 Alisa E. Lee
Alisa E. Lee Iris R. Hartley1
Iris R. Hartley1 Chaim Vanek
Chaim Vanek- 1National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD, United States
- 2Division of Endocrinology, Diabetes and Clinical Nutrition, Oregon Health and Science University, Portland, OR, United States
While dental pulp calcifications and root anomalies may be inconsequential incidental findings in dental radiographs, they can, especially in combination, represent a clue, hidden in plain sight, for the diagnosis of hyperphosphatemic familial tumoral calcinosis (HFTC). HFTC is an autosomal recessive disease of mineral metabolism characterized by sometimes massive, painful calcification around large joints, systemic inflammation, dental pulp calcification, and thistle-shaped roots. This paper describes a woman with HFTC who endured not only the symptoms of HFTC for decades, but also the frustration of not knowing the cause. The diagnosis was finally made at the age of 73 years, when the connection between a large right shoulder calcification and hyperphosphatemia was made. The dental findings were likely present on her initial radiographs taken in childhood. Increased awareness of the association between characteristic dental findings and HFTC may allow for earlier diagnosis and interventions to improve the care of patients with this rare condition.
Introduction
In 2000, the U.S. Surgeon General's Report on Oral Health in America reported numerous ways in which oral and general health are interconnected (1). A thorough oral examination can reveal signs and symptoms of endocrinopathies, systemic infections, immunologic disorders, and nutritional deficiencies (2). Identifying these oral manifestations of systemic disorders may enable early diagnosis and treatment.
Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare autosomal recessive disease characterized by high blood phosphate, calcific masses, and dental anomalies (OMIM 211900) (3). The dental anomalies are the most penetrant phenotypic finding in the condition (4). The extra-dental clinical symptoms cover a broad spectrum, ranging from no significant involvement to lesions that are large, painful, and debilitating (5). HFTC is caused by deficiency of, or resistance to the phosphorus regulating hormone fibroblast growth factor 23 (FGF23) (6). Absence of FGF23 promotes renal tubule phosphate reabsorption that leads to hyperphosphatemia, which promotes ectopic calcifications in tissues exposed to trauma (7). Pathogenic variants in FGF23, GALNT3, or KLOTHO, have been found to cause HFTC (6); an acquired form due to FGF23 autoantibodies has also been described (8). Current treatment interventions focus on managing blood phosphate, reducing pain and inflammation, and addressing calcifications and their complications (6).
The unique dental phenotype of HFTC has been recently described in detail by our group (4). Internal pulp calcification and thistle-shaped, short roots with midroot bulging were most commonly observed in almost all of our cohort of 17 patients. The pulp can be partially to completely obliterated. Maxillary and mandibular premolars are most severely affected. In addition, primary dentition from pediatric patients with HFTC had findings similar to the permanent teeth.
Here we present a patient with hyperphosphatemic familial tumoral calcinosis who, despite numerous symptoms throughout her life, was not diagnosed with HFTC until the age of 73. Her diagnostic evaluation and treatment are presented and discussed.
Case Report
A 73-year-old Caucasian woman was referred in December 2019 to the National Institute of Dental and Craniofacial Research at the National Institutes of Health by her endocrinologist at Oregon Health and Science University for evaluation of recently diagnosed HFTC. Review of the patient's medical history revealed several symptoms associated with HFTC that arose throughout her life since childhood (Figure 1). She had been in her usual state of health until age 10, when she experienced unusual pain in her left leg, which was evaluated with a biopsy and diagnosed as osteomyelitis. She received IV antibiotics for 10 days. As a teenager, she had no major health issues and participated in sports such as basketball and volleyball. During her regular dental checkups, she was told that she has abnormal pulp stones, but was not further evaluated (Figure 2).
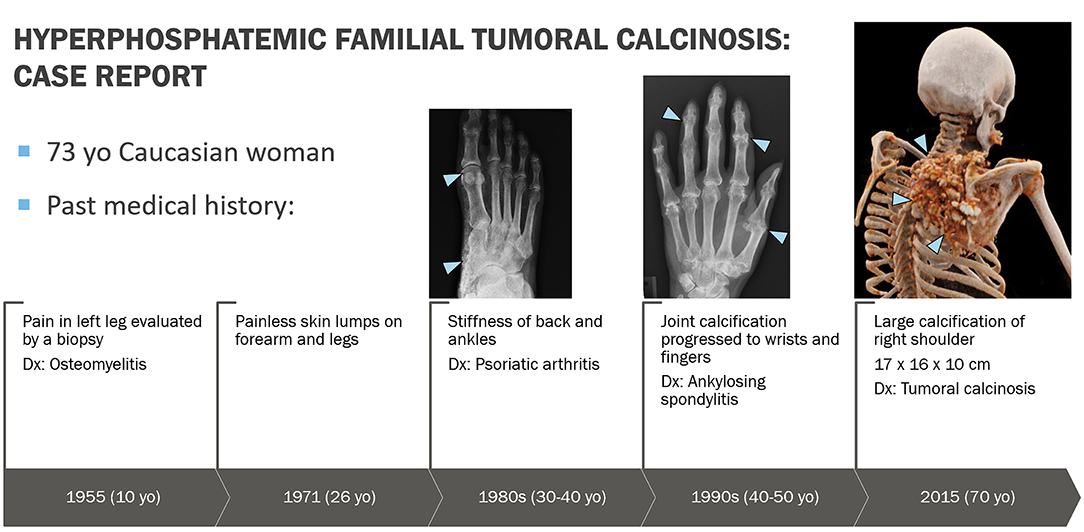
Figure 1. Graphic representation of clinical course. At age 10 years, patient experienced unusual pain in her left leg that represented hyperostosis-hyperphosphatemia syndrome, which was evaluated with a biopsy and misdiagnosed as osteomyelitis. At age 26 years, she started noticing painless skin lumps on her forearm and legs. In her 30s, she started experiencing back pain and joint stiffness in her ankles and fingers. She was then evaluated by a rheumatologist and was diagnosed with an ill-defined rheumatologic disorder, possibly psoriatic arthritis. Over the next 30 years, the arthritis slowly progressed to involve almost all joints of her body. Eventually this was determined to be an aggressive form of osteoarthritis, currently felt not to be related to underlying HFTC. In 2015, a large bump appeared around her right shoulder, which was initially thought to be a sarcoma. Upon biopsy, bone calcifications were found with the possibility of tumoral calcinosis.
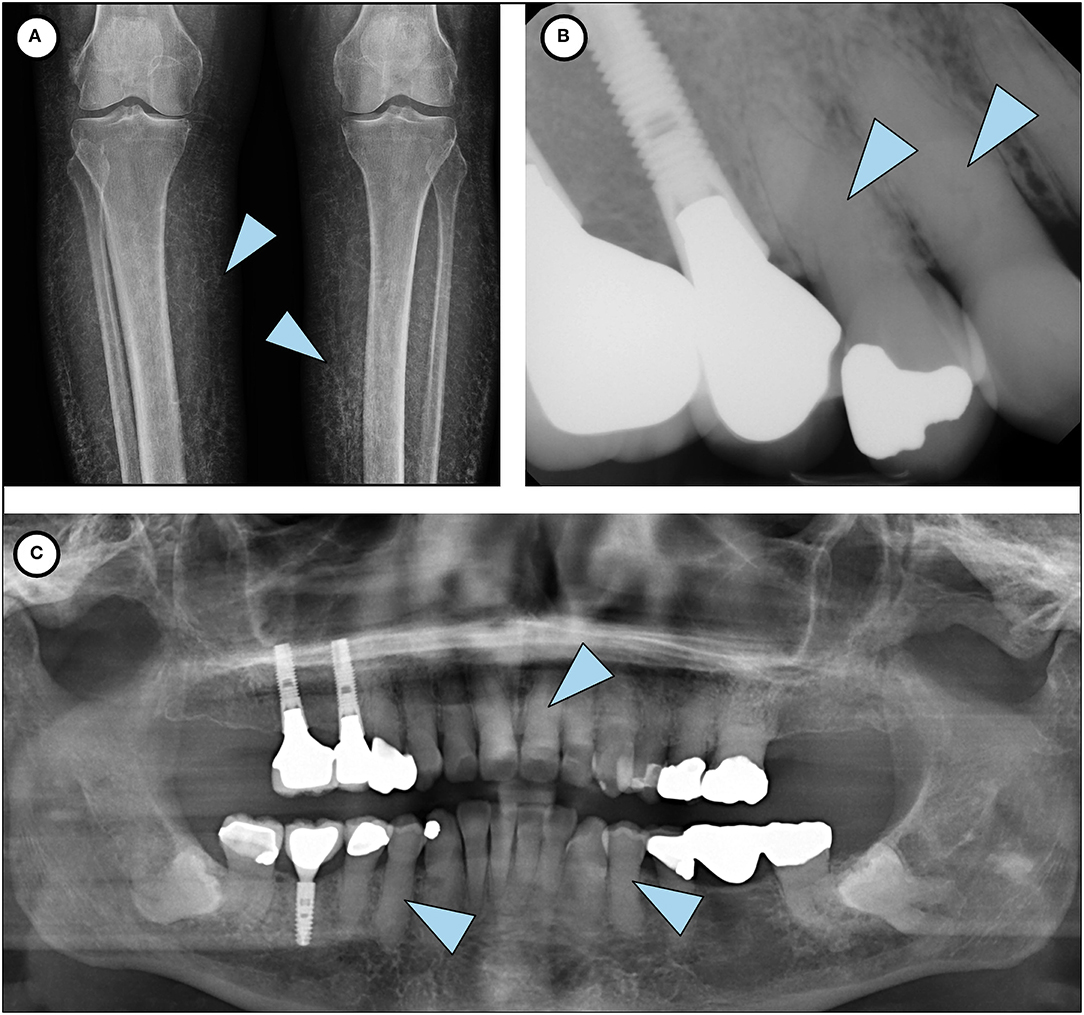
Figure 2. Phenotypic features of HFTC. (A) Lower limb radiograph shows subcutaneous calcification of her legs, an unusual and rare finding seen in older patients with HFTC. (B) Periapical radiograph of teeth # 5 and 6 shows complete pulp obliteration and thistle-shaped roots. (C) Panoramic radiograph shows short bulbous root with complete pulp obliteration in all teeth.
At age 26, she started noticing painless skin lumps on her forearm and legs. In her 30s, she experienced back pain and joint stiffness in her ankles and fingers. She was then evaluated by a rheumatologist and was diagnosed with an ill-defined rheumatologic disorder. Over the next 30 years, the arthritis slowly progressed to involve almost all joints of her body. Her finger joints have fused, and she is unable to make a fist or pick up small items. She has been treated with prednisone, methotrexate, and infliximab. Multiple steroid injections in her back have provided temporary relief. On re-evaluation, the prevailing diagnosis of her rheumatologic findings is aggressive osteoarthritis, which is not believed to be related to the diagnosis of HFTC. In 2015, a large bump was noted around her right shoulder (Figure 3). Initial concern was that it might represent a sarcoma. On biopsy, paucicellular calcific material was seen, characteristic of tumoral calcinosis. In 2019, she was diagnosed with peripheral vascular disease requiring angioplasty.
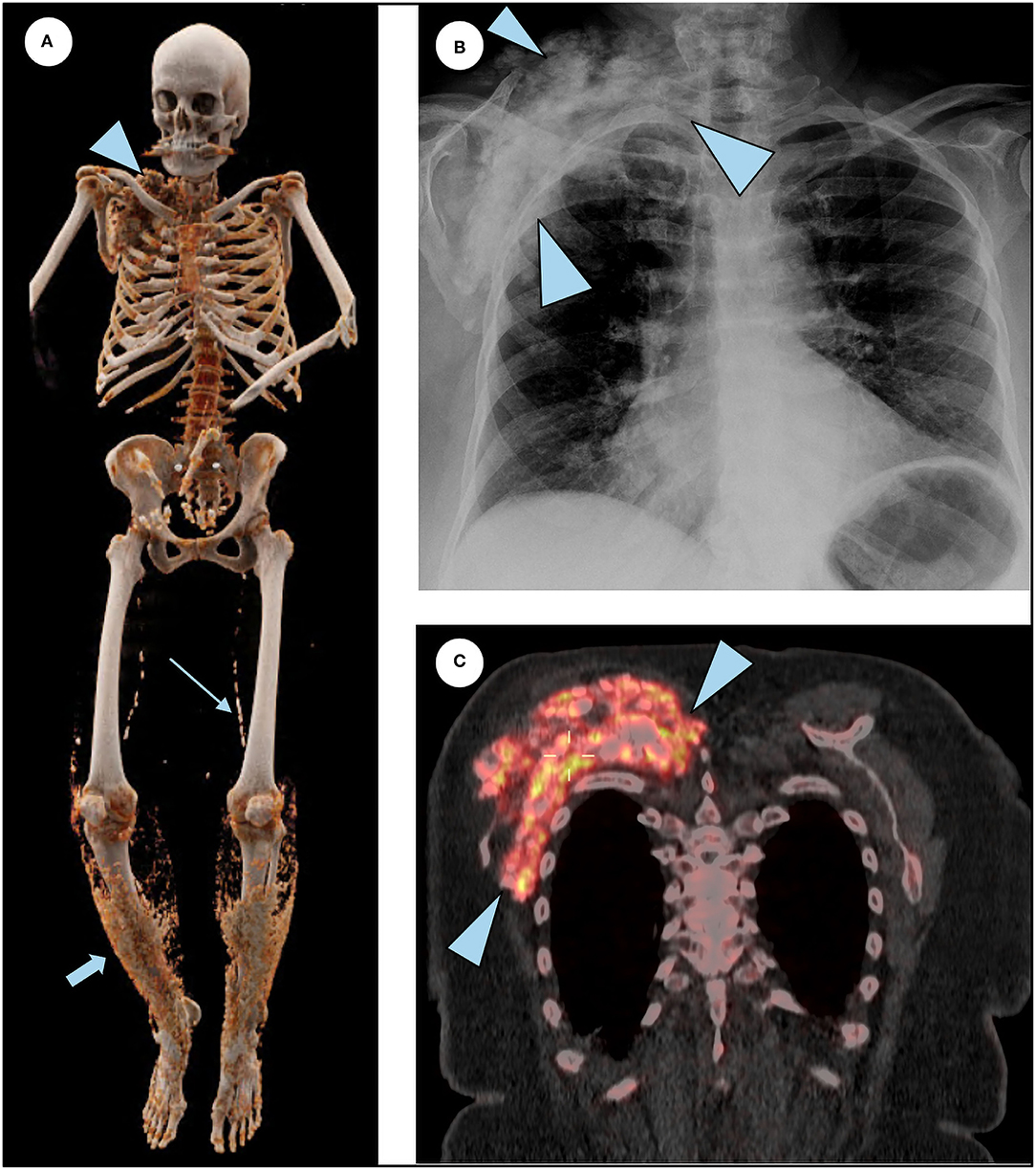
Figure 3. Additional features of HFTC. (A) 3D reconstruction of a total body CT scan illustrates subcutaneous calcifications surrounding the right shoulder (arrowhead), superficial lacey calcification in the lower extremities bilaterally (short arrow), and calcifications of femoral arteries (long arrow). (B) Chest radiograph shows right shoulder calcification (arrowheads). (C) Coronal slice of the merged 18F NaF PET/CT scan demonstrating intense tracer uptake in the actively forming calcific lesion seen in the chest radiograph (arrowheads).
Biochemical evaluation for causes of tumoral calcinosis revealed hyperphosphatemia and led to a referral to an endocrinologist in April 2019. HFTC was suspected and genetic testing identified two heterozygous variants in GALNT3 (c.746_749del and c.926T>G), consistent with HFTC. The first change (c.746_749del) is in exon 4 of the GALNT3 gene. This four-nucleotide deletion causes a frameshift and results in aberrant mRNA processing. This change has been previously reported as a variant associated with HFTC (9). The second change (c.926T>G) is in exon 5 of the GALNT3 gene. This variant converts an isoleucine to arginine. To our knowledge, this change has not been previously reported as a disease-causing variant. Pathogenic variants in GALNT3 often result in excessive cleavage of the active, intact FGF23 molecule, causing deficiency of functional FGF23 (10).
Following the diagnosis, the patient was referred to the NIH for further evaluation including blood and urine assessments and skeletal imaging. Laboratory studies showed elevated phosphorus 5.4 mg/dL (normal 2.5–4.5) and markers of systemic inflammation such as erythrocyte sedimentation rate 71 mm/h (0–42) and C-reactive protein 44 mg/L (0.00–4.99). Intact FGF23 was inappropriately low-normal 31 pg/mL (22–63) for the degree of hyperphosphatemia, while C-terminal FGF23 was markedly elevated 2,450 RU/mL ( ≤ 180), consistent with HFTC.
Extraoral and intraoral examination revealed healthy mucosal tissues and well-restored permanent dentition. Panoramic radiograph showed moderate alveolar bone loss around mandibular anterior teeth, fully impacted # 17 and 32 with dilacerated roots, and changes in the roots consistent with HFTC (Figure 2). Complete pulp obliteration with short, thistle shaped roots are observed in most of the teeth. Radiographs provided by her general dentist (taken 2008) also showed the altered tooth structure.
Discussion
The earliest clinical features of HFTC can include characteristic dental findings, painful cortical hyperostosis of certain long bones (especially the tibiae), subcutaneous masses, and calcification around joints (especially the hips and/or shoulders) (Table 1) (6). Most diagnosed patients develop symptoms by 13 years of age (11). The patient in this case developed clinical symptoms at age 10, but it was not until the patient presented with a much larger calcification of her shoulder in combination with hyperphosphatemia that prompted genetic testing confirming the diagnosis of HFTC at age 73. The painful tibial lesions this patient experienced in childhood and misdiagnosed as osteomyelitis represented hyperostosis-hyperphosphatemia syndrome, one of the early manifestations and part of the spectrum of phenotypic findings of HFTC (OMIM 211900) (10). Despite the numerous symptoms throughout her life, the patient was not correctly diagnosed for many years partly due to the fact the mutation in GALNT3 was not discovered until 2005 (12) and due to the rarity of the disorder.
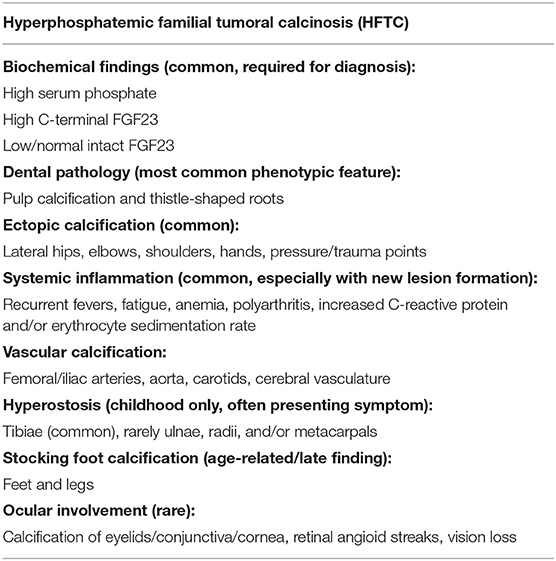
Table 1. The diagnostic criteria of hyperphosphatemic familial tumoral calcinosis (HFTC).
Patients present with a moderate to severe dental phenotype with root bulging, pulp calcification and shortened thistle-shaped roots. It is believed that the severity of the dental phenotype in HFTC does not appear to progress over time, but there is insufficient longitudinal data to know definitively (4). Based on her current dental radiographs and review of past dental history, it is likely that the patient had similar dental findings in her primary dentition. Early identification and recognition of the dental phenotype of the disorder by an astute dentist may have contributed to a timelier diagnosis in this case.
In this case, we did not observe mineralization in the PDL space. The alveolar bone appears to be within normal limits as well. The patient has had multiple implants placed without complications. In addition, patients with HFTC have been able to get extractions without any complications and have had successful orthodontic treatment. In histological analysis of a previous study, widening of the cellular cementum layer was observed (4).
Upon review of the biochemical studies and confirmation of the diagnosis, the patient was encouraged to have routine evaluation of phosphorous and calcium metabolism. She was recommended to follow a low-phosphate diet, limiting meat, nuts, beans, and dairy, as well as avoiding excessive calcium and vitamin D supplements. The patient was encouraged to maintain physical activity as tolerated.
The patient was instructed to continue treatment with sevelamer, which reduces hyperphosphatemia by binding to dietary phosphate in the gut. The patient was started on anakinra, an interleukin-1 receptor antagonist, to address the marked degree of systemic inflammation. Infliximab was discontinued, but she remained on low dose prednisone and methotrexate. She had a significant degree of overall improvement in well-being on this regimen, probably related to the decrease in inflammation brought about by the anakinra.
In conclusion, this report describes the oldest known living patient with HFTC and highlights the importance of recognizing dental phenotypes that may be a sign of systemic disorders. In patients with HFTC, dental changes are often the first sign of disease. Dentists can contribute to early diagnosis and interdisciplinary care of systemic conditions that manifest in the oral cavity.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by IRB of the NIDCR. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors contributed to manuscript preparation, revision, read, and approved the submitted version.
Funding
Work in the authors' laboratory was sponsored by the Intramural Research Program of the NIDCR, NIH (Grant# Z01 DE000649 25). This work was made possible through the National Institute of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, Genentech, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors (AEL). The funders were not involved in preparing this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, without collaboration, with the authors at the time of review.
Acknowledgments
We thank the patient and her family for their participation in the study. We thank Dr. Pamela Gardner for conducting the dental evaluation. We thank Dr. Martha Somerman for the invitation to contribute to this important research topic. We thank Dr. Emily Chu for providing guidance in manuscript preparation.
References
2. Chi AC, Neville BW, Krayer JW, Gonsalves WC. Oral manifestations of systemic disease. Am Fam Physician. (2010) 82:1381–8.
3. Ramnitz MS, Gafni RI, Collins MT. Hyperphosphatemic familial tumoral calcinosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al. editors. GeneReviews((R)). Seattle, WA: University of Washington (2018).
4. Lee AE, Chu EY, Gardner PJ, Duverger O, Saikali A, Wang SK, et al. A cross-sectional cohort study of the effects of FGF23 deficiency and hyperphosphatemia on dental structures in hyperphosphatemic familial tumoral calcinosis. JBMR Plus. (2021) 5:e10470. doi: 10.1002/jbm4.10470
5. Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, et al. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. (2010) 152A:896–903. doi: 10.1002/ajmg.a.33337
6. Boyce AM, Lee AE, Roszko KL, Gafni RI. Hyperphosphatemic tumoral calcinosis: pathogenesis, clinical presentation, and challenges in management. Front Endocrinol (Lausanne). (2020) 11:293. doi: 10.3389/fendo.2020.00293
7. Bhattacharyya N, Chong WH, Gafni RI, Collins MT. Fibroblast growth factor 23: state of the field and future directions. Trends Endocrinol Metab. (2012) 23:610–8. doi: 10.1016/j.tem.2012.07.002
8. Roberts MS, Burbelo PD, Egli-Spichtig D, Perwad F, Romero CJ, Ichikawa S, et al. Autoimmune hyperphosphatemic tumoral calcinosis in a patient with FGF23 autoantibodies. J Clin Invest. (2018) 128:5368–73. doi: 10.1172/JCI122004
9. Ramnitz MS, Gourh P, Goldbach-Mansky R, Wodajo F, Ichikawa S, Econs MJ, et al. Phenotypic and genotypic characterization and treatment of a cohort with familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome. J Bone Miner Res. (2016) 31:1845–54. doi: 10.1002/jbmr.2870
10. Ichikawa S, Guigonis V, Imel EA, Courouble M, Heissat S, Henley JD, et al. Novel GALNT3 mutations causing hyperostosis-hyperphosphatemia syndrome result in low intact fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. (2007) 92:1943–7. doi: 10.1210/jc.2006-1825
11. Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet. (2014) 15:98. doi: 10.1186/s12863-014-0098-3
Keywords: fibroblast growth factor 23, dental pulp calcification, root anomaly, phosphate metabolism, hyperphosphataemia
Citation: Lee AE, Hartley IR, Roszko KL, Vanek C, Gafni RI and Collins MT (2021) Hyperphosphatemic Familial Tumoral Calcinosis Hidden in Plain Sight for 73 Years: A Case Report. Front. Dent. Med. 2:719752. doi: 10.3389/fdmed.2021.719752
Received: 03 June 2021; Accepted: 28 June 2021;
Published: 22 July 2021.
Edited by:
Jacqueline W. Mays, National Institutes of Health (NIH), United StatesReviewed by:
Maiko Suzuki, Augusta University, United StatesMichael Baker Chavez, The Ohio State University, United States
Copyright © 2021 Lee, Hartley, Roszko, Vanek, Gafni and Collins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael T. Collins, bWNvbGxpbnNAbWFpbC5uaWguZ292