases. However, we also have to recognize that they could also be manifestations of common underlying pathophysiological mechanisms. This said, these two concepts are not mutually exclusive, and therefore we must emphasize that both putative mechanisms could be involved in those associations, as demonstrated by studies that show bidirectionality of association. For instance, early epidemiological studies have demonstrated the bidirectional adverse interrelationship between an altered host inflammatory response in PD and the metabolic imbalance in diabetes (37) while more recently, a causal association was demonstrated between periodontitis and chronic kidney diseases mediated via oxidative stress (38), which seems highly relevant to this argument. We also point out that oxidative stress is a key element of PMN hyperfunctionality related to overproduction of ROS and downregulation of endogenous antioxidants such as NrF2 mediated expression of superoxide dismutase (39). Inflammation is therefore the common factor amongst periodontitis and the chronic diseases of aging, or simply “the disease” (40). Insofar the individual's susceptibility to systemic diseases, our research support the hypothesis that PD sensitizes or primes the peripheral innate immune system, and predominantly the PMNs in such a manner as to allow those cells to trigger and/or exacerbate inflammatory diseases in distant organ systems (41).
Periodontal Disease-Induced Immunopathology and COVID-19
The oral cavity is unique in that the teeth are the only structures in the body that de facto protrude through the lining epithelium, in this case the gingival tissues. As such a unique seal exists between the gingiva and tooth surfaces and therefore between the oral cavity and its contents thereby preventing ingress of microbial or other pathogens into the body (42). This biologic seal, specifically a connective tissue and epithelial attachment to cementum, is not perfect and is permeable even in health but moreso in states of inflammation. To enhance protection from pathogens, cells of the innate immune system such as PMNs, are recruited constantly to the oral cavity as part of a healthy and self-limiting inflammatory response against the challenges imposed by the oral microorganisms found in the dental biofilm (43). Interestingly, while bacteria or their by-products may lead to periodontal tissue damage, the host immunoinflammatory response to microorganisms in dental biofilms, when uncontrolled, is considered the main cause of periodontal pathogenesis (40, 44), something akin to destruction of lung tissues observed in ARDS. In parallel to what is seen in ARDS, the initial host immune response starts when PRRs expressed in the membrane of epithelial cells and gingival fibroblasts interact with PAMPs, including lipopolysaccharide (LPS) found in the cell wall of specific periodontal bacteria (45). LPS is considered a potent ligand for TLR4 (46) and activation of both the TLR2 and TLR4 pathways has been described in studies with Porphyromonas gingivalis (47). PAMP-TLRs binding and MyD88 signaling results in the activation of the downstream signaling pathways associated with inflammation and upregulation of pro-inflammatory transcription factors, such as NFκB (48), leading to the release of inflammatory cytokines and chemokines (49, 50). The most common cytokines involved in this process are TNF-α, IL- 1β, IL-6, and IL-8 (51, 52), while chemokines include CXCL8/IL-8, CCL2, CCL3, and CCL5 (49, 50) and their release causes vasodilation and chemical gradients that facilitate the migration of leukocytes, mostly PMNs from the vasculature to the site of injury (53). Infiltration of such inflammatory cells leads to release of ROS, MMPs and NETs, as well as to chemotaxis and phagocytosis as defense mechanisms against infection and inflammation (54–58). However, as periodontal diseases are not considered as a classic bacterial infection but rather a dysbiotic disease such mechanisms are necessary but possibly not sufficient to cause disease (59). Periodontal dysbiosis leads to a disturbance of the local homeostasis and immune subversion that increases microbial colonization, virulence, and persistence to disease, and result in persistent recruitment of PMNs (60) with hyperfunctional or hyperactive phenotypes (43, 54–56, 61–63). Similar to what happens in patients with COVID-19, these now hyperactivated PMNs pour out high levels of ROS and degradative enzymes along with ever increasing levels of proinflammatory cytokines (55, 56, 62). These actions lead to severe destruction of the connective tissues about the affected teeth leading to pain, bleeding and ultimately tooth loss (58, 64).
This phenotype of hyperinflammatory PMNs has also been observed to play an important role in the pathogenesis of systemic diseases such as diabetes and cardiovascular disease, suggesting an epidemiological association between periodontal diseases and systemic conditions (65, 66). More importantly, these hyperactivated phenotypes have been observed in severe cases of COVID-19 (67, 68), as well as in aging-related conditions (69). Therefore, the presence of periodontitis in patients who are infected with SARS-CoV-2 could represent an as yet unrecognized comorbidity that could contribute to more severe symptoms of COVID-19. While there are now emerging scientific publications that align with this suggestion (70, 71), it still stands in the grounds of scientific inference. Two plausible mechanisms may explain this association: one being related to the periodontitis-induced inflammatory response; a pre-existing pro-inflammatory state. This could act synergistically and therefore amplify the systemic inflammatory response induced by infection with SARS-CoV-2. Another possibility includes the notion there could be a genetic predisposition of the host to develop hyperinflammatory conditions that are favorable to both the development of PD or COVID-19. Regarding the former, our team's previous research has shown that an increase in the level of hyperactivated PMNs in bone marrow and blood can be caused by periodontal inflammation and that this predisposes to an exacerbated PMN response to distant inflammatory conditions. In other words, PD primes the immune system and thus intensifies the overall innate immune response, thus exacerbating general inflammatory disease (41) including COVID-19.
Similarly, in an experimental study of the respiratory mucosa before, during, and after respiratory syncytial virus (RSV) infection in humans, participants who succumbed to infection had more activated PMNs in their airways before exposure to the virus than those who staved off infection. After viral exposure, a reduction in antiviral response in the neutrophilic mucosal environment was observed, more specifically suppression of interleukin-17 (IL-17), followed by disease onset. The authors hypothesized that primed PMNs, typically associated with immune response to previous bacterial infections might increase the individual's susceptibility to symptomatic viral infections and potentially even COVID-19 (72). A strong hyperactivation phenotype in peripheral PMNs has already been directly associated with severe cases of COVID-19, including increased phagocytosis, degranulation and chemotaxis, and increased expression of genes involved in pro-inflammatory cytokine release. Within these severe cases, the emergence of an immature PMN population, characteristic of emergency myelopoiesis, was the main difference observed between the immune responses in fatal and non-fatal cases of COVID-19 (73). NET formation in tissue injury and thrombotic complications are additional pathogenic mechanisms whereby circulating PMNs can lead to more severe COVID-19 (6, 28, 31, 32, 34). This highlights another potential mechanisms linking PD as a potential comorbidity in COVID-19 cases: the pro-thrombotic state as a result of PD-associated haemodynamic, endothelial, and inflammatory triggers that may lead to an abnormality in the coagulation or fibrinolysis system (74).
From Bench to Chairside With a Bridge to the Bedside—Host Modulation Therapy (HMT)
We suggest that the SARS-CoV-2 pandemic has highlighted the need for a greater understanding of the role of PMNs in combating viral infections, as COVID-19-related PMN-mediated inflammation in the lungs can be life-threatening (6). While supporting the potential role of PD-related innate immune response in systemic inflammatory conditions, our team proposes the use of host modulation therapy (HMT), as designed initially for treatment of PD for treatment of systemic inflammatory diseases that interact with PD (41), as well as in the prevention and treatment of ARDS, given the similarity of the underlying inflammatory mechanisms. Hereunder we describe the tenets of HMT.
HMT has been established in periodontology as a successful therapeutic approach for management of chronic periodontal and refractory periodontal diseases, all of which are PMN-mediated disorders. This therapy, pioneered by our group (notably Dr. Golub's group), was a paradigm shift in periodontal therapy for using tetracycline-based molecules and not reliant on their antimicrobial properties, to downregulate the activities of PMN-derived MMPs, suppression of inflammatory cytokines, and for quenching of ROS (75–78). In relation to periodontitis, work has focused on the use of subantimicrobial dose doxycycline (Periostat®), but higher dose use over a short term is certainly feasible when treating extreme cases of inflammation as in the acute stage of COVID-19. This has also led to the development of effective treatment for rosacea using sub-antimicrobial-dose doxycycline slow release form (Oracea®/Aprillon®) (79) More recently, this concept was boosted by Serhan's studies on pro-resolving lipid mediators in which he argues that a failure in resolution of inflammation rather than its hyperactivation leads to chronic inflammation (80, 81). Pre-clinical studies have shown that treatment with lipid mediators after experimentally induced periodontitis in animals was associated with bone loss prevention, regeneration of periodontal tissues and bacterial shifts in the subgingival microbiota (82–84). In humans, differences in pro-resolving lipid mediator profiles were observed between periodontally healthy and periodontitis participants and thus associated with the state of periodontal inflammation (85). These targets are important factors contributing to the breakdown of periodontal tissues, but also to other tissues being attacked by dysregulated inflammation-mediated destruction observed in periodontitis and ARDS (including stimulation of the vasculitides). Along similar lines, our group has shown that the flavonoids, resveratrol, and curcumin, downregulate ROS-mediated oxidative stress, inhibit ROS production/activity, and inhibit pro-inflammatory cytokine formation in animal model studies of periodontitis, which should protect tissues under inflammatory attack (86, 87). In animals subjected to cigarette smoke inhalation, we showed that resveratrol effectively blocks the harmful effects of aryl hydrocarbons found in cigarette smoke and the environment, which could be very important inasmuch as smoking represents a significant comorbidity for COVID-19 and is also a major risk factor for periodontitis and favor healing (88, 89). There is also evidence animal model data showing that by using HMT, the development of ARDS can be blocked (90, 91).
We suggest that the effectiveness of HMT could be independent of the type of infectious virus because it targets the host's cellular mechanisms that propagate ARDS (and of course PD) and not only the virus itself. Therefore, we suggest that mutations of the virus should be equally less material insofar as the putative effectiveness of HMT in prevention and treatment of ARDS. Recent evidence showed that tetracyclines have in vitro activity in post-entry stages of the infection with SARS-CoV-2 (92) and resveratrol blocks replication of coronavirus and other respiratory viruses (93, 94). We propose that the use of drug/nutraceutical HMT described initially for periodontitis could reduce morbidity, mortality, and possibly longer-term sequelae of COVID-19.
The concept of HMT emerged for the treatment of periodontitis almost 40 years ago, after the identification of host-response mechanisms as the mediators of the destruction of the collagen-rich periodontal tissues and subsequent experiments with systemic drugs that inhibited collagen- and bone-destructive enzymes. Around the same time HMT was shown to be effective for downregulation of pathologically elevated levels of inflammation in systemic conditions such as arthritis, cancer, lung and cardiovascular diseases and rosacea (95). Based on this concept, we have presented evidence, described initially in the periodontal research literature, about the protective properties of a new approach to therapy, HMT, that fits precisely the treatment needs of patients with COVID-19/SARS-CoV-2 infection. And, unlike other medications being investigated for the treatment of COVID-19-mediated lung disease there are virtually no concerns about potential toxicity.
The rationale for this proposed treatment approach is based on the use of some or all the compounds identified above to inhibit the cytokine storm/ARDS, including PMN-mediated hyperinflammatory responses and tissue destruction and including the development of thromboembolic disorders. This approach should reduce hospitalization, ICU admissions, and death associated with COVID-19 markedly as suggested in Figure 2.
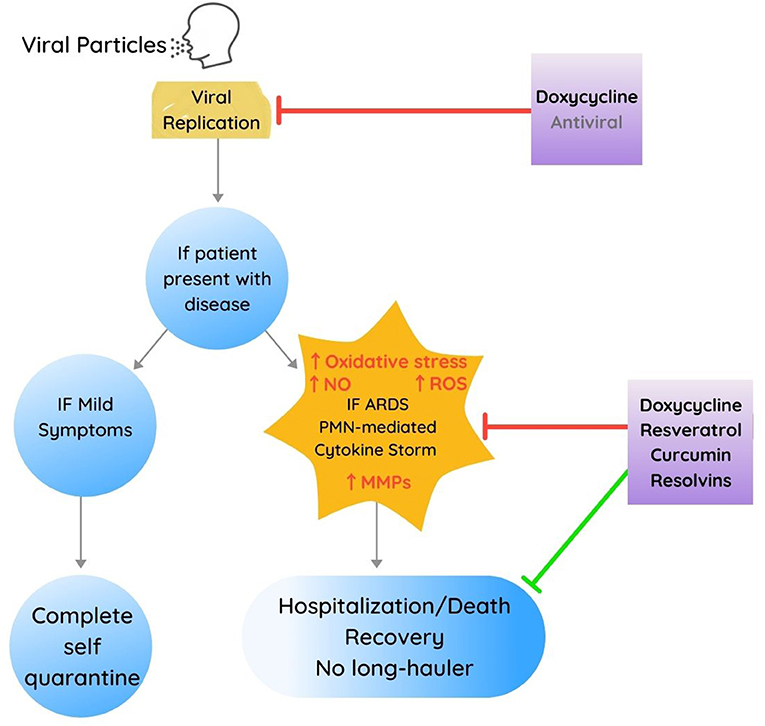
Figure 2. Proposed multidrug treatment approach, developed in part from therapeutic targets identified for management of periodontal diseases for each phase of Covid-19 presenting both inhibitory (in red) and stimulating or improvement-associated effects (in green). Note also that the use of antivirals (other than doxycycline) is depicted in a slightly faded font to delineate that these other therapeutics were not developed from or based on periodontal research investigations.
Conclusion
We suggest that we've demonstrated how research focused initially on oral inflammatory diseases has illuminated therapeutic targets that can be attacked by relatively simple and safe compounds, thereby reducing hospitalization, morbidity and mortality associated with COVID-19.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author Contributions
HT, LG, MGo, and MGl contributed to conception and design of the study. NF, LG, HT, and MGl, contributed to the experimental analyses and performance. EC and HT wrote the first draft of the manuscript while all others wrote or edited sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
3. WHO Director-General's Remarks at the Media Briefing on 2019-nCoV on 11 February 2020. World Health Organization. Available online at: https://www.who.int/director-general/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020
4. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
5. Wang QW, Su Y, Sheng JT, Gu LM, Zhao Y, Chen XX, et al. Anti-influenza A virus activity of rhein through regulating oxidative stress, TLR4, Akt, MAPK, and NF-κB signal pathways. PLoS ONE. (2018) 13:e0191793. doi: 10.1371/journal.pone.0191793
6. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. (2020) 217:e20200652. doi: 10.1084/jem.20200652
7. Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Natl Acad Sci USA. (2014) 111:3799–804. doi: 10.1073/pnas.1400593111
8. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. (2020) 71:762–8. doi: 10.1093/cid/ciaa248
9. Amor S, Fernández Blanco L, Baker D. Innate immunity during SARS-CoV-2: evasion strategies and activation trigger hypoxia and vascular damage. Clin Exp Immunol. (2020) 202:193–209. doi: 10.1111/cei.13523
10. Domingo P, Mur I, Pomar V, Corominas H, Casademont J, de Benito N. The four horsemen of a viral apocalypse: the pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine. (2020) 58:102887. doi: 10.1016/j.ebiom.2020.102887
11. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. (2020) 93:250–6. doi: 10.1002/jmv.26232
12. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
13. Sun X, Wang T, Cai D, Hu Z, Chen J, Liao H, et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. (2020) 53:38–42. doi: 10.1016/j.cytogfr.2020.04.002
14. Hussman JP. Cellular and molecular pathways of Covid-19 and potential points of therapeutic intervention. Front Pharmacol. (2020) 11:1169. doi: 10.3389/fphar.2020.01169
15. Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: the anger of inflammation. Cytokine. (2020) 133:155151. doi: 10.1016/j.cyto.2020.155151
16. Soy M, Keser G, Atagündüz P, Tabak F, Atagündüz I, Kayhan S. Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol. (2020) 39:2085–94. doi: 10.1007/s10067-020-05190-5
17. Ingraham NE, Barakat AG, Reilkoff R, Bezdicek T, Schacker T, Chipman JG, et al. Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: a comprehensive review. Eur Respir J. (2020) 56:2000912. doi: 10.1183/13993003.00912-2020
18. Devaux CA, Rolain JM, Raoult D. ACE2 receptor polymorphism: susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J Microbiol Immunol Infect. (2020) 53:425–35. doi: 10.1016/j.jmii.2020.04.015
19. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm' in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
20. Jagroop IA, Mikhailidis DP. Angiotensin II can induce and potentiate shape change in human platelets: effect of losartan. J Hum Hypertens. (2000) 14:581–5. doi: 10.1038/sj.jhh.1001102
21. Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. (2008) 359:1261–70. doi: 10.1056/NEJMra0800887
22. Roy SK, Kendrick D, Sadowitz BD, Gatto L, Snyder K, Satalin JM, et al. Jack of all trades: pleiotropy and the application of chemically modified tetracycline-3 in sepsis and the acute respiratory distress syndrome (ARDS). Pharmacol Res. (2011) 64:580–9. doi: 10.1016/j.phrs.2011.06.012
23. Zhou X, Wang D, Ballard-Croft CK, Simon SR, Lee HM, Zwischenberger JB. A tetracycline analog improves acute respiratory distress syndrome survival in an ovine model. Ann Thorac Surg. (2010) 90:419–26. doi: 10.1016/j.athoracsur.2010.04.052
24. Galani IE, Andreakos E. Neutrophils in viral infections: current concepts and caveats. J Leukoc Biol. (2015) 98:557–64. doi: 10.1189/jlb.4VMR1114-555R
25. Abraham E. Neutrophils and acute lung injury. Crit Care Med. (2003) 31(4 Suppl.):S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8
26. Sideras P, Apostolou E, Stavropoulos A, Sountoulidis A, Gavriil A, Apostolidou A, et al. Activin, neutrophils, and inflammation: just coincidence? Semin Immunopathol. (2013) 35:481–99. doi: 10.1007/s00281-013-0365-9
27. Potey PM, Rossi AG, Lucas CD, Dorward DA. Neutrophils in the initiation and resolution of acute pulmonary inflammation: understanding biological function and therapeutic potential. J Pathol. (2019) 247:672–85. doi: 10.1002/path.5221
28. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
29. Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. (2020) 18:206. doi: 10.1186/s12967-020-02374-0
30. Meizlish ML, Pine AB, Bishai JD, Goshua G, Nadelmann ER, Simonov M, et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. (2021) 5:1164–77. doi: 10.1182/bloodadvances.2020003568
31. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. (2020) 5:e138999. doi: 10.1172/jci.insight.138999
32. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d'Emal C, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med. (2020) 217:e20201012. doi: 10.1084/jem.20201012
33. Gong J, Dong H, Xia QS, Huang ZY, Wang DK, Zhao Y, et al. Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19: a retrospective study. BMC Infect Dis. (2020) 20:963. doi: 10.1186/s12879-020-05681-5
34. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217:e20201129. doi: 10.1084/jem.20201129
35. Aschenbrenner AC, Mouktaroudi M, Krämer B, Oestreich M, Antonakos N, Nuesch-Germano M, et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. (2021) 13:7. doi: 10.1186/s13073-020-00823-5
36. U.S. Department of Health and Human Services. Oral Health in America: A Report of the Surgeon General. Services USDoHaH, editor. Rockville, MD: National Institute of Dental and Craniofacial Research; National Institutes of Health (2000).
37. Taylor GW. Bidirectional interrelationships between diabetes and periodontal diseases: an epidemiologic perspective. Ann Periodontol. (2001) 6:99–112. doi: 10.1902/annals.2001.6.1.99
38. Sharma P, Fenton A, Dias IHK, Heaton B, Brown CLR, Sidhu A, et al. Oxidative stress links periodontal inflammation and renal function. J Clin Periodontol. (2021) 48:357–67. doi: 10.1111/jcpe.13414
39. Chiu AV, Saigh MA, McCulloch CA, Glogauer M. The role of NrF2 in the regulation of periodontal health and disease. J Dent Res. (2017) 96:975–83. doi: 10.1177/0022034517715007
40. Van Dyke TE. Inflammation and periodontal diseases: a reappraisal. J Periodontol. (2008) 79(8 Suppl):1501–2. doi: 10.1902/jop.2008.080279
41. Fine N, Chadwick JW, Sun C, Parbhakar KK, Khoury N, Barbour A, et al. Periodontal inflammation primes the systemic innate immune response. J Dent Res. (2020) 100:318–25. doi: 10.1177/0022034520963710
42. Nanci A. Ten Cate's Oral Histology - E-Book: Development, Structure, and Function. Philadelphia, PA: Elsevier (2017).
43. Fine N, Hassanpour S, Borenstein A, Sima C, Oveisi M, Scholey J, et al. Distinct oral neutrophil subsets define health and periodontal disease states. J Dent Res. (2016) 95:931–8. doi: 10.1177/0022034516645564
44. Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol. (2008) 79(8 Suppl):1601–8. doi: 10.1902/jop.2008.080173
45. Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. (2002) 13:413–21. doi: 10.1016/S1359-6101(02)00026-6
46. Zeng XZ, Zhang YY, Yang Q, Wang S, Zou BH, Tan YH, et al. Artesunate attenuates LPS-induced osteoclastogenesis by suppressing TLR4/TRAF6 and PLCγ1-Ca. Acta Pharmacol Sin. (2020) 41:229–36. doi: 10.1038/s41401-019-0289-6
47. Hajishengallis G, Sojar H, Genco RJ, DeNardin E. Intracellular signaling and cytokine induction upon interactions of Porphyromonas gingivalis fimbriae with pattern-recognition receptors. Immunol Invest. (2004) 33:157–72. doi: 10.1081/IMM-120030917
48. Song B, Zhang YL, Chen LJ, Zhou T, Huang WK, Zhou X, et al. The role of Toll-like receptors in periodontitis. Oral Dis. (2017) 23:168–80. doi: 10.1111/odi.12468
49. Cavalla F, Araujo-Pires AC, Biguetti CC, Garlet GP. Cytokine networks regulating inflammation and immune defense in the oral cavity. Curr Oral Health Rep. (2014) 1:104–13. doi: 10.1007/s40496-014-0016-9
50. Souto GR, Queiroz CM, Costa FO, Mesquita RA. Relationship between chemokines and dendritic cells in human chronic periodontitis. J Periodontol. (2014) 85:1416–23. doi: 10.1902/jop.2014.130662
51. Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. (2010) 89:1349–63. doi: 10.1177/0022034510376402
52. Han X, Lin X, Seliger AR, Eastcott J, Kawai T, Taubman MA. Expression of receptor activator of nuclear factor-kappaB ligand by B cells in response to oral bacteria. Oral Microbiol Immunol. (2009) 24:190–6. doi: 10.1111/j.1399-302X.2008.00494.x
53. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
54. Fredriksson M, Gustafsson A, Asman B, Bergström K. Periodontitis increases chemiluminescence of the peripheral neutrophils independently of priming by the preparation method. Oral Dis. (1999) 5:229–33. doi: 10.1111/j.1601-0825.1999.tb00306.x
55. Matthews JB, Wright HJ, Roberts A, Cooper PR, Chapple IL. Hyperactivity and reactivity of peripheral blood neutrophils in chronic periodontitis. Clin Exp Immunol. (2007) 147:255–64. doi: 10.1111/j.1365-2249.2006.03276.x
56. Matthews JB, Wright HJ, Roberts A, Ling-Mountford N, Cooper PR, Chapple IL. Neutrophil hyper-responsiveness in periodontitis. J Dent Res. (2007) 86:718–22. doi: 10.1177/154405910708600806
57. Gustafsson A, Asman B. Increased release of free oxygen radicals from peripheral neutrophils in adult periodontitis after Fc delta-receptor stimulation. J Clin Periodontol. (1996) 23:38–44. doi: 10.1111/j.1600-051X.1996.tb00502.x
58. Chapple IL, Matthews JB. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol 2000. (2007) 43:160–232. doi: 10.1111/j.1600-0757.2006.00178.x
59. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. (2012) 27:409–19. doi: 10.1111/j.2041-1014.2012.00663.x
60. Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. (2015) 21:172–83. doi: 10.1016/j.molmed.2014.11.004
61. Fredriksson M, Gustafsson A, Asman B, Bergström K. Hyper-reactive peripheral neutrophils in adult periodontitis: generation of chemiluminescence and intracellular hydrogen peroxide after in vitro priming and FcgammaR-stimulation. J Clin Periodontol. (1998) 25:394–8. doi: 10.1111/j.1600-051X.1998.tb02461.x
62. Ling MR, Chapple IL, Matthews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. (2015) 21:714–25. doi: 10.1177/1753425915589387
63. Wright HJ, Matthews JB, Chapple IL, Ling-Mountford N, Cooper PR. Periodontitis associates with a type 1 IFN signature in peripheral blood neutrophils. J Immunol. (2008) 181:5775–84. doi: 10.4049/jimmunol.181.8.5775
64. Figueredo CM, Fischer RG, Gustafsson A. Aberrant neutrophil reactions in periodontitis. J Periodontol. (2005) 76:951–5. doi: 10.1902/jop.2005.76.6.951
65. Hatanaka E, Monteagudo PT, Marrocos MS, Campa A. Neutrophils and monocytes as potentially important sources of proinflammatory cytokines in diabetes. Clin Exp Immunol. (2006) 146:443–7. doi: 10.1111/j.1365-2249.2006.03229.x
66. Yasunari K, Watanabe T, Nakamura M. Reactive oxygen species formation by polymorphonuclear cells and mononuclear cells as a risk factor of cardiovascular diseases. Curr Pharm Biotechnol. (2006) 7:73–80. doi: 10.2174/138920106776597612
67. Reyes L, Sanchez-Garcia MA, Morrison T, Howden AJM, Watts ER, Arienti S, et al. A type I IFN, prothrombotic hyperinflammatory neutrophil signature is distinct for COVID-19 ARDS. Wellcome Open Res. (2021) 6:38. doi: 10.12688/wellcomeopenres.16584.1
68. Morrissey S, Geller A, Hu X, Tieri D, Cooke E, Ding C, et al. Emergence of low-density inflammatory neutrophils correlates with hypercoagulable state and disease severity in COVID-19 patients. medRxiv, (2020). doi: 10.1101/2020.05.22.20106724
69. Tseng CW, Liu GY. Expanding roles of neutrophils in aging hosts. Curr Opin Immunol. (2014) 29:43–8. doi: 10.1016/j.coi.2014.03.009
70. Aquino-Martinez R, Hernández-Vigueras S. Severe COVID-19 lung infection in older people and periodontitis. J Clin Med. (2021) 10:279. doi: 10.3390/jcm10020279
71. Marouf N, Cai W, Said KN, Daas H, Diab H, Chinta VR, et al. Association between periodontitis and severity of COVID-19 infection: a case-control study. J Clin Periodontol. (2021) 48:483–91. doi: 10.1111/jcpe.13435
72. Habibi MS, Thwaites RS, Chang M, Jozwik A, Paras A, Kirsebom F, et al. Neutrophilic inflammation in the respiratory mucosa predisposes to RSV infection. Science. (2020) 370:eaba9301. doi: 10.1126/science.aba9301
73. Wilk AJ, Lee MJ, Wei B, Parks B, Pi R, Martínez-Colón GJ, et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. bioRxiv. (2020). doi: 10.1101/2020.12.18.423363
74. Bizzarro S, van der Velden U, ten Heggeler JM, Leivadaros E, Hoek FJ, Gerdes VE, et al. Periodontitis is characterized by elevated PAI-1 activity. J Clin Periodontol. (2007) 34:574–80. doi: 10.1111/j.1600-051X.2007.01095.x
75. Golub LM, McNamara TF, D'Angelo G, Greenwald RA, Ramamurthy NS. A non-antibacterial chemically-modified tetracycline inhibits mammalian collagenase activity. J Dent Res. (1987) 66:1310–4. doi: 10.1177/00220345870660080401
76. Golub LM, Ramamurthy NS, McNamara TF, Greenwald RA, Rifkin BR. Tetracyclines inhibit connective tissue breakdown: new therapeutic implications for an old family of drugs. Crit Rev Oral Biol Med. (1991) 2:297–321. doi: 10.1177/10454411910020030201
77. Golub LM, Suomalainen K, Sorsa T. Host modulation with tetracyclines and their chemically modified analogues. Curr Opin Dent. (1992) 2:80–90.
78. Golub LM, Wolff M, Roberts S, Lee HM, Leung M, Payonk GS. Treating periodontal diseases by blocking tissue-destructive enzymes. J Am Dent Assoc. (1994) 125:163–9; discussion: 9–71. doi: 10.14219/jada.archive.1994.0261
79. Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. (2011) 63:130–45. doi: 10.1016/j.phrs.2010.10.007
80. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8:349–61. doi: 10.1038/nri2294
81. Serhan CN. Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators. J Periodontol. (2008) 79(8 Suppl):1520–6. doi: 10.1902/jop.2008.080231
82. Lee CT, Teles R, Kantarci A, Chen T, McCafferty J, Starr JR, et al. Resolvin E1 reverses experimental periodontitis and dysbiosis. J Immunol. (2016) 197:2796–806. doi: 10.4049/jimmunol.1600859
83. Van Dyke TE, Hasturk H, Kantarci A, Freire MO, Nguyen D, Dalli J, et al. Proresolving nanomedicines activate bone regeneration in periodontitis. J Dent Res. (2015) 94:148–56. doi: 10.1177/0022034514557331
84. Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. (2007) 179:7021–9. doi: 10.4049/jimmunol.179.10.7021
85. Ferguson B, Bokka NR, Maddipati KR, Ayilavarapu S, Weltman R, Zhu L, et al. Distinct profiles of specialized pro-resolving lipid mediators and corresponding receptor gene expression in periodontal inflammation. Front Immunol. (2020) 11:1307. doi: 10.3389/fimmu.2020.01307
86. Corrêa MG, Pires PR, Ribeiro FV, Pimentel SZ, Casarin RC, Cirano FR, et al. Systemic treatment with resveratrol and/or curcumin reduces the progression of experimental periodontitis in rats. J Periodontal Res. (2017) 52:201–9. doi: 10.1111/jre.12382
87. Ikeda E, Ikeda Y, Wang Y, Fine N, Sheikh Z, Viniegra A, et al. Resveratrol derivative-rich melinjo seed extract induces healing in a murine model of established periodontitis. J Periodontol. (2018) 89:586–95. doi: 10.1002/JPER.17-0352
88. Ribeiro FV, Pino DS, Franck FC, Benatti BB, Tenenbaum H, Davies JE, et al. Resveratrol inhibits periodontitis-related bone loss in rats subjected to cigarette smoke inhalation. J Periodontol. (2017) 88:788–98. doi: 10.1902/jop.2017.170025
89. Corrêa MG, Absy S, Tenenbaum H, Ribeiro FV, Cirano FR, Casati MZ, et al. Resveratrol attenuates oxidative stress during experimental periodontitis in rats exposed to cigarette smoke inhalation. J Periodontal Res. (2019) 54:225–32. doi: 10.1111/jre.12622
90. Steinberg J, Halter J, Schiller H, Gatto L, Carney D, Lee HM, et al. Chemically modified tetracycline prevents the development of septic shock and acute respiratory distress syndrome in a clinically applicable porcine model. Shock. (2005) 24:348–56. doi: 10.1097/01.shk.0000180619.06317.2c
91. Steinberg J, Halter J, Schiller HJ, Dasilva M, Landas S, Gatto LA, et al. Metalloproteinase inhibition reduces lung injury and improves survival after cecal ligation and puncture in rats. J Surg Res. (2003) 111:185–95. doi: 10.1016/S0022-4804(03)00089-1
92. Gendrot M, Andreani J, Jardot P, Hutter S, Delandre O, Boxberger M, et al. In vitro antiviral activity of doxycycline against SARS-CoV-2. Molecules. (2020) 25:5064. doi: 10.3390/molecules25215064
93. Campagna M, Rivas C. Antiviral activity of resveratrol. Biochem Soc Trans. (2010) 38(Pt 1):50–3. doi: 10.1042/BST0380050
94. Lin SC, Ho CT, Chuo WH, Li S, Wang TT, Lin CC. Effective inhibition of MERS-CoV infection by resveratrol. BMC Infect Dis. (2017) 17:144. doi: 10.1186/s12879-017-2253-8
Keywords: COVID-19, SARS-CoV-2, periodontal research, ARDS, PMN hyperactivation
Citation: Cardoso EOC, Fine N, Glogauer M, Johnson F, Goldberg M, Golub LM and Tenenbaum HC (2021) The Advent of COVID-19; Periodontal Research Has Identified Therapeutic Targets for Severe Respiratory Disease; an Example of Parallel Biomedical Research Agendas. Front. Dent. Med. 2:674056. doi: 10.3389/fdmed.2021.674056
Received: 28 February 2021; Accepted: 26 April 2021;
Published: 20 May 2021.
Edited by:
Martha J. Somerman, National Institutes of Health (NIH), United StatesReviewed by:
Iain Chapple, University of Birmingham, United KingdomCarina Maciel Silva-Boghossian, Federal University of Rio de Janeiro, Brazil
Copyright © 2021 Cardoso, Fine, Glogauer, Johnson, Goldberg, Golub and Tenenbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Howard C. Tenenbaum, aG93YXJkLnRlbmVuYmF1bUBzaW5haWhlYWx0aC5jYQ==