 Ines Kapferer-Seebacher
Ines Kapferer-Seebacher Lena Foradori1
Lena Foradori1 Johannes Zschocke
Johannes Zschocke Reinhard Schilke
Reinhard Schilke- 1Department of Dental and Oral Medicine and Cranio-Maxillofacial and Oral Surgery, University Hospital for Dental Prosthetics and Restorative Dentistry, Medical University of Innsbruck, Innsbruck, Austria
- 2Division of Human Genetics, Medical University of Innsbruck, Innsbruck, Austria
- 3Department of Conservative Dentistry, Periodontology and Preventive Dentistry, Hannover Medical School, Hannover, Germany
In adolescents periodontal destruction may be the primary manifestation of an as yet unrecognized rare systemic disease, and it may be up to the periodontist to make the correct tentative diagnosis. Many genetic diseases that present with primary periodontal manifestations in adolescence affect immune function, sometimes with only mild or absent systemic features. They include periodontal Ehlers-Danlos syndrome (lack of attached gingiva, various connective tissue abnormalities), Papillon-Lefèvre syndrome (palmoplantar hyperkeratosis), and plasminogen deficiency (fibrin deposition within mucous membranes). Other immune disorders with severe periodontitis manifesting in adolescence are usually diagnosed in early childhood due to unmistakeable systemic features. They include Cohen syndrome (developmental disorder, truncal obesity, and microcephaly), Hermansky-Pudlak Syndrome (oculocutaneous albinism, bleeding diathesis, and other systemic manifestations), glycogen storage disease type 1b, and Chediak-Higashi syndrome (pyogenic infections, albinism, and neuropathy). The structural integrity of periodontal tissue is affected in genodermatoses such as Kindler syndrome, a type of epidermolysis bullosa. In primary hyperoxaluria, inflammatory periodontal destruction is associated with renal calculi. Breakdown of periodontal tissues independent of dental plaque biofilm-induced periodontitis is found in hypophosphatasia (highly variable skeletal hypomineralization) or isolated odontohypophosphatasia, hypophosphatemic rickets and primary hyperparathyroidism. Finally, alveolar osteolysis mimicking localized periodontitis may be due to neoplastic processes, e.g., in neurofibromatosis type 1 (typical skin features including café au lait macules and neurofibromas), Langerhans cell histiocytosis (locally destructive proliferation of bone marrow-derived immature myeloid dendritic cells), and Gorham-Stout disease (diffuse cystic angiomatosis of bone).
Introduction
The current “2017” classification of periodontal and peri-implant diseases and conditions distinguishes “periodontitis” from necrotizing periodontal diseases, periodontitis as a manifestation of systemic disease, and non-inflammatory periodontal manifestation of systemic disease (1, 2). Periodontitis in general is a highly prevalent condition characterized by inflammatory destruction of periodontal tissues (2). In most affected individuals it is a multifactorial disorder caused by complex interactions between various environmental and genetic factors that in combination determine the variability of disease onset, progression and severity. Different forms of the disease previously recognized in the classification of 1999 as “chronic” or “aggressive,” and in the classification of 1989 as “adult,” “refractory” and “early-onset periodontitis” —further including “prepubertal,” “juvenile,” and “rapidly progressive periodontitis” —are now grouped under a single category (“periodontitis”) (2). Genetic predisposition in this context means that an individual has a constitutional susceptibility to develop periodontitis which only becomes manifest in conjunction with other pathogenic factors. Not all persons harboring the genetic risk factors will develop the disease (3).
In contrast, there are a number of specific rare syndromes that are associated with periodontitis in most or all affected individuals irrespective of other risk factors. Many of them are monogenic diseases caused by alterations of specific genes. They are usually rare diseases, i.e., they have a prevalence of <1 case per 2,000 in the population (4). Approximately 8,000 different rare diseases have been identified so far, with up to 14% showing periodontal involvement (5). In the 2017 classification, conditions associated with presentation of severe periodontitis are grouped in the category “Periodontitis as a Manifestation of Systemic Disease.” Other systemic conditions that affect the structural integrity of the periodontal apparatus independently of biofilm-induced inflammatory destruction—including neoplastic disorders but also rare genetic diseases—are grouped as “Systemic Diseases or Conditions Affecting the Periodontal Supporting Tissues” (1).
In adolescents, i.e., the age range of 10–19 years (6), the finding of the correct periodontal diagnosis is quite challenging. Slowly advancing multifactorial periodontitis has to be differentiated from rapidly progressing forms with early onset as well as rare systemic diseases and conditions affecting the periodontal supporting structures (Figure 1). Incipient loss of periodontal support due to general/multifactorial periodontitis is common in most teenage populations and is causally linked to the presence of local irritants. Usually only minor loss of periodontal attachment is found (8). Botero et al. summarized several clinical studies from Latin America and concluded that periodontitis affects up to 10% of young Latin Americans, with large fluctuations of prevalence and severity within the studies (9). Among Chilean adolescents aged 12–21 years, clinical attachment loss ≥1 mm was seen in 69.2%, ≥2 mm in 16%, and ≥3 mm in 4.5% of cases (10).
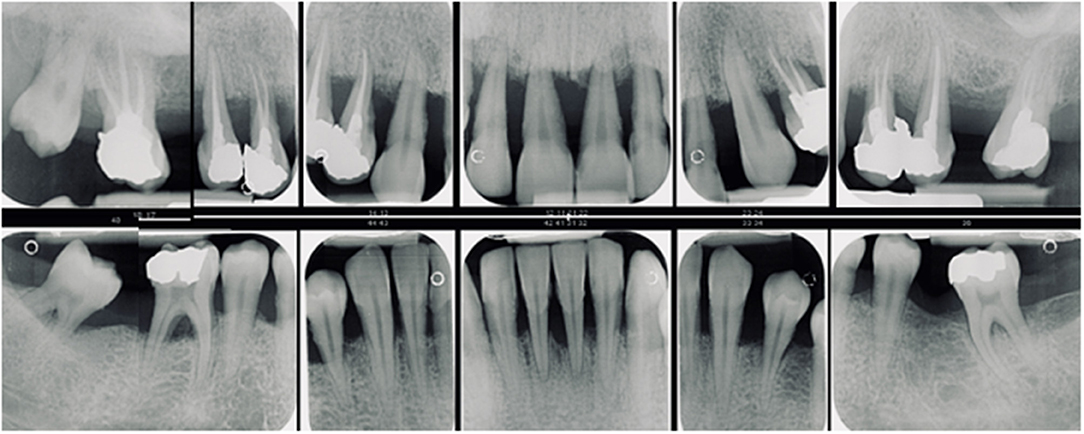
Figure 1. Generalized periodontitis with early onset in a female patient aged 18 years. With a prevalence of 0.1% in white populations it is more likely to see young people with generalized complex periodontitis than an individual with a rare genetic disease. However, the detection of rare diseases increases with physicians awareness (7) and although the prevalence of each individual orphan disease may be low, taken together they affect up to 5% of the general population (7).
Around puberty a subset of otherwise healthy individuals may experience rapidly progressing periodontal destruction, previously called early-onset or aggressive periodontitis, which may be localized or generalized. This has been estimated to affect ~0.1% of white populations and up to 2.6% of black populations (8).
In some adolescents, periodontal attachment loss is a manifestation of defined monogenic conditions. In case of mild systemic features the underlying cause may not yet been identified, and it may be the task of the periodontist to recognize the abnormality in order to make a tentative diagnosis.
Here we present an overview of rare genetic diseases which still were not be diagnosed in adolescents and are associated with an initial diagnosis of periodontal involvement in this age group (Table 1). Genetic diseases that typically present with perpubertal onset of periodontal destruction are excluded or mentioned only briefly.
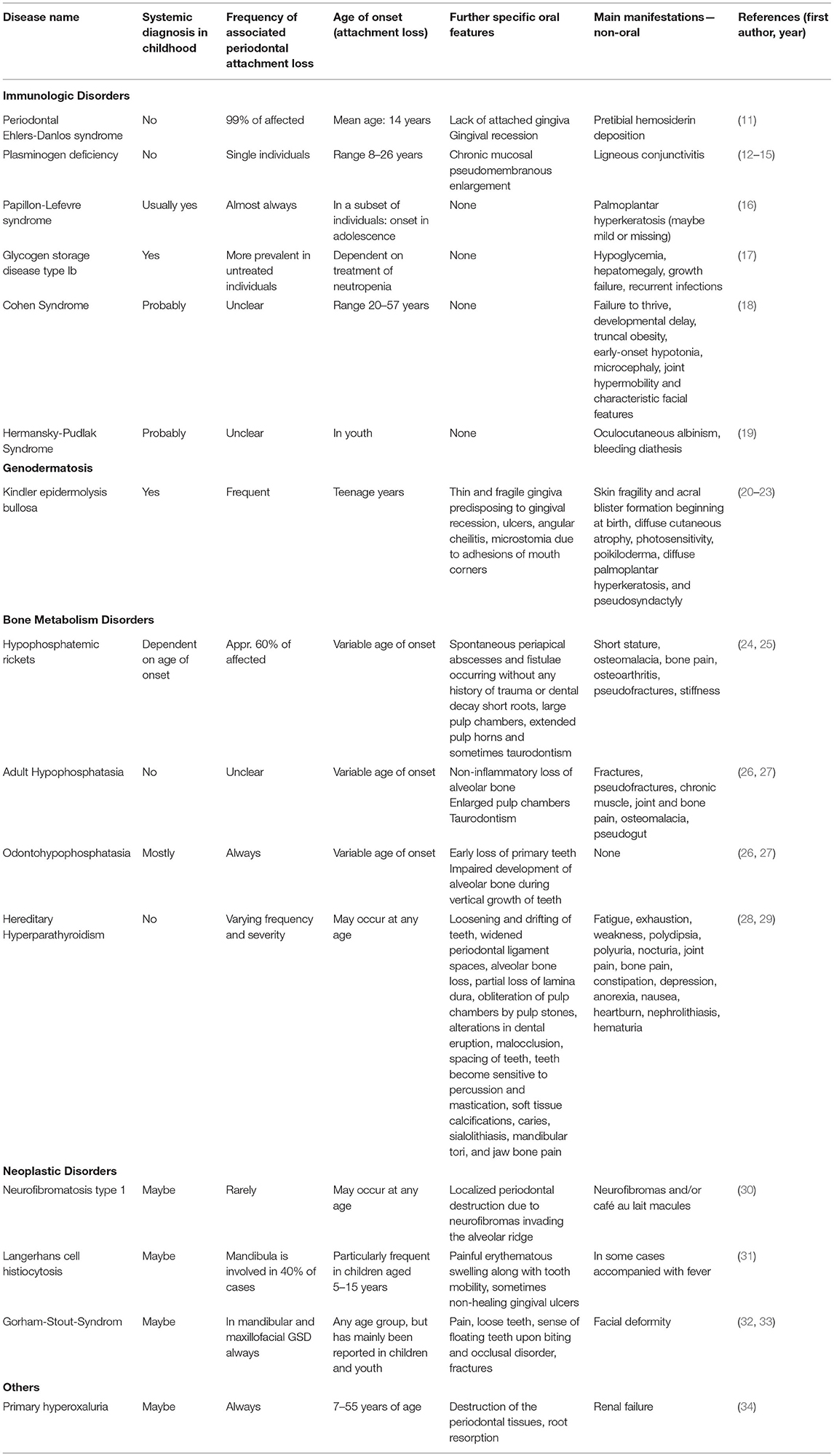
Table 1. Periodontitis as manifestation of systemic diseases with primary onset in adolescence.
Periodontitis As Manifestation of Systemic Diseases
Immunologic Disorders
Many of the systemic disorders that typically present periodontitis at an early age affect immune function. They include primary immune deficiencies with congenital defects of phagocyte number and/or function, diseases of immune dysregulation, and other well-defined immunodeficiency syndromes (19, 35). Severe immune deficiencies are usually diagnosed by pediatricians in early childhood and are not included in this review. Some disorders of immune function—although hereditary—may have their onset in adolescence or youth, and affected individuals may therefore still lack systemic diagnosis. These disorders are described below.
Periodontal Ehlers-Danlos syndrome (OMIM 130080, 617174) is a specific Ehlers-Danlos entity with autosomal-dominant inheritance caused by heterozygous mutations in complement 1 subunits r and s (C1S, C1R). The defining clinical feature is early severe periodontitis diagnosed at a mean age of 14 years (11). Systemic features range from mild or absent to severe with organ or vascular ruptures. Pretibial hyperpigmentation found in 80% of affected individuals is the second defining feature and is caused by hemosiderin deposition (36). Bruising out of proportion to trauma (>90%) and joint hypermobility of mostly distal joints (~40%) have been reported. The dentist can distinguish periodontal Ehlers-Danlos syndrome from other types of periodontitis by the striking lack of attached gingiva causing oral tissue fragility and predisposing to gingival recession (Figure 2). Pattern of alveolar bone loss was described as localized to any region, proceeding in a domino effect from one tooth to the next. Some individuals report on early loss of some primary teeth. For details see (37, 38). The diagnosis of pEDS is reached by molecular genetic analysis with identification of specific heterozygous mutations in the C1S or C1R genes.
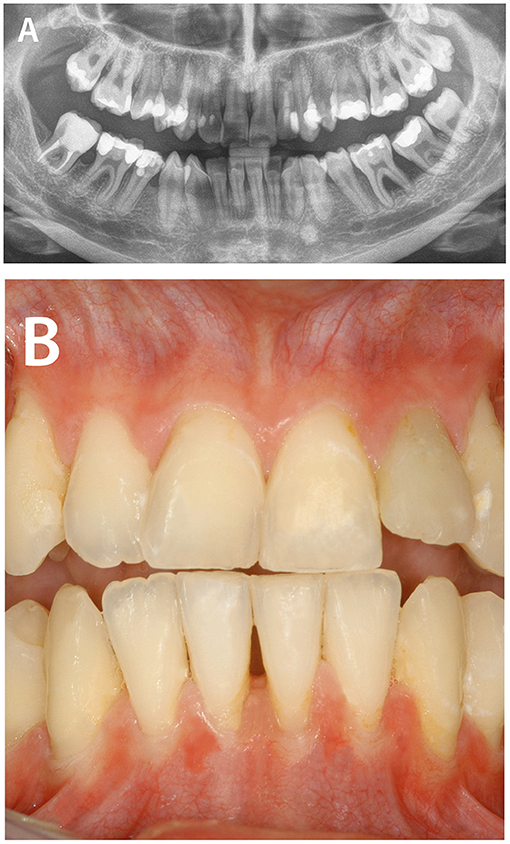
Figure 2. Periodontitis as manifestation of systemic diseases. Periodontal Ehlers-Danlos syndrome is caused by heterozygous mutations in complement 1 subunit genes C1R and C1S leading to hyperactivation of the classical complement pathway. The defining clinical feature is early severe periodontitis diagnosed at a mean age of 14 years. (A) Pattern of alveolar bone loss was described as localized to any region, proceeding in a domino effect from one tooth to the next. (B) The clinician can distinguish periodontal Ehlers-Danlos syndrome from other types of periodontitis by the lack of attached gingiva causing oral tissue fragility and predisposing to severe gingival recession.
Papillon-Lefèvre syndrome (OMIM 245000) is caused by homozygous or compound heterozygous mutations in the cathepsin C gene (CTSC) encoding a cysteine protease involved in a variety of immune and inflammatory pathways. Papillon-Lefèvre syndrome usually presents with severe periodontitis of primary and secondary teeth in early childhood in association with palmoplantar hyperkeratosis. In a subset of individuals mutations in CTSC present as an isolated finding of periodontitis with onset in adolescence (16). The diagnosis is again reached by molecular genetic analysis.
Autosomal-recessive plasminogen deficiency (OMIM 217090) is caused by homozygous or compound heterozygous mutations in the gene encoding plasminogen (PLG). Plasminogen is not only a complement inhibitor but also a broad-spectrum proteolytic factor either by directly degrading extracellular matrix proteins, e.g., laminin, fibronectin and proteoglycans, and indirectly by activating latent metalloproteinases (39). Plasminogen deficiency is associated with massive fibrin deposition within mucous membranes due to the absence of fibrin clearance by plasmin, leading to a so-called “ligneous” conjunctivitis (80% of patients), ligneous gingivitis (34%) and ligneous periodontitis, characterized by extensive generalized periodontal breakdown in association with pseudomembranous gingival enlargement (12–15). The diagnosis is confirmed by the identification of biallelic pathogenic PLG mutations in molecular genetic analysis.
Other immunologic disorders described in the literature with onset of severe periodontitis in adolescence are usually associated with typical systemic manifestations diagnosed in early childhood. For example, autosomal-recessive Cohen syndrome (MIM 216550) predisposes to periodontitis with early onset in adolesence or youth with alveolar bone loss of up to 10 mm (18). Due to characteristic systemic features—failure to thrive, developmental delay, truncal obesity, early-onset hypotonia, microcephaly, joint hypermobility and characteristic facial features—it is usually diagnosed in early childhood (40). The same is true for Hermansky-Pudlak Syndrome (OMIM 203300), comprising a group of rare autosomal recessive disorders with oculocutaneous albinism, bleeding diathesis, and in some individuals, granulomatous colitis, severe chronic neutropenia and immunodeficiency, and sometimes fatal pulmonary fibrosis (41). It is due to defects in lysosome-related organelles and may be caused by pathogenic variants in at least 10 different genes (42). Hermansky-Pudlak syndrome is confirmed by molecular analysis of the candidate genes by massive parallel sequencing.
Some children affected by monogenic diseases associated with severe chronic neutropenia such as glycogen storage disease type 1b, Chediak-Higashi syndrome, and others are typically treated with granulocyte colony stimulating factor (G-CSF) which prevents periodontitis as manifestation of the disease in the permanent dentition. Non-regular administration by the patient or failure to adjust the G-CSF dose to the body weight or the individual response can lead to periodontal destruction in adolescence. For patients with severe chronic neutropenia who do not respond to G-CSF treatment, hematopoietic stem-cell transplantation is currently considered the only treatment available (43).
Genodermatoses
A second category of genetic diseases manifesting with severe periodontitis in adolescence are conditions affecting the structural integrity of periodontal tissues. An example is Kindler syndrome, a type of epidermolysis bullosa characterized by impaired adhesion of gingival junctional epithelium to the tooth which predisposes to severe generalized periodontitis in teenage years (44, 45). Due to striking systemic features with acral blisters and skin atrophy the syndrome is usually diagnosed in early childhood. Kindler syndrome is inherited as an autosomal recessive disease caused by mutations in the FERMIT1 gene.
Others
Primary hyperoxaluria (MIM 259900) is an autosomal-recessive deficiency of peroxisomal alanine glyoxylate aminotransferase encoded by the AGXT gene resulting in reduced or absent conversion of glyoxylate to glycine and leading to accumulation of calcium oxalate in various bodily tissues, especially the kidney. Inflammatory periodontal destruction with primary hyperoxaluria has been attributed to a foreign body reaction caused by deposition of calcium oxalate crystals in periodontal tissues (46–48). External root resorptions were common in all case reports. Reported age of onset ranges from 7 to 55 years (34). The diagnosis is confirmed by increased urinary concentrations of oxalate and glycate or by molecular genetic studies.
Rare Diseases or Conditions Affecting The Periodontal Supporting Tissues
Some systemic conditions mimic the clinical presentation of periodontitis although breakdown of periodontal tissues is independent of dental plaque biofilm-induced periodontitis (1, 49). Some may manifest in adolescence.
Disorders of Bone Metabolism
Hypophosphatasia (MIM 146300) is a rare genetic disorder caused by mutations in ALPL encoding tissue-non-specific alkaline phosphatase. It is usually inherited as an autosomal-recessive trait, some mutations may cause an attenuated late onset form. The disease is characterized by mineralization defects of bone and teeth. Early tooth loss is not caused by inflammatory bone loss but rather by cement dysplasia resulting in compromised periodontal attachment (50). In most cases, the reduced height of the bony alveolar attachment in the first dentition and early mixed dentition is a consequence of the absence of alveolar process development during the vertical growth of the teeth. The severity and age of onset is highly variable ranging from severe intrauterine onset skeletal hypomineralization and deformities to odontohypophosphatasia, i.e., isolated tooth loss without skeletal alterations that may present at any age (51). Independent of the highly variable clinical presentation, most affected individuals show premature loss of deciduous teeth at <3 years of age, affecting all primary teeth or just those in the incisor–canine region (26). The condition may be recognized by reduced serum concentrations of alkaline phosphatase (AP) and is confirmed by mutation studies. For reviews see (26, 27).
Genetic disorders of bone metabolism like hypophosphatemic rickets and primary hyperparathyroidism are associated with varying frequency, severity, and age of onset of periodontal involvement, depending on etiology, systemic treatment, and as yet unknown factors (52). Increased risk for severe periodontal breakdown in hypophosphatemic rickets is probably caused by cementum hypoplasia and altered alveolar bone repair (53, 54). The most frequent type is X-linked hypophosphataemia due to mutations in the X-chromosomal PHEX gene, with manifestation in both males and females, but hypophosphatemic rickets can also be caused by mutations in a range of autosomal genes with dominant or recessive inheritance. For primary hyperparathyroidism the evidence is somehow contradictory. Hyperparathyroidism per se does not seem to be associated with a higher prevalence of periodontitis (55, 56). However, high serum levels of Ca2+ (≥1.51 mmol/L) are associated with an increased probability of tooth extractions (56). Loosening and drifting of teeth due to widened periodontal ligament spaces and partial loss of lamina dura may be misdiagnosed as periodontitis (28). Other intraoral manifestations of hyperparathyroidism include pulp calcifications, dental sensitivity to percussion and mastication, soft tissue calcifications, mandibular tori, and jawbone pain (28, 29). Primary hyperparathyroidism is associated with pathogenic variants in a range of genes.
Neoplastic Disorders
Rare genetic disorders causing neoplastic proliferation may present in adolescence as alveolar osteolysis mimicking localized periodontitis or other neoplastic processes like squamous cell carcinomas (Figure 3).
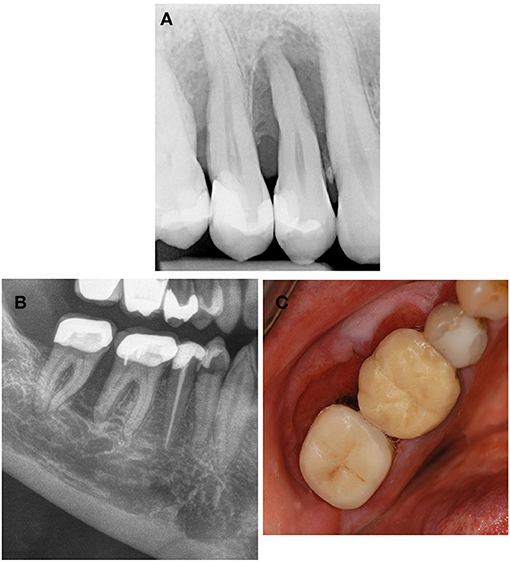
Figure 3. Systemic diseases and conditions affecting the periodontal supporting tissues—neoplastic disorders. (A) Neurofibromatosis type 1 is a neurocutaneous disorder caused by mutation or deletion in the NF-1 gene and affecting the cell growth of neuronal tissues. Plexiform neurofibromas can also invade the alveolar ridge mimicking localized periodontitis. (B,C) Squamous cell carcinoma invading the alveolar bone is a differential diagnosis to benign neoplastic disorders but also to localized periodontitis.
Neurofibromatosis type 1 (MIM 162200) (Von Recklinghausen disease) is an autosomal-dominant neurocutaneous disorder affecting the cell growth of neuronal tissues. It is caused by heterozygous mutations in the NF1 gene. Typical skin features include café au lait macules that increase in number and size, axillary and/or inguinal freckles, and the development of dermal neurofibromas which usually have led to the diagnosis before adolescence. There is a range of other manifestations but individuals with only mild neurocutaneous features may escape diagnosis in childhood. Oral manifestations are found in about 72% of the patients; they include impacted or ankylosed teeth, abnormalities in form or number of teeth, as well as oral plexiform neurofibromas (30). The latter commonly affect the tongue, palate, buccal mucosa, floor of the mouth, gingiva or lips, and can also invade the alveolar ridge mimicking localized periodontitis (30) (Figure 3).
Langerhans cell histiocytosis is a benign but locally destructive entity that represents true neoplastic proliferation of bone marrow-derived immature myeloid dendritic cells but lacks malignant changes histologically. It is a non-hereditary disorder associated with somatic Langerhans cell mutations that activate the cellular MAPK/ERK signaling pathway and is particularly frequent in children aged 5–15 years (57). The mandibula is involved in 40% of cases, presenting as painful erythematous swelling along with tooth mobility, in some cases accompanied with non-healing gingival ulcers and fever (31). From a radiographic point of view, a similarity of bone lesions to a wide spectrum of diseases was described, such as odontogenic cysts, certain benign and malignant tumors, periapical lesions, and alveolar bone loss (58).
Gorham-Stout disease (synonyms: diffuse cystic angiomatosis of bone; disappearing / vanishing / phantom bone disease) (OMIM 123880) is a rare disorder with unknown etiology, characterized by progressive osteolysis associated with angiomatous proliferation and dilation of lymphatic vessels. It can affect any bone of the human body leading to partial or complete resorption (59). Gorham-Stout disease may start at any age but is most commonly diagnosed in children and young adults (average age of diagnosis is 25 years) (33). Mandibular or maxillofacial involvement is associated with pain, loose teeth, sense of floating teeth upon biting and occlusal disorder, spontaneous fractures, and facial deformity (32, 33, 60).
Discussion
Due to the rarity and diversity of genetic disorders manifesting in the oral and maxillofacial region, even specialist dentists find it difficult to remember and diagnose them (61). In youth, severe multifactorial periodontitis may be difficult to differentiate from rare genetic diseases with sometimes only mild systemic manifestations. Using a systematic approach—as discussed below for adolescents aged 10–19 years—may assist in finding the correct periodontal diagnosis.
An adolescent with severe periodontal destruction should first of all receive detailed clinical and radiologic periodontal diagnostics followed by a critical assessment of the oral soft tissue quality. Thin, translucent and fragile gums or missing frenula may point to connective tissue disease like Ehlers-Danlos syndromes. Pseudomembranous gingival hypertrophy is a typical sign of plasminogen deficiency. Missing inflammatory signs in association with generalized periodontal destruction points to cement dysplasia. Radiologic analysis may elucidate further dental features giving a hint to the underlying systemic diseases, e.g., taurodontism has been reported as additional feature of CTSC mutations (16), pulp calcifications are associated with renal disorders and connective tissue diseases like Ehlers-Danlos syndromes (62, 63).
Histologic analysis of extracted teeth or soft-tissues and in case of osteolytic processes bone marrow biopsies will point to the right diagnosis.
Periodontists have to take a detailed medical history with special focus on immunologic diseases, systemic manifestations of bone metabolism disorders like bone, joint or muscle pain, features of connective tissue diseases like bruising out of proportion to trauma and joint hypermobility, and dermatologic problems. An accurate family health history as well as drawing a pedigree are valuable tools to illustrate how conditions are passed down through generations and to define the pattern of inheritance (autosomal-recessive, autosomal-dominant, X-linked, and mitochondrial).
Inspection of hair and skin may give a hint to ectodermal syndromes, connective tissue diseases or genodermatoses like Papillon-Lefèvre and Haim-Munk syndrome. Specialized pediatricians, dermatologists and/or geneticists may be helpful with the further clarification of the diagnosis. However, the periodontist has to be aware of that competent genetic analysis relies on precise clinical diagnosis.
Conditions that increase the risk of complex and biofilm-associated periodontitis were not included in the present review. For example, it was previously shown that systemic lupus erythematosus and familial scleroderma are associated with an increased risk of biofilm-induced periodontitis, with a risk ratio of 1.76 (95% CI 1.29–2.41) and 1.84 (95%CI 1.29–2.41), respectively (64, 65). In the authors' opinion the diagnosis in these cases should be (multifactorial) “periodontitis” as the systemic disease represents a risk factor for periodontitis rather than a specific monogenic cause. In the end this is a purely semantic discussion which neglects the fact that monogenic and multifactorial conditions represent a pathogenetic spectrum which cannot be forced into simple clearly separated categories. The majority of clinical phenotypes are due to a variety of (often as yet unknown) genetic variants of variable penetrance and population frequencies in conjunction with various non-genetic influences in the individual case.
This does not negate the need to make a rapid, precise diagnosis in individuals with a specific monogenic disease. Most of these conditions can now be confirmed with widely available massive parallel (next generation) DNA sequencing approaches. Nevertheless, surveys of patients and physicians found that it takes more than a decade to reach a diagnosis for a rare disease after onset of symptoms (66, 67). Specialist dentists have to be aware of their responsibility in diagnosing rare diseases as young people with orphan diseases may still be undiagnosed especially when systemic features are mild. With precise clinical descriptions of oral features and basic knowledge on pathomechanisms they can provide valuable information to pediatricians and geneticists for finding the right systemic diagnosis.
Author Contributions
IK-S designed and directed the project, did the primary analysis of the results, and wrote the first version of the manuscript. LF and JZ substantially contributed to the literature search and revised the manuscript. RS aided in interpreting the results and worked on the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Caton JG, Armitage G, Berglundh T, Chapple ILC, Jepsen S, Kornman KS, et al. A new classification scheme for periodontal and peri-implant diseases and conditions - introduction and key changes from the 1999 classification. J Periodontol. (2018) 89(Suppl.1):S1–8. doi: 10.1002/JPER.18-0157
2. Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions. J Clin Periodontol. (2018) 45(Suppl.20):S162–70. doi: 10.1111/jcpe.12946
3. Schaefer AS. Genetics of periodontitis: discovery, biology, and clinical impact. Periodontol. (2018) 78:162–73. doi: 10.1111/prd.12232
4. Richter T, Nestler-Parr S, Babela R, Khan ZM, Tesoro T, Molsen E, et al. Rare disease terminology and definitions-a systematic global review: report of the ISPOR rare disease special interest group. Value Health. (2015) 18:906–14. doi: 10.1016/j.jval.2015.05.008
5. Hanisch M, Hoffmann T, Bohner L, Hanisch L, Benz K, Kleinheinz J, et al. Rare diseases with periodontal manifestations. Int J Environ Res Public Health. (2019) 1:508676. doi: 10.3390/ijerph16050867
6. WHO. Adolescent Health. (2021). Available online at: https://www.who.int/southeastasia/health-topics/adolescent-health (accessed March 1, 2021).
7. Vanakker O, Callewaert B, Malfait F, Coucke P. The genetics of soft connective tissue disorders. Ann Rev Genom Hum Genet. (2015) 16:229–55. doi: 10.1146/annurev-genom-090314-050039
8. Jenkins WM, Papapanou PN. Epidemiology of periodontal disease in children and adolescents. Periodontol. (2001) 26:16–32. doi: 10.1034/j.1600-0757.2001.2260102.x
9. Botero JE, Rosing CK, Duque A, Jaramillo A, Contreras A. Periodontal disease in children and adolescents of Latin America. Periodontol. (2015) 67:34–57. doi: 10.1111/prd.12072
10. Lopez R, Frydenberg M, Baelum V. Clinical features of early periodontitis. J Periodontol. (2009) 80:749–58. doi: 10.1902/jop.2009.080463
11. Kapferer-Seebacher I, Pepin M, Werner R, Aitman TJ, Nordgren A, Stoiber H, et al. Periodontal ehlers-danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1r and C1s of complement. Am J Hum Gen. (2016) 99:1005–14. doi: 10.1016/j.ajhg.2016.08.019
12. Neering SH, Adyani-Fard S, Klocke A, Ruttermann S, Flemmig TF, Beikler T. Periodontitis associated with plasminogen deficiency: a case report. BMC Oral Health. (2015) 15:59. doi: 10.1186/s12903-015-0045-3
13. Kurtulus Waschulewski I, Gokbuget AY, Christiansen NM, Ziegler M, Schuster V, Wahl G, et al. Immunohistochemical analysis of the gingiva with periodontitis of type I plasminogen deficiency compared to gingiva with gingivitis and periodontitis and healthy gingiva. Arch Oral Biol. (2016) 72:75–86. doi: 10.1016/j.archoralbio.2016.07.013
14. Ertas U, Saruhan N, Gunhan O. Ligneous periodontitis in a child with plasminogen deficiency. Niger J Clin Pract. (2017) 20:1656–8. doi: 10.4103/1119-3077.164357
15. Sadasivan A, Ramesh R, Mathew DG. Ligneous periodontitis in a patient with type 1 plasminogen deficiency: a case report and review of the literature. Case Rep Dent. (2020) 2020:5680535. doi: 10.1155/2020/5680535
16. Molitor A, Prud'homme T, Miao Z, Conrad S, Bloch-Zupan A, Pichot A, et al. Exome sequencing identifies a novel missense variant in CTSC causing nonsyndromic aggressive periodontitis. J Hum Genet. (2019) 64:689–94. doi: 10.1038/s10038-019-0615-3
17. Mortellaro C, Garagiola U, Carbone V, Cerutti F, Marci V, Bonda PL. Unusual oral manifestations and evolution in glycogen storage disease type Ib. J Craniofac Surg. (2005) 16:45–52. doi: 10.1097/00001665-200501000-00010
18. Alaluusua S, Kivitie-Kallio S, Wolf J, Haavio ML, Asikainen S, Pirinen S. Periodontal findings in Cohen syndrome with chronic neutropenia. J Periodontol. (1997) 68:473–8. doi: 10.1902/jop.1997.68.5.473
19. Jung S, Gies V, Korganow AS, Guffroy A. Primary immunodeficiencies with defects in innate immunity: focus on orofacial manifestations. Front Immunol. (2020) 11:1065. doi: 10.3389/fimmu.2020.01065
20. Yıldırım TT, Kaya FA, Taşkesen M, Dündar S, Bozoğlan A, Tekin GG, et al. Aggressive periodontitis associated with Kindler syndrome in a large Kindler syndrome pedigree. Turk J Pediatr. (2017) 59:56–61. doi: 10.24953/turkjped.2017.01.009
21. Suman N, Kaur S, Kaur S, Sarangal V. Kindler's syndrome: A rare case report. Contemp Clin Dent. (2014) 5:217–20. doi: 10.4103/0976-237X.132342
22. Wiebe CB, Petricca G, Häkkinen L, Jiang G, Wu C, Larjava HS. Kindler syndrome and periodontal disease: review of the literature and a 12-year follow-up case. J Periodontol. (2008) 79:961–6. doi: 10.1902/jop.2008.070167
23. Wiebe CB, Penagos H, Luong N, Slots J, Epstein E Jr, Siegel D, et al. Clinical and microbiologic study of periodontitis associated with Kindler syndrome. J Periodontol. (2003) 74:25–31. doi: 10.1902/jop.2003.74.1.25
24. Ye L, Liu R, White N, Alon US, Cobb CM. Periodontal status of patients with hypophosphatemic rickets: a case series. J Periodontol. (2011) 82:1530–5. doi: 10.1902/jop.2011.100736
25. Croonenborghs TM, Grisar K, Politis C. Hereditary hypophosphatemic rickets and spontaneous dental abscesses: a case report. Oral Health Care. 3:1–4. doi: 10.15761/OHC.1000137
26. Bloch-Zupan A. Hypophosphatasia: diagnosis and clinical signs - a dental surgeon perspective. Int J Paediatr Dent. (2016) 26:426–38. doi: 10.1111/ipd.12232
27. Bloch-Zupan A, Vaysse F. Hypophosphatasia: oral cavity and dental disorders. Arch Pediatr. (2017) 24:5S80–85S84. doi: 10.1016/S0929-693X(18)30020-4
28. Khalekar Y, Zope A, Brahmankar U, Chaudhari L. Hyperparathyroidism in dentistry: issues and challenges!! Indian J Endocrinol Metab. (2016) 20:581–2. doi: 10.4103/2230-8210.183452
29. Kakade SP, Gogri AA, Umarji HR, Kadam SG. Oral manifestations of secondary hyperparathyroidism: a case report. Contemp Clin Dent. (2015) 6:552–8. doi: 10.4103/0976-237X.169840
30. Javed F, Ramalingam S, Ahmed HB, Gupta B, Sundar C, Qadri T, et al. Oral manifestations in patients with neurofibromatosis type-1: a comprehensive literature review. Crit Rev Oncol Hematol. (2014) 91:123–9. doi: 10.1016/j.critrevonc.2014.02.007
31. Chugh A, Kaur A, Kumar Patnana A, Kumar P, Chugh VK. Unisystem Langerhans cell histiocytosis in maxillofacial region in pediatrics: comprehensive and systematic review. Oral Maxillofac Surg. (2021). doi: 10.1007/s10006-021-00949-9. [Epub ahead of print].
32. Liu M, Liu W, Qiao C, Han B. Mandibular Gorham-Stout disease: a case report and literature review. Medicine. (2017) 96:e8184. doi: 10.1097/MD.0000000000008184
33. Jagtap R, Gupta S, Lamfon A, Ruprecht A, Schlott B, Hardeman J, et al. Gorham-Stout disease of the mandible: case report and review of literature of a rare type of osteolysis. Oral Radiol. (2020) 36:389–94. doi: 10.1007/s11282-019-00417-x
34. Mitsimponas KT, Wehrhan T, Falk S, Wehrhan F, Neukam FW, Schlegel KA. Oral findings associated with primary hyperoxaluria type I. J Craniomaxillofac Surg. (2012) 40:e301–6. doi: 10.1016/j.jcms.2012.01.009
35. Silva LM, Brenchley L, Moutsopoulos NM. Primary immunodeficiencies reveal the essential role of tissue neutrophils in periodontitis. Immunol Rev. (2019) 287:226–35. doi: 10.1111/imr.12724
36. Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. (2017) 175:8–26. doi: 10.1002/ajmg.c.31552
37. Kapferer-Seebacher I, Lundberg P, Malfait F, Zschocke J. Periodontal manifestations of Ehlers-Danlos syndromes: a systematic review. J Clin Periodontol. (2017) 44:1088–100. doi: 10.1111/jcpe.12807
38. Kapferer-Seebacher I, Oakley-Hannibal E, Lepperdinger U, Johnson D, Ghali N, Brady AF, et al. Prospective clinical investigations of children with periodontal Ehlers-Danlos syndrome identify generalized lack of attached gingiva as a pathognomonic feature. Genet Med. (2021) 23:316–22. doi: 10.1038/s41436-020-00985-y
39. Barthel D, Schindler S, Zipfel PF. Plasminogen is a complement inhibitor. J Biol Chem. (2012) 287:18831–42. doi: 10.1074/jbc.M111.323287
40. Wang H, Falk MJ, Wensel C, Traboulsi EI. Cohen syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2006).
41. Huizing M, Malicdan MCV, Gochuico BR, Gahl WA. Hermansky-Pudlak Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2000).
42. Huizing M, Malicdan MCV, Wang JA, Pri-Chen H, Hess RA, Fischer R, et al. Hermansky-Pudlak syndrome: mutation update. Hum Mutat. (2020) 41:543–80. doi: 10.1002/humu.23968
43. Skokowa J, Germeshausen M, Zeidler C, Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hematol. (2007) 14:22–8. doi: 10.1097/00062752-200701000-00006
44. Siegel DH, Ashton GH, Penagos HG, Lee JV, Feiler HS, Wilhelmsen KC, et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am J Hum Genet. (2003) 73:174–87. doi: 10.1086/376609
45. Petricca G, Leppilampi M, Jiang G, Owen GR, Wiebe C, Tu Y, et al. Localization and potential function of kindlin-1 in periodontal tissues. Eur J Oral Sci. (2009) 117:518–27. doi: 10.1111/j.1600-0722.2009.00651.x
46. Fantasia JE, Miller AS, Chen SY, Foster WB. Calcium oxalate deposition in the periodontium secondary to chronic renal failure. Oral Surg Oral Med Oral Pathol. (1982) 53:273–9. doi: 10.1016/0030-4220(82)90303-6
47. Boyce BF, Prime SS, Halls D, Johnston E, Critchlow H, Macdonald DG, et al. Does osteomalacia contribute to development of oral complications of oxalosis? Oral Surg Oral Med Oral Pathol. (1986) 61:272–7. doi: 10.1016/0030-4220(86)90374-9
48. Panis V, Tosios KI, Gagari E, Griffin TJ, Damoulis PD. Severe periodontitis in a patient with hyperoxaluria and oxalosis: a case report and review of the literature. J Periodontol. (2010) 81:1497–504. doi: 10.1902/jop.2010.100092
49. Jepsen S, Caton JG, Albandar JM, Bissada NF, Bouchard P, Cortellini P, et al. Periodontal manifestations of systemic diseases and developmental and acquired conditions: consensus report of workgroup 3 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. (2018) 89(Suppl.1):S237–48. doi: 10.1002/JPER.17-0733
50. Foster BL, Ramnitz MS, Gafni RI, Burke AB, Boyce AM, Lee JS, et al. Rare bone diseases and their dental, oral, and craniofacial manifestations. J Dent Res. (2014) 93:7S−19S. doi: 10.1177/0022034514529150
51. Whyte MP, Zhang F, Wenkert D, Mcalister WH, Mack KE, Benigno MC, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. (2015) 75:229–39. doi: 10.1016/j.bone.2015.02.022
52. Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, et al. Phosphate and vitamin D prevent periodontitis in X-linked hypophosphatemia. J Dent Res. (2017) 96:388–95. doi: 10.1177/0022034516677528
53. Coyac BR, Falgayrac G, Baroukh B, Slimani L, Sadoine J, Penel G, et al. Tissue-specific mineralization defects in the periodontium of the Hyp mouse model of X-linked hypophosphatemia. Bone. (2017) 103:334–46. doi: 10.1016/j.bone.2017.07.026
54. Zhang H, Chavez MB, Kolli TN, Tan MH, Fong H, Chu EY, et al. Dentoalveolar defects in the Hyp mouse model of X-linked hypophosphatemia. J Dent Res. (2020) 99:419–28. doi: 10.1177/0022034520901719
55. Padbury AD Jr, Tozum TF, Taba M. Jr., Ealba EL, West BT, Burney RE, et al. The impact of primary hyperparathyroidism on the oral cavity. J Clin Endocrinol Metab. (2006) 91:3439–45. doi: 10.1210/jc.2005-2282
56. Koman A, Nasman P, Discacciati A, Ekbom A, Nilsson IL, Sandborgh-Englund G. Increased risk for tooth extraction in primary hyperparathyroidism and hypercalcemia: a population study. Clin Oral Investig. (2020) 24:2755–61. doi: 10.1007/s00784-019-03137-y
57. Reisi N, Raeissi P, Harati Khalilabad T, Moafi A. Unusual sites of bone involvement in Langerhans cell histiocytosis: a systematic review of the literature. Orphanet J Rare Dis. (2021) 16:1. doi: 10.1186/s13023-020-01625-z
58. Difloe-Geisert JC, Bernauer SA, Schneeberger N, Bornstein MM, Walter C. Periodontal manifestations of Langerhans cell histiocytosis: a systematic review. Clin Oral Invest. (2021) 25:3341–9. doi: 10.1007/s00784-021-03873-0
59. Hardegger F, Simpson LA, Segmueller G. The syndrome of idiopathic osteolysis. Classification, review, and case report. J Bone Joint Surg Br. (1985) 67:88–93. doi: 10.1302/0301-620X.67B1.3968152
60. Thompson AA, Patrawala S. Gorham-Stout disease of the mandible, manubrium and cervical spine presenting as bilateral chylothorax. BMJ Case Rep. (2021) 14:237638. doi: 10.1136/bcr-2020-237638
61. Kuhne A, Kleinheinz J, Jackowski J, Koppe J, Hanisch M. Study to investigate the knowledge of rare diseases among dentists, orthodontists, periodontists, oral surgeons and craniomaxillofacial surgeons. Int J Environ Res Public Health. (2020) 18:10139. doi: 10.3390/ijerph18010139
62. Ashkenazi M, Rafe Z, Sarnat H, Levin L. Nephrocalcinosis associated with continuous enamel hypoplasia and severe alveolar bone loss: a case report and literature review. Pediatr Dent. (2014) 36:250–3.
63. Kapferer-Seebacher I, Schnabl D, Zschocke J, Pope FM. Dental manifestations of ehlers-danlos syndromes: a systematic review. Acta Derm Venereol. (2020) 100:adv00092. doi: 10.2340/00015555-3428
64. Baron M, Hudson M, Tatibouet S, Steele R, Lo E, Gravel S, et al. The Canadian systemic sclerosis oral health study: orofacial manifestations and oral health-related quality of life in systemic sclerosis compared with the general population. Rheumatology. (2014) 53:1386–94. doi: 10.1093/rheumatology/ket441
65. Rutter-Locher Z, Smith TO, Giles I, Sofat N. Association between systemic lupus erythematosus and periodontitis: a systematic review and meta-analysis. Front Immunol. (2017) 8:1295. doi: 10.3389/fimmu.2017.01295
66. Boice N, Kishnani PS, Philipson T, Levin D, Hendriksz C, Drummond M. Rare Disease Impact Report. (2013). Available online at: https://globalgenes.org/wp-content/uploads/2013/04/ShireReport-1.pdf:Shire (accessed March 25, 2021).
Keywords: orphan diseases, periodontitis, adolescence, osteolysis, immune deficiency, bone metabolism
Citation: Kapferer-Seebacher I, Foradori L, Zschocke J and Schilke R (2021) Rare Genetic Disorders Affecting the Periodontal Supporting Tissues in Adolescence. Front. Dent. Med. 2:687510. doi: 10.3389/fdmed.2021.687510
Received: 29 March 2021; Accepted: 27 May 2021;
Published: 09 July 2021.
Edited by:
Patrick R. Schmidlin, University of Zurich, SwitzerlandReviewed by:
Nurcan Buduneli, Ege University, TurkeyPriscila Casado, Fluminense Federal University, Brazil
Copyright © 2021 Kapferer-Seebacher, Foradori, Zschocke and Schilke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes Zschocke, am9oYW5uZXMuenNjaG9ja2VAaS1tZWQuYWMuYXQ=; Reinhard Schilke, c2NoaWxrZS5yZWluaGFyZEBtaC1oYW5ub3Zlci5kZQ==
†These authors have contributed equally to this work and share senior authorship