 Marzia Di Donato
Marzia Di Donato Pia Giovannelli
Pia Giovannelli Gustavo Cernera
Gustavo Cernera Annalisa Di Santi
Annalisa Di Santi Irene Marino
Irene Marino Antonio Bilancio
Antonio Bilancio Giovanni Galasso
Giovanni Galasso Ferdinando Auricchio
Ferdinando Auricchio Antimo Migliaccio
Antimo Migliaccio Gabriella Castoria*
Gabriella Castoria*- Department of Biochemistry, Biophysics and General Pathology, II University of Naples, Naples, Italy
Prostate cancer (PCa) is the major cause of cancer-related death among the male population of Western society, and androgen-deprivation therapy (ADT) represents the first line in PCa treatment. However, although androgen receptor (AR) expression is maintained throughout the various stages of PCa, ADT frequently fails. Clinical studies have demonstrated that different androgen/AR signaling pathways operate in target tissues. AR stimulates growth and transformation of target cells, but under certain conditions slows down their proliferation. In this review, we discuss the role of AR in controlling different functions of mesenchymal and transformed mesenchymal cells. Findings here presented support the role of AR in suppressing proliferation and stimulating migration of stromal cells, with implications for current approaches to cancer therapy.
Introduction
Prostate cells are the primary targets of androgens, which regulate development, growth, and function of prostate. In this organ, androgens act through the androgen receptor (AR), which is expressed in both epithelium and stroma, and studies with recombinant tissues indicate that prostatic development is mediated by stromal, but not epithelial AR (1). Sex steroids influence prostate cancer (PCa) initiation and/or progression (2), whereas androgen removal increases PCa cell death and inhibits cell proliferation. Hence, androgen-deprivation therapy (ADT) is the standard therapy for men with PCa. However, despite extensive efforts in the clinical development of PCa therapies, current treatments only control tumor growth initially, and fail to achieve long-term efficacy since most PCa patients eventually relapse.
The role of epithelial AR in PCa has been extensively studied in the last few decades. There are, however, many other AR target tissues throughout the body (3), though specific information on the role of AR in these cells is still minimal and few investigations have been undertaken to elucidate the role of stromal AR. It has been suggested that AR may direct stromal cells toward epithelial PCa cells upon an increase in local androgen levels, as frequently occurs in PCa (4). Stromal AR has also been shown to mediate PCa metastasis (5) and induce prostatic intraepithelial neoplasia (6). Nonetheless, data on the role of stromal AR still remain controversial.
AR paradoxically fosters or inhibits proliferation of target cells depending on cell type, microenvironment, and hormone levels (7). Reciprocal AR responses in epithelium and stroma regulate prostate development and homeostasis. Aberrant responses might result in tumorigenesis. Androgen binding to AR induces proliferation in PCa epithelial cells and differentiation in normal prostate epithelial cells (8). Prostate stromal cells express AR, but their growth is insensitive to androgen stimulation (9). Fibroblasts and human fibrosarcoma HT1080 cells do not grow in response to 10 nM androgen. At this hormone concentration, these cells undergo migration as a consequence of a bipartite AR/filamin A (FlnA) complex assembly (10, 11).
The dichotomous response (migration/proliferation) of fibroblasts to different androgen concentrations mimics that of growth factors, which trigger motility or proliferation [reviewed in Ref. (12)]. A flurry of reports has investigated the molecular basis for a cell decision to “go or grow.” EGF, VEGF, and PDGF trigger motility or proliferation depending on cell type, receptor distribution or internalization, ligand concentration, and dynamic regulation of signaling networks (13–17). In NIH3T3 cells, for instance, low PDGF concentration induces migration, whereas high concentration triggers proliferation. Cells make these different decisions as a consequence of the different endocytic routes (clathrin- and non-clathrin-mediated) engaged by the PDGF receptor (18). A G-alpha i-GIV complex binds EGF-R and decides whether cancer cells migrate or proliferate (16). Again, interaction of the EphB2 tyrosine kinase receptor with focal adhesion kinase (FAK) promotes invasiveness and inhibits proliferation in glioblastoma multiforme (19).
Here, we discuss the opposite (proliferative or migratory) functions of stromal AR and the role of the upstream AR/FlnA complex in stromal cell decision to “go or grow.” Since the AR/FlnA complex drives androgen signaling toward migration and halts cell cycle, this complex might be specifically targeted, with implications in the therapy of AR-related human diseases. The potential role of non-genomic signaling activated by androgen/AR axis in cancer-associated fibroblasts (CAFs) is also discussed.
Androgen Signaling in Stromal Cells
AR mRNA levels are very similar in the stroma and epithelium, although AR-mediated transcription is differentially regulated in each, with a more active co-regulator recruitment by epithelial AR.
In the early 2000s, we made the exciting finding that NIH3T3 cells and mouse embryo fibroblasts (MEFs) harbor a transcriptionally incompetent AR (20). Further expansion of the study led to the discovery that AR plays a dual and opposite role in mesenchymal and transformed mesenchymal cells. At sub-optimal androgen concentration (1 pM) AR mediates cell cycle progression and proliferation, while at optimal androgen concentration (10 nM) the receptor halts mitogenesis and fosters migration (10, 11, 20). The dichotomous effect is not restricted to immortalized (NIH3T3 cells) or transformed (HT1080 cells) fibroblasts, but can also be observed in primary fibroblasts from adult or embryonic mouse (11).
A few years ago, by recombination of prostate stromal WPMY-1 cells with PCa epithelial PC-3 cells in a mouse model, Niu and colleagues showed that stromal but not epithelial AR promotes tumor proliferation at very early stage (21). Noteworthy, as occurs in fibroblasts and transformed fibroblasts, prostate stromal WPMY-1 cells harbor a transcriptionally incompetent AR, which is permanently localized in cytoplasm (22). Thus, the receptor expressed in WPMY-1 cells likely promotes growth of epithelial PC-3 cells through a non-transcriptional mechanism.
Prostate CAFs express AR, although the role of this receptor in prostate tumorigenesis is still unclear. Loss of AR expression in the tumor stroma, but not in the surrounding normal prostatic stroma tissue, increases the risk of relapse following radical prostatectomy (23, 24). Stromal AR expression progressively decreases during PCa progression (22, 23). Thus, loss of stromal AR might enable the growth, progression, and response to the therapy in PCa. In apparent contrast with these findings, tissue-specific knockout approach showed that deletion of AR in fibroblasts and fibro-muscular cells (dARKO) inhibits the growth of PCa in mouse model (6). A similar conclusion was reached using immortalized CAFs from PCa biopsies (25). By using these experimental settings, it has been shown that stromal AR modulates the release of pro-inflammatory chemokines by CAFs, thereby promoting recruitment of inflammatory and immune cells in tumor microenvironment (6, 25). These events may result in extracellular matrix remodeling and angiogenesis, as well as proliferation and invasion of neighboring epithelial PCa cells. In conclusion, conflicting findings on the role of stromal AR can be obtained using different experimental settings (i.e., tissue recombination in immune-depressed mouse or AR tissue-specific knockout in immune-intact mouse). Thus, further analysis of stromal AR should be undertaken to gain valuable preclinical findings. Moreover, the contribution of AR transcriptional and/or non-transcriptional activity to biological responses in CAFs still remains elusive.
We have dissected in recent years the pathways activated by different androgen concentrations in fibroblasts, and this approach has revealed some fascinating aspects. Low androgen concentration (1 pM) induces proliferation in NIH3T3 fibroblasts as a consequence of AR association with Src and p85, the regulatory subunit of PI3-K (20). This complex activates MAPKs and Akt, which then increase cyclin D1 expression and re-localize p27 to cytoplasm for its subsequent degradation (11, 20). Conversely, stimulation of NIH3T3 fibroblasts with optimal (nanomolar) androgen concentration does not induce AR association with Src and p85, but triggers AR association with the actin-binding protein, FlnA. Ten nM androgen stabilizes AR/FlnA co-localization at intermediate cytoskeletal filaments and induces a complex including AR/FlnA/integrin beta1 in NIH3T3 fibroblasts. This complex triggers FAK and paxillin tyrosine phosphorylation. Furthermore, the AR/FlnA/integrin beta1 complex activates Rac. These events lead to cytoskeleton reorganization, adhesion changes, and cell migration [(10); Figure 1].
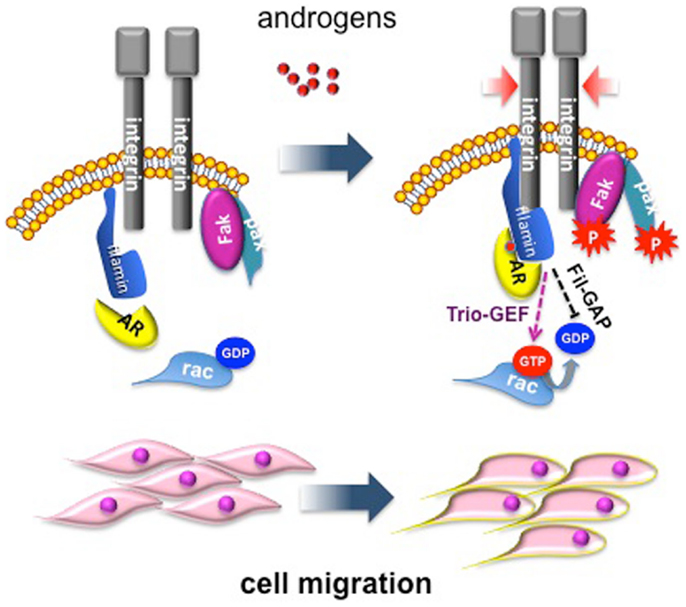
Figure 1. Migratory pathway activated by androgens in mesenchymal and transformed mesenchymal cells. Androgen (10 nM) enhances AR/FlnA co-localization at intermediate cytoskeleton filaments and induces a tripartite complex including AR/FlnA/integrin beta1 in NIH3T3 fibroblasts. This complex, likely through Trio-GEF, activates Rac. The Fln-associated GTPase activating protein (Fil-GAP) might switch off Rac-GTP. The AR/FlnA/integrin beta1 complex also triggers FAK and paxillin tyrosine phosphorylation. Activation of this machinery modifies cytoskeleton actin and cell adhesion, thus increasing motility of target cells (10).
On this basis, we may hypothesize that AR-mediated activation of MAPK induces the release of chemokines and growth factors by CAFs when androgen levels are low. Indeed, MAPK activation increases growth factor release in various cancer cell types [(26) and refs therein; (27)]. Again, by activating PI3-K at low androgen concentration, stromal AR may act in concert with NF-kappa-B to induce the release of macrophage inflammatory protein-1 beta (MIP-1beta) in tumor microenvironment, or it may activate IKK-alpha-dependent gene transcription. Stromal AR co-operates with NF-Kappa-B to modulate the gene expression of MIP-1beta (6), and nuclear receptors induce IKK-alpha-dependent epigenetic modifications and gene expression changes through activation of PI3-K pathway (28, 29). When androgen levels increase, CAFs might move toward epithelial PCa cells as a consequence of the AR/FlnA complex assembly and activation of molecular machinery involved in cell motility.
In conclusion, non-genomic functions of stromal AR might either sustain the proliferation and invasiveness of epithelial PCa cells or induce the recruitment of CAFs toward neighboring epithelial PCa cells. Androgen levels in tumor microenvironment may thus dictate the type of biological outcome in CAFs.
We recently showed that by increasing androgen concentration from 1 pM to 10 nM, AR shifts from Src/PI3-K toward association with FlnA, which in turns mediates the ligand-dependent activation of a different set of effectors (e.g., Trio-GEF, Rac) in stromal cells. Thus, AR-protein interactions regulate the type of hormonal responses, and these interactions are in turn controlled by ligand concentration. Different androgen levels might, for instance, affect post-translational modifications of AR. Phosphorylation, sumoylation, acetylation, and ubiquitination are potential reversible mechanisms affecting AR stability, localization, and interactions of the receptor with other proteins [reviewed in Ref. (30, 31)]. By enhancing the receptor interaction with signaling effectors, post-translational modifications might impact the internalization route of AR and receptor functions. In sum, there are different potential routes for androgens to orchestrate association of AR with Src/PI3-K or FlnA, thus providing a mechanism for downstream pathway regulation.
Migration rarely occurs in proliferating cells, and signals stimulating migration inhibit cell proliferation [reviewed in Ref. (32)]. We recently identified androgen-activated Rac as the switch regulating transition from proliferative to migratory phenotype in NIH3T3 fibroblasts and HT1080 fibrosarcoma cells. In these cells, Rac activation by 10 nM androgen halts cell cycle progression and triggers cell motility (11). Molecular studies have shown that these cells make the decision to halt cell cycle by triggering activation of the Rac-dependent DYRK 1B kinase upon challenging with 10 nM androgen. Once activated, DYRK 1B kinase triggers Ser10 phosphorylation of p27 and its stabilization. This pathway, which is controlled by the upstream AR/FlnA complex, fosters cell quiescence, and even inhibits transformation induced by oncogenic Ras [(11); Figure 2]. Thus, AR mediates growth suppression by specifically targeting Ras-driven growth-promoting pathways, highlighting the role of androgen/AR axis in human cancers. Oncogenic Ras mutations have been described in carcinomas of the pancreas, colon, lung, and thyroid, as well as in myeloid leukemia (33). Interestingly, most of these tumors (pancreas, colon, and lung) express AR (3, 34). Proliferation of various pancreatic cancer-derived cells, which express AR, is insensitive to androgens (35), and androgens inhibit survival signals in colon cancer in vitro and in vivo (36). Again, small-cell lung carcinoma H1184 cell line exhibits significant growth upon stimulation with sub-optimal androgen concentrations, but is growth-inhibited at higher androgen concentrations. Androgens also impair the growth of non-small-cell lung carcinoma H1993 cell line (37). Human fibrosarcoma HT1080 cells, exhibiting an activated N-Ras, do not grow in response to high androgen concentrations (11, 34). Hence, the arguments put forward here raise the possibility that androgens elicit anti-proliferative signals in human cancers bearing oncogenic Ras mutations through activation of DYRK 1B. Noteworthy, DYRK 1B is an active kinase in various human cancers and regulates Ras-driven transformation and tumor progression (38–40). Thus, by activating DYRK 1B kinase, androgen/AR axis might restrain Ras-driven transformation.
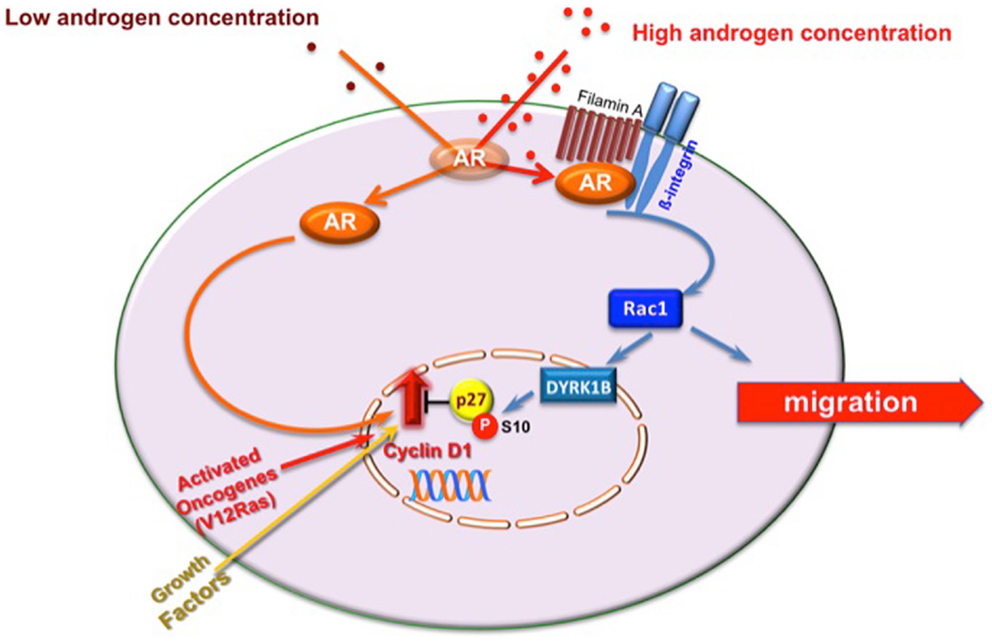
Figure 2. Model of proliferative/migratory pathways activated by androgens in mesenchymal and transformed mesenchymal cells. One pM androgen triggers cell proliferation through the AR/Src/p85alpha complex assembly (20). Ten nM androgen triggers the AR/FlnA complex assembly that in turn activates Rac. Rac enhances cell migration (10) and triggers DYRK 1B activation (11). This results in p27 Ser10 phosphorylation and p27 stabilization. Quiescence of fibroblasts and human fibrosarcoma cells then follows (11). In AR-expressing cells, oncogenic Ras or excessive growth factor stimuli increase cyclin D1 expression and cell proliferation. By stimulating DYRK 1B activation, 10 nM androgen triggers Ser10 phosphorylation and stabilization of p27 (11). This event likely counteracts the effect of activated Ras or growth factors on cyclin D1 and cell proliferation.
Stromal AR promotes the growth and differentiation of developing prostate, while it appears to inhibit the growth of PCa under certain conditions (22). We recently observed that treatment of androgen-stimulated mouse and MEFs, as well as NIH3T3 and HT1080 cells with the anti-androgen bicalutamide (Casodex) increases cell proliferation. By displacing the androgen binding to AR, bicalutamide inhibits AR-elicited migratory signals and enables AR-mediated cell proliferation through the prevention of AR/FlnA complex assembly (11). This finding is of interest, since bicalutamide is frequently used in human PCa as ADT and often promotes PCa progression [reviewed in Ref. (7)]. Therefore, our recent results in migration/proliferation of mesenchymal and transformed mesenchymal cells might partly clarify the action of bicalutamide in enhancing PCa progression through activation of undesired pathways in stromal cells. These data further suggest that novel approaches are urgently needed for PCa treatment.
In summary, our results show that upon 10 nM androgen-triggered AR/FlnA complex assembly, FlnA acts as a scaffold for Rac and effectors of its dependent pathway, thereby enabling Rac activation and recruitment by Rac of specific signaling proteins (e.g., DYRK 1B). These events lead to cell motility and simultaneous cell cycle arrest in mesenchymal and transformed mesenchymal cells. Interference in AR/FlnA complex assembly by new molecules, such as AR-derived peptides (11), may represent a promising approach to the therapy of PCa by specifically modulating signaling pathways activated by AR in stromal cells.
Conflict of Interest Statement
The Review Editor Daniela Pasquali declares that, despite being affiliated to the same institution as the authors, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Italian Ministry of University and Scientific Research (P.R.I.N. 2010-2011; 2010NFEB9L_002 to Gabriella Castoria) and the Italian Association for Cancer Research (IG11520 to Antimo Migliaccio) supported this work. Pia Giovannelli and Marzia Di Donato are supported by a fellowship of the Italian Ministry of University and Scientific Research. We thank C. Fisher for linguistic editing.
References
1. Cunha GR, Lung B. The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J Exp Zool (1978) 205:181–93. doi: 10.1002/jez.1402050203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Ricke WA, Ishii K, Ricke EA, Simko J, Wang Y, Hayward SW, et al. Steroid hormones stimulate human prostate cancer progression and metastasis. Int J Cancer (2006) 118(9):2123–31.
3. Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene (2014) 33:3225–34. doi:10.1038/onc.2013.274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res (2008) 68:4447–54. doi:10.1158/0008-5472.CAN-08-0249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Ricke EA, Williams K, Lee YF, Couto S, Wang Y, Hayward SW, et al. Androgen hormone action in prostatic carcinogenesis: stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis (2012) 33:1391–8. doi:10.1093/carcin/bgs153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Lai KP, Yamashita S, Huang CK, Yeh S, Chang C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol Med (2012) 4:791–807. doi:10.1002/emmm.201101140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Niu Y, Chang TM, Yeh S, Ma WL, Wang YZ, Chang C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene (2010) 29:3593–604. doi:10.1038/onc.2010.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle (2007) 6:647–51. doi:10.4161/cc.6.6.4028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Sun X, He H, Xie Z, Qian W, Zhau HE, Chung LW, et al. Matched pairs of human prostate stromal cells display differential tropic effects on LNCaP prostate cancer cells. In vitro Cell Dev Biol Anim (2010) 46:538–46. doi:10.1007/s11626-010-9309-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Castoria G, D’Amato L, Ciociola A, Giovannelli P, Giraldi T, Sepe L, et al. Androgen-induced cell migration: role of androgen receptor/filamin A association. PLoS One (2011) 6:e17218. doi:10.1371/journal.pone.0017218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Castoria G, Giovannelli P, Di Donato M, Ciociola A, Hayashi R, Bernal F, et al. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis (2014) 5:e1548. doi:10.1038/cddis.2014.497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Migliaccio A, Castoria G, Bilancio A, Giovannelli P, Di Donato M, Auricchio F. Non-genomic action of steroid hormones: more questions than answers. In: Castoria G, Migliaccio A, editors. Advances in Rapid Sex-Steroid Action: New Challenges and New Chances in Breast and Prostate Cancers. New York, NY: Springer (2012). p. 1–19.
13. Chen P, Gupta K, Wells A. Cell movement elicited by epidermal growth factor receptor requires kinase and autophosphorylation but is separable from mitogenesis. J Cell Biol (1994) 124:547–55. doi:10.1083/jcb.124.4.547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Chen P, Xie H, Sekar MC, Gupta K, Wells A. Epidermal growth factor receptor-mediated cell motility: phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J Cell Biol (1994) 127:847–57. doi:10.1083/jcb.127.3.847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol (2003) 161:1163–77. doi:10.1083/jcb.200302047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, et al. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell (2010) 21:2338–54. doi:10.1091/mbc.E10-01-0028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature (2013) 499:166–71. doi:10.1038/nature12308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. De Donatis A, Comito G, Buricchi F, Vinci MC, Parenti A, Caselli A, et al. Proliferation versus migration in platelet-derived growth factor signaling: the key role of endocytosis. J Biol Chem (2008) 283:19948–56. doi:10.1074/jbc.M709428200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Wang SD, Rath P, Lal B, Richard JP, Li Y, Goodwin CR, et al. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene (2012) 31:5132–43. doi:10.1038/onc.2012.16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Castoria G, Lombardi M, Barone MV, Bilancio A, Di Domenico M, Bottero D, et al. Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a non-transcriptional receptor action. J Cell Biol (2003) 161:547–56. doi:10.1083/jcb.200211099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Niu Y, Altuwaijri S, Yeh S, Lai KP, Yu S, Chuang KH, et al. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc Natl Acad Sci U S A (2008) 105:12188–93. doi:10.1073/pnas.0804701105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Singh M, Jha R, Melamed J, Shapiro E, Hayward SW, Lee P. Stromal androgen receptor in prostate development and cancer. Am J Pathol (2014) 184:2598–607. doi:10.1016/j.ajpath.2014.06.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Henshall SM, Quinn DI, Lee CS, Head DR, Golovsky D, Brenner PC, et al. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res (2001) 61(2):423–7.
24. Ricciardelli C, Choong CS, Buchanan G, Vivekanandan S, Neufing P, Stahl J, et al. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate (2005) 63:19–28. doi:10.1002/pros.20154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Yu S, Xia S, Yang D, Wang K, Yeh S, Gao Z, et al. Androgen receptor in human prostate cancer-associated fibroblasts promotes prostate cancer epithelial cell growth and invasion. Med Oncol (2013) 30:674–80. doi:10.1007/s12032-013-0674-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev (2001) 15:981–94. doi:10.1101/gad.191101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Phan VT, Wu X, Cheng JH, Sheng RX, Chung AS, Zhuang G, et al. Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF-induced neutrophil recruitment. Proc Natl Acad Sci U S A (2013) 110:6079–84. doi:10.1073/pnas.1303302110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Perillo B, Di Santi A, Cernera G, Ombra MN, Castoria G, Migliaccio A. Phosphorylation of H3 serine 10 by IKKα governs cyclical production of ROS in estrogen-induced transcription and ensures DNA wholeness. Cell Death Differ (2014) 9:1503. doi:10.1038/cdd.2014.91
29. Perillo B, Di Santi A, Cernera G, Ombra MN, Castoria G, Migliaccio A. Nuclear receptor-induced transcription is driven by spatially and timely restricted waves of ROS: the role of Akt, IKKα, and DNA damage repair enzymes. Nucleus (2014) 5:5. doi:10.4161/nucl.36274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol (2012) 352:70–8. doi:10.1016/j.mce.2011.07.004
31. van der Steen T, Tindall DJ, Huang H. Posttranslational modification of the androgen receptor in prostate cancer. Int J Mol Sci (2013) 14:14833–59. doi:10.3390/ijms140714833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Fedotov S, Iomin A. Migration and proliferation dichotomy in tumor-cell invasion. Phys Rev Lett (2007) 98:118101. doi:10.1103/PhysRevLett.98.118101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Castoria G, Giovannelli P, Di Donato M, Hayashi R, Arra C, Appella E, et al. Targeting androgen receptor/Src complex impairs the aggressive phenotype of human fibrosarcoma cells. PLoS One (2013) 8:e76899. doi:10.1371/journal.pone.0076899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Okitsu K, Kanda T, Imazeki F, Yonemitsu Y, Ray RB, Chang C, et al. Involvement of interleukin-6 and androgen receptor signaling in pancreatic cancer. Genes Cancer (2010) 1:859–67. doi:10.1177/1947601910383417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Gu S, Papadopoulou N, Nasir O, Föller M, Alevizopoulos K, Lang F, et al. Activation of membrane androgen receptors in colon cancer inhibits the prosurvival signals Akt/bad in vitro and in vivo and blocks migration via vinculin/actin signaling. Mol Med (2011) 17:48–58. doi:10.2119/molmed.2010.00120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Jeong Y, Xie Y, Lee W, Bookout AL, Girard L, Raso G, et al. Research resource: diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Mol Endocrinol (2012) 26:1443–54. doi:10.1210/me.2011-1382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Jin K, Park S, Ewton DZ, Friedman E. The survival kinase Mirk/Dyrk1B is a downstream effector of oncogenic K-ras in pancreatic cancer. Cancer Res (2007) 1:7247–55. doi:10.1158/0008-5472.CAN-06-4099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Lauth M, Bergström A, Shimokawa T, Tostar U, Jin Q, Fendrich V, et al. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat Struct Mol Biol (2010) 17:718–25. doi:10.1038/nsmb.1833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: androgen receptor, filamin A, migration, growth suppression, fibroblasts, fibrosarcoma
Citation: Di Donato M, Giovannelli P, Cernera G, Di Santi A, Marino I, Bilancio A, Galasso G, Auricchio F, Migliaccio A and Castoria G (2015) Non-genomic androgen action regulates proliferative/migratory signaling in stromal cells. Front. Endocrinol. 5:225. doi:10.3389/fendo.2014.00225
Received: 17 October 2014; Paper pending published: 13 November 2014;
Accepted: 08 December 2014; Published online: 19 January 2015.
Edited by:
Rosamaria Lappano, University of Calabria, ItalyReviewed by:
Maria Marino, University Roma Tre, ItalyDaniela Pasquali, II University of Naples, Italy
Longfei Huo, M.D. Anderson Cancer Center, USA
Laura Corbo, UMR INSERM 1052-CNRS 5286, France
Copyright: © 2015 Di Donato, Giovannelli, Cernera, Di Santi, Marino, Bilancio, Galasso, Auricchio, Migliaccio and Castoria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriella Castoria, Department of Biochemistry, Biophysics and General Pathology, II University of Naples, Via L. De Crecchio 7, Naples 80138, Italy e-mail: gabriella.castoria@unina2.it