 Bin-Sheng He1
Bin-Sheng He1 Jia Qu
Jia Qu Qi Zhao
Qi Zhao- 1The First Affiliated Hospital, Changsha Medical University, Changsha, China
- 2School of Information and Control Engineering, China University of Mining and Technology, Xuzhou, China
- 3School of Mathematics, Liaoning University, Shenyang, China
- 4Research Center for Computer Simulating and Information Processing of Bio-Macromolecules of Liaoning Province, Shenyang, China
With the rapid development of biological research, microRNAs (miRNA) have become an attractive topic because lots of experimental studies have revealed the significant associations between miRNAs and diseases. However, considering that experiments are expensive and time-consuming, computational methods for predicting associations between miRNAs and diseases have become increasingly crucial. In this study, we proposed a neighborhood regularized logistic matrix factorization method for miRNA-disease association prediction (NRLMFMDA) by integrating miRNA functional similarity, disease semantic similarity, Gaussian interaction profile kernel similarity, and experimentally validation of disease-miRNA association. We used Gaussian interaction profile kernel similarity to cover the shortage of the traditional similarity to make it more reasonable and complete. Furthermore, NRLMFMDA also considered the important influences of the neighborhood information and took full advantage of them to improve the accuracy of the miRNA-disease association prediction. We also improved the accuracy by giving higher weights to the known association data in the process of calculating the potential association probabilities. In the global and the local leave-one-out cross validation, NRLMFMDA got the AUCs of 0.9068 and 0.8239, respectively. Moreover, the average AUC of NRLMFMDA in 5-fold cross validation was 0.8976 ± 0.0034. All the three kinds of cross validations have shown significant advantages to a number of previous models. In the case studies of breast neoplasms, esophageal neoplasms and lymphoma according to known miRNA-disease associations in the recent version of HMDD database, there were 78, 80, and 74% of top 50 predicted related miRNAs verified to have associations with these three diseases, respectively. In the further case studies for new disease without any known related miRNAs and the previous version of HMDD database, there were also high proportions of the predicted miRNAs verified by experimental reports. All the validation experiment results have demonstrated the effectiveness and practicability of NRLFMDA to predict the potential miRNA-disease associations.
Introduction
MicroRNAs (miRNAs) are a category of endogenous and short non-coding single-stranded RNAs (21~24 nucleotides) which could regulate the gene expression by targeting mRNAs for cleavage or translational repression at the posttranscriptional level (Ambros, 2001, 2004; Bartel, 2004; Meister and Tuschl, 2004). The first miRNA was found 20 years ago. And since then, people have discovered thousands of miRNAs in a wide variety of species (Jopling et al., 2005; Kozomara and Griffiths-Jones, 2011). Furthermore, more and more studies have found that the miRNAs play crucial roles at multiple stages of the biological processes (Lee et al., 1993; Chen et al., 2017b; Li et al., 2017), such as early cell growth, proliferation (Cheng et al., 2005), differentiation (Miska, 2005), development (Karp and Ambros, 2005), aging (Bartel, 2009), apoptosis (Xu et al., 2004), and so on. Additionally, the key regulatory roles of miRNAs have increasingly been paid attention to in the abnormal gene expression of biological cells. For example, the dysregulation of the miRNAs has been confirmed as a main reason of aberrant cell behavior by many studies (Griffiths-Jones et al., 2006). In the recent years, more and more experiments have been implemented to show that miRNAs have great connections with the various development processes of many human complex diseases (Lynam-Lennon et al., 2009; Meola et al., 2009; Huang et al., 2016b). For example, researches have implicated that miRNA-7a has clinical significance of high mobility group A2 in human gastric cancer. And Schulte et al. reported the capacity of miRNA-197 and miRNA-223 in predicting cardiovascular death and burden of future cardiovascular events in a large cohort of Coronary artery disease patients (Schulte et al., 2015). Besides, Thomas Thum et al. (Thum et al., 2008) showed that miR-21 affects the global cardiac structure and function through regulating the ERK–MAP kinase signaling pathway in cardiac fibroblasts. Therefore, identifying disease-related miRNAs is important and beneficial to the treatment, diagnosis, and prevention of a variety of clinically important disease. Nevertheless, identifying the associations between miRNAs and diseases with experimental methods is expensive and time-consuming. With the development of biological technology, lots of experiments have been implemented to produce vast numbers of miRNA-associated datasets. There is an urgent need for us to make further efforts to develop novel computational models for potential miRNAs-disease association prediction. In fact, many computational methods are well behaved in predicting miRNA-disease associations (Chen and Yan, 2013; Chen, 2015b; Chen et al., 2016a,g; Chen et al., 2018c). Therefore, further experimental studies can be more efficiently implemented by selecting the most promising associated miRNAs predicted by computational models.
Based on the assumption that functionally similar miRNAs are more likely to have associations with phenotypically similar diseases, many computational approaches have been introduced for the identification of miRNA-disease associations (Bandyopadhyay et al., 2010; Jiang et al., 2010; Liu et al., 2016b; Pasquier and Gardès, 2016; Zeng et al., 2016b; Zou et al., 2016; Chen and Huang, 2017; Chen et al., 2017a,c,d; You et al., 2017; Chen et al., 2018a,b,d,e,f; Tang et al., 2018). A hypergeometric distribution-based model was proposed by Jiang et al. (2010). Through using the human known disease-miRNA association network, disease phenotype similarity network and miRNA functional similarity network, this model gave the prediction of miRNA-disease associations. But there was a high proportion of false positive and false negative samples in the miRNA-target associations set on which this method extremely depended. Shi et al. (2013) proposed a random walk algorithm-based model in protein-protein interaction (PPI) network under the assumption that miRNAs have closer associations with the diseases that are more correlated to the miRNA targets. They obtained potential miRNA-disease associations by the comprehensive consideration of miRNA–target interactions, disease–gene associations and PPIs. Mørk et al. (2014) presented a miRPD method by integration of miRNA-protein association scores, protein-disease association scores and the shared proteins between miRNAs and diseases to obtain the best scoring protein connections between miRNA-disease pairs. Xu et al. (2014) introduced a miRNA prioritization model by the integrationof known disease–gene associations and miRNA-target interactions. It is worthy mentioning that the model is independent of the experimentally verified miRNA-disease associations. Instead, they need to calculate the similarity between miRNA targets and disease genes. Nonetheless, the aforementioned methods could not provide sufficiently accurate prediction results due to the incomplete disease-gene association network or/and the miRNA-target interactions with high false positive and false negative samples.
Xuan et al. (2013a) constructed a computational method called HDMP for the identification of miRNA-disease associations based on the experimentally verified miRNA-disease associations, miRNA functional similarity, disease semantic similarity and disease phenotype similarity. According to miRNAs with similar functions are normally related to similar diseases and vice versa, they used the k nearest neighbors of miRNAs for estimating more reliable relevance scores of the unlabeled miRNAs. To overcome the shortages of the previous methods, it assigned higher weights to members in the same miRNA cluster when they calculated the miRNA functional similarity. However, the HDMP cannot prioritize miRNAs(diseases) for diseases(miRNAs) that have no known related miRNAs(diseases). Additionally, the performance of HDMP could not better than most of previous models which were calculated based on the global network similarity measure. A global network similarity-based computational model was proposed by Chen et al. (2012b) called RWRMDA, which used the random walk method based on the dataset of human known miRNA–disease associations and miRNA functional similarity. We can see that RWRMDA has excellent prediction performance through cross-validation and case studies of several important human complex cancers. However, there is a non-negligible limitation that it could not work for diseases without any known associated miRNAs. Chen et al. (2016f) developed another computational approach of WBSMDA by integrating the Gaussian interaction profile kernel similarity, miRNA functional similarity, disease semantic similarity, and miRNA-disease associations for the prediction of potential miRNAs-diseases associations. WBSMDA could effectively predict disease(miRNA)-related miRNAs(diseases) that without known related miRNAs(diseases). Recently, Chen et al. (2016d) developed a novel computational model named HGIMDA, which had superior performance compared with four classical methods (WBSMDA, RLSMDA, RWRMDA, and HDMP).
Nowadays, machine learning has been applied in extensive scientific fields, and it is highly effective for most of the research problems (Chen et al., 2012a, 2015c, 2016c; Wong et al., 2015; Huang et al., 2016b). Therefore, more and more studies have focused on it. For instance, Xu et al. (2011) proposed a computational model, named miRNA-target dysregulated network (MTDN), which combined miRNA-target interactions and expression pattern of miRNAs and mRNAs. In the model, the support vector machine (SVM) classifier was constructed to distinguish positive miRNA-disease associations from negative ones by extracting the feature of network topologic information. It is known that negative miRNA-disease associations are difficult to obtain, and the ambiguity caused by negative samples usually affects the accuracy of the supervised. Chen et al. (Chen and Yan, 2014) provided RLSMDA, a computational model in which they used semi-supervised learning to predict potential disease-related miRNAs by the consideration of disease semantic similarity, miRNA functional similarity, and known miRNA-disease associations. Furthermore, RLSMDA could also predict disease(miRNA)-related miRNAs(diseases) without any known miRNAs(diseases) and avoid the problem of using negative miRNA-disease associations. However, the ways of combining the classifiers in different spaces together and the selection of parameters for RLSMDA would greatly influence the prediction result. Based on known miRNA-disease associations, Chen et al. (2015b) further developed a computational model of RBMMMDA by presenting restricted Boltzmann machine (RBM). RBMMMDA is a two-layer (visible and hidden) undirected graphical model, which can not only obtain new miRNA-disease associations, but also corresponding association types. Nevertheless, it is difficult to make decision on the parameter values.
In our proposed method, we introduced a novel matrix factorization computational approach, namely neighborhood regularized logistic matrix factorization for miRNA-disease association prediction (NRLMFMDA). In consideration of the effectiveness of the classical method with integrated similarities, we combined the Gaussian interaction profile kernel similarity and the modified matrix factorization to get a more accuracy prediction result. Based on the known miRNA-disease associations, disease semantic similarity, miRNA functional similarity, and Gaussian interaction profile kernel similarity, the proposed method focuses on predicting the probability that a miRNA would be associated with a disease by mapping a miRNA and a disease to a shared low dimensional latent space as two latent vectors. Additionally, we also studied the local structure of the association data to further improve the prediction accuracy by exploiting the influences of the neighbors which were from the most similar miRNAs and most similar diseases. Moreover, the proposed approach assigned higher importance level to the nearest neighbors for avoiding noisy information. Furthermore, we used global LOOCV, local LOOCV, and 5-fold cross validation to evaluate the effectiveness of NRLMFMDA. As a result, the AUCs of global and local LOOCV are 0.9068 and 0.8239, respectively. By adopting 5-fold cross validation, NRLMFMDA model obtained the average AUC of 0.8976 ± 0.0034. In three types of case studies, we tested the prediction effect of NRLMFMDA for known diseases in the recent version of HMDD database, new diseases without any known related miRNAs and known disease based on previous version of HMDD database, respectively. As a result, most of the predicted miRNAs have been confirmed by recent experimental reports. Thus, we can conclude that NRLMFMDA is a useful tool in predicting potential miRNA-disease associations.
Materials and Methods
Human miRNA-Disease Association
For convenience, we have built an adjacency matrix Y ∈ Rm×n to formalize the known miRNA-disease associations that acquired from the HMDD v2.0 database (Li et al., 2014). The known miRNA-disease associations dataset used in this paper includes 5430 distinct experimentally confirmed miRNA-disease between 383 diseases and 495 miRNAs, m and n were expressed as the miRNAs and diseases numbers in the dataset. Then we stored the known miRNA-disease association information into the matrix Y. If a miRNA ri has been experimentally verified to be associated with a diseasedj, then yij equals to 1, otherwise 0.
miRNA Functional Similarity
The miRNA functional similarity was calculated according to the method proposed by Wang et al. (2010) by the consideration of miRNAs with functional similar tend to be interacted with semantic similar diseases, and vice versa (Goh et al., 2007; Lu et al., 2008). Owing to their excellent work, we can download the miRNA functional similarity data from http://www.cuilab.cn/files/images/cuilab/misim.zip. The matrix MS was constructed to represent the miRNA functional similarity. The element MS(ri, rj) represented the value of similarity between the miRNA ri and the miRNArj.
Disease Semantic Similarity Model 1
We constructed a Directed Acyclic Graph (DAG) to describe the diseases according to the MeSH descriptors downloaded from the National Library of Medicine (http://www.nlm.nih.gov/) (Chen, 2015a; Chen et al., 2015a, 2016a,e; Huang et al., 2016a). Then we defined the contribution of disease d in DAG(D) to the semantic value of disease D as follows:
where Δ is the semantic contribution decay factor and we set the value of Δ to 0.5 (Xuan et al., 2013b). The self-semantic value of disease D is defined as follows:
where T(D) represents D itself and all its ancestral nodes. According to the observation that two diseases with larger shared part of their DAGs have larger similarity score, the semantic similarity score between disease di and dj are defined as follows:
Disease Semantic Similarity Model 2
Different from disease semantic similarity model 1, we considered that assigning the same contribution value to the diseases in the same layer of DAG(D) was not reasonable. Actually, a more specific disease which appears in less DAGs contributes to the semantic similarity of disease D at a higher contribution level. So we made definition for the contribution of disease d in DAG(D) to the semantic value of disease D as follows:
We gave definition of the semantic similarity between disease di and dj are the proportion of the summing contributions of their shared ancestor nodes and themselves to them in all the contributions of their ancestor nodes and themselves defined as the disease semantic similarity model 1.
Gaussian Interaction Profile Kernel Similarity
Considering that Gaussian kernel function is one of the Radial Basis function whose value depends only on the distance from the origin, we constructed Gaussian interaction profile kernel similarity as another similarity algorithm that different from disease semantic similarity and miRNA functional similarity (Van et al., 2011; Chen et al., 2016b). Our definition of vector IV(di) and IV(rj) are the ith row and jth column of adjacent matrix Y which represents whether the disease or the miRNA associated with each of the miRNAs or the diseases. Accordingly, the Gaussian interaction profile kernel similarity of diseases and miRNAs can be computed as follows:
where adjustment coefficient βd and βr for the kernel bandwidth can be denoted as follows:
where and are the original bandwidths and both of them were set 1 according to the previous literature (Chen and Yan, 2013).
Integrated Similarity for MiRNAs and Diseases
As mentioned above, a Directed Acyclic Graph (DAG) was introduced to describe a disease based on the MeSH descriptors. Disease semantic similarity was calculated according to the assumption that the two diseases with larger shared area of their DAGs may have greater similarity score. In fact, for the specific disease that without DAG, we cannot calculate the semantic similarity between the specific disease and other diseases. Thus, for disease pairs that have no semantic similarity, we used Gaussian interaction profile kernel similarity score to define their similarity. We gave a definition of integrated disease similarity by the combination of disease semantic similarity and Gaussian interaction profile kernel similarity for disease. Specifically, if disease di and dj have semantic similarity, the integrated disease similarity can be defined as the average of SS1 and SS2, otherwise we would attach the value of Gaussian interaction profile kernel similarity for disease to the integrated disease similarity. The formulations show as follows:
In the same way, we made a definition for integrated miRNA similarity through combining miRNA functional similarity and Gaussian interaction profile kernel similarity for miRNA. we obtained the integrated miRNA similarity as follows:
NRLMFMDA
In this study, we proposed a neighborhood regularized logistic matrix factorization method for miRNA-disease association prediction (NRLMFMDA) by integrating known miRNA-disease associations, miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity (see Figure 1). As far as we have known, the matrix factorization has been applied to recommender systems and obtained successful association prediction results currently. For example, logistic matrix factorization (LMF) (Johnson, 2014) has been demonstrated to be effective for personalized recommendations. Therefore, the probability of the association between a miRNA and a disease can be computed based on it. In details, we mapped the diseases and the miRNAs into a shared latent space with a dimensionality r which is far lower than the minimum of m and n. The latent space vectors and are used to represent the properties of the miRNA ri and the diseasedj, respectively. For simplicity, we further denote the latent vectors of all miRNAs and all diseases by U ∈ Rm×r and V ∈ Rn×r respectively, where ui is the ith row in U and vj is the jth row in V. Simultaneously, the probability distributions of U and V are assumed as Gaussian distributions with zero-means and their variances are set as and , respectively. Their formulations are shown as follows:
where I denotes the identity matrix. Afterwards, based on the Bayesian theorem, we know that
Based on the assumption that all the training examples are independent, we denoted the probability of associations under the condition of U and V as follows:
where we denote the probability pij of the association between miRNA ri and disease djas follows:
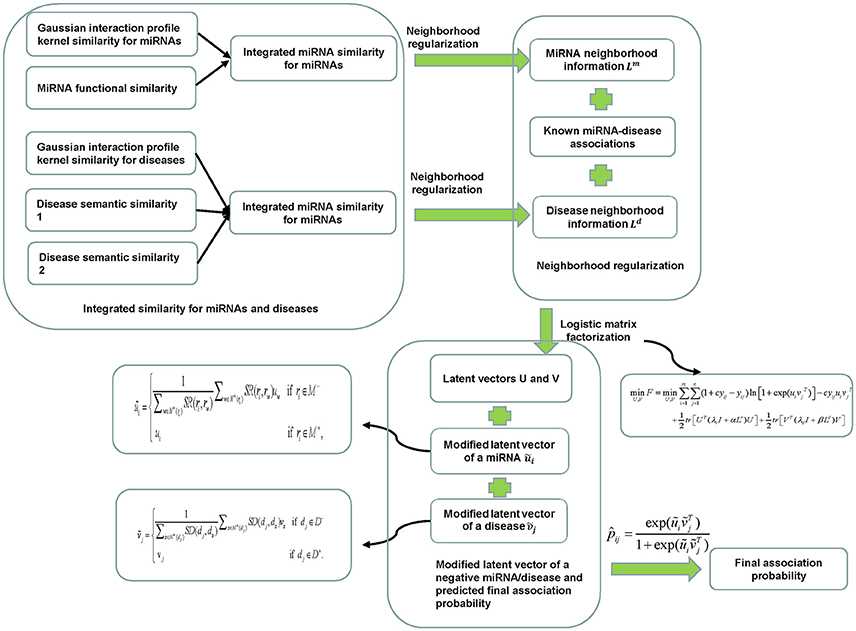
Figure 1. Flowchart of NRLMFMDA model to predict the potential miRNA-disease associations based on the known associations in HMDD database.
And the known associations between diseases and miRNAs are assigned with higher importance levels of c (c > 1) which is empirically set to 5 in experiment so that we could get more accurate predictions with the help of the trustworthy data. Then, we made the log form on the both side of the formula (13) as follows:
where C is a constant. We maximized the posterior distribution to obtain the most possible U and V. And it is equivalent to the problem as follows:
where, and ||•||F is the Frobenius norm of a matrix. We solved this searching minimum problem with an alternating gradient descent method (Johnson, 2014). Because the neighborhoods of a miRNA or a disease have strong associations, the nearest miRNAs and diseases can provide the most useful information about how to find the reasonable way to factorize the logical matrix. Therefore, our object is to minimize the distances between di and its nearest neighbors in set N(di) which is formed by K1 nearest neighbors of the diseasedi. The same to miRNArj, N(rj)is the set formed by K1 nearest neighbors of the miRNArj. K1is empirically set to 5 in experiment. We used the adjacency matrix A and B to represent the neighborhood information, and their elements aiu and bjv are defined as follows:
Based on them, we aimed to minimize the following functions:
where and . In the two formulations, Dr, , Dd,and are diagonal matrices and their diagonal elements are , , , and , respectively. According to the analysis above, the integrated formulation to minimize the objective function F is as follows:
However, the alternating gradient descent method needs the partial differential of F with respect to U and V, so they are computed and simplified as follows:
where P ∈ Rm×n is the matrix with elements pij in equation (10) and ∗ represents the Hadamard product. The gradient step size is chosen based on the AdaGrad algorithm (Duchi et al., 2011). In the experiments, we selected the dimensionality of the latent space r from {50, 100}. Simultaneously, we set λr = λd and chose the values from {2−5, 2−4, …, 21}. Neighborhood regularization parameters α and β were selected from {2−5, 2−4, …, 22} and {2−5, 2−4, …, 20}. The optimal learning rate γ was selected from {2−3, 2−2, …, 20}.
In the training procedure, the new diseases and new miRNAs are learned based on the mixed negative samples (including potential positive miRNA-disease associations) which will lead to a bias on the prediction results. Therefore, before obtaining the final probabilities with the learned U and V above, we further improved the prediction accuracy for new diseases or new miRNAs by replacing the latent vectors of negative samples with the linear combination of its nearest positive neighbors. For a miRNA ri in negative set M− which is the set of new miRNAs without any known related diseases, we denoted its K2 nearest neighbors in positive set M+ by . And for a disease dj in negative set D− which is the set of new diseases without any known related miRNAs, we denoted its K2 nearest neighbors in positive set D+ by , where K2 is empirically set to 5 in experiment. Hence, the modified association probability is represented as follows:
Where,
The modified latent vectors are helpful to overcome the bias due to using the uncertain negative samples to train the latent vectors of miRNAs and diseases in negative sets.
Results
Performance Evaluation
Leave-one-out cross validation (LOOCV) and 5-fold cross validation were applied to evaluate the performance of NRLMFMDA. And the LOOCV was implemented in two ways. (1) Based on the experimentally confirmed miRNA-disease associations in HMDD v2.0 database, Global LOOCV was used to evaluate the performance of NRLMFMDA. The “global” means that each one of the known miRNA-disease associations will be left out in turn to be considered as candidate association which are the unconfirmed miRNA-disease associations. Then after calculating prediction association scores of all the miRNA-disease pairs by NRLMFMDA, we compared the score of each test sample with all the candidate ones to observe whether its rank was above the threshold which was given in advance. (2) Unlike the Global LOOCV, Local LOOCV only compared the score of each test sample with the candidate samples composed of all the miRNA-disease pairs whose miRNAs did not have any known associations with the investigated disease. And if the rank of the test association exceeded the threshold which was given ahead of time, the model was considered to successfully predict this miRNA-disease association. Further, we drew Receiver operating characteristics (ROC) curve by plotting the true positive rate (TPR, sensitivity) vs. the false positive rate (FPR, 1-specificity) at different thresholds. Sensitivity refers to the percentage of the positive samples correctly identified among all the positives. Meanwhile, specificity denotes the percentage of negative samples correctly identified among all the negatives. After that, the prediction ability of NRLMFMDA would be evaluated by Area under the ROC curve (AUC). AUC = 1 indicates the prediction performance of NRLMFMDA is perfect; AUC = 0.5 indicates the prediction performance of NRLMFMDA is random. The results showed that NRLMFMDA obtained the AUC of 0.9068 and 0.8239 in global and local LOOCV, respectively (see Figure 2). The AUC results implied that the NRLMFMDA had shown reliable and effective prediction performance for potential miRNA–disease association prediction. However, HGIMD, RLSMDA, HDMP, and WBSMDA obtained the AUC of 0.8781, 0.8426, 0.8366 and 0.8030 in global LOOCV, respectively. In local LOOCV, their AUCs are 0.8077, 0.6953, 0.7702, and 0.8031, respectively. Differently, RWRMDA only has AUC of local LOOCV (0.7891) which is one of its defects because it cannot uncover the missing associations for all the diseases simultaneously. Therefore, in comparison with the previous methods, we can intuitively observe the improvement of predicting the miRNA-disease associations with NRLMFMDA.
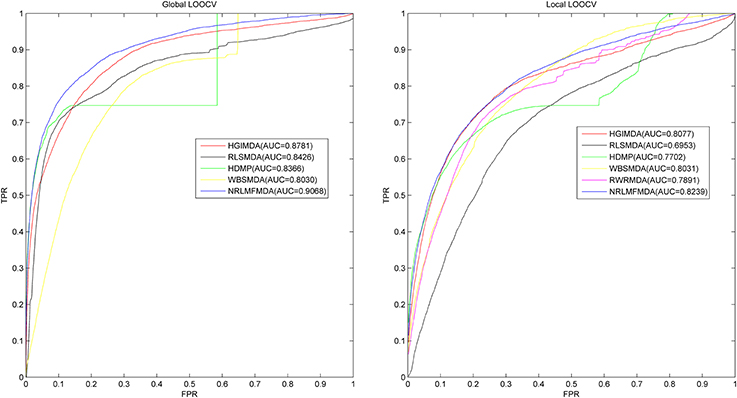
Figure 2. AUC of global LOOCV (left) compared with HGIMDA, RLSMDA, HDMP, and WBSMDA; AUC of local LOOCV (right) compared with HGIMDA, RLSMDA, HDMP, WBSMDA, and RWRMDA. As a result, NRLMFMDA achieved AUCs of 0.9068 and 0.8239 in the global and local LOOCV, which exceed all the previous classical models.
Additionally, we also implemented 5-fold cross validation to evaluate the prediction effectiveness of NRLMFMDA. We firstly divided the known miRNA-disease associations into five parts randomly. Then, one of the five parts was treated as test samples and the remaining four parts were regarded as training samples in turn. In the same way as LOOCV, the miRNA-disease pairs without known evidence of association were regarded as candidate samples. Afterwards, the scores of test samples were taken out to compare with the scores of candidate samples, and we finally acquired their rankings. This procedure was repeated 100 times randomly to make validation more accuracy. In comparison with RLSMDA, HDMP, and WBSMDA whose average AUCs were 0.8569 ± 0.0020, 0.8342 ± 0.0010 and 0.8185 ± 0.0009 respectively, the average AUC of NRLMFMDA in 5-fold cross validation was 0.8976 ± 0.0034 which further confirmed the effectiveness and superiority for predicting potential miRNA-disease associations. At last, in order to obtain a clear knowledge of the predictability performance of NRLMFMDA. We listed evaluation result of NRLMFMDA and other several typical models in global LOOCV, local LOOCV as well as 5-fold cross validation by using tabular format (see Table 1).
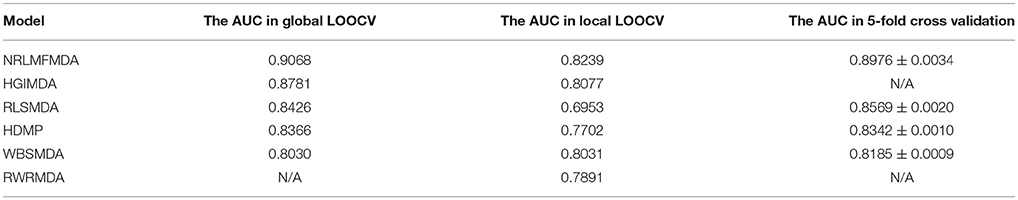
Table 1. Performance evaluation comparison between NRLMFMDA and other several typical models in global LOOCV, local LOOCV and 5-fold cross validation based on known miRNA-disease associations.
Case Studies
Based on another two miRNA-disease association databases, namely dbDEMC (Yang et al., 2010) and miR2Disease (Jiang et al., 2009), we studied three common major diseases of human beings to verify the prediction results of NRLMFMDA. The dataset of 5430 known miRNA-disease associations from HMDD v2.0 was treated as training set. For each disease, all candidate miRNAs would be ranked in the light of their predicted scores and the top 50 predicted miRNAs would be confirmed using another two miRNA-disesase association databases (i.e., dbDEMC and miR2Disease). It is worth noting that only candidate miRNAs that without known associations with investigated disease were ranked and confirmed. Therefore, there is no overlap between the training samples and the prediction lists and none of the top 50 predicted miRNAs existed in HMDD v2.0. We ulteriorly observed the number of the verified miRNAs in the top 10, top 20 and top 50 ones which are related with the three diseases respectively in the two databases.
Breast cancer is the worldwide women's health threatening, and it has caused large quantity of death in female all over the world. More than 80% of breast cancers are hormone-receptor positive in the western world (Van et al., 2014). About 232,340 new cases of invasive breast cancer including 39,620 breast cancer deaths occurred among women of America in 2013. At present, more and more researchers have paid attention to the original etiology of miRNAs in breast cancers and increasing number of evidences show that several miRNAs are closely related to breast cancer and play important roles in the tumorigenesis of breast cancer. For example, among the differentially expressed miRNAs, miR-10b, miR-125b, miR145, miR-21, and miR-155 showed as the most consistently deregulated in breast cancer. It is worthy noting that miR-10b, miR-125b, and miR-145, were down-regulated and the other two, miR-21 and miR-155, were up-regulated, which means that they can be treated as tumor suppressor genes or oncogenes, respectively (Iorio et al., 2005). After implementing NRLMFMDA, we can obtained all the rankings for potential miRNA-disease associations from the HMDD v2.0. The final results showed that 8, 16 and 39 of the top 10, 20 and 50 potential miRNAs associated with breast cancer were confirmed, respectively (see Table 2).

Table 2. Prediction of the top 50 predicted miRNAs associated with breast neoplasms based on known associations in HMDD database.
Esophageal Neoplasms is a cancer generated from the esophagus which runs between the throat and the stomach. It is still a common cancer happened among the public. The estimated number of new esophageal cancer cases and deaths were 291238 and 218957, respectively. The crude incidence and mortality rates for esophageal cancer were 21.62/100000 and 16.25/100000, respectively(Zeng et al., 2016a). Researches have showed that low expression of let-7b and let-7c associated with poor response to chemotherapy both clinically and histopathologically, which was observed from 74 patients as the training set in before-treatment biopsies (Sugimura et al., 2012). NRLMFMDA was implemented to identify esophageal neoplasms-associated miRNAs. As a result, 9 out of the top 10 and 40 out of the top 50 predicted esophageal neoplasms related miRNAs were experimentally confirmed by reports (see Table 3).
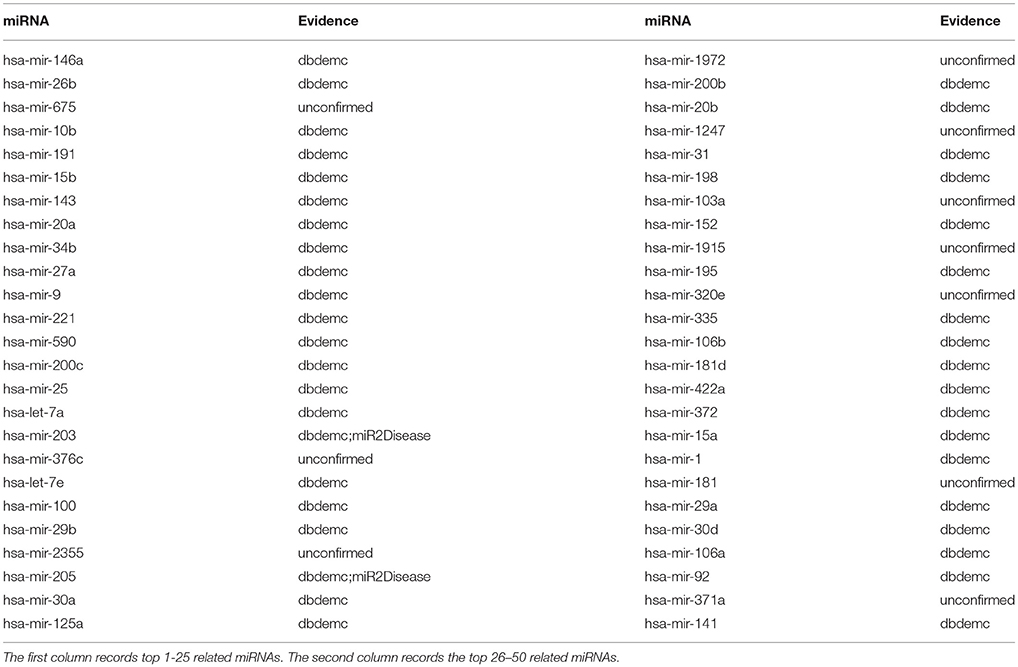
Table 3. Prediction of the top 50 predicted miRNAs associated with esophageal neoplasms based on known associations in HMDD database.
Lymphoma is a group of blood cell tumors developed from lymphocytes that is a type of white blood cell. It's also worth mentioning that Hodgkin lymphoma and non-Hodgkin lymphoma are the two main types, among which the proportion of patients with non-Hodgkin lymphoma (NHL) is about 90%. (Alizadeh et al., 2000). Experimental studies showed that the miR155 is significantly up-regulated in some Burkitt's lymphoma and several other types of lymphomas (Metzler, 2004). In canine B-cell lymphomas, compared with normal canine peripheral blood mononuclear cells (PBMC) and normal lymph nodes (LN), the expression of miRNA hsa-mir-19a was increased. After the implementation of NRLMFMDA, we took lymphomas as a case study for the identification of potential miRNA-disease association. The results showed that 8 out of top 10 and 37 out of 50 potential lymphoma-associated miRNAs in the prediction result list have been verified based on recent experimental reports (see Table 4).

Table 4. Prediction of the top 50 predicted miRNAs associated with lymphoma based on known associations in HMDD database.
To demonstrate the result of ranking completely, we have provided the prediction list of the whole potential miRNA-disease associations in HMDD v2.0 database and their association scores predicted by NRLMFMDA (see Supplementary Table 1).
In addition, we want to test the prediction ability of NRLMFMDA for the new diseases, namely the ones that have no known association with any miRNA. Therefore, we hid the association information between the miRNAs and the test disease by setting any of the known associations between them as unknown ones. After implementing the NRLMFMDA, we obtained the ranking of the miRNA-disease association prediction scores. We showed the result of hepatocellular carcinoma ranking in Table 5, in which we can see that 9, 18 and 42 related miRNAs out of the top 10, 20, and 50 had been confirmed by at least one of the three databases HMDD, dbDEMC and miR2Disease. Moreover, hsa-mir-146a was ranked first in the top 50 and the recent research has confirmed that a functional polymorphism (rs2910164) in the miR-146a gene is associated with the risk for hepatocellular carcinoma (Xu et al., 2008).
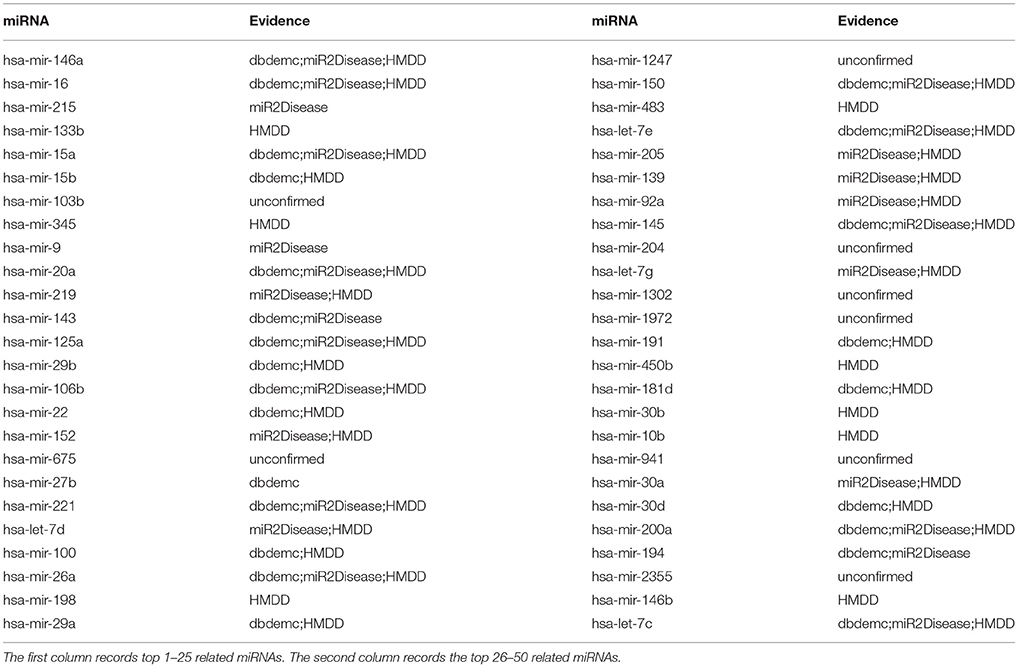
Table 5. Prediction of the top 50 predicted miRNAs associated with carcinoma, hepatocellular based on known associations in HMDD database.
Finally, we implemented NRLMFMDA on the old version of the database HMDD to observe whether the model still performs well on it. After implementing the experiment with the proposed method, it had shown the effectiveness on predicting potential miRNA-disease associations based on the previous dataset. For instance, there are 5, 11, and 31 respectively out of top 10, 20, and 50 miRNAs related with the lung neoplasms have been confirmed (see Table 6). As we can see, hsa-mir-96 was ranked first in the top 50 and research has confirmed that the expression of miR-96 in tumors was positively related to its expression in sera. Besides, high expression of tumor and serum miRNAs of the miR-183 family were associated with overall poor survival in patients with lung cancer, which was demonstrated by Log-rank and Cox regression analyses (Zhu et al., 2011).
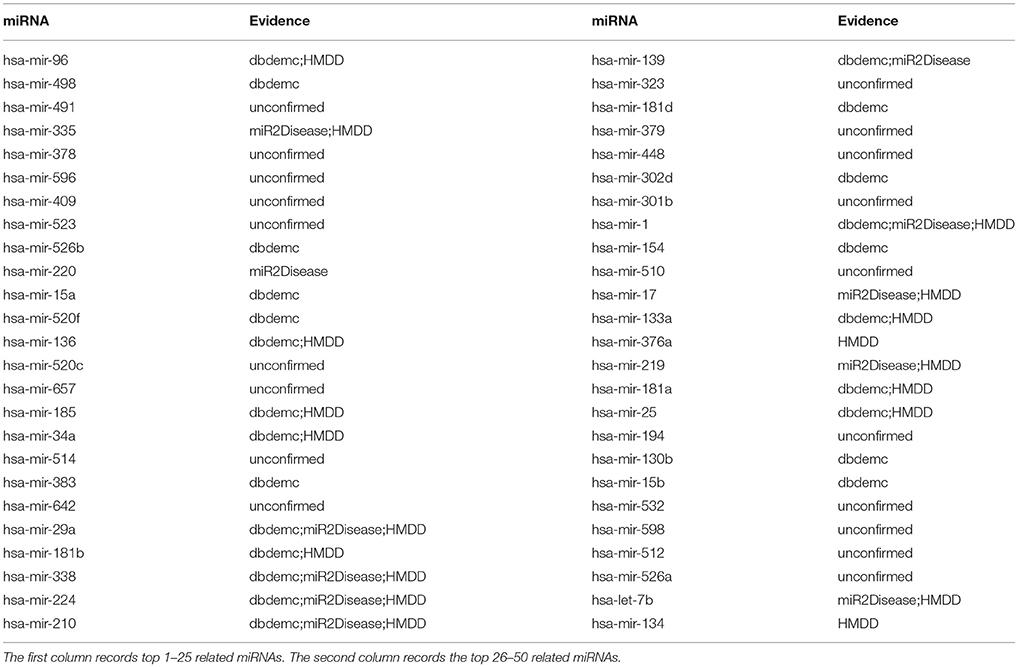
Table 6. Prediction of the top 50 predicted miRNAs associated with lung neoplasms based on known associations in old version HMDD database.
According to the result of case studies on the five major human diseases, excellent prediction performance of NRLMFMDA has been presented. With the development of experimental tools and the improvement of experimental measures, we look forward that more and more miRNA-disease association data verified by experiment will spring up. At that time, increasing portion of the predictions with NRLMFMDA can be verified by researches in the future.
Discussion
Nowadays, researchers have made progress not only in discovering miRNAs, but also in discovering the important roles that miRNAs play in physiological and pathophysiological processes (Liu and Olson, 2010). For example, aberrant expression of miRNAs has been related with various neurological disorders (NDs) in the central nervous system such as Huntington disease, amyotrophic lateral sclerosis, schizophrenia and autism, Alzheimer disease, Parkinson's disease. If dysregulated miRNAs are discoveried in patients with NDs, this may be used as a biomarker for the earlier diagnosis and monitoring of disease progression (Kamal et al., 2015). MiRNA can also be transcriptional regulators participated in pulmonary sarcoidosis and packaged in extracellular vesicles (EV) during cellular communication (Kishore et al., 2018). In biomedical research, identification of disease-associated miRNAs has become an important filed, which will accelerate people's understanding of disease pathogenesis at the molecular level and disease diagnosis, treatment and prevention in medical(Chen et al., 2017d).
This paper introduced the computational method called NRLMFMDA in which we combined the novel method of logistic matrix factorization with the similarity computational method of Gaussian interaction profile kernel similarity and further assigned higher importance level to the known associations in the process of calculating the potential miRNA-disease association probabilities to assure the larger positive influence of the known data. Additionally, we also took full advantage of the information of nearest neighbor diseases and miRNAs to improve the accuracy of the miRNA-disease association prediction (Liu et al., 2016a). As is known, the logistic matrix factorization technique has been applied in many early work of predicting associations. And it has shown remarkable effectiveness. Taking the neighborhood principle into consideration, we modified it in a more reasonable way to improve the accuracy of prediction. Due to the introduction of the Gaussian interaction profile kernel similarity, the information of the disease similarity and the miRNA similarity was fully excavated to improve the accuracy of the prediction. To verify the accuracy of the NRLMFMDA, three types of cross validation which contains Global LOOCV, Local LOOCV, and 5-fold cross validation have been implemented. As a result, the excellent performance of NRLMFMDA has been showed both from the cross validation and the case studies with several crucial diseases.
Several important factors contribute to the excellent performance of NRLMFMDA. First of all, more and more association pairs between miRNAs and diseases have been discovered and confirmed till now. Due to the data-dependent property of NRLMFMDA, the increasing of known associations assuredly improved the predicting accuracy. Secondly, NRLMFMDA can take full advantage of the similarity information by introducing the Gaussian interaction profile kernel similarity. Thirdly, NRLMFMDA pays attention to the neighborhood information which provides more reliable associations by using the neighborhood regularization method in the training procedure and the neighborhood smoothing method in the final prediction. What's more, some machine learning-based model randomly selected negative samples as training data, this inaccurate chosen process would affect the model's prediction accuracy. The modified latent vectors used in NRLMFMDA can overcome the bias because of using the uncertain negative samples to train the latent vectors of miRNAs and diseases in negative sets, which would helpful to the improvement of prediction accuracy for NRLMFMDA. Last but not least, searching the optimal solution with an alternating gradient ascent procedure made sure the reliability of the disease eigenvectors and the miRNA eigenvectors. In view of above-mentioned, NRLMFMDA has greatly improved the accuracy in prediction association between miRNA and disesase.
Some limitations have been noted in this study. Firstly, though current studies benefit from the increased known data, it is never a finished work to expand data. Numerous excellent methods were proposed just to cover the shortage of the data (Liu et al., 2014; You et al., 2014). Secondly, in the iterative process, we have five parameters that are difficult to choose as the optimal combination. Actually, we have some ranges for the five parameters. However, even using grid search strategy, it wastes a lot of time and resources due to the limitation of current situation. Therefore, we expect to use some optimized search strategy to improve the accuracy of prediction method in the future.
Author Contributions
JQ implemented the experiments, analyzed the result, and wrote the paper. B-SH conceived the project, designed the experiments, analyzed the result, and revised the paper. QZ conceived the project, implemented the experiments, and analyzed the result, and revised the paper. All authors read and approved the final manuscript.
Funding
B-SH was supported by Key Program of Hunan Provincial Education Department (Grant No. 15A026), General Program of Hunan Provincial Philosophy and Social Science Planning Fund office (Grant No. 15YBA035). QZ was supported by Innovation Team Project from the Education Department of Liaoning Province under Grant No. LT2015011 and the Doctor Startup Foundation from Liaoning Province under Grant No. 20170520217.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00303/full#supplementary-material
References
Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. doi: 10.1038/35000501
Ambros, V. (2001). microRNAs: tiny regulators with great potential. Cell 107, 823–826. doi: 10.1016/S0092-8674(01)00616-X
Bandyopadhyay, S., Mitra, R., Maulik, U., and Zhang, M. Q. (2010). Development of the human cancer microRNA network. Silence 1:6. doi: 10.1186/1758-907X-1-6
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Chen, X. (2015a). KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 5:16840. doi: 10.1038/srep16840
Chen, X. (2015b). Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 5:13186. doi: 10.1038/srep13186
Chen, X., Guan, N. N., Li, J. Q., and Yan, G. Y. (2018a). GIMDA: graphlet interaction-based MiRNA-disease association prediction. J. Cell. Mol. Med. 22, 1548–1561. doi: 10.1111/jcmm.13429
Chen, X., and Huang, L. (2017). LRSSLMDA: laplacian regularized sparse subspace learning for mirna-disease association prediction. PLoS Comput. Biol. 13:e1005912. doi: 10.1371/journal.pcbi.1005912
Chen, X., Huang, L., Xie, D., and Zhao, Q. (2018b). EGBMMDA: extreme gradient boosting machine for mirna-disease association prediction. Cell Death Dis. 9:3. doi: 10.1038/s41419-017-0003-x
Chen, X., Huang, Y. A., Wang, X. S., You, Z. H., and Chan, K. C. (2016a). FMLNCSIM: fuzzy measure-based lncRNA functional similarity calculation model. Oncotarget 7, 45948–45958. doi: 10.18632/oncotarget.10008
Chen, X., Huang, Y. A., You, Z. H., Yan, G. Y., and Wang, X. S. (2016b). A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 33, 733–739. doi: 10.1093/bioinformatics/btw715
Chen, X., Liu, M. X., Cui, Q. H., and Yan, G. Y. (2012a). Prediction of disease-related interactions between microRNAs and environmental factors based on a semi-supervised classifier. PLoS ONE 7:e43425. doi: 10.1371/journal.pone.0043425
Chen, X., Liu, M. X., and Yan, G. Y. (2012b). RWRMDA: predicting novel human microRNA-disease associations. Mol. Biosyst. 8, 2792–2798. doi: 10.1039/c2mb25180a
Chen, X., Niu, Y. W., Wang, G. H., and Yan, G. Y. (2017a). HAMDA: hybrid approach for MiRNA-disease association prediction. J. Biomed. Inform. 76, 50–58. doi: 10.1016/j.jbi.2017.10.014
Chen, X., Ren, B., Chen, M., Wang, Q., Zhang, L., and Yan, G. (2016c). NLLSS: Predicting synergistic drug combinations based on semi-supervised learning. PLoS Comput. Biol. 12:e1004975. doi: 10.1371/journal.pcbi.1004975
Chen, X., Sun, Y. Z., Liu, H., Zhang, L., Li, J. Q., and Meng, J. (2017b). RNA methylation and diseases: experimental results, databases, web servers and computational models. Brief Bioinformatics doi: 10.1093/bib/bbx142. [Epub ahead of print].
Chen, X., Wang, L., Qu, J., Guan, N. N., and Li, J. Q. (2018c). Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics. doi: 10.1093/bioinformatics/bty503. [Epub ahead of print].
Chen, X., Wu, Q. F., and Yan, G. Y. (2017c). RKNNMDA: ranking-based KNN for MiRNA-disease association prediction. RNA Biol. 14, 952–962. doi: 10.1080/15476286.2017.1312226
Chen, X., Xie, D., Wang, L., Zhao, Q., You, Z. H., and Liu, H. (2018d). BNPMDA: bipartite network projection for mirna-disease association prediction. Bioinformatics doi: 10.1093/bioinformatics/bty333. [Epub ahead of print].
Chen, X., Xie, D., Zhao, Q., and You, Z. H. (2017d). MicroRNAs and complex diseases: from experimental results to computational models. Brief Bioinformatics doi: 10.1002/ajmg.a.38607. [Epub ahead of print].
Chen, X., Yan, C. C., Luo, C., Ji, W., Zhang, Y., and Dai, Q. (2015a). Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 5:11338. doi: 10.1038/srep11338
Chen, X., Yan, C. C., Zhang, X., Li, Z., Deng, L., Zhang, Y., et al. (2015b). RBMMMDA: predicting multiple types of disease-microRNA associations. Sci. Rep. 5:13877. doi: 10.1038/srep13877
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2016e). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinformatics 18, 558–576. doi: 10.1093/bib/bbw060
Chen, X., Yan, C. C., Zhang, X., You, Z. H., Deng, L., Liu, Y., et al. (2016f). WBSMDA: within and Between Score for MiRNA-disease association prediction. Sci. Rep. 6:21106. doi: 10.1038/srep21106
Chen, X., Yan, C. C., Zhang, X., You, Z. H., Huang, Y. A., and Yan, G. (2016d). HGIMDA: heterogeneous graph inference for MiRNA-disease association prediction. Oncotarget 7, 65257–65269. doi: 10.18632/oncotarget.11251
Chen, X., Yan, C. C., Zhang, X., Zhang, X., Dai, F., Yin, J., et al. (2015c). Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinformatics 17, 696–712. doi: 10.1093/bib/bbv066
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Chen, X., and Yan, G. Y. (2014). Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 4:5501. doi: 10.1038/srep05501
Chen, X., Yang, J. R., Guan, N. N., and Li, J. Q. (2018e). GRMDA: graph regression for MiRNA-disease association prediction. Front. Physiol. 9:92. doi: 10.3389/fphys.2018.00092
Chen, X., You, Z. H., Yan, G. Y., and Gong, D. W. (2016g). IRWRLDA: improved random walk with restart for lncRNA-disease association prediction. Oncotarget 7, 57919–57931. doi: 10.18632/oncotarget.11141
Chen, X., Zhou, Z., and Zhao, Y. (2018f). ELLPMDA: Ensemble learning and link prediction for miRNA-disease association prediction. RNA Biol. 1–12. doi: 10.1080/15476286.2018.1460016. [Epub ahead of print].
Cheng, A. M., Byrom, M. W., Shelton, J., and Ford, L. P. (2005). Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 33, 1290–1297. doi: 10.1093/nar/gki200
Duchi, J., Hazan, E., and Singer, Y. (2011). Adaptive subgradient methods for online learning and stochastic optimization. J. Mach. Learn. Res. 12, 2121–2159.
Goh, K. I., Cusick, M. E., Valle, D., Childs, B., Vidal, M., Barabási, A., L., and Goh, A., L. (2007). The human disease network. Proc. Natl. Acad. Sci. U.S.A. 104, 8685–8690. doi: 10.1073/pnas.0701361104
Griffiths-Jones, S., Grocock, R. J., Van Dongen, S., Bateman, A., and Enright, A. J. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144. doi: 10.1093/nar/gkj112
Huang, Y. A., Chen, X., You, Z. H., Huang, D. S., and Chan, K. C. C. (2016a). ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget 7, 25902–25914. doi: 10.18632/oncotarget.8296
Huang, Y. A., You, Z. H., Chen, X., Chan, K., and Luo, X. (2016b). Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinformatics 17:184. doi: 10.1186/s12859-016-1035-4
Iorio, M. V., Ferracin, M., Liu, C. G., Veronese, A., Spizzo, R., Sabbioni, S., et al. (2005). MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 65, 7065–7070. doi: 10.1158/0008-5472.CAN-05-1783
Jiang, Q., Hao, Y., Wang, G., Juan, L., Zhang, T., Teng, M., et al. (2010). Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 4 (Suppl. 1):S2. doi: 10.1186/1752-0509-4-S1-S2
Jiang, Q., Wang, Y., Hao, Y., Juan, L., Teng, M., Zhang, X., et al. (2009). miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 37, D98–D104. doi: 10.1093/nar/gkn714
Johnson, C. C. (2014). Logistic matrix factorization for implicit feedback data. Adv. Neural Inf. Process. Syst. 27.
Jopling, C. L., Yi, M., Lancaster, A. M., Lemon, S. M., and Sarnow, P. (2005). Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581. doi: 10.1126/science.1113329
Kamal, M. A., Mushtaq, G., and Greig, N. H. (2015). Current update on synopsis of miRNA dysregulation in neurological disorders. CNS Neurol. Disord. Drug Targets 14, 492–501. doi: 10.2174/1871527314666150225143637
Karp, X., and Ambros, V. (2005). Encountering microRNAs in cell fate signaling. Science 310, 1288–1289. doi: 10.1126/science.1121566
Kishore, A., Navratilova, Z., Kolek, V., Novosadova, E., Cépe, K., Du Bois, R. M., et al. (2018). Expression analysis of extracellular microRNA in bronchoalveolar lavage fluid from patients with pulmonary sarcoidosis. Respirology. doi: 10.1111/resp.13364. [Epub ahead of print].
Kozomara, A., and Griffiths-Jones, S. (2011). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–157. doi: 10.1093/nar/gkq1027
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The, C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 89, 1828–1835. doi: 10.1016/0092-8674(93)90529-Y
Li, J. Q., You, Z. H., Li, X., Ming, Z., and Chen, X. (2017). PSPEL: In Silico prediction of self-interacting proteins from amino acids sequences using ensemble learning. IEEE/ACM Trans. Comput. Biol. Bioinformatics 14, 1165–1172. doi: 10.1109/TCBB.2017.2649529
Li, Y., Qiu, C. X., Tu, J., Geng, B., Yang, J. C., Jiang, T. Z., et al. (2014). HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 42, D1070–D1074. doi: 10.1093/nar/gkt1023
Liu, N., and Olson, E. N. (2010). MicroRNA regulatory networks in cardiovascular development. Dev. Cell 18, 510–525. doi: 10.1016/j.devcel.2010.03.010
Liu, Y. P., Tennant, D. A., Zhu, Z. X., Heath, J. K., Yao, X., and He, S. (2014). DiME: a scalable disease module identification algorithm with application to glioma progression. PLoS ONE 9:e0086693. doi: 10.1371/journal.pone.0086693
Liu, Y., Wu, M., Miao, C., Zhao, P., and Li, X. L. (2016a). Neighborhood regularized logistic matrix factorization for drug-target interaction prediction. PLoS Comput. Biol. 12:e1004760. doi: 10.1371/journal.pcbi.1004760
Liu, Y., Zeng, X., He, Z., and Zou, Q. (2016b). Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Trans. Comput Biol. Bioinformatics 14, 905–915. doi: 10.1109/TCBB.2016.2550432
Lu, M., Zhang, Q. P., Deng, M., Miao, J., Guo, Y. H., Gao, W., et al. (2008). An analysis of human MicroRNA and disease associations. PLoS ONE 3:e0003420. doi: 10.1371/journal.pone.0003420
Lynam-Lennon, N., Maher, S. G., and Reynolds, J. V. (2009). The roles of microRNA in cancer and apoptosis. Biol. Rev. 84, 55–71. doi: 10.1111/j.1469-185X.2008.00061.x
Meister, G., and Tuschl, T. (2004). Mechanisms of gene silencing by double-stranded RNA. Nature 431, 343–349. doi: 10.1038/nature02873
Meola, N., Gennarino, V. A., and Banfi, S. (2009). microRNAs and genetic diseases. PathoGenetics 2, 1–14. doi: 10.1186/1755-8417-2-7
Metzler, M. (2004). High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer 39, 167–169. doi: 10.1002/gcc.10316
Miska, E. A. (2005). How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 15, 563–568. doi: 10.1016/j.gde.2005.08.005
Mørk, S., Pletscherfrankild, S., Caro, A., Gorodkin, J., and Jensen, L. J. (2014). Protein-driven inference of miRNA–disease associations. Bioinformatics 30, 392–397. doi: 10.1093/bioinformatics/btt677
Pasquier, C., and Gardès, J. (2016). Prediction of miRNA-disease associations with a vector space model. Sci. Rep. 6:27036. doi: 10.1038/srep27036
Schulte, C., Molz, S., Appelbaum, S., Karakas, M., Ojeda, F., Lau, D. M., et al. (2015). miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS ONE 10:e0145930. doi: 10.1371/journal.pone.0145930
Shi, H., Xu, J., Zhang, G., Xu, L., Li, C., Li, W., et al. (2013). Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 7:101. doi: 10.1186/1752-0509-7-101
Sugimura, K., Miyata, H., Tanaka, K., Hamano, R., Takahashi, T., Kurokawa, Y., et al. (2012). Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin. Cancer Res. 18, 5144–5153. doi: 10.1158/1078-0432.CCR-12-0701
Tang, W., Wan, S., Yang, Z., Teschendorff, A. E., and Zou, Q. (2018). Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 34, 398–406. doi: 10.1093/bioinformatics/btx622
Thum, T., Gross, C., Fiedler, J., Fischer, T., Kissler, S., Bussen, M., et al. (2008). MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456, 980–984. doi: 10.1038/nature07511
Van, A. K., Neven, P., Lintermans, A., Wildiers, H., and Paridaens, R. (2014). Aromatase inhibitors in the breast cancer clinic: focus on exemestane. Endocr. Relat. Cancer 21, R31–R49. doi: 10.1530/ERC-13-0269
Van, L. T., Nabuurs, S. B., and Marchiori, E. (2011). Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 27, 3036–3043. doi: 10.1093/bioinformatics/btr500
Wang, D., Wang, J. A., Lu, M., Song, F., and Cui, Q. (2010). Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26, 1644–1650. doi: 10.1093/bioinformatics/btq241
Wong, L., You, Z. H., Ming, Z., Li, J., Chen, X., and Huang, Y. A. (2015). Detection of interactions between proteins through rotation forest and local phase quantization descriptors. Int. J. Mol. Sci. 17:21. doi: 10.3390/ijms17010021
Xu, C., Ping, Y., Li, X., Zhao, H., Wang, L., Fan, H., et al. (2014). Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol. Biosyst. 10, 2800–2809. doi: 10.1039/C4MB00353E
Xu, J., Li, C. X., Lv, J. Y., Li, Y. S., Xiao, Y., Shao, T. T., et al. (2011). Prioritizing candidate disease miRNAs by topological features in the miRNA target-dysregulated network: case study of prostate cancer. Mol. Cancer Ther. 10, 1857–1866. doi: 10.1158/1535-7163.MCT-11-0055
Xu, P. Z., Guo, M., and Hay, B. A. (2004). MicroRNAs and the regulation of cell death. Trends Genet. 20, 617–624. doi: 10.1016/j.tig.2004.09.010
Xu, T., Zhu, Y., Wei, Q. K., Yuan, Y., Zhou, F., Ge, Y. Y., et al. (2008). A functional polymorphism in the miR-146a gene is associated with the risk for hepatocellular carcinoma. Carcinogenesis 29, 2126–2131. doi: 10.1093/carcin/bgn195
Xuan, P., Han, K., Guo, M., Guo, Y., Li, J., Ding, J., et al. (2013a). Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 8:e70204. doi: 10.1371/journal.pone.0070204
Xuan, P., Han, K., Guo, M., Guo, Y., Li, J., Ding, J., et al. (2013b). Correction: prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 8:e70204. doi: 10.1371/annotation/a076115e-dd8c-4da7-989d-c1174a8cd31e
Yang, Z., Ren, F., Liu, C., He, S., Sun, G., Gao, Q., et al. (2010). dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genomics 11:S5. doi: 10.1186/1471-2164-11-S4-S5
You, Z. H., Huang, Z. A., Zhu, Z., Yan, G. Y., Li, Z. W., Wen, Z., et al. (2017). PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 13:e1005455. doi: 10.1371/journal.pcbi.1005455
You, Z. H., Zhu, L., Zheng, C. H., Yu, H. J., Deng, S. P., and Ji, Z. (2014). Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinformatics 15:S9. doi: 10.1186/1471-2105-15-S15-S9
Zeng, H., Zheng, R., Zhang, S., Zuo, T., Xia, C., Zou, X. et al. (2016a). Esophageal cancer statistics in China, 2011: Estimates based on 177 cancer registries. Thoracic Cancer 7, 232–237. doi: 10.1111/1759-7714.12322
Zeng, X., Zhang, X., and Zou, Q. (2016b). Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinformatics 17, 193–203. doi: 10.1093/bib/bbv033
Zhu, W., Liu, X., He, J., Chen, D., Hunag, Y., and Zhang, Y. (2011). Overexpression of members of the microRNA-183 family is a risk factor for lung cancer: a case control study. BMC Cancer 11:393. doi: 10.1186/1471-2407-11-393
Keywords: microRNA, disease, association prediction, neighborhood regularized, matrix factorization
Citation: He B-S, Qu J and Zhao Q (2018) Identifying and Exploiting Potential miRNA-Disease Associations With Neighborhood Regularized Logistic Matrix Factorization. Front. Genet. 9:303. doi: 10.3389/fgene.2018.00303
Received: 25 June 2018; Accepted: 18 July 2018;
Published: 07 August 2018.
Edited by:
Quan Zou, Tianjin University, ChinaReviewed by:
Pengwei Hu, Hong Kong Polytechnic University, Hong KongYuangen Yao, Huazhong Agricultural University, China
Copyright © 2018 He, Qu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Qu, dGIxNzA2MDAxNWI0QGN1bXQuZWR1LmNu
Qi Zhao, emhhb3FpQGxudS5lZHUuY24=