 D.S. Kishor
D.S. Kishor Jeonghwan Seo
Jeonghwan Seo Joong Hyoun Chin2*
Joong Hyoun Chin2* Hee-Jong Koh
Hee-Jong Koh- 1Department of Plant Science, Plant Genomics and Breeding Institute, and Research Institute of Agriculture and Life Science, Seoul National University, Seoul, South Korea
- 2Department of Integrative Bio-industrial Engineering, Sejong University, Seoul, South Korea
Basmati is considered a unique varietal group of rice (Oryza sativa L.) because of its aroma and superior grain quality. Previous genetic analyses of rice showed that most of the Basmati varieties are classified into the aromatic group. Despite various efforts, genomic relationship of Basmati rice with other varietal groups and genomic variation in Basmati rice are yet to be understood. In the present study, we resequenced the whole genome of three traditional Basmati varieties at a coverage of more than 25X using Illumina HiSeq2500 and mapped the obtained sequences to the reference genome sequences of Nipponbare (japonica rice), Kasalath (aus rice), and Zhenshan 97 (indica rice). Comparison of these sequences revealed common single nucleotide polymorphisms (SNPs) in the genic regions of three Basmati varieties. Analysis of these SNPs revealed that Basmati varieties showed fewer sequence variations compared with the aus group than with the japonica and indica groups. Gene ontology (GO) enrichment analysis indicated that SNPs were present in genes with various biological, molecular, and cellular functions. Additionally, functional annotation of the Basmati mutated gene cluster shared by Nipponbare, Kasalath, and Zhenshan 97 was found to be associated with the metabolic process involved in the cellular aromatic compound, suggesting that aroma is an important specific genomic feature of Basmati varieties. Furthermore, 30 traditional Basmati varieties were classified into three different groups, aromatic (22 varieties), aus (four varieties), and indica (four varieties), based on genome-wide SNPs. All 22 aromatic Basmati varieties harbored the fragrant-inducing Badh2 allele. We also performed comparative analysis of 13 key agronomic and grain quality traits of Basmati rice and other rice varieties. Three traits including length-to-width ratio of grain (L/W ratio), panicle length (PL), and amylose content (AC) showed significant (P < 0.05 and P < 0.01) differences between the aromatic and indica/aus groups. Comparative analysis of genome structure, based on genome sequence variation and GO analysis, revealed that the Basmati genome was derived mostly from the aus and japonica groups. Overall, whole-genome sequence data and genetic diversity information obtained in this study will serve as an important resource for molecular breeding and genetic analysis of Basmati varieties.
Introduction
Rice (Oryza sativa L.) is an important cereal crop and represents the staple food of more than half of the global population (Wang and Li, 2005). O. sativa is classified into two distinct subspecies, japonica and indica (Kato, 1928), and into five groups including indica, aus, aromatic, temperate japonica, and tropical japonica (Garris et al., 2005). O. sativa was domesticated more than 10,000 years ago from Asian wild rice species, O. rufipogon and O. nivara (Kovach et al., 2007; Sang and Ge, 2007; Chen et al., 2019). Both japonica and indica rice have undergone significant phenotypic changes compared with O. rufipogon (proto-japonica) and O. nivara (proto-indica), respectively, and have expanded their geographical distribution during domestication (Fuller et al., 2010).
Basmati rice is considered a unique varietal group because of its aroma and superior grain quality (Ahuja et al., 1995; Siddiq et al., 2012). These unique varietal group occupies a special status among the consumers due to its unique quality traits such as extra-long slender grain, lengthwise excessive kernel elongation upon cooking, soft and fluffy texture after cooking, and aroma. Therefore, Basmati varieties are designated as the most highly produced and economically successful group (Civáň et al., 2019). The term Basmati is derived from two Sanskrit words, “Vas” meaning “aroma” and “matup” meaning “possessing.” The combination of the two Sanskrit words, “Vaasmati,” is pronounced as “Basmati” (Siddiq et al., 2012). Studies suggest that Basmati rice varieties represent the aromatic group from indica and japonica subspecies (Glaszmann, 1987; Garris et al., 2005).
From the decades, less attention has given at the origin of Basmati group. This is mainly due to the conflicting phylogenetic relationships were observed among Basmati and other rice groups (Choi et al., 2017). Furthermore, genome-wide polymorphism analysis in Asian cultivated rice showed that Basmati rice varieties share a close phylogenetic relationship with japonica varieties (Huang et al., 2012; Wang et al., 2018). Recent findings of Choi et al. (2018) and Civáň et al. (2019) providing more evidence that Basmati genome was genetically close to japonica and aus rice. However, these studies were carried out using single Basmati genome, which has limited information on Basmati genome variation. Although some progresses have been made in understanding of origin of Basmati genome, further study is needed to identify the Basmati-specific genome features and genome variation by assembling the traditional Basmati varieties compared with japonica, indica, and aus groups. Next-generation sequencing (NGS) technologies are important for genomic analysis and molecular breeding (Chen et al., 2014), and enable the identification of functional genomic variation, and unique SNPs, and insertion-deletion polymorphisms (InDels) across the genome, which offer an exciting opportunity to genetic diversity studies in the crop plants (Jimenez et al., 2013; Serba et al., 2019).
In Basmati rice, molecular mapping and cloning of the fgr gene, which encodes betaine aldehyde dehydrogenase homologue 2 (Badh2), revealed an 8-bp deletion and three single nucleotide polymorphisms (SNPs) in the 7th exon, resulting in the fragrant trait (Bradbury et al., 2005). Haplotype analysis of the Badh2 gene showed that the 8-bp deletion in the majority of fragrant Basmati varieties causes a loss-of-function mutation, which enhances the biosynthesis of 2-acetyl-1-pyroline (2-AP); this haplotype is identical to the ancestral japonica haplotypes, suggesting that introgression between japonica accessions and Basmati varieties is responsible for the fragrant trait in Basmati rice (Kovach et al., 2009). A recent study by Daygon et al. (2017) reported that four other amine heterocycles: 6-methyl, 5-oxo-2,3,4,5-tetrahydropyridine (6M5OTP), 2-acetylpyrrole, pyrrole, and 1-pyrroline, that correlate strongly with the production of 2AP, and are present in consistent proportions in a collection of recombinant inbred lines derived from Basmati-type rice, and these compounds were also co-localized with a single QTL that harbors the fgr gene. Although genetic basis of fragrant trait in Basmati rice seems to be complicated, most researchers proposed that grain aroma in Basmati rice is controlled by a single recessive gene (Badh2) (Bradbury et al., 2005; Kovach et al., 2009). However, some researchers also think that fragrant trait in Basmati rice is controlled by major and minor-effective genes (Daygon et al., 2017), and by several QTLs (Amarawathi et al., 2008; Pachauri et al., 2014; Vemireddy et al., 2015). Overall, the molecular genetic mechanism of fragrant trait is not clearly understood, more studies is needed on the functional allelic variation of aroma gene and number of genes controlling the grain aroma in Basmati rice.
In this study, we analyzed the differences between Basmati rice genome vs. indica, japonica, and aus rice genomes through whole-genome sequencing and marker analysis. The main objective is to identify the genomic features and genetic variation in Basmati rice that can be utilized for genetic studies and marker development for breeding. We also identified unique SNPs and Indel marker sets, and evaluated the key agronomic and grain quality traits of Basmati rice with other rice groups for varietal improvement.
Materials and Methods
Plant Materials
A total of 60 rice varieties belonging to indica, aus, aromatic, temperate japonica, and tropical japonica groups were used in this study (Table 1). Among the 60 rice varieties, seeds of 30 traditional Basmati varieties [International Rice GenBank Collection (IRGC) designated] were obtained from the International Rice Research Institute (IRRI), while the other 30 rice varieties were from the Crop Molecular Breeding Lab, Seoul National University. Among the 30 traditional Basmati varieties, Basmati 370, Rato Basmati, and Dahrdun Basmati were selected for whole-genome resequencing, based on their geo-location (Figure 1). Seeds from each accession were surface sterilized and sown in pots containing wet soil. The pots were placed in an experimental greenhouse for 30 days. Then, 30-day-old seedlings were transplanted in an experimental field at Seoul National University.
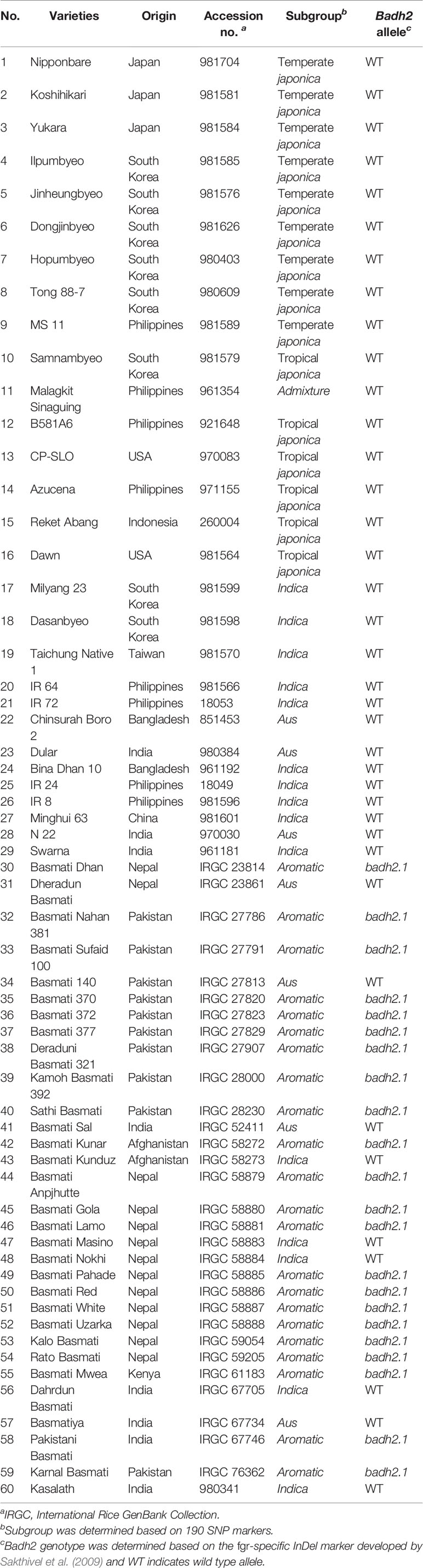
Table 1 List of rice varieties used in this study.

Figure 1 Phenotypic comparison of Basmati 370, Dahrdun Basmati, and Rato Basmati. (A) Plant phenotype. (B) Spikelet and mature grain.
Genome Sequencing
Supplementary Figure 1 provides an overview of the work plan used in this study. To perform whole-genome resequencing, shotgun DNA libraries were prepared from high molecular weight genomic DNA of three traditional Basmati varieties using the NEXTflex™ Rapid DNA-Seq kit (Bioo Scientific Corporation, Austin, TX, USA). Then, the libraries were used for cluster generation and sequenced for 250 cycles on the Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions, at the National Instrumentation Center for Environmental Management (NICEM) of Seoul National University.
Mapping and SNP Discovery
Raw sequence reads were subjected to quality trimming using FastQC v0.11.3 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and reads with a Phred quality (Q) score <20 were discarded. Adapter trimming was carried out by using Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic). The clean reads were mapped to the reference genomes of the temperate japonica cultivar Nipponbare (Os-Nipponbare-Reference-IRGSP-1.0; Kawahara et al., 2013), indica cultivar Zhenshan 97 (Os-Zhenshan 97-Reference; Zhang et al., 2016), and aus cultivar Kasalath (Os-Kasalath-Reference; Sakai et al., 2014) using the Burrows–Wheeler Aligner (BWA) program (Li and Durbin, 2010). The alignment results were merged and converted into binary alignment map (BAM) files (Barnett et al., 2011). The BAM files were used to calculate the sequencing depth and to identify SNPs and InDels using the GATK program, with default parameters (McKenna et al., 2010).
Genomic Analysis
The genic and intergenic distribution of SNPs and InDels was determined relative to Nipponbare, Zhenshan 97, and Kasalath reference genomes. The distribution of genic SNPs and InDels common to the three Basmati genomes were presented using Circos (Krzywinski et al., 2009).
In silico analysis was performed to identify Basmati-specific SNPs and InDels using resequencing data of 54 diverse rice varieties in the Crop Molecular Breeding Lab, Seoul National University database (unpublished data) and Rice Variation Map v2.0 public database (http://ricevarmap.ncpgr.cn/v2/). InDel in nine traditional Basmati varieties and 11 indica, aus, and japonica check varieties were verified by gel electrophoresis, based on in silico analysis, using primers designed with Primer-3 (http://bioinfo.ut.ee/primer3-0.4.0/).
GO Analysis
The annotated Nipponbare, Zhenshan 97, and Kasalath reference genes were classified based on the pattern of common SNPs in the three Basmati genomes. Functional annotation of genes was investigated with “Oryza sativa” as the background species. GO analysis was performed using the BLAST2GO software (www.blast2go.com) (Conesa et al., 2005). Whole-genome orthologous gene comparison, annotation, and clustering were performed using the Orthovenn program (Wang et al., 2015).
DNA Extraction and Genome-Wide SNP Marker Analysis
Genomic DNA was isolated from the leaf tissues of plants at the 3–4 leaf stage using the modified cetyltrimethylammonium bromide (CTAB) method (McCouch et al., 1988). DNA concentration and quality were determined using the NanoDrop spectrophotometer (Thermo Scientific, Wilmington, NC, USA).
On the basis of differences in DNA sequences between indica and japonica genomes, 190 subspecies-specific SNP markers, representing all 12 rice chromosomes, were developed in the Crop Molecular Breeding Lab, Seoul National University (unpublished data). SNP genotyping was conducted on Fluidigm 96.96 Dynamic Arrays using the BioMark HD System (Fluidigm Corp, San Francisco, CA), according to the manufacturer’s instructions, and genotypes were determined using the Fluidigm SNP Genotyping Analysis software.
Phylogenetic and Population Structure Analyses
Phylogenetic analysis was performed using PowerMarker v3.25 (Liu and Muse, 2005). Cavalli-Sforza and Edwards (1967) genetic distance was used to construct an unweighted pair group method with an arithmetic average (UPGMA) dendrogram, which was visualized in Molecular Evolutionary Genetics Analysis 7 (MEGA7) (Kumar et al., 2016). The population structure of 60 rice varieties was determined using a model-based approach available in the STRUCTURE 2.3.4 software (Falush et al., 2003). The number of genetically distinct populations (K) was adjusted from 1 to 10, and the model was repeated three times for each K. The burn in-period was adjusted with 100,000 iterations, followed by 100,000 Markov Chain Monte Carlo (MCMC) per run. The best K value was determined based on delta K (ΔK) using the Evanno method in the web-based python program, STRUCTURE HARVESTER (Earl and Vonholdt, 2012).
Badh2 Marker Analysis
All 60 rice varieties were classified as badh2.1 and wild Badh2 allele harboring genotypes by PCR-based genotyping of the Badh2 InDel marker using the forward primer 5′-TGTTTTCTGTTAGGTTGCATT-3′ and reverse primer 5′-ATCCACAGAAATTTGGAAAC-3′ (Sakthivel et al., 2009). PCR was conducted using the following conditions: initial denaturation at 94°C for 2 min, followed by 35 cycles of denaturation at 95°C for 20 s, annealing at 54°C for 30 s, and extension at 72°C for 30 s, and a final extension at 72°C for 1 min. The amplified products were separated by electrophoresis on 3.5% agarose gel.
Agronomic and Grain Quality Trait Analyses
Passport data on 13 agronomic and grain quality traits of 30 traditional Basmati varieties, including days to heading (DH), leaf width (LW), days to maturity (DM), culm length (CL), culm number (CN), culm diameter (CD), grain length (GL), grain width (GW), length-to-width ratio of grain (L/W ratio), 1,000 grain weight (KGW), panicle length (PL), spikelet fertility count (SFC), and amylose content (AC), were obtained from Genesys (https://www.genesys-pgr.org). Cluster analysis and Student’s t-test were performed using SPSS 16.0 (https://www.ibm.com/analytics/spss-statistics-software).
Results
Basmati Genome Sequencing
High-throughput sequencing of three traditional Basmati varieties was performed to facilitate downstream analysis. A total of 43,024,210 reads were generated from Basmati 370; 43,263,296 reads from Dahrdun Basmati; and 44,099,730 reads from Rato Basmati, each corresponding to more than 10 GB read length, and more than 90% of these reads were clean reads (Table 2). The clean reads were mapped to the reference genomes of Nipponbare (japonica rice), Zhenshan 97 (indica rice), and Kasalath (aus rice). The mapping results indicated that all genomes were sequenced at a depth ranging from 26.02X to 30.75X, with more than 90% coverage.
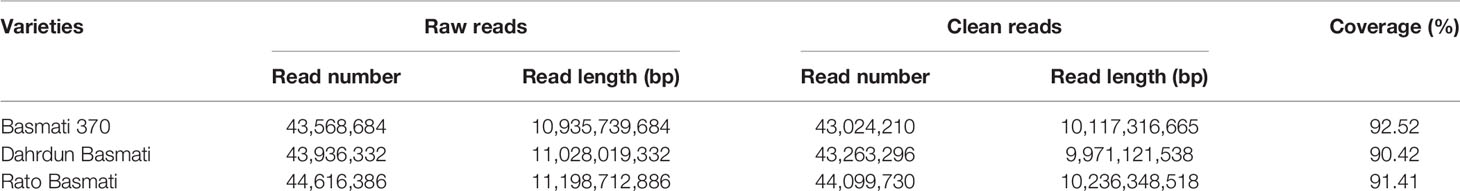
Table 2 Data generated from whole-genome resequencing of three Basmati varieties.
The number of SNPs in each Basmati variety were determined relative to each reference genome. Compared with Nipponbare, we identified 1,544,399 SNPs in Basmati 370; 2,105,019 SNPs in Dahrdun Basmati; and 1,229,155 SNPs in Rato Basmati. Similarly, comparison with the Kasalath reference genome revealed 1,453,259 SNPs in Basmati 370; 1,336,541 in Dahrdun Basmati; and 1,627,481 SNPs in Rato Basmati, whereas comparison with the Zhenshan 97 reference genome revealed 1,409,129 SNPs in Basmati 370; 793,929 SNPs in Dahrdun Basmati; and 1,659,254 SNPs in Rato Basmati. Thus, Dahrdun Basmati showed the highest number of SNPs compared with Nipponbare and the lowest number of SNPs compared with Zhenshan 97 (Table 3).
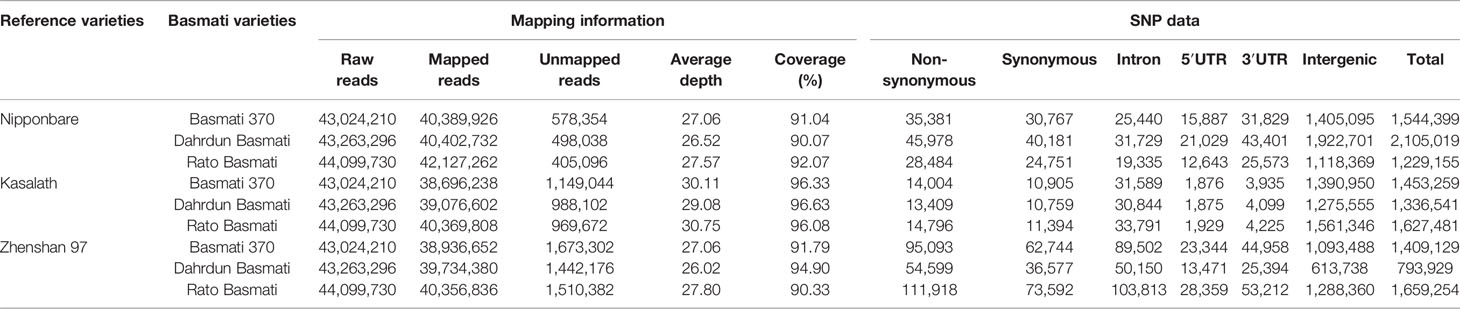
Table 3 Mapping and SNP summary of three traditional Basmati varieties.
In comparison with the Nipponbare reference genome, relatively high numbers of SNPs were detected on chromosomes 1, 3, 6, and 11 in Basmati genomes, while the lowest numbers of SNPs were detected on chromosomes 9 and 5. Compared with Kasalath, Basmati varieties showed a high proportion of SNPs on chromosomes 1, 2, 3, 6, and 7, and the lowest numbers of SNPs on chromosome 10. Compared with Zhenshan 97, we found a high proportion SNPs on chromosomes 1, 2, 6, and 7 in Basmati varieties and lower SNP numbers on chromosomes 5 and 9. The distribution of SNPs on all 12 chromosomes of the three Basmati varieties in comparison with all three reference genomes is summarized in Supplementary Table 1.
Furthermore, we also determined the number of SNPs and InDels in each Basmati variety against the three reference genomes. Accordingly, in Supplementary Table 2. In comparison with Nipponbare, InDels were abundant on chromosomes 3 and 6 in Basmati varieties, while the number of SNPs was the highest on chromosome 1. In comparison with Kasalath and Zhenshan 97 reference genomes, chromosomes 1, 2, and 3 of Basmati varieties contained a high proportion of InDels, while chromosomes 1, 2, and 6 showed the highest number of substitutions.
Distribution of Common SNPs and InDels in Genic Regions
Common SNPs in genic regions, functional SNPs [non-synonymous SNPs and SNPs in untranslated regions (UTRs)], and InDels (5–30 bp) in genic regions were identified by comparing all three Basmati genomes with all three reference genomes. The total number of common SNPs identified in Basmati varieties were 52,204 compared with Nipponbare; 19,207 compared with Kasalath; and 73,219 compared with Zhenshan 97. The extracted common SNPs were plotted within the Nipponbare (Figure 2A), Kasalath (Figure 2B), and Zhenshan 97 (Figure 2C) reference genomes.
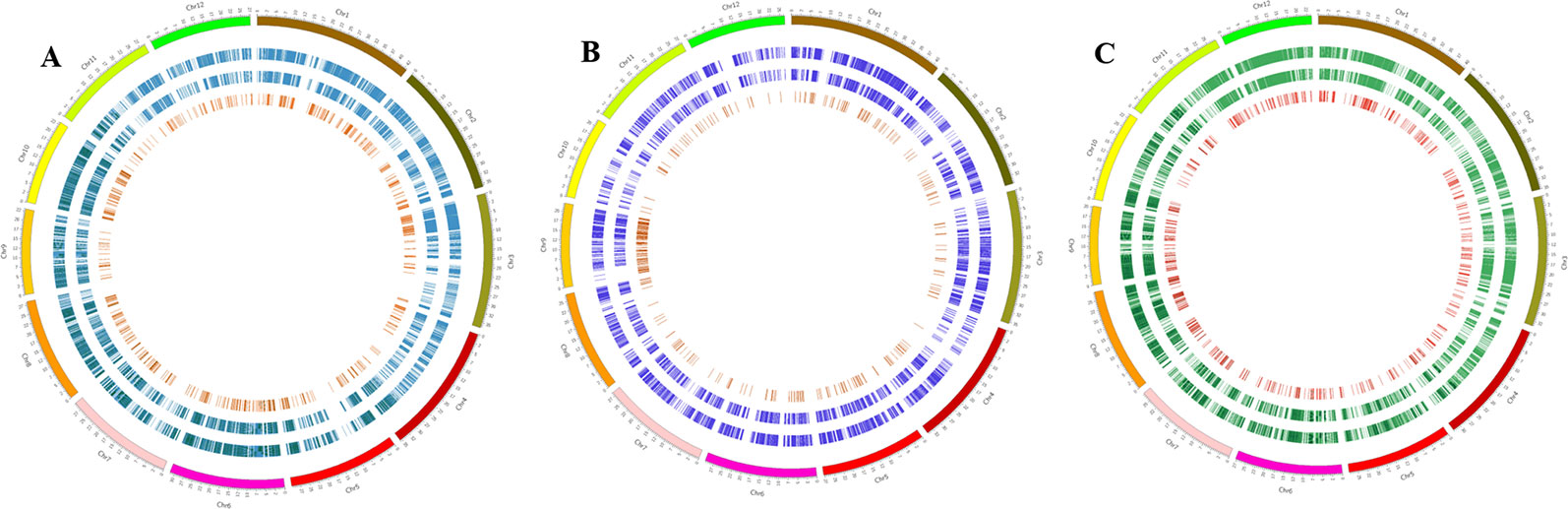
Figure 2 Circos plots showing the distribution pattern of SNPs and InDels in the genic regions of three Basmati varieties. (A–C) Distribution of SNPs and InDels in Basmati varieties in comparison with Nipponbare (A), Kasalath (B), and Zhenshan 97 (C) reference genomes. The outermost circle represents 12 chromosomes of the rice genome. The second circle from the outside represents common SNPs. The third circle from the outside represents functional SNPs. The innermost circle with red bars shows the distribution of InDels ranging in size from 5 to 30 bp.
In addition, in silico analysis using resequencing data of 54 varieties revealed 20 novel unique SNPs in genic regions of the Basmati genomes. These unique SNPs were also confirmed using the public rice database (http://ricevarmap.ncpgr.cn/v2/). Additionally, we identified 11 unique InDels in the Basmati genomes. The unique SNPs and InDels, and the functions of genes containing these polymorphisms, are listed in Supplementary Table 3. PCR amplification of 289 bp fragments using gene-specific primers (forward primer, 5′-CTGTTTATACGTAGTACGGGTTG-3′; reverse primer, 5′-TGTTTGTAGGGGGATGCAAT-3′), which confirmed that the 25 bp insertion in the intron of the gene involved in seed development regulation (Os10g0139300; IRGSP-1.0; position: 2,425,049 bp) was only specific to the Basmati and aus groups, and could be discriminated among 20 rice varieties (Supplementary Figure 2). We further examined the spatiotemporal expression pattern of Os10g0139300 in the RiceXpro database (Sato et al., 2011); this gene showed high expression in the embryo and endosperm after flowering, indicating a possible role in seed development during ripening.
GO Analysis of Basmati Varieties
We investigated the functions of genes containing common SNPs and InDels among the three Basmati genomes via GO analysis. Genes were assigned to three categories, namely, biological process (BP), molecular function (MF), and cellular component (CC). The major GO associations were found in metabolic process, cellular process, biological regulation for BP terms (Figure 3A). For the MF terms, binding and catalytic activity (Figure 3B). Whereas, cell, cell part, and membrane were associated with CC terms (Figure 3C).
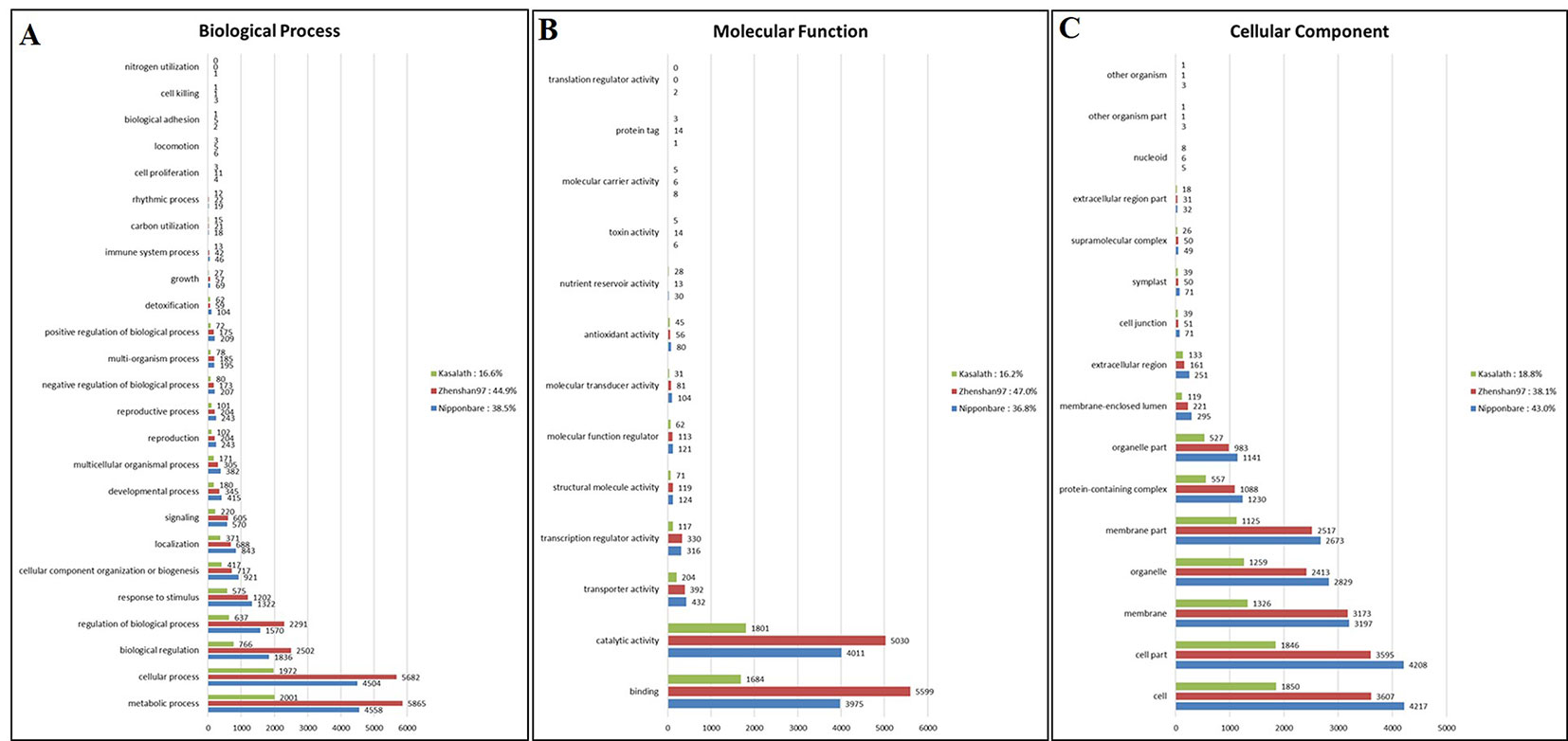
Figure 3 Gene ontology (GO) analysis of Basmati genomes in comparison with Zhenshan 97, Kasalath, and Nipponbare genomes. (A–C) GO categories including biological process (A), molecular function (B), and cellular component (C) are shown.
Furthermore, we analyzed genome-wide orthologous clusters of genes from Basmati varieties using common SNPs by comparison with Nipponbare, Zhenshan 97, and Kasalath reference genomes. The analysis revealed 5,395 orthologous clusters based on protein sequences of the three reference genomes (Figure 4A). The Venn diagram showed that 1,132 gene clusters were shared by all three reference genomes, suggesting their conservation in the lineage after speciation (Figure 4B). Additionally, 348, 354, and 51 clusters specific to Nipponbare, Zhenshan 97, and Kasalath reference genomes, respectively, were identified. Additionally, cluster analysis of the mutated genes in the three Basmati varieties revealed 4,415 clusters in comparison with Nipponbare; 2,721 clusters in comparison with Kasalath; and 4,033 clusters in comparison with Zhenshan 97 reference genomes. The presence of 2,721 clusters in comparison with Kasalath suggests that Basmati varieties show less genetic variation compared with the aus group.
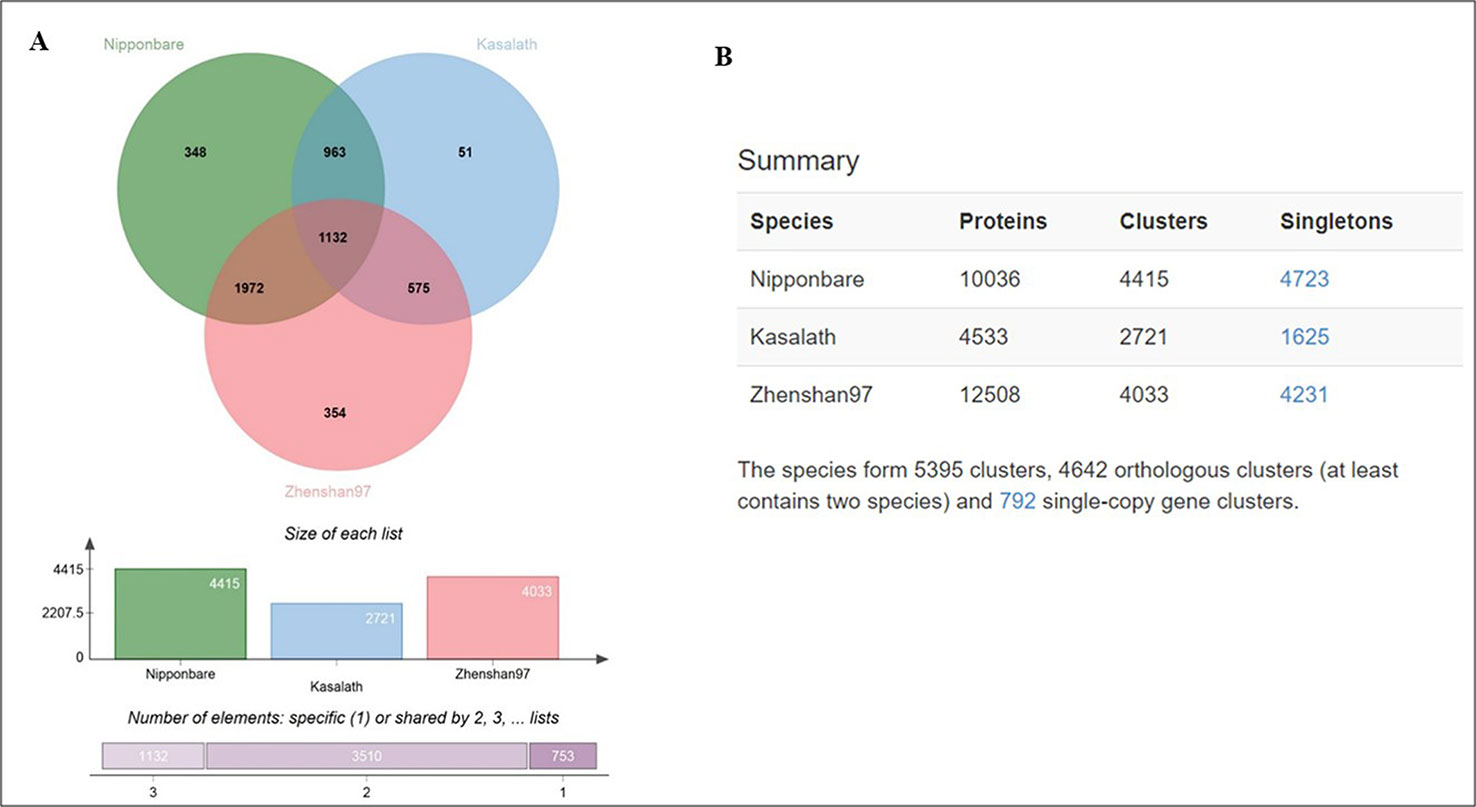
Figure 4 Ortho Venn diagram. (A) Venn diagram showing the distribution of shared gene families among Nipponbare, Kasalath, and Zhenshan 97. Specific gene clusters are indicated within the three reference genomes. (B) Counts of clusters in each genome.
In phylogenetic studies, the identification of single-copy orthologs is critical in any species (Creevey et al., 2011). Orthologous cluster analysis revealed 792 clusters representing single-copy genes, which were shared by all three reference genomes, suggesting that the single-copy status of genes was maintained during evolution after species divergence.
Furthermore, 1,132 gene clusters shared by Nipponbare, Kasalath, and Zhenshan 97 reference genomes harbored unique SNPs from all three Basmati varieties, and functional annotation of the genes harboring these unique SNPs showed that the majority of these genes were involved in biological regulation, metabolic process, and cellular process (Figure 5A); binding and catalytic activity (Figure 5B); and membrane, cell parts, and cellular component (Figure 5C). We also detected mutated gene clusters associated with the metabolic process involved in the cellular aromatic compound (Figure 5A). Further, a total of 35 genes including Badh2 gene were found to be involved in aromatic compound biosynthesis based on biological process and molecular functional annotation. While, genomic regions from three Basmati varieties compared to Nipponbare reference genome showed functional variation across the 35 genes involved in aromatic compound biosynthesis (data not shown). Whereas, in sillico analysis of 35 genes using Rice Variation Map v2.0 revealed that only nine genes including Badh2 gene having alternative alleles in 96 varieties of aromatic group with more than 80% of frequency.
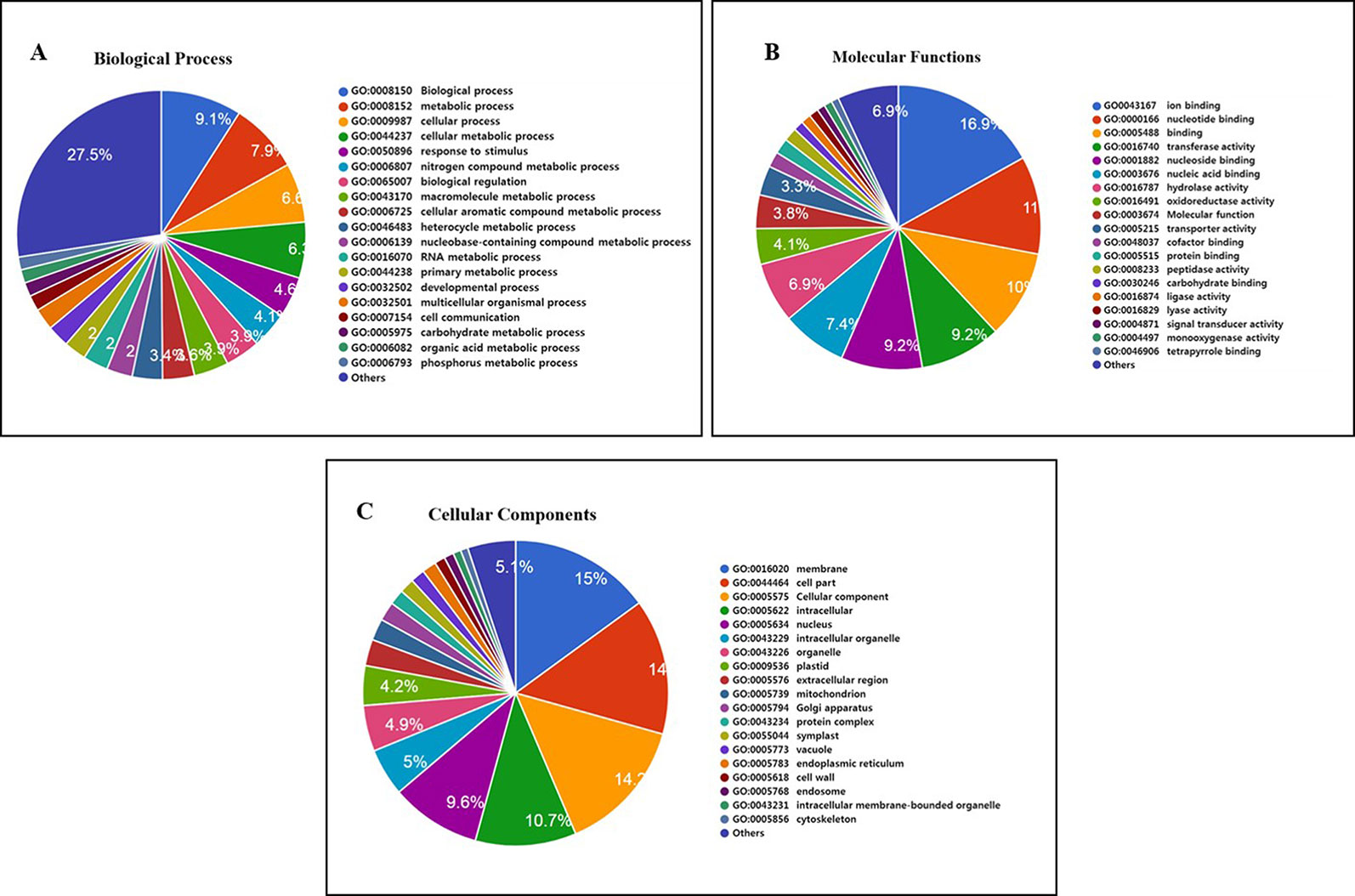
Figure 5 Functional annotation of 1,132 mutated gene cluster of Basmati genome shared by Nipponbare, Kasalath, and Zhenshan 97 reference genomes. (A) Biological process. (B) Molecular function. (C) Cellular component.
SNP Genotyping and Genetic Relationship
To determine the genetic relationship of 30 traditional Basmati varieties, including three resequenced Basmati varieties, with rice varieties belonging to other groups, a total of 60 varieties were genotyped with two sets of 96-plex indica/japonica SNPs. Two of these SNP markers were excluded from the analysis because of their low quality. The number of SNP markers, average physical interval between SNPs per chromosome, and coverage percentage are summarized in Supplementary Table 4.
All 190 SNP markers were biallelic between indica and japonica varieties, and the average allele number was 2.12. In addition, the average value of major allele frequency (MAF) was 0.681, and almost all SNPs showed no heterozygosity (average heterozygosity = 0.020). Consistent with these data, the average polymorphic information content (PIC) was 0.33 (Supplementary Table 5).
The UPGMA dendrogram based on Cavalli-Sforza and Edwards (1967) genetic distance (Figure 6A) classified all 60 varieties into two subspecies, indica and japonica. Additionally, the japonica group showed two distinct subgroups, aromatic and japonica. The 30 Basmati varieties were divided into two groups, indica (comprising Dahrdun Basmati) and japonica (comprising Rato Basmati and Basmati 370). To identify the population structure of all 60 rice varieties, STRUCTURE analysis was carried out. The value of delta K was maximum at K = 2 (Supplementary Figure 3). At K = 2, 60 varieties were classified into indica and japonica, as expected based on marker characteristics; however, more than half of the varieties in the japonica group showed admixture with indica ancestry. At K = 3, the aromatic group along with Rato Basmati and Basmati 370 grouped at the japonica group, and at K = 4, the aromatic group was divided into two clear subgroups and one admixed group. All nine varieties, including Rato Basmati, in the upper yellow subgroup within the aromatic group (Figure 6B), were from Nepal. At K = 6, five subgroups were evident among the 60 varieties including indica, aus, aromatic, tropical japonica, and temperate japonica, except one variety, which showed less than 65% of estimated ancestry derived from any single subgroup (Figure 6B). The results of phylogenetic and population structure analyses were consistent. Among the 30 traditional Basmati varieties, four varieties, including Dahrdun Basmati, were classified into the indica group; four into the aus group; and 22, including Rato Basmati and Basmati 370, into the aromatic group.
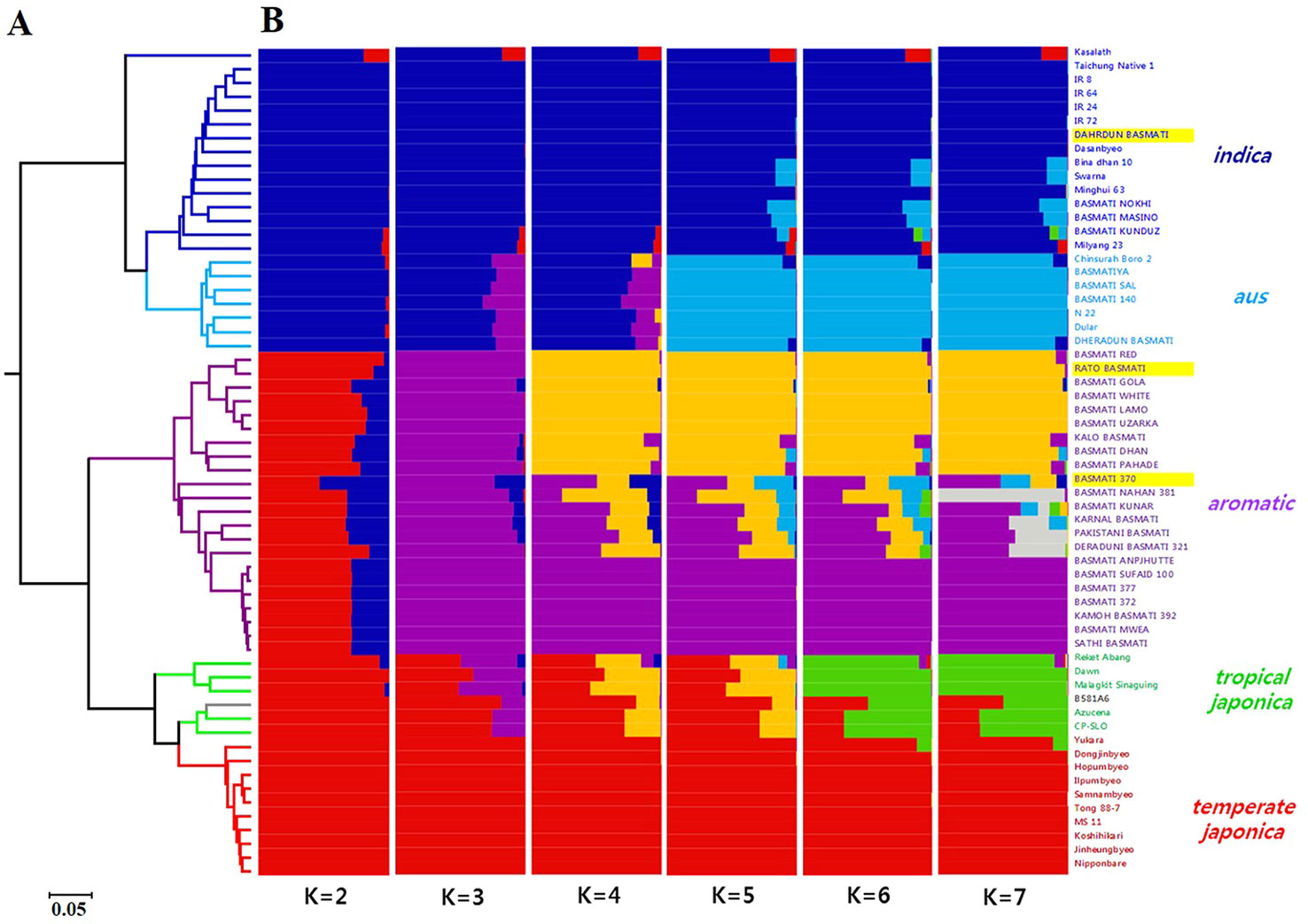
Figure 6 Genetic diversity analysis of 60 rice varieties using 190 SNPs. (A) UPGMA dendrogram. The branches are colored according to the subpopulation assessment in (B) based on K = 6, except for the aromatic group, which is based on K = 3. Gray branches indicate admixture. (B) Population structure analysis using the STRUCTURE software for K values ranging from 2 to 7. Three varieties used for genomic analysis are highlighted in yellow.
Badh2 Marker Analysis
Among 60 rice varieties, 30 traditional Basmati varieties were further investigated on the basis of the 8 bp deletion in the Badh2 gene to classified into badh2.1 and wild Badh2 allele harboring genotypes. PCR-based genotyping of the Badh2 InDel marker divided the traditional Basmati varieties into two groups: badh2.1 (22 varieties; 95 bp PCR product) and wild Badh2 allele carrying genotypes (8 varieties; 103 bp PCR product) (Supplementary Figure 4, Table 1). The remaining 30 non-Basmati rice varieties were classified in the wild Badh2 allele group (Table 1).
Agronomic and Grain Quality Trait Analysis
The mean performance of 13 agronomic and grain quality traits of 30 traditional Basmati varieties is presented in Supplementary Table 6. The coefficient of variation of CN was the highest (25.49), followed by that of SFC (24.87). Comparison of the mean performance between aromatic and indica/aus groups revealed significant differences in only L/W ratio, PL, and AC; the aromatic group showed significantly longer panicles, longer and slender grains, and lower AC than the indica/aus group (Supplementary Table 7).
Next, hierarchical cluster analysis was performed to elucidate the relationship among the 30 traditional Basmati varieties. These varieties were divided into two major clusters (I and II), and each cluster was further divided into three subclusters (Supplementary Figure 5, Supplementary Table 6). Cluster I contained 20 moderate duration varieties from diverse geographical regions with superior grain quality. Cluster II consisted of ten late duration varieties, mostly from Nepal, with poor grain quality; thus cluster II showed less genetic diversity than cluster I.
Discussion
Basmati rice varieties, considered a unique varietal group, have been generally classified into the aromatic group (Glaszmann, 1987; Garris et al., 2005; Civáň et al., 2015). Recent findings suggest that Basmati rice was derived mostly from aus and japonica varietal groups (Civáň et al., 2015). Recently, the genome assembly of Basmati rice was performed using “Basmati Surkh 89-15,” an improved cultivar from Pakistan (Zhao et al., 2018). However, a higher level of introgression from other rice populations in improved varieties of Basmati makes it difficult to define the genome structure. A latest preprint of phylogenomic analysis involving “Basmati 334” proposed admixture events between Basmati rice, aus, and O. rufipogon; this study concluded that Basmati rice has a hybrid origin and is closely related to both japonica and aus rice (Choi et al., 2018). However, phylogenomic analysis using a single genome cannot provide detailed information about the Basmati genome structure, when referring to the entire Basmati group, irrespective of the Badh2 allele type. Therefore, defining Basmati-specific genome features is important to understand the domestication of Asian rice.
In this study, we performed whole-genome resequencing and analysis of three traditional Basmati varieties. The identification of genome-wide nucleotide polymorphisms, including SNPs and InDels, using NGS has gained importance in the rice genome (Markkandan et al., 2018) and has enabled researchers to identify genome-specific features in rice varieties. Therefore, we performed NGS data analysis of Basmati 370, Dahrdun Basmati, and Rato Basmati to characterize the Basmati genome in detail. We found that millions of SNPs in all Basmati varieties in comparison with Nipponbare, Kasalath, and Zhenshan 97 reference genomes (Table 3), thus providing an opportunity to identify Basmati-specific features. Additionally, the genome-wide common SNPs and InDels identified in this study would serve as a useful resource for the development of SNP and InDel markers for the Basmati genome, specific to japonica, aus, and indica varietal groups (Figures 2A–C). Similarly, in silico analysis of the three Basmati rice genomes along with 54 rice varieties revealed high-quality Basmati-specific features.
Basmati rice varieties showed less genomic variation compared with the aus group and was phylogenetically close to the japonica group; these results are consistent with those of previous studies (Choi et al., 2018; Civáň et al., 2019). GO enrichment analysis also showed less genomic variation between the Basmati genome and the aus group in terms of GO categories. Most of the genes assigned to the three GO categories were mainly involved in metabolic process, cellular process, binding, catalytic activity, cell, and cell part. This functional annotation of genes is consistent with previous findings in rice (Kim et al., 2014; Liu et al., 2017). Additionally, our data showed that the metabolic process involved in the cellular aromatic compound was associated with the common mutated gene cluster (Figure 5A) and further analyses revealed that nine genes including Badh2 gene having alternative allele’s among aromatic group of rice varieties with more than 80% of frequency (Supplementary Table 8). However, possible involvement of these genes except Badh2 remains to be determined for cellular aromatic biosynthesis.
A recent genomic analysis of a population of over 1,000 wild and cultivated rice accessions using genome-wide polymorphisms showed that Basmati rice arose from hybridization between japonica and wild rice related to the aus group (Civáň et al., 2019). Similarly, our comparative analysis of genome structure, based on genomic variation and GO analysis, showed that the Basmati genome is probably derived mostly from the aus and japonica groups.
Previously, it was shown that the recessive fgr allele encoding Badh2 carries an 8 bp deletion and three SNPs in the seventh exon, resulting in the fragrant trait in Basmati varieties (Bradbury et al., 2005). Recently, haplotype analysis of the Badh2 gene and analysis of 2-AP using 242 rice accessions classified two Basmati varieties harboring the wild Badh2 allele under the aus and indica groups (Kovach et al., 2009). In this study, our comparative analysis found that both Basmati 370 and Rato Basmati carrying the badh2.1 allele was consistent with the badh2.1 allele reported by Kovach et al. (2009). Further, we genotyped the Badh2 allele in the 30 Basmati varieties using the Badh2 InDel maker developed by Sakthivel et al. (2009). The results indicated that 22 of the 30 traditional Basmati varieties belonging to the aromatic group carry the fragrant-inducing badh2.1 allele and are more closely related to the japonica group. However, eight of the 30 Basmati varieties were harboring the wild Badh2 allele under the aus and indica groups (Supplementary Figure 4, Table 1). Thus, the results of Badh2 allele genotyping were consistent with those of phylogenetic analysis. We propose that classification of these wild Badh2 allele carrying Basmati varieties under the indica and aus groups might results from either natural selection or human error during varietal diversification or germplasm collection.
The success of any crop breeding program depends on the magnitude of genetic variability within the germplasm (Kishor et al., 2016). In this study, although efforts were made to evaluate the agronomic and grain quality traits of 30 traditional Basmati varieties in the experimental field of Seoul National University, most of the Basmati varieties failed to flower in the rice growing season of the temperate region. By contrast, Basmati 370 and a few other wild Badh2 allele carrying Basmati varieties were flowered, and their agronomic traits were evaluated for further studies in temperate regions (data not shown). Furthermore, agronomic and grain quality trait passport data obtained from the public database Genesys showed wide variation in most of the traits among the 30 traditional Basmati varieties (Supplementary Table 6). These finding are in agreement with previous genetic diversity studies in Basmati varieties (Lingaiah et al., 2014; Nirmaladevi et al., 2015). Most of the agronomic and grain quality traits, except L/W ratio, PL, and AC, did not show significant differences among Basmati varieties belonging to the aromatic and indica/aus groups (Supplementary Table 7). AC is an important factor affecting the palatability and grain quality of cooked rice (Tian et al., 2009). Rice grains with low AC (12–20%) are usually glossy, soft, and sticky after cooking, whereas those with high AC (> 25%), generally found in Basmati varieties belonging to the indica group, exhibit a dry texture, remain separate, and are less tender upon cooking and become hard upon cooling (Bao et al., 2006).
Hierarchical cluster analysis revealed two major clusters (I and II) among the 30 traditional Basmati varieties, based on agronomic and grain quality traits (Supplementary Figure 5). Cluster I comprised varieties from diverse geographical regions, with moderate duration and superior grain qualities. By contrast, cluster II comprised of varieties with late duration and poor grain qualities. These findings are in accordance with a previous study where traditional Basmati varieties with superior agronomic and grain quality traits were grouped in a separate cluster (Roy et al., 2012). Accessions in cluster I with superior agronomic and grain quality could be exploited for the development of improved Basmati varieties in breeding programs.
In conclusion, our study provides a detailed analysis of the Basmati genome structure in comparison with indica, japonica, and aus genomes via whole-genome resequencing and genome-wide SNP marker analysis. This data will serve as an important resource for molecular breeding and genetic studies in Basmati rice.
Data Availability Statement
All relevant raw sequence data are available in the NCBI Short Read Archive (SRA) database under the following BioProject accession numbers: Basmati [PRJNA551546], Dahrdun Basmati [PRJNA551547], and Rato Basmati [PRJNA551548].
Author Contributions
DK and JS conceptualized the study, conducted formal analysis, determined the software for data analysis, and performed data visualization. DK and H-JK curated the data and determined the methodology and resources for this study. JS and JC performed data validation. H-JK acquired the funding and supervised the study. DK, JS, and JC wrote the first draft of the manuscript. All authors reviewed, edited, and approved the final manuscript.
Funding
This research was funded by the Rural Development Administration through the Next-generation BioGreen 21 Program (Grant No. PJ013165).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Crop Molecular Breeding Lab at Seoul National University for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00086/full#supplementary-material
References
Ahuja, S. C., Panwar, D. V. S., Uma, A., Gupta, K. R. (1995). Basmati rice: the scented pearl (Hisar: Directorate of Publications, CCS Haryana Agricultural University). doi: 10.13140/RG.2.2.26828.87681
Amarawathi, Y., Singh, R., Singh, A. K., Singh, V. P., Mohapatra, T., Sharma, T. R., et al. (2008). Mapping of quantitative trait loci for basmati quality traits in rice (Oryza sativa L.). Mol. Breed. 21, 49–65. doi: 10.1007/s11032-007-9108-8
Bao, J., Shen, S., Sun, M., Corke, H. (2006). Analysis of genotypic diversity in the starch physicochemical properties of nonwaxy rice: apparent amylose content, pasting viscosity and gel texture. Starch - Stärke 58 (6), 259–267. doi: 10.1002/star.200500469
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P., Marth, G. T. (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27 (12), 1691–1692. doi: 10.1093/bioinformatics/btr174
Bradbury, L. M. T., Fitzgerald, T. L., Henry, R. J., Jin, Q., Waters, D. L. E. (2005). The gene for fragrance in rice. Plant Biotechnol. J. 3 (3), 363–370. doi: 10.1111/j.1467-7652.2005.00131.x
Cavalli-Sforza, L. L., Edwards, A. W. (1967). Phylogenetic analysis. models and estimation procedures. Am. J. Hum. Genet. 19 (3 Pt 1), 233–257.
Chen, H., Xie, W., He, H., Yu, H., Chen, W., Li, J., et al. (2014). A high-density SNP genotyping array for rice biology and molecular breeding. Mol. Plant 7 (3), 541–553. doi: 10.1093/mp/sst135
Chen, E., Huang, X., Tian, Z., Wing, R. A., Han, B. (2019). The genomics of Oryza species provides insights into rice domestication and heterosis. Annu. Rev. Plant Biol. 70, 639–665. doi: 10.1146/annurev-arplant-050718-100320
Choi, J. Y., Platts, A. E., Fuller, D. Q., Hsing, Y.-I., Wing, R. A., Purugganan, M. D. (2017). The rice paradox: multiple origins but single domestication in Asian rice. Mol. Biol. Evol. 34 (4), 969–979. doi: 10.1093/molbev/msx049
Choi, J. Y., Groen, S. C., Zaaijer, S., Purugganan, M. D. (2018). Nanopore sequence-based genome assembly of the basmati rice. bioRxiv, 396515. doi: 10.1101/396515
Civáň, P., Craig, H., Cox, C. J., Brown, T. A. (2015). Three geographically separate domestications of Asian rice. Nat. Plants 1, 15164–15164. doi: 10.1038/nplants.2015.164
Civáň, P., Ali, S., Batista-Navarro, R., Drosou, K., Ihejieto, C., Chakraborty, D., et al. (2019). Origin of the aromatic group of cultivated rice (Oryza sativa L.) traced to the Indian Subcontinent. Genome Biol. Evol. 11 (3), 832–843. doi: 10.1093/gbe/evz039
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 (18), 3674–3676. doi: 10.1093/bioinformatics/bti610
Creevey, C. J., Muller, J., Doerks, T., Thompson, J. D., Arendt, D., Bork, P. (2011). Identifying single copy orthologs in Metazoa. PloS Comput. Biol. 7 (12), e1002269. doi: 10.1371/journal.pcbi.1002269
Daygon, V. D., Calingacion, M., Forster, L. C., et al. (2017). Metabolomics and genomics combine to unravel the pathway for the presence of fragrance in rice. Sci. Rep. 7, 8767. doi: 10.1038/s41598-017-07693-9
Earl, D. A., Vonholdt, B. M. (2012). Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Cons Genet. Res. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Falush, D., Stephens, M., Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 (4), 1567–1587.
Fuller, D. Q., Sato, Y.-I., Castillo, C., Qin, L., Weisskopf, A. R., Kingwell-Banham, E. J., et al. (2010). Consilience of genetics and archaeobotany in the entangled history of rice. Archaeol. Anthropological Sci. 2 (2), 115–131. doi: 10.1007/s12520-010-0035-y
Garris, A. J., Tai, T. H., Coburn, J., Kresovich, S., McCouch, S. (2005). Genetic structure and diversity in Oryza sativa L. Genetics 169 (3), 1631–1638. doi: 10.1534/genetics.104.035642
Glaszmann, J. C. (1987). Isozymes and classification of Asian rice varieties. Theor. Appl. Genet. 74 (1), 21–30. doi: 10.1007/bf00290078
Huang, X., Kurata, N., Wei, X., Wang, Z.-X., Wang, A., Zhao, Q., et al. (2012). A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497. doi: 10.1038/nature11532
Jimenez, F. R., Maughan., P. J., Alvarez., A., Kietlinski., K. D., Smith., S. M., Pratt, D. B., et al (2013). Assessment of genetic diversity in Peruvian Amaranth (Amaranthus caudatus and A. hybridus) germplasm using single nucleotide polymorphism markers. Crop Sci. 53, 532–541. doi: 10.2135/cropsci2012.07.0413
Kato, S. (1928). On the affinity of rice varieties as shown by fertility of hybrid plants. Bull. Sci. Fac. Agric. Kyushu Univ. 3, 132–147.
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 6 (1), 4. doi: 10.1186/1939-8433-6-4
Kim, B., Kim, D.-G., Lee, G., Seo, J., Choi, I.-Y., Choi, B.-S., et al. (2014). Defining the genome structure of ‘Tongil’ rice, an important cultivar in the Korean “Green Revolution”. Rice (New York N.Y.) 7 (1), 22–22. doi: 10.1186/s12284-014-0022-5
Kishor, D. S., Krishnan, A., Kebbahalli Jayaramu, Y., Kumar, V., Yashoda, Jong Koh, H. (2016). Genotypic variation among okra (Abelmoschus esculentus (L.) Moench) germplasms in South India. Plant Breed. Biotechnol. 1, 234–241. doi: 10.9787/PBB.2016.4.2.234
Kovach, M. J., Sweeney, M. T., McCouch, S. R. (2007). New insights into the history of rice domestication. Trends In Genet. 23 (11), 578–587. doi: 10.1016/j.tig.2007.08.012
Kovach, M. J., Calingacion, M. N., Fitzgerald, M. A., McCouch, S. R. (2009). The origin and evolution of fragrance in rice (Oryza sativa L.). Proc. Natl. Acad. Sci. U S A 106 (34), 14444–14449. doi: 10.1073/pnas.0904077106
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5), 589–595. doi: 10.1093/bioinformatics/btp698
Lingaiah, N., Venkanna, V., Cheralu, C. (2014). Genetic variability analysis in rice (Oryza sativa L.). Int. J. Pure App. Biosci. 2 (5), 203–204.
Liu, K., Muse, S. V. (2005). PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21 (9), 2128–2129. doi: 10.1093/bioinformatics/bti282
Liu, W., Ghouri, F., Yu, H., Li, X., Yu, S., Shahid, M. Q., et al. (2017). Genome wide re-sequencing of newly developed Rice Lines from common wild rice (Oryza rufipogon Griff.) for the identification of NBS-LRR genes. PloS One 12 (7), e0180662. doi: 10.1371/journal.pone.0180662
Markkandan, K., Yoo, S.-I., Cho, Y.-C., Lee, D. (2018). Genome-wide identification of insertion and deletion markers in Chinese commercial rice cultivars, based on next-generation sequencing data. Agronomy 8 (4), 36. doi: 10.3390/agronomy8040036
McCouch, S. R., Kochert, G., Yu, Z. H., Wang, Z. Y., Khush, G. S., Coffman, W. R., et al. (1988). Molecular mapping of rice chromosomes. Theor. Appl. Genet. 76 (6), 815–829. doi: 10.1007/bf00273666
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi: 10.1101/gr.107524.110
Nirmaladevi, G., Padmavathi, G., Kota, S., Babu, V. (2015). Genetic variability, heritability and correlation coefficients of grain quality characters in rice (Oryza sativa L.). SABRAO J. Breed. Genet. 47 (4), 424–433.
Pachauri, V., Mishra, V., Mishra, P. P., Singh, A., Singh, S., Singh, R., et al. (2014). Identification of candidate genes for rice grain aroma by combining QTL mapping and transcriptome profiling approaches. Cereal Res. Commun. 42 (3), 376–388. doi: 10.1556/CRC.42.2014.3.2
Roy, S., Banerjee, A., K. Senapati, B., Sarkar, G. (2012). Comparative analysis of agro-morphology, grain quality and aroma traits of traditional and Basmati-type genotypes of rice, Oryza sativa L. Plant Breed. 131, 486–492. doi: 10.1111/j.1439-0523.2012.01967.x
Sakai, H., Kanamori, H., Arai-Kichise, Y., Shibata-Hatta, M., Ebana, K., Oono, Y., et al. (2014). Construction of pseudomolecule sequences of the aus rice cultivar Kasalath for comparative genomics of Asian cultivated rice. DNA Res. 21 (4), 397–405. doi: 10.1093/dnares/dsu006
Sakthivel, K., Shobha Rani, N., Pandey, M. K., Sivaranjani, A. K. P., Neeraja, C. N., Balachandran, S. M., et al. (2009). Development of a simple functional marker for fragrance in rice and its validation in Indian Basmati and non-Basmati fragrant rice varieties. Mol. Breed. 24 (2), 185–190. doi: 10.1007/s11032-009-9283-x
Sang, T., Ge, S. (2007). Genetics and phylogenetics of rice domestication. Curr. Opin. In Genet. Dev. 17 (6), 533–538. doi: 10.1016/j.gde.2007.09.005
Sato, Y., Antonio, B. A., Namiki, N., Takehisa, H., Minami, H., Kamatsuki, K., et al. (2011). RiceXPro: a platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res. 39 (Database issue), D1141–D1148. doi: 10.1093/nar/gkq1085
Serba, D., Muleta, K., Amand, P., Bernardo, A., Perumal, R., Bashir, E. (2019). Genetic diversity, population structure, and linkage disequilibrium of pearl millet. Plant Genome. 12, 180091. doi: 10.3835/plantgenome2018.11.0091
Siddiq, E. A., Vemireddy, L. R., Nagaraju, J. (2012). Basmati rices: genetics, breeding and trade. Agric. Res. 1 (1), 25–36. doi: 10.1007/s40003-011-0011-5
Tian, Z., Qian, Q., Liu, Q., Yan, M., Liu, X., Yan, C., et al. (2009). ). Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc. Natl. Acad. Sci. U S A 106 (51), 21760–21765. doi: 10.1073/pnas.0912396106
Vemireddy, L. R., Noor, S., Satyavathi, V., et al. (2015). Discovery and mapping of genomic regions governing economically important traits of Basmati rice. BMC Plant Biol. 15, 207. doi: 10.1186/s12870-015-0575-5
Wang, Y., Li, J. (2005). The plant architecture of rice (Oryza sativa). Plant Mol. Biol. 59 (1), 75–84. doi: 10.1007/s11103-004-4038-x
Wang, Y., Coleman-Derr, D., Chen, G., Gu, Y. Q. (2015). OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43 (W1), W78–W84. doi: 10.1093/nar/gkv487
Wang, W., Mauleon, R., Hu, Z., Chebotarov, D., Tai, S., Wu, Z., et al. (2018). Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557 (7703), 43–49. doi: 10.1038/s41586-018-0063-9
Zhang, J., Chen, L.-L., Sun, S., Kudrna, D., Copetti, D., Li, W., et al. (2016). Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Sci. Data 3, 160076. doi: 10.1038/sdata.2016.76
Keywords: Basmati rice, aromatic, SNPs, NGS, gene ontology
Citation: Kishor DS, Seo J, Chin JH and Koh H-J (2020) Evaluation of Whole-Genome Sequence, Genetic Diversity, and Agronomic Traits of Basmati Rice (Oryza sativa L.). Front. Genet. 11:86. doi: 10.3389/fgene.2020.00086
Received: 10 October 2019; Accepted: 27 January 2020;
Published: 21 February 2020.
Edited by:
Felipe Klein Ricachenevsky, Universidade Federal de Santa Maria, BrazilReviewed by:
Gireesh Channappa, Indian Institute of Rice Research (ICAR), IndiaRaju Bheemanahalli Rangappa, Kansas State University, United States
Copyright © 2020 Kishor, Seo, Chin and Koh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joong Hyoun Chin, amhjaGluQHNlam9uZy5hYy5rcg==; Hee-Jong Koh, aGVlamtvaEBzbnUuYWMua3I=
†These authors have contributed equally to this work