 Kento Tazawa1,2,†
Kento Tazawa1,2,† Hajime Sasaki
Hajime Sasaki- 1Department of Cariology, Restorative Sciences and Endodontics, University of Michigan School of Dentistry, Ann Arbor, MI, United States
- 2Department of Pulp Biology and Endodontics, Division of Oral Health Sciences, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 3Department of Oral and Maxillofacial Pathobiology, Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan
- 4Department of Translational Dental Medicine and Department of Endodontics, Boston University Goldman School of Dental Medicine, Boston, MA, United States
Apical periodontitis (AP) develops as a result of an immune response to pulpal bacterial infection, and various cytokines are involved in the pathogenesis of AP, with Interleukin (IL)-1 being considered a key cytokine. The role of IL-1 in the pathogenesis of AP has been well studied. It is known that IL-1 expression in periapical lesions correlates closely with the development of AP. IL-1 is a potent bone-resorptive cytokine that induces osteoclast formation and activation. Hence, inhibiting its signaling with IL-1 receptor antagonist (IL-1RA) results in a reduction in periapical lesion size. On the other hand, IL-1 is also a central cytokine that combats bacterial infection by activating innate immune responses. Therefore, a complete loss of IL-1 signaling leads to a failure to limit bacterial dissemination and consequently exacerbates AP. In vivo, IL-1 expression is tightly regulated and its signaling is modulated to optimize the immune response. Obesity causes systemic low-grade chronic inflammation and increases the risk of cardiovascular, renal, and other disorders. In experimentally induced AP, obesity significantly increases periapical bone loss, albeit the underlying mechanism remains unclear. Recent technological innovations have enabled more comprehensive and detailed analyses than previously, leading to new insights into the role of IL-1RA in regulating IL-1 signaling, and modulating apical lesion progression in obesity. In this review, we provide a brief overview of the function of IL-1 in AP development, with special emphasis on the latest findings in normal weight and obese states.
Introduction
Apical periodontitis (AP) involves chronic inflammation and alveolar bone loss. Kakehashi et al. demonstrated for the first time that AP is caused by pulpal infection. Rats maintained in a conventional microbial environment developed pulp necrosis and periapical inflammation after pulp exposure. In a germ-free environment, the pulps remained vital without periapical bone destruction, and dentin bridges formed over the exposed pulp, demonstrating the capacity for tissue regeneration in the absence of infection (1).
In response to infection, complexly mixed immune cells migrate to the infected site. First neutrophils infiltrate, followed by monocytes/macrophages, and subsequently by lymphocytes [T, B, and natural killer (NK) cells] (2, 3). These cells play critical roles in innate and adaptive immunity. Innate immunity comprises nonspecific responses that do not require prior sensitization to an antigen. Phagocytes are key to innate responses; neutrophils and macrophages engulf bacteria; and NK cells eliminate infected cells. Innate cells also produce inflammatory cytokines, which mediate immune and connective tissue cell activity (4–6). To eliminate pathogens and establish immune memory, the adaptive response activates antigen-specific CD4 + helper and CD8 + cytotoxic T cells, as well as B cells and plasma cells that produce antibodies (7, 8). The innate immune system also eliminates bacteria, apoptotic/dead cells, and debris. These responses are precisely regulated by the complex cytokine network.
Cytokines thus primarily protect the pulp and periodontal tissue from infection; however, cytokine-activated immune and inflammatory responses induce tissue destruction, particularly bone resorption (9, 10). Regarding bone resorption, Horton et al. first reported in 1972 that immune cells can influence osteoclast activity. Osteoclast-activating factor (OAF), a powerful stimulator of osteoclastic bone resorption, was released from human peripheral blood leukocytes stimulated by the mitogen phytohemagglutinin, or by antigenic material present in human dental plaque (11). In 1985, OAF was subsequently purified to homogeneity and sequenced, and shown to be identical to interleukin-1-beta (IL-1β). It was later shown that macrophage-derived IL-1 is a prominent mediator in developing bone destructive periapical lesions (12–15). These and other basic studies on the interactions between the immune system and bone following pulpal infections have been important in establishing the field of osteoimmunology. These basic studies have provided a rationale of clinical research on IL-1/IL-1 signaling in AP and foundation for interpreting their outcomes (16–22).
Obesity is one of the most prevalent non-communicable diseases and predisposes to various disorders, including hypertension, type 2 diabetes mellitus (DM), dyslipidemia, and coronary heart disease (23, 24). The increased morbidity associated with obesity is a worldwide public health issue (25). Besides, obese people are more susceptible to infections than their non-obese counterparts as well as to developing serious complications from common infections (26). AP is one of the most prevalent oral infectious diseases. In DM subjects, where obesity is the greatest risk factor, the success of root canal treatment is decreased in teeth with AP (27, 28). Moreover, studies in the rodent diet-induced obesity (DIO) model have revealed that obesity promotes the progression and severity of experimental AP (29–31). However, the underlying mechanism(s) by which obesity alters the immune response in AP remain unclear.
As the background for future basic and clinical research, this mini review aims first to reaffirm the role of IL-1 signaling in the development of AP in the lean state, and then to provide new insights into the possible mechanisms underlying the expansion of periapical bone destruction associated with obesity, based on the latest experimental findings.
IL-1 signaling is the central pathway in periapical lesion development
The IL-1 family comprises 11 cytokines: 7 pro-inflammatory mediators (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ), and 4 anti-inflammatory cytokines [IL-1 receptor antagonist (RA), IL-36RA, IL-37, and IL-38] (32). Each family member binds to a specific primary receptor, which combines with co-receptors to transduce pro-inflammatory or anti-inflammatory activity. The primary receptors include IL-1 receptor type 1 (IL-1R1), IL-1R2, IL-1R4, IL-1R5, and IL-1R6. The co-receptors include IL-1R3, IL-1R7, IL-1R8, IL-1R9, and IL-1R10 (32, 33). IL-1α, IL-1β, and IL-1RA are the primary members that regulate the progression of periapical lesions, and their roles have been well studied. In contrast, the role of the other family members in the development of AP has not been systematically evaluated.
IL-1α and IL-1β are encoded by IL1A and IL1B, respectively, in humans (34). Both isoforms bind to IL-1R1 and show similar biologic activities, including immune cell activation (33, 35). IL-1 is also closely involved in both bone formation (36) and resorption (12, 15). IL-1 inhibits nodule formation by osteoblasts in a dose-dependent manner (36). IL-1 strongly promotes osteoclast differentiation indirectly by inducing the expression of receptor activator of NF-κB ligand (RANKL; tumor necrosis factor ligand superfamily member 11) in osteoblasts (37). IL-1 directly induces the fusion of mononuclear pre-fusion osteoclasts and enhances osteoclast function (resorption pit-forming activity) (38–40). Moreover, activation of NF-κB promoted by IL-1 prolongs osteoclast survival (41, 42). However, IL-1α and IL-1β differ in several ways. First, species differences are found in their expression in periapical lesions. In rodent lesions, the predominant isoform is IL-1α rather than IL-1β (43, 44). In contrast, the protein level of IL-1β in human periapical exudate is double that of IL-1α (45). Furthermore, the bone resorption potency of IL-1β is 13-fold that of IL-1α in a rat assay system (10). Second, the expression level after root canal treatment is different. Following treatment, the level of IL-1β in the periapical exudates decreased, while the level of IL-1α increased. This suggests that IL-1α and IL-1β may play different biological roles in the healing process (45, 46). In this regard, a finding that bacteria-induced IL-1β and IL-1RI-myeloid differentiation factor 88 (MyD88) signaling are necessary and sufficient for efficient wound healing and tissue regeneration (47) is interesting. Third, the IL-1β cannot bind to IL-1R1 unless it is cleaved into its biologically active mature form. Conversely, IL-1α precursor can bind to and activate the IL-1 receptor without proteolysis (48).
The expression level of IL-1 positively correlates to the extension of bone destruction and severity of AP. IL-1α mRNA and protein expression was identified in murine periapical lesions from the early stage of development, with increased levels found on day 7 after pulp infection (43, 44, 49). Higher levels of IL-1α and IL-1β were detected in human periapical lesions with severe inflammation than mild inflammation (50, 51). In periapical lesions, IL-1 is produced by various cells, including macrophages, fibroblasts, polymorphonuclear leukocytes, endothelial cells, osteoblasts, and osteoclasts in response to infections (44, 49). Among these cells, macrophages are the major source of IL-1. Macrophage-derived IL-1 plays a critical role in periapical immunity. IL-1β and IL-1α are 1000- and 75-fold more potent, respectively, in stimulating bone resorption than TNFα or TNFβ (lymphotoxin) in vitro (10). Besides, IL-1 neutralization significantly reduced bone resorptive activity in extracts from periapical tissue explants, whereas TNF-α neutralization had no effect (13, 15).
These studies focused on the bone-destructive effects of IL-1, but IL-1 also protects the host early after bacterial challenge. Antibody-mediated neutralization of both IL-1α and IL-1β leads to a failure to contain pulpal infection in male but not female mice, resulting in orofacial abscesses and sepsis (52). Ovariectomized mice also developed sepsis, but were protected by an estrogen implant. Accordingly, IL-1 signaling synergizes with estrogen signaling to prime phagocytic cells for enhanced anti-microbial activity resulting in infection localization. IL-1R1 deficient mice identically showed severe bone destruction and sepsis after pulpal infection (53, 54). Taken together, a severe deficiency of IL-1 signaling leads to poor infection control, dissemination of infection, and elevated bone destruction.
Subsequent studies using IL-1RA have confirmed the correlation between IL-1 and bone resorption. IL-1RA, produced by macrophages and monocytes (55), competitively blocks the action of IL-1. IL-1RA binds to IL-1R1 with equal or greater affinity than IL-1α and IL-1β but does not activate downstream signaling (34, 55, 56). IL-1RA has a significant impact by suppressing periapical lesion development. Stashenko et al. demonstrated a 14-day IL-1RA treatment inhibited lesion development by approximately 60% (57). Maintaining IL-1 and IL-1RA in balance prevents excess inflammation and bone destruction. Once this balance is upset, inflammation and tissue damage may deteriorate (58). To block IL-1-mediated bone resorption ex vivo, rat fetal long bones and mouse newborn calvariae require approximately 10-fold and 100–1000-fold IL-1RA to IL-1, respectively (59). In periapical lesions, the level of IL-1RA is more abundant than IL-1 (mean IL-1RA: IL-1β ratio = 128: 7). Interestingly, exudates from symptomatic human lesions contained a significantly lower ratio of IL-1RA to IL-1β than exudates from asymptomatic human lesions (22). Taken together, the local balance of IL-1 and IL-1RA is crucially important in the periapical lesion development.
The cytokine network in periapical lesions centered on IL-1 signaling
Macrophages are major players involved in the cytokine network, and secrete various immunoregulatory mediators, including IL-1 (35, 60). TNF-α is another pro-inflammatory cytokine expressed by macrophages (61) and increased in periapical lesions (44, 49). TNF-α promotes IL-1 secretion from murine resident peritoneal macrophages in vitro (62) and increases osteoclastogenesis by upregulating RANKL (63, 64). However, as noted above, TNF-α itself is not much bone resorptive as IL-1 isoforms, and TNF-α deficient mice exhibited similar periapical lesion size to wild-type controls (65).
The role of type-1 T-helper (Th1) cytokines [gamma interferon (IFN-γ), IL-12, IL-18] and Th2 cytokines (IL-4, IL-6, IL-10) on periapical bone destruction has also been evaluated. IFN-γ, IL-12, and IL-18 potentiate pro-inflammatory signaling (66–68) and their expression is increased in periapical lesions (43, 69, 70). IFN-γ modulates macrophage-derived IL-1 expression, but its effect is not consistent. IFN-γ promotes secretion of IL-1 from LPS-stimulated human macrophages in vitro (71), whereas suppresses it IL-1 in mouse RAW 264.7 macrophages (72). IL-12 induces Th1 cell development, and IL-18, with IL-12, activates established-Th1 cells to produce IFN-γ. Thus, IL-12 and IL-18 are considered pro-inflammatory cytokines that facilitate type-1 responses (67, 73). However, previous studies demonstrated that gene knockouts of IL-12, IL-18, and IFN-γ all exhibited similar lesion sizes as wild-type controls (65, 74). Recombinant IL-12-infused wild-type mice also showed similar bone resorption to controls. The findings with IFN-γ were not confirmed in another study, which reported that IFN-γ-deficient(−/−) mice presented with periapical lesions larger than those in wild-type animals (75). The expression level of IL-1 in periapical lesions was unchanged in these mice (74). Taken together, these results indicate that none of these cytokines has a non-redundant function in mediating periapical bone resorption.
IL-6, another macrophage-derived cytokine, was also detected in inflamed periapical tissue (76, 77). Its expression was found to be transiently increased on day 14 after infection and decreased in the chronic phase (43). IL-6 is a well-known pro-inflammatory cytokine, promoting bone resorption via osteoclastogenesis (78–80). Recent research has demonstrated that IL-6 also has anti-inflammatory effects by promoting macrophage IL-1RA secretion (81) and bone-forming effects by enhancing osteoblast differentiation (82–84). Previously, the protective role of IL-6 in periapical lesions was shown in vivo. Bone destruction was significantly increased in IL-6−/− mice versus in wild-type mice (69, 85). IL-6 antibody-mediated neutralization also increased bone resorption compared to untreated controls. In IL-6−/− mice, increased bone resorption importantly correlated with osteoclast count and IL-1 expression in periapical lesions, and inversely with anti-inflammatory IL-10 expression (69).
Both IL-4 and IL-10 are increased in periapical lesions (69). IL-4 is an anti-inflammatory cytokine playing pleiotropic roles in inflammation (86, 87). IL-10, a potent anti-inflammatory cytokine produced by regulatory T cells (Treg), macrophages, dendritic cells, Th 2 cells, and Th1 cells, among other immune cells (88–90). However, IL-4 and IL-10 have different anti-inflammatory effects on macrophages. In macrophages stimulated by oral pathogens, recombinant IL-10 inhibited IL-1α production, whereas recombinant IL-4 had no significant suppressive effect (91). Consistent with these in vitro findings, IL-10−/− mice exhibited significantly greater infection-stimulated bone resorption than wild-type mice, as well as markedly elevated IL-1 production in periapical inflammatory tissues (91). In contrast, there was no difference in periapical lesion size between IL-4−/− and wild-type mice (75, 91).
IL-17 is a pleiotropic cytokine produced by Th17 cells that induces a myriad of pro-inflammatory mediators (92). The expression of IL-17 was increased in infection-induced periapical lesions (65) and was significantly higher in symptomatic versus asymptomatic lesions (93). IL-17 induces human macrophages to produce and secrete pro-inflammatory cytokines IL-1β and TNF-α in vitro (94). IL-17A−/− mice were resistant to periapical lesions versus wild-type controls (65). However, IL-17 receptor type A-deficient (IL-17RA−/−) mice conversely exhibited significantly increased bone destruction and inflammation. The expression of IL-1 was significantly upregulated in IL-17RA−/− lesions in vivo and IL-17RA−/− macrophages in vitro. The lesion size of IL-17RA−/− mice was decreased by IL-1β neutralization (95). IL-17A utilizes two IL-17 receptors, and IL-17RA has four ligands (96); therefore, this system must be meticulously dissected to comprehend these data. Nevertheless, IL-17RA signaling likely plays a protective role in periapical lesions via IL-1 signaling and neutrophil priming.
Table 1 Summarizes the effect of cytokine or receptor deficiency/neutralization on periapical lesions. Although it is difficult to evaluate the effect of each cytokine because of their complex interactions (97), the above reviewed experimental models suggest that anti-inflammatory cytokines such as IL-10 and, to a lesser extent, IL-6 are dominant and have non-redundant functions, compared to inflammatory cytokines in the immunomodulation of AP. In addition, the positive correlation between the IL-1 level and lesion size implies IL-1 is a principal cytokine in periapical lesion expansion and a useful biomarker for assessing inflammation.
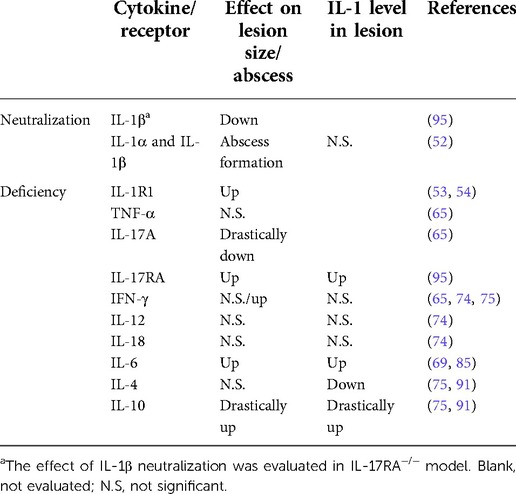
Table 1. The effect of deficiency/neutralization of each cytokine or receptor on periapical lesion.
The impact of obesity and diabetes mellitus on periapical lesions
It is now widely accepted that obesity causes systemic low-grade chronic inflammation (98). As noted above, obesity increases the risk of severe inflammation (26), and predisposes to the development of postoperative and nosocomial infections, as well as serious complications of common infections (98, 99). Obesity also increases the risk for severe symptoms and poor prognosis in viral infections, including coronavirus disease 2019 (100). In the oral cavity, obesity correlates with the prevalence and severity of periodontitis (101). Deshpande et al. reported that obesity worsens all gingival index, probing depth, gingival recession, and clinical attachment levels than non-obese patients (102).
Diabetes, as an obesity complication, also has negative effects on AP. Diabetes decreases the success rate of endodontic treatment in teeth with AP preoperatively, and increases the risk of post-treatment tooth loss (27, 28, 103–105). According to previous in vivo rodent studies, obesity significantly increases bone destruction in experimentally induced AP (29–31). As discussed in the following section, several potential mechanisms underlying obesity-induced inflammation have been proposed, but the actual mechanism is not yet fully understood.
Potential mechanism of obesity-exacerbating periapical bone destruction
Many studies provide evidence that obesity alters immune responses. In obesity, macrophages significantly accumulate in the white adipose tissue (106, 107); and the phenotype of accumulated macrophages possesses a pro-inflammatory M1-polarized state, whereas resident macrophages in lean mice have a pro-resolving M2 phenotype (108–111). The M1-dominant adipose macrophages likely develop an inflammatory milieu (112). The circulating levels of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β were elevated in obese subjects (113, 114). Chronic exposure to these cytokines potentially causes insulin resistance resulting in hyperglycemia (115, 116). In addition, the serum levels of adipose tissue-derived cytokines, adipokines and adiponectin are also altered in the obese state. Obese adipose tissue increases inflammatory adipokines, including leptin, resistin, visfatin, IL-6, TNF-α, and monocyte chemoattractant protein-1, while decreasing anti-inflammatory adipokines, including adiponectin, omentin, IL-10, and IL-4. The dysregulation of adipokine production may alter cellular immune function and contribute to chronic low-grade inflammation and disease pathology (117–119). Obesity also increases the populations of activated CD4+ and CD8+ T cells in adipose tissue (120) and significantly reduces circulating Treg cells (121–123) which may sustain low-grade chronic inflammation. Furthermore, obesity induces thymic involution and convergent T cell repertoire, impairing immune responses and increasing the risk and severity of infections (124).
As noted above, the effects of obesity on immune function are manifold. However, it remains unclear how obesity is associated with the expansion of periapical bone destruction. Therefore, our group examined possible pathways involved in bone loss in obesity using bulk-mRNA next-generation sequencing analysis. Comprehensive gene expression analysis revealed that, among a total 15,029 expressed genes, only 51 were differentially expressed in periapical lesions in DIO-B6 mice versus lean controls. Among them, Il1rn encoding IL-1RA was remarkably down-regulated (Log2 fold change = −1.18, false discovery rate (q-value) = 0.0002). At the same time, Il1a, but not Il1b, was also decreased (−0.994-fold, q = 0.046) (31). These results suggest that DIO impairs IL-1RA-dependent homeostatic suppression of IL-1 signaling, at least in the local environment.
Systemically, significantly increased IL-1 serum levels (114, 125) likely contribute to worsening of insulin resistance under obese conditions (116). However, given the lack of significant changes in the expression of IL-1 signaling genes, including NF-κB, in AP (31), systemically increased IL-1 may have little effect on AP. Interestingly, IL-1RA serum levels are also elevated in obesity (126). However, the concentration of IL-1RA is likely insufficient to block the effects of elevated IL-1. Indeed, administration of IL-1RA improves insulin sensitivity in animal models of obesity (116), suggesting that IL-1RA-dependent homeostatic regulation of IL-1 signaling is not fully functional in obesity. We therefore examined if a decrease or loss of IL-1RA contributes to obesity-associated periapical inflammation by IL-1RA administration in infected DIO-B6 mice. Remarkably, periapical bone destruction was inhibited by 41.2% by IL-1RA (Figure 1A, p < 0.05). Histological analysis revealed that IL-1RA-treated mice showed less inflammatory cell infiltration and well-developed fibrosis (Figure 1B). These results indicate that inflammation was down-regulated by IL-1RA, and that the lesion was composed mainly of mature granulation tissue compared to the immune granulomas in the controls. Therefore, immunomodulation by IL-1RA is likely important for the control of AP, even in obesity.
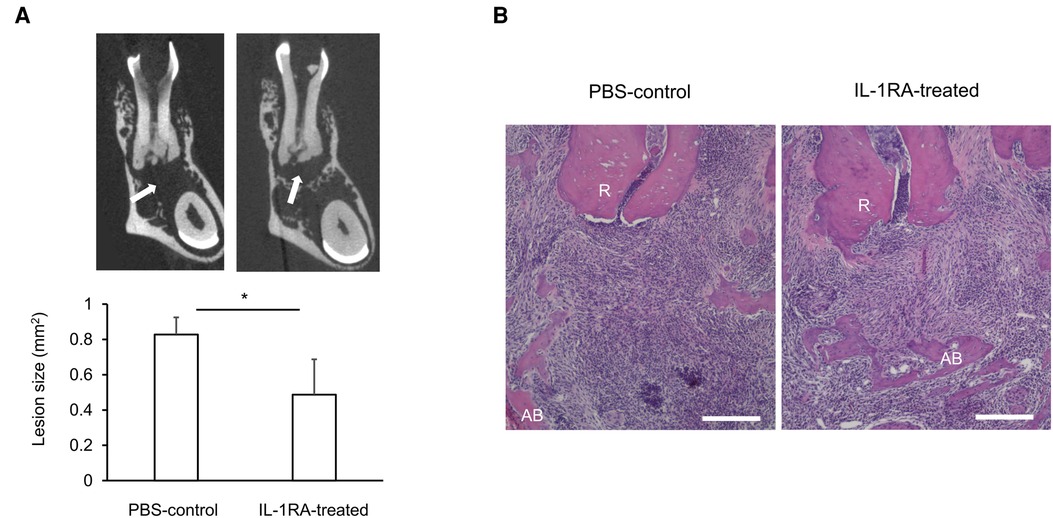
Figure 1. (A) Representative microCT images and periapical lesion size in phosphate buffered saline (PBS)-control and IL-1RA administration on day 42 after endodontic infection. Values are mean and SD; *p < 0.05. Arrow: periapical lesion. (B) Histology of periapical lesions on day 42 after endodontic infection. Representative images of hematoxylin and eosin (HE) staining for each group are shown. PBS, phosphate buffered saline; AB, alveolar bone; R, dental root; Scale bars = 200 µm.
Appropriate regulation of IL-1 signaling according to the host and infection status may lead to an optimal immune/inflammatory response in terms of timely onset/resolution and adequate host defense. In the first section, we explained that excessive IL-1 and its signaling cause exacerbation of AP in the non-obese state. At the same time, IL-1RA homeostatically regulates IL-1 signaling, suppressing excessive IL-1-mediated responses. In the second section, we described that obesity dysregulates IL-1RA-dependent homeostatic IL-1 signaling regulation and causes chronic elevation of inflammation, tissue destruction, and prolonged healing. Endodontic infection in DIO may exacerbate bone destruction in the long term via chronically elevating IL-1 signaling at a low level due to downregulation of Il1rn. However, the role of IL-1 signaling is diverse and complex. The impact of IL-1 signaling on both systemic and local conditions has not been fully understood. Thus, further studies are essential for the changes in IL-1 signaling associated with various systemic conditions, the underlying mechanisms, and infection-stimulated bone destruction.
Author contributions
PS and HS designed the study. KT and MMAP performed experiments in DIO mice. HF contributed to histological analysis. KT, MMAP, HF, and HS analyzed the data. KT, HS, and PS prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported in part by a grant from National Institute of Dental and Craniofacial Research/National Institutes of Health (R01DE024796 to HS).
Acknowledgments
We thank Dr. Mutsumi Miyauchi (Department of Oral and Maxillofacial pathobiology Graduate School of Biomedical and Health Sciences Hiroshima University) for co-operating histological analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AP, apical periodontitis; DM, diabetes mellitus; DIO, diet-induced obesity; IFN-γ, gamma interferon; IL, interleukin; IL-1RA, interleukin-1 receptor antagonist; IL-1R1, interleukin-1 receptor type 1; IL-17RA, interleukin-17 receptor type A; MyD88, myeloid differentiation factor 88; NK cell, natural killer cell; OAF, osteoclast-activating factor; RANKL, receptor activator of NF-κB ligand; Treg cell, regulatory T cell; Th cell, thelper cell; TNF, tumor necrosis factor
References
1. Kakehashi S, Stanley HR, Fitzgerald RJ. The effects of surgical exposures of dental pulps in germ-free and conventional laboratory rats. Oral Surg Oral Med Oral Pathol. (1965) 20:340–9. doi: 10.1016/0030-4220(65)90166-0
2. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2017) 9:7204–18. doi: 10.18632/oncotarget.23208
3. Kawashima N, Okiji T, Kosaka T, Suda H. Kinetics of macrophages and lymphoid cells during the development of experimentally induced periapical lesions in rat molars: A quantitative immunohistochemical study. J Endod. (1996) 22:311–6. doi: 10.1016/S0099-2399(96)80266-4
4. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
5. Stashenko P, Wang CY, Riley E, Wu Y, Ostroff G, Niederman R. Reduction of infection-stimulated periapical bone resorption by the biological response modifier PGG glucan. J Dent Res. (1995) 74:323–30. doi: 10.1177/00220345950740010701
6. Kawashima N, Niederman R, Hynes RO, Ullmann-Cullere M, Stashenko P. Infection-stimulated infraosseus inflammation and bone destruction is increased in P-/E-selectin knockout mice. Immunology. (1999) 97:117–23. doi: 10.1046/j.1365-2567.1999.00754.x
7. Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. (2002) 91:281–91. doi: 10.1161/01.res.0000029784.15893.10
8. Graunaite I, Lodiene G, Maciulskiene V. Pathogenesis of apical periodontitis: A literature review. J Oral Maxillofac Res. (2012) 2:e1. doi: 10.5037/jomr.2011.2401
9. Stashenko P, Teles R, D'Souza R. Periapical inflammatory responses and their modulation. Crit Rev Oral Biol Med. (1998) 9:498–521. doi: 10.1177/10454411980090040701
10. Stashenko P, Dewhirst FE, Peros WJ, Kent RL, Ago JM. Synergistic interactions between interleukin 1, tumor necrosis factor, and lymphotoxin in bone resorption. J Immunol. (1987) 138:1464–8.3492553
11. Horton JE, Raisz LG, Simmons HA, Oppenheim JJ, Mergenhagen SE. Bone resorbing activity in supernatant fluid from cultured human peripheral blood leukocytes. Science. (1972) 177:793–5. doi: 10.1126/science.177.4051.793
12. Dewhirst FE, Stashenko PP, Mole JE, Tsurumachi T. Purification and partial sequence of human osteoclast-activating factor: Identity with interleukin 1 beta. J Immunol. (1985) 135:2562–8.3875658
13. Wang CY, Stashenko P. Characterization of bone-resorbing activity in human periapical lesions. J Endod. (1993) 19:107–11. doi: 10.1016/S0099-2399(06)80503-0
14. Wang CY, Stashenko P. Kinetics of bone-resorbing activity in developing periapical lesions. J Dent Res. (1991) 70:1362–6. doi: 10.1177/00220345910700100901
15. Wang CY, Stashenko P. The role of interleukin-1 alpha in the pathogenesis of periapical bone destruction in a rat model system. Oral Microbiol Immunol. (1993) 8:50–6. doi: 10.1111/j.1399-302x.1993.tb00543.x
16. Popovska L, Dimova C, Evrosimovska B, Stojanovska V, Muratovska I, Cetenović B, et al. Relationship between IL-1β production and endodontic status of human periapical lesions. Vojnosanitski Pregled. (2017) 74:1134–9. doi: 10.2298/VSP151228309P
17. Miller GA, DeMayo T, Hutter JW. Production of interleukin-1 by polymorphonuclear leukocytes resident in periradicular tissue. J Endod. (1996) 22:346–51. doi: 10.1016/S0099-2399(96)80215-9
18. Gomes BPFA, Berber VB, Marinho ACS, Louzada LM, Arruda-Vasconcelos R, Passini MRZ, et al. Chemomechanical preparation influences the microbial community and the levels of LPS, LTA and cytokines in combined endodontic-periodontal lesions: A clinical study. J Periodontal Res. (2022) 57:341–56. doi: 10.1111/jre.12964
19. Maia LM, Espaladori MC, Diniz JMB, Tavares WLF, de Brito LCN, Vieira LQ, et al. Clinical endodontic procedures modulate periapical cytokine and chemokine gene expressions. Clin Oral Investig. (2020) 24:3691–7. doi: 10.1007/s00784-020-03247-y
20. Espaladori MC, Diniz JMB, de Brito LCN, Tavares WLF, Kawai T, Vieira LQ, et al. Selenium intracanal dressing: Effects on the periapical immune response. Clin Oral Investig. (2021) 25:2951–8. doi: 10.1007/s00784-020-03615-8
21. Martinho FC, Chiesa WM, Leite FR, Cirelli JA, Gomes BP. Correlation between clinical/radiographic features and inflammatory cytokine networks produced by macrophages stimulated with endodontic content. J Endod. (2012) 38:740–5. doi: 10.1016/j.joen.2012.02.021
22. Seckinger P, Klein-Nulend J, Alander C, Thompson RC, Dayer JM, Raisz LG. Natural and recombinant human IL-1 receptor antagonists block the effects of IL-1 on bone resorption and prostaglandin production. J Immunol. (1990) 145:4181–4.2147937
23. Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. (1999) 282:1523–9. doi: 10.1001/jama.282.16.1523
24. Centers for Disease Control and Prevention. The health effects of overweight and obesity (2021). Available at: https://www.cdc.gov/healthyweight/effects/index.html (Accessed May 05, 2021).
25. World Health Organization. Obesity and overweight (2021). Available at: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (Accessed May 05, 2021).
26. Pugliese G, Liccardi A, Graziadio C, Barrea L, Muscogiuri G, Colao A. Obesity and infectious diseases: Pathophysiology and epidemiology of a double pandemic condition. Int J Obes. (2022) 46:449–65. doi: 10.1038/s41366-021-01035-6
27. Laukkanen E, Vehkalahti MM, Kotiranta AK. Impact of systemic diseases and tooth-based factors on outcome of root canal treatment. Int Endod J. (2019) 52:1417–26. doi: 10.1111/iej.13143
28. Segura-Egea JJ, Cabanillas-Balsera D, Jiménez-Sánchez MC, Martín-González J. Endodontics and diabetes: Association versus causation. Int Endod J. (2019) 52:790–802. doi: 10.1111/iej.13079
29. Tibúrcio-Machado CDS, Lang PM, Campos MM, Wolle CFB, Barcelos RCS, Zago NB, et al. High-fat diet effect on periapical lesions and hepatic enzymatic antioxidant in rats. Life Sci. (2021) 264:118637. doi: 10.1016/j.lfs.2020.118637
30. Brasil SC, Santos RMM, Fernandes A, Lima RS, Costa CAS, Pinto KMMDC, et al. Influence of a high-fat diet in the progression of apical periodontitis. J Endod. (2021) 47:600–5. doi: 10.1016/j.joen.2020.12.015
31. Azuma MM, Tazawa K, Furusho H, Sasaki H. Mechanisms of elevated periapical bone loss in diet-induced obesity. 2021 IADR/AADR/CADR General Session; 2021 July 21–24. Virtual J Dent Res. (2021) 100(Special Issue A):0380.
32. Sims JE, Smith DE. The IL-1 family: Regulators of immunity. Nat Rev Immunol. (2010) 10:89–102. doi: 10.1038/nri2691
33. Dinarello CA. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat Rev Rheumatol. (2019) 15:612–32. doi: 10.1038/s41584-019-0277-8
34. Fields JK, Günther S, Sundberg EJ. Structural basis of IL-1 family cytokine signaling. Front Immunol. (2019) 10:1412. doi: 10.3389/fimmu.2019.01412
35. Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. (1991) 77:1627–52. doi: 10.1182/blood.V77.8.1627.1627
36. Stashenko P, Dewhirst FE, Rooney ML, Desjardins LA, Heeley JD. Interleukin-1 beta is a potent inhibitor of bone formation in vitro. J Bone Miner Res. (1987) 2:559–65. doi: 10.1002/jbmr.5650020612
37. Amarasekara DS, Yun H, Kim S, Lee N, Kim H, Rho J. Regulation of osteoclast differentiation by cytokine networks. Immune Netw. (2018) 18:e8. doi: 10.4110/in.2018.18.e8
38. Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, et al. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res. (1999) 247:84–93. doi: 10.1006/excr.1998.4320
39. Nakamura I, Jimi E. Regulation of osteoclast differentiation and function by interleukin-1. Vitam Horm. (2006) 74:357–70. doi: 10.1016/S0083-6729(06)74015-8
40. Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, et al. The mechanism of osteoclast differentiation induced by IL-1. J Immunol. (2009) 183:1862–70. doi: 10.4049/jimmunol.0803007
41. Jimi E, Nakamura I, Ikebe T, Akiyama S, Takahashi N, Suda T. Activation of NF-kappaB is involved in the survival of osteoclasts promoted by interleukin-1. J Biol Chem. (1998) 273:8799–805. doi: 10.1074/jbc.273.15.8799
42. Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, et al. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J Cell Biol. (2000) 148:333–42. doi: 10.1083/jcb.148.2.333
43. Kawashima N, Stashenko P. Expression of bone-resorptive and regulatory cytokines in murine periapical inflammation. Arch Oral Biol. (1999) 44:55–66. doi: 10.1016/s0003-9969(98)00094-6
44. Wang CY, Tani-Ishii N, Stashenko P. Bone-resorptive cytokine gene expression in periapical lesions in the rat. Oral Microbiol Immunol. (1997) 12:65–71. doi: 10.1111/j.1399-302x.1997.tb00619.x
45. Matsuo T, Ebisu S, Nakanishi T, Yonemura K, Harada Y, Okada H. Interleukin-1 alpha and interleukin-1 beta periapical exudates of infected root canals: Correlations with the clinical findings of the involved teeth. J Endod. (1994) 20:432–5. doi: 10.1016/s0099-2399(06)80032-4
46. Martinez ZR, Naruishi K, Yamashiro K, Myokai F, Yamada T, Matsuura K, et al. Gene profiles during root canal treatment in experimental rat periapical lesions. J Endod. (2007) 33:936–43. doi: 10.1016/j.joen.2007.04.016
47. Wang G, Sweren E, Liu H, Wier E, Alphonse MP, Chen R, et al. Bacteria induce skin regeneration via IL-1β signaling. Cell Host Microbe. (2021) 29:777–791.e6. doi: 10.1016/j.chom.2021.03.003
48. Mosley B, Urdal DL, Prickett KS, Larsen A, Cosman D, Conlon PJ, et al. The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J Biol Chem. (1987) 262:2941–4. doi: 10.1016/S0021-9258(18)61450-4
49. Tani-Ishii N, Wang CY, Stashenko P. Immunolocalization of bone-resorptive cytokines in rat pulp and periapical lesions following surgical pulp exposure. Oral Microbiol Immunol. (1995) 10:213–9. doi: 10.1111/j.1399-302x
50. Jakovljevic A, Knezevic A, Karalic D, Soldatovic I, Popovic B, Milasin J, et al. Pro-inflammatory cytokine levels in human apical periodontitis: Correlation with clinical and histological findings. Aust Endod J. (2015) 41:72–7. doi: 10.1111/aej.12072
51. Yang NY, Zhou Y, Zhao HY, Liu XY, Sun Z, Shang JJ. Increased interleukin 1α and interleukin 1β expression is involved in the progression of periapical lesions in primary teeth. BMC Oral Health. (2018) 18:124. doi: 10.1186/s12903-018-0586-3
52. Youssef H, Stashenko P. Interleukin-1 and estrogen protect against disseminating dentoalveolar infections. Int J Oral Sci. (2017) 9:16–23. doi: 10.1038/ijos.2016.61
53. Chen CP, Hertzberg M, Jiang Y, Graves DT. Interleukin-1 and tumor necrosis factor receptor signaling is not required for bacteria-induced osteoclastogenesis and bone loss but is essential for protecting the host from a mixed anaerobic infection. Am J Pathol. (1999) 155:2145–52. doi: 10.1016/S0002-9440(10)65532-0
54. Graves DT, Chen CP, Douville C, Jiang Y. Interleukin-1 receptor signaling rather than that of tumor necrosis factor is critical in protecting the host from the severe consequences of a polymicrobe anaerobic infection. Infect Immun. (2000) 68:4746–51. doi: 10.1128/IAI.68.8.4746-4751.2000
55. Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. (2000) 117:1162–72. doi: 10.1378/chest.117.4.1162
56. Svenson M, Hansen MB, Heegaard P, Abell K, Bendtzen K. Specific binding of interleukin 1 (IL-1) beta and IL-1 receptor antagonist (IL-1ra) to human serum. High-affinity binding of IL-1ra to soluble IL-1 receptor type I. Cytokine. (1993) 5:427–35. doi: 10.1016/1043-4666(93)90032-z
57. Stashenko P, Wang CY, Tani-Ishii N, Yu SM. Pathogenesis of induced rat periapical lesions. Oral Surg Oral Med Oral Pathol. (1994) 78:494–502. doi: 10.1016/0030-4220(94)90044-2
58. Arend WP, Guthridge CJ. Biological role of interleukin 1 receptor antagonist isoforms. Ann Rheum Dis. (2000) 59(Suppl 1):i60–4. doi: 10.1136/ard.59.suppl_1.i60
59. Shimauchi H, Takayama S, Imai-Tanaka T, Okada H. Balance of interleukin-1 beta and interleukin-1 receptor antagonist in human periapical lesions. J Endod. (1998) 24:116–9. doi: 10.1016/s0099-2399(98)80089-7
60. Cavaillon JM. Cytokines and macrophages. Biomed Pharmacother. (1994) 48:445–53. doi: 10.1016/0753-3322(94)90005-1
61. Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. (2010) 20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10
62. Bachwich PR, Chensue SW, Larrick JW, Kunkel SL. Tumor necrosis factor stimulates interleukin-1 and prostaglandin E2 production in resting macrophages. Biochem Biophys Res Commun. (1986) 136:94–101. doi: 10.1016/0006-291x(86)90881-8
63. Luo G, Li F, Li X, Wang ZG, Zhang B. TNF-Α and RANKL promote osteoclastogenesis by upregulating RANK via the NF-κB pathway. Mol Med Rep. (2018) 17:6605–11. doi: 10.3892/mmr.2018.8698
64. Marahleh A, Kitaura H, Ohori F, Kishikawa A, Ogawa S, Shen WR, et al. TNF-α Directly enhances osteocyte RANKL expression and promotes osteoclast formation. Front Immunol. (2019) 10:2925. doi: 10.3389/fimmu.2019.02925
65. Oseko F, Yamamoto T, Akamatsu Y, Kanamura N, Iwakura Y, Imanishi J, et al. IL-17 is involved in bone resorption in mouse periapical lesions. Microbiol Immunol. (2009) 53:287–94. doi: 10.1111/j.1348-0421.2009.00123.x
66. Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol. (2017) 8:873. doi: 10.3389/fimmu.2017.00873
67. Nakanishi K. Unique action of interleukin-18 on T cells and other immune cells. Front Immunol. (2018) 9:763. doi: 10.3389/fimmu.2018.00763
68. Bohn E, Autenrieth IB. IL-12 is essential for resistance against Yersinia enterocolitica by triggering IFN-gamma production in NK cells and CD4+ T cells. J Immunol. (1996) 156:1458–68.8568248
69. Balto K, Sasaki H, Stashenko P. Interleukin-6 deficiency increases inflammatory bone destruction. Infect Immun. (2001) 69:744–50. doi: 10.1128/IAI.69.2.744-750.2001
70. Liu S, Li Q, Liu Y. Immunohistochemical localization of NALP3 inflammasome in experimental periapical lesions. Int Endod J. (2014) 47:949–57. doi: 10.1111/iej.12240
71. Haq AU, Rinehart JJ, Maca RD. The effect of gamma interferon on IL-1 secretion of in vitro differentiated human macrophages. J Leukoc Biol. (1985) 38:735–46. doi: 10.1002/jlb.38.6.735
72. Chujor CS, Klein L, Lam C. Selective inhibition of interleukin-1 beta gene expression in activated RAW 264.7 macrophages by interferon-gamma. Eur J Immunol. (1996) 26:1253–9. doi: 10.1002/eji.1830260611
73. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. (1993) 260:547–9. doi: 10.1126/science.8097338
74. Sasaki H, Balto K, Kawashima N, Eastcott J, Hoshino K, Akira S, et al. Gamma interferon (IFN-gamma) and IFN-gamma-inducing cytokines interleukin-12 (IL-12) and IL-18 do not augment infection-stimulated bone resorption in vivo. Clin Diagn Lab Immunol. (2004) 11:106–10. doi: 10.1128/cdli.11.1.106-110.2004
75. De Rossi A, Rocha LB, Rossi MA. Interferon-gamma, interleukin-10, intercellular adhesion molecule-1, and chemokine receptor 5, but not interleukin-4, attenuate the development of periapical lesions. J Endod. (2008) 34:31–8. doi: 10.1016/j.joen.2007.09.021
76. Prso IB, Kocjan W, Simić H, Brumini G, Pezelj-Ribarić S, Borcić J, et al. Tumor necrosis factor-alpha and interleukin 6 in human periapical lesions. Mediators Inflamm. (2007):38210. doi: 10.1155/2007/38210
77. Bando Y, Henderson B, Meghji S, Poole S, Harris M. Immunocytochemical localization of inflammatory cytokines and vascular adhesion receptors in radicular cysts. J Oral Pathol Med. (1993) 22:221–7. doi: 10.1111/j.1600-0714.1993.tb01060.x
78. Ishimi Y, Miyaura C, Jin CH, Akatsu T, Abe E, Nakamura Y, et al. IL-6 is produced by osteoblasts and induces bone resorption. J Immunol. (1990) 145:3297–303.2121824
79. Tat S K, Padrines M, Théoleyre S, Heymann D, Fortun Y. IL-6, RANKL, TNF-alpha/IL-1: Interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. (2004) 15:49–60. doi: 10.1016/j.cytogfr.2003.10.005
80. Hughes FJ, Howells GL. Interleukin-6 inhibits bone formation in vitro. Bone Miner. (1993) 21:21–8. doi: 10.1016/s0169-6009(08)80117-1
81. Tilg H, Trehu E, Atkins MB, Dinarello CA, Mier JW. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: Induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. (1994) 83:113–8. doi: 10.1182/blood.V83.1.113.113
82. Blanchard F, Duplomb L, Baud'huin M, Brounais B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. (2009) 20:19–28. doi: 10.1016/j.cytogfr.2008.11.004
83. Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, et al. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone. (2006) 39:505–12. doi: 10.1016/j.bone.2006.02.074
84. Sims NA. Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int J Biochem Cell Biol. (2016) 79:14–23. doi: 10.1016/j.biocel.2016.08.003
85. Huang GT, Do M, Wingard M, Park JS, Chugal N. Effect of interleukin-6 deficiency on the formation of periapical lesions after pulp exposure in mice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (2001) 92:83–8. doi: 10.1067/moe.2001.115025
86. Chatterjee P, Chiasson VL, Bounds KR, Mitchell BM. Regulation of the anti-inflammatory cytokines interleukin-4 and interleukin-10 during pregnancy. Front Immunol. (2014) 5:253. doi: 10.3389/fimmu.2014.00253
87. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: A cytokine to remember. J Immunol. (2012) 189:4213–9. doi: 10.4049/jimmunol.1202246
88. Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev. (2010) 21:331–44. doi: 10.1016/j.cytogfr.2010.09.002
89. Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J Exp Med. (2007) 204:239–43. doi: 10.1084/jem.20070104
90. Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. (1993) 150:353–60.8419468
91. Sasaki H, Hou L, Belani A, Wang CY, Uchiyama T, Müller R, et al. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J Immunol. (2000) 165:3626–30. doi: 10.4049/jimmunol.165.7.3626
92. Cua DJ, Tato CM. Innate IL-17-producing cells: The sentinels of the immune system. Nat Rev Immunol. (2010) 10:479–89. doi: 10.1038/nri2800
93. Colić M, Vasilijić S, Gazivoda D, Vucević D, Marjanović M, Lukić A. Interleukin-17 plays a role in exacerbation of inflammation within chronic periapical lesions. Eur J Oral Sci. (2007) 115:315–20. doi: 10.1111/j.1600-0722.2007.00460.x
94. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. (1998) 160(7):3513–219531313
95. AlShwaimi E, Berggreen E, Furusho H, Rossall JC, Dobeck J, Yoganathan S, et al. IL-17 receptor A signaling is protective in infection-stimulated periapical bone destruction. J Immunol. (2013) 191:1785–91. doi: 10.4049/jimmunol.1202194
96. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
97. Elenkov IJ, Iezzoni DG, Daly A, Harris AG, Chrousos GP. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation. (2005) 12:255–69. doi: 10.1159/000087104
98. De Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. (2012) 71:332–8. doi: 10.1017/S0029665112000092
99. Falagas ME, Kompoti M. Obesity and infection. Lancet Infect Dis. (2006) 6:438–46. doi: 10.1016/S1473-3099(06)70523-0
100. Petrakis D, Margină D, Tsarouhas K, Tekos F, Stan M, Nikitovic D, et al. Obesity - a risk factor for increased COVID-19 prevalence, severity and lethality (review). Mol Med Rep. (2020) 22:9–19. doi: 10.3892/mmr.2020.11127
101. Martinez-Herrera M, Silvestre-Rangil J, Silvestre FJ. Association between obesity and periodontal disease. A systematic review of epidemiological studies and controlled clinical trials. Med Oral Patol Oral Cir Bucal. (2017) 22:e708–15. doi: 10.4317/medoral.21786
102. Deshpande NC, Amrutiya MR. Obesity and oral health - is there a link? An observational study. J Indian Soc Periodontol. (2017) 21:229–33. doi: 10.4103/jisp.jisp_305_16
103. Segura-Egea JJ, Martín-González J, Cabanillas-Balsera D, Fouad AF, Velasco-Ortega E, López-López J. Association between diabetes and the prevalence of radiolucent periapical lesions in root-filled teeth: Systematic review and meta-analysis. Clin Oral Investig. (2016) 20:1133–41. doi: 10.1007/s00784-016-1805-4
104. Gupta A, Aggarwal V, Mehta N, Abraham D, Singh A. Diabetes mellitus and the healing of periapical lesions in root filled teeth: A systematic review and meta-analysis. Int Endod J. (2020) 53:1472–84. doi: 10.1111/iej.13366
105. Fouad AF, Burleson J. The effect of diabetes mellitus on endodontic treatment outcome: Data from an electronic patient record. J Am Dent Assoc. (2003) 134:43–51; quiz 117–8. doi: 10.14219/jada.archive.2003.0016.12555956
106. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI19246
107. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI19451
108. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
109. Li P, Lu M, Nguyen MTA, Bae EJ, Chapman J, Feng D, et al. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J Biol Chem. (2010) 285(20):15333–45. doi: 10.1074/jbc.M110.100263
110. Prieur X, Mok CY, Velagapudi VR, Núñez V, Fuentes L, Montaner D, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. (2011) 60:797–809. doi: 10.2337/db10-0705
111. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. (2008) 57:3239–46. doi: 10.2337/db08-0872
112. Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: Phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care. (2011) 14:341–6. doi: 10.1097/MCO.0b013e328347970b
113. Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract. (2005) 69:29–35. doi: 10.1016/j.diabres.2004.11.007
114. Palomera LF, Gómez-Arauz AY, Villanueva-Ortega E, Meléndez-Mier G, Islas-Andrade SA, Escobedo G. Serum levels of interleukin-1 beta associate better with severity of simple steatosis than liver function tests in morbidly obese patients. J Res Med Sci. (2018) 23:93. doi: 10.4103/jrms.JRMS_142_18
115. Rodríguez-Hernández H, Simental-Mendía LE, Rodríguez-Ramírez G, Reyes-Romero MA. Obesity and inflammation: Epidemiology, risk factors, and markers of inflammation. Int J Endocrinol. (2013):678159. doi: 10.1155/2013/678159
116. Ballak DB, Stienstra R, Tack CJ, Dinarello CA, van Diepen JA. IL-1 family members in the pathogenesis and treatment of metabolic disease: Focus on adipose tissue inflammation and insulin resistance. Cytokine. (2015) 75:280–90. doi: 10.1016/j.cyto.2015.05.005
117. Tilg H, Moschen AR. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. (2006) 6:772–83. doi: 10.1038/nri1937
118. Taylor EB. The complex role of adipokines in obesity, inflammation, and autoimmunity. Clin Sci. (2021) 135:731–52. doi: 10.1042/CS20200895
119. Carbone F, La Rocca C, Matarese G. Immunological functions of leptin and adiponectin. Biochimie. (2012) 94:2082–8. doi: 10.1016/j.biochi.2012.05.018
120. Kiran S, Kumar V, Murphy EA, Enos RT, Singh UP. High fat diet-induced CD8+ T cells in adipose tissue mediate macrophages to sustain low-grade chronic inflammation. Front Immunol. (2021) 12:680944. doi: 10.3389/fimmu.2021.680944
121. Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S, et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity. (2013) 21:461–8. doi: 10.1002/oby.20087
122. Agabiti-Rosei C, Trapletti V, Piantoni S, Airò P, Tincani A, De Ciuceis C, et al. Decreased circulating T regulatory lymphocytes in obese patients undergoing bariatric surgery. PLoS One. (2018) 13:e0197178. doi: 10.1371/journal.pone.0197178
123. Yun JM, Jialal I, Devaraj S. Effects of epigallocatechin gallate on regulatory T cell number and function in obese v. Lean volunteers. Br J Nutr. (2010) 103:1771–7. doi: 10.1017/S000711451000005X
124. Yang H, Youm YH, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood. (2009) 114:3803–12. doi: 10.1182/blood-2009-03-213595
125. Um JY, Rim HK, Kim SJ, Kim HL, Hong SH. Functional polymorphism of IL-1 alpha and its potential role in obesity in humans and mice. PLoS One. (2011) 6:e29524. doi: 10.1371/journal.pone.0029524
Keywords: obesity, diabetes, apical periodontitis, cytokines, interleukin-1 signaling, immune response
Citation: Tazawa K, Azuma Presse MM, Furusho H, Stashenko P and Sasaki H (2022) Revisiting the role of IL-1 signaling in the development of apical periodontitis. Front. Dent. Med 3:985558. doi: 10.3389/fdmed.2022.985558
Received: 4 July 2022; Accepted: 28 July 2022;
Published: 11 August 2022.
Edited by:
Francisco Nociti, Universidade Estadual de Campinas, BrazilReviewed by:
Brenda P. Gomes, Campinas State University, Brazil© 2022 Tazawa, Azuma Presse, Furusho, Stashenko and Sasaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hajime Sasaki, aGFqaW1lc0B1bWljaC5lZHU=; Philip Stashenko, cHN0YXNoQGJ1LmVkdQ==
†These authors share corresponding authorship.
Specialty Section: This article was submitted to Endodontics, a section of the journal Frontiers in Dental Medicine