 Sergei V. Shekhovtsov1,2,3*
Sergei V. Shekhovtsov1,2,3* Aleksandra A. Shipova1,2Tatiana V. Poluboyarova1,2,3Gennady V. Vasiliev1
Aleksandra A. Shipova1,2Tatiana V. Poluboyarova1,2,3Gennady V. Vasiliev1 Elena V. Golovanova4Anna P. Geraskina5
Elena V. Golovanova4Anna P. Geraskina5 Nina A. Bulakhova3,6Tímea Szederjesi7
Nina A. Bulakhova3,6Tímea Szederjesi7 Sergei E. Peltek1,2
Sergei E. Peltek1,2- 1Department of Molecular Biotechnology, Institute of Cytology and Genetics of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia
- 2Kurchatov Genomic Center, Institute of Cytology and Genetics of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia
- 3Laboratory of Biocenology, Institute of Biological Problems of the North of the Far Eastern Branch of the Russian Academy of Sciences, Magadan, Russia
- 4Laboratory of Systematics and Ecology of Invertebrates, Omsk State Pedagogical University, Omsk, Russia
- 5Center for Forest Ecology and Productivity of the Russian Academy of Sciences, Moscow, Russia
- 6Laboratory of Biodiversity and Ecology, Tomsk State University, Tomsk, Russia
- 7Department of Zoology, Hungarian Natural History Museum, Budapest, Hungary
Eisenia nordenskioldi (Eisen, 1879) is the only autochthonous Siberian earthworm with a large distribution that ranges from tundra to steppe and broadleaved forests. This species has a very high morphological, ecological, karyological, and genetic diversity, so it was proposed that E. nordenskioldi should be split into several species. However, the phylogeny of the complex was unclear due to the low resolution of the methods used and the high diversity that should have been taken into account. We investigated this question by (1) studying the diversity of the COI gene of E. nordenskioldi throughout its range and (2) sequencing transcriptomes of different genetic lineages to infer its phylogeny. We found that E. nordenskioldi is monophyletic and is split into two clades. The first one includes the pigmented genetic lineages widespread in the northern and western parts of the distribution, and the second one originating from the southern and southeastern part of the species' range and representing both pigmented and non-pigmented forms. We propose to split the E. nordenskioldi complex into two species, E. nordenskioldi and Eisenia sp. 1 (aff. E. nordenskioldi), corresponding to these two clades. The currently recognized non-pigmented subspecies E. n. pallida will be abolished as a polyphyletic and thus a non-natural taxon, while Eisenia sp. 1 will be expanded to include several lineages earlier recognized as E. n. nordenskioldi and E. n. pallida.
Introduction
Eisenia nordenskioldi (Eisen, 1879) is the only autochthonous Siberian earthworm with a large distribution, which includes the whole Siberia from the steppe zone in the south to certain Arctic islands, as well as the East European plain, parts of Kazakhstan, China, Korea, and Mongolia (Perel, 1979; Vsevolodova-Perel, 1997). This species was found to contain a very high morphological, ecological, and karyotypic diversity (Viktorov, 1997; Vsevolodova-Perel and Bulatova, 2008; Blakemore, 2013). E. nordenskioldi is traditionally divided into two subspecies, E. n. nordenskioldi and E. n. pallida, the latter differing solely by the absence of pigmentation (Malevich, 1956; Vsevolodova-Perel, 1997). Blakemore (2013) recently added two more subspecies, E. n. mongol and E. n. onon, based on single locations in Mongolia.
DNA sequence data (Blakemore, 2013; Shekhovtsov et al., 2013, 2015, 2016a,b, 2017b, 2018a,b; Hong and Csuzdi, 2016) demonstrated that genetic diversity within E. nordenskioldi is extremely high. Different genetic lineages of E. nordenskioldi can differ by as much as 22% of nucleotide substitutions in the mitochondrial COI gene (Shekhovtsov et al., 2013). Such level of divergence is typically characteristic for distinct congeneric species, or even different genera within a family (Hebert et al., 2003). This diversity is not only found in geographically remote populations: individuals identified as E. nordenskioldi, but highly diverged on the DNA level are often found in the same soil sample (Shekhovtsov et al., 2013, 2018b).
This newly discovered genetic diversity was characterized either as new subspecies (Blakemore, 2013) or as “genetic lineages” (Shekhovtsov et al., 2013, 2016a). Genetic lineage is an established term among earthworm scientists (King et al., 2008; Martinsson and Erséus, 2017; Marchán et al., 2018) that refers to intraspecific clades that are highly genetically diverged from other clades within the species. At least 15 distinct lineages are currently recognized within E. nordenskioldi, and deep genetic divergence between some of them were verified by the phylogenomic approach (Shekhovtsov et al., 2019). On the whole, the gathered evidence implies that this taxon encompasses several distinct species. However, phylogenetic relationships among the recognized subspecies and genetic lineages of E. nordenskioldi remain unclear.
Studies based on single mitochondrial and nuclear genes (Shekhovtsov et al., 2013, 2016a) suggested that both of the subspecies of E. nordenskioldi may be polyphyletic. However, this evidence could not be considered decisive due to the few DNA loci involved in the analysis. An attempt to resolve the phylogeny of the complex using mitochondrial genomes (Shekhovtsov et al., 2020) proved to be insufficient to clarify some questions. When using different datasets (i.e., protein sequences, protein-coding genes, rRNAs, total genomes, etc.), E. nordenskioldi was recovered either as mono- or as paraphyletic. Relationships among different lineages were also inconsistent. Extensive multigene data on nuclear sequences is obviously needed to resolve these issues.
The aim of this study was to determine the phylogeny of the E. nordenskioldi complex and to clarify its systematics. To do this, we sequenced transcriptomes of 12 E. nordenskioldi specimens belonging to different subspecies and genetic lineages. We assembled protein and DNA sequence datasets to construct a multigene phylogeny of the complex. In addition, we sequenced the COI gene of multiple geographically remote populations and combined those with the data obtained earlier in order to estimate the diversity within the complex throughout its distribution. We also investigated the validity of the closely related species E. atlavinyteae and the unpigmented subspecies E. n. pallida. Based on this data, we propose the ways this complex could be split into natural taxa.
Materials and Methods
Earthworm Sampling and DNA Sequencing
For phylogeographic analysis, we took the ethanol-fixed specimens we collected in 2011–2020, as well as those provided by colleagues (Figures 1, 2). Earthworms were identified according to the key of Vsevolodova-Perel (1997). A section of the posterior body end was excised for DNA isolation using the BioSilica columns (BioSilica, Russia) according to the manufacturer's instructions. The fragment of the mitochondrial COI gene was amplified using the universal LCO and HCO primers (Folmer et al., 1994). The obtained amplicons were purified with the exonuclease I—shrimp alkaline phosphatase mix (New England Biolabs, USA) and sequenced using the BigDye 3.1 (Applied Biosystems, USA). Only the new and unique haplotypes (a total of 62) were submitted to the GenBank under accession nos. MT863028–MT863089.
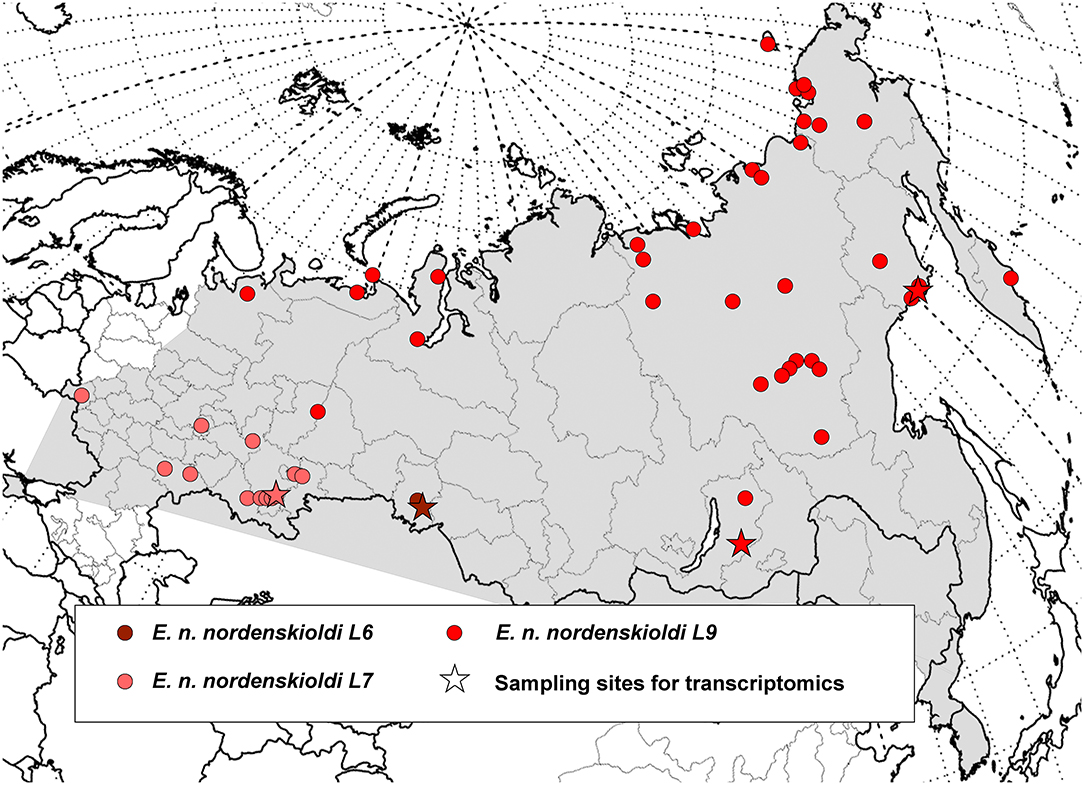
Figure 1. Geographic distribution of the genetic lineages of E. nordenskioldi. Distribution of the E. nordenskioldi complex is shown in gray. Color and symbol code refers to Figures 2–4.
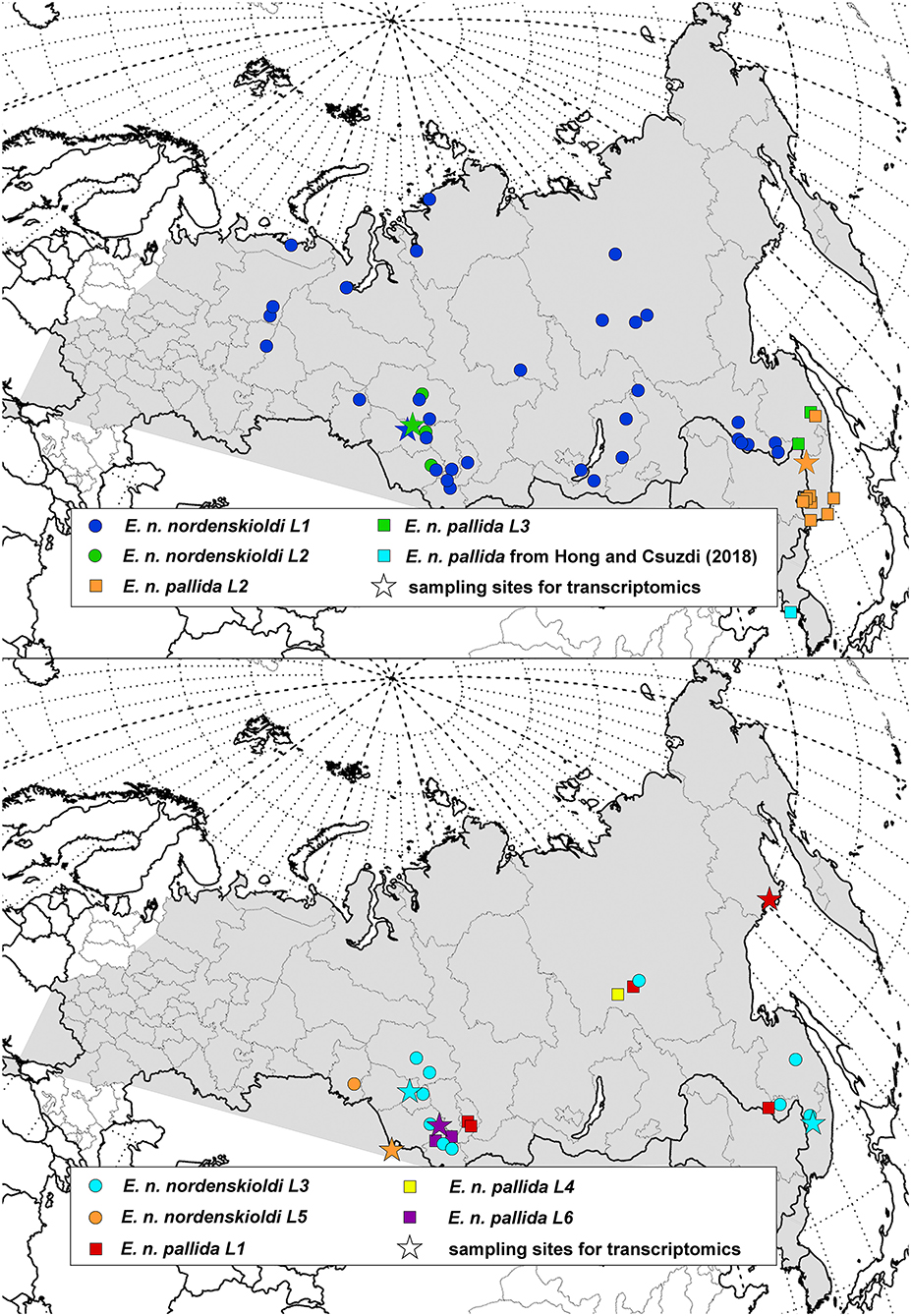
Figure 2. Geographic distribution of the genetic lineages of Eisenia sp. 1. Distribution of the E. nordenskioldi complex is shown in gray. Color and symbol code refers to Figures 1, 3, 4.
For transcriptomic analysis, live earthworm individuals were collected in 2016–2019 (see Table 1). RNA was extracted by the Trizol method as described in Shekhovtsov et al. (2019). For large worms, RNA was extracted from a section of the body containing the clitellum; for small individuals, the whole body was used. A small part of the tail was fixed in ethanol and used for DNA extraction and COI fragment sequencing as described above. The COI barcode was used to identify the genetic affinity of the specimens against our dataset of Eisenia sequences. Individuals belonging to the desired genetic lineage were taken for transcriptome analysis.
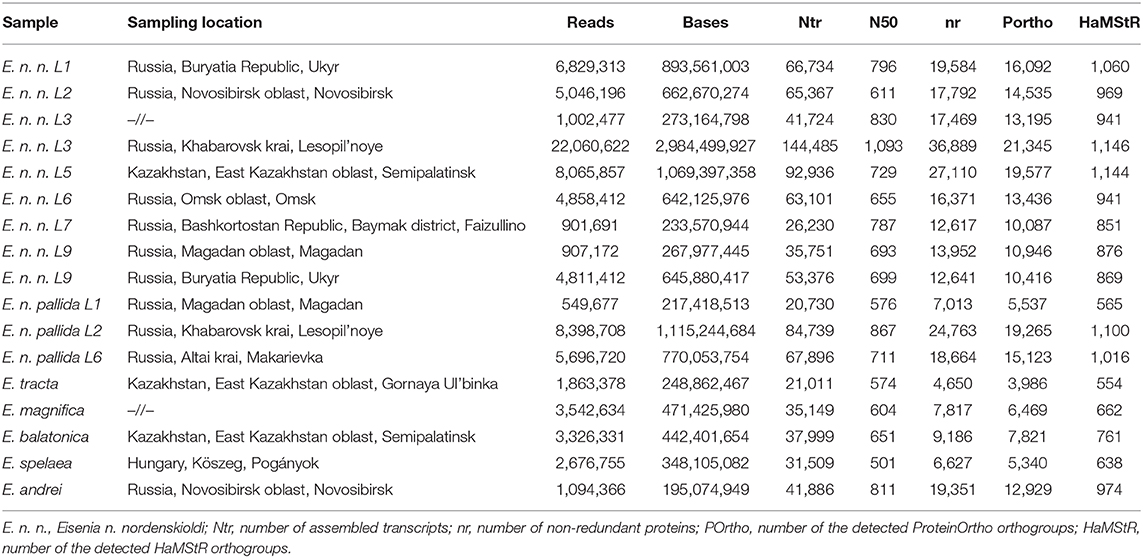
Table 1. Samples taken for transcriptomic analysis.
Transcriptome Sequencing and Assembly
RNA was purified using the PureLink RNA Micro Kit (Invitrogen, USA). RNASeq library preparations were carried out with 1 μg of total RNA using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, USA) according to the manufacturer's instructions with small modifications (4 min RNA fragmentation time and 12 PCR cycles). The quantity and quality of the libraries were assessed using the Agilent Bioanalyzer 2100 System and DNA High sensitivity kit (Agilent Technologies, USA). After normalization, barcoded libraries were pooled and sequenced on the NextSeq550 sequencer using the NextSeq 550 Mid Output v2 Kit 150 cycles (Illumina, USA).
Transcriptome de novo assembly was performed using the Trinity v.2.4.0 (Grabherr et al., 2011) with the default parameters. Protein coding sequences were predicted by the TransDecoder v.5.0.0 (http://transdecoder.github.io). Redundant proteins were removed by the CD-HIT v.4.6 (Fu et al., 2012) at the 99% similarity threshold. Mitochondrial sequences were filtered out using a custom script (https://github.com/sashulkaSh/worms/blob/main/script.sh).
Dataset Construction and Phylogenetic Analysis
Datasets of orthologous groups were constructed using two programs that employ different algorithms. ProteinOrtho v.5.16b (Lechner et al., 2011) was used to search for related proteins present in all studied transcriptomes with the 1e−30 e-value threshold. HaMStR v.13.2.6 (Ebersberger et al., 2009) detected only the proteins annotated in two reference genomes, Helobdella robusta and Capitella sp. Functional annotation of the predicted orthologous groups was performed by mapping the abovementioned Pfam and Swiss-Prot hits to the Gene Ontology (GO) categories using the built-in Swiss-Prot information and pfam2go data (http://www.geneontology.org/external2go/pfam2go). In both cases, only the proteins present in all 17 studied transcriptomes were retained in the final datasets.
The detected protein sequences were aligned by the MAFFT v.7.394 (Katoh and Standley, 2013). Gaps and ambiguously aligned portions of the alignment were discarded using the TrimAl v.1.4.rev22 (Capella-Gutiérrez et al., 2009) with the “strictplus” and “nogaps” options. Protein alignments shorter than 20 amino acids were removed. To avoid the impact of outliers, we discarded alignments with dissimilarities between any pair of sequences outside the upper 95th percentile. The rest were filtered using the PhyloTreePruner v.1.0 (Kocot et al., 2013) on the corresponding ML trees generated with the FastTreeMP v.2.1.7 (Price et al., 2010).
The obtained protein datasets were taken into phylogenetic analysis. We also created nucleotide datasets by extracting transcript data for each of the proteins from the datasets described above. For each gene, the transcripts were aligned by the MAFFT with the “-adjustdirection” option. When several isoforms were detected, the longest one was used. Alignments shorter than 30 bp were discarded.
For phylogenetic tree construction, the Maximum Likelihood (ML) analysis was performed in RAxML (Stamatakis, 2014). The most appropriate substitution models were selected based on the Bayesian information criterion as implemented in the MEGA X (Kumar et al., 2018): JTT for the ProteinOrtho amino acid dataset, JTT+I+G for the HaMStR amino acid dataset, and GTR+I+G for all nucleotide datasets. For each dataset, 1,000 bootstrap replicates were performed. Bayesian inference was done by the MrBayes v. 3.4 (Ronquist et al., 2012) with the same substitution models. For each dataset, 10,000,000 generations were run in two independent analyses from different random starting trees; 25% of generations were discarded as burn-in.
For automated species delimitation, the ABGD algorithm (Puillandre et al., 2012) was used. This analysis was run online at https://bioinfo.mnhn.fr/abi/public/abgd/. Another method used was the multi-rate Poisson Tree Process (Kapli et al., 2017; http://mptp.h-its.org/#/tree) with the ML phylogenetic tree as the input. The ancestral area reconstruction analysis was performed using the S-DIVA (Statistical dispersal-vicariance analysis) implemented in the RASP v. 4 (Yu et al., 2015, 2020).
Results
Transcriptome Sequencing
In this study, we performed low-depth Illumina singe-end transcriptomes for 12 specimens of E. nordenskioldi and outgroup species, and combined this data with five Ion Torrent transcriptomes from our earlier study (Shekhovtsov et al., 2019). The final dataset contained transcriptomes for 12 E. nordenskioldi lineages, as well as five outgroup species of the genus Eisenia. Sequence statistics on the studied transcriptomes is given in Table 1. The number of reads for different libraries ranged from 0.5 to 22 million reads. Sequences statistics are summarized in Table 1. De novo assembly by Trinity produced 21–144 thousand transcripts with N50 ranging from 500 to 1,000.
Two different algorithms were used to construct datasets for phylogenetic analysis. The algorithm implemented in ProteinOrtho found 287 common genes for the 17 specimens (212 after filtering). The final amino acid alignment contained 29,097 amino acids; nucleotide alignment contained 107,394 nucleotide positions. HaMStR detected 227 common genes, but only 91 remained after filtration. The resulting alignments contained 7,309 amino acids and 35,917 nucleotides, respectively.
The average pairwise distances between E. nordenskioldi lineages are provided in Supplementary Table 1. For the COI dataset, these distances ranged from 15 to 19%, negligibly lower than between them and E. andrei. For the ProteinOrtho nucleotide dataset, distances between the lineages were 2.5–5.5%, which was significantly lower than between them and E. andrei (8.2–8.6%).
Multigene Phylogeny of the E. nordenskioldi Complex
Phylogenetic trees constructed using both algorithms had similar topologies (Figure 3). The studied species of the genus Eisenia were divided into two groups, one containing European species [E. spelaea (Rosa, 1901) and E. andrei (Bouché, 1972)], and the other, specimens from Siberia [E. nordenskioldi, E. magnifica (Svetlov, 1957), E. tracta (Perel, 1985), and E. balatonica (Pop, 1943)]. E. tracta was found to be the sister species of E. nordenskioldi. E. nordenskioldi invariably split into two groups. One of those comprised E. n. nordenskioldi lineages 6, 7, and 9, and the other, all E. n. pallida lineages and the rest of E. n. nordenskioldi.
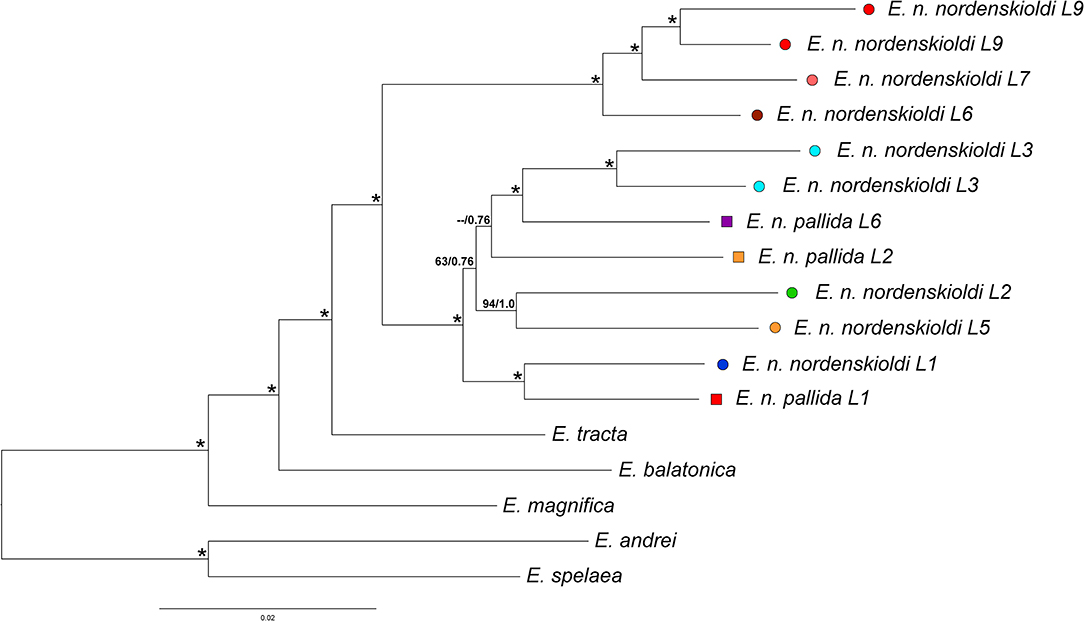
Figure 3. Phylogenetic tree for the nucleotide ProteinOrtho dataset constructed using the ML algorithm. Numbers near branches indicate ML bootstrap support/Bayesian posterior probabilities; asterisks stand for 100/1.0. Lineage symbols refer to Figures 1, 2, 4.
Within the second group, the recovered phylogenetic relationships were not completely resolved. E. n. nordenskioldi lineage 1 and E. n. pallida lineage 1 clustered together. Another groups that were invariably recovered were E. n. nordenskioldi lineages 2 and 5, and E. n. nordenskioldi lineage 3 and E. n. pallida lineage 6. However, relationships among those clades was unstable; E. n. nordenskioldi lineage 1/E. n. pallida lineage 1 was recovered as a sister group to the rest of the lineages, but with low statistical support (Figure 3).
E. nordenskioldi Haplotype Distribution and Automated Species Delimitation
We assembled a dataset containing 927 COI barcode sequences of E. nordenskioldi specimens. Of those, 252 were newly sequenced, and the rest was taken from previous studies (Shekhovtsov et al., 2013, 2015, 2016a,b, 2018a, 2020). Haplotypes of E. n. pallida lineage 5 were excluded from the analysis because it was demonstrated that this is probably a species of the Aporrectodea genus similar in morphology to E. n. pallida (Shekhovtsov et al., 2016a) and it did not group together with the rest of E. nordenskioldi on phylogenetic trees (not shown).
The final sample contained 373 full-length unique haplotypes. No indels were detected. Of the total 658 nucleotide positions, 284 were variable and 263 were parsimony-informative. Phylogenetic tree for this dataset (Figure 4) confirmed the division of E. nordenskioldi into two groups. One can also see that the majority of genetic lineages reported in earlier studies were detected, as well as several new ones. E. n. nordenskioldi lineages 7 and 9 are usually recovered as separate clades, but here, lineage 7 was nested within lineage 9. E. n. nordenskioldi lineage 1 and E. n. pallida lineage 1 also merged into a single clade.

Figure 4. Phylogenetic tree for the COI dataset constructed using the ML algorithm. Lineage symbols refer to Figures 1–3.
The obtained data was used to construct distribution maps. Short COI sequences, as well as those with poorly read fragments were assigned to one of the detected E. nordenskioldi lineages. This was also applied to the data from Blakemore (2013) and Hong and Csuzdi (2016). The sequences of E. n. mongol and E. n. onon (Blakemore, 2013) were nested within E. n. nordenskioldi lineage 1, close to certain haplotypes from the Buryatia Republic, while those of E. n. pallida from Korea (Hong and Csuzdi, 2016) formed a separate branch within Eisenia sp. 1 (not shown).
We attempted to divide E. nordenskioldi into putative species by automated species delimitation using various algorithms. The multi-rate Poisson tree processes analysis (Kapli et al., 2017) detected 77 putative species. The ABGD algorithm (Puillandre et al., 2012) suggested 29 to 46 species with the default parameters (relative gap width = 1.5X) and 8–12 species with the 4X relative gap width. Boundaries of these species were consistent with those drawn manually for E. nordenskioldi (Figure 4; Shekhovtsov et al., 2013, 2018a,b). Pairwise differences between the studied lineages for the COI gene are high, in most cases ranging from 15 to 19% (Supplementary Table 1).
The ancestral area reconstruction analysis (Supplementary Figure 1) suggested that the areas of the E. nordenskioldi complex and of the two main clades found within were located in the south of West Siberia, most probably in the Ob and Irtysh basins.
Discussion
Phylogeny of the E. nordenskioldi Complex
Phylogenomic analysis performed in this study allowed us to provide definitive answers to several issues. Although complete mitochondrial genomes suggested E. nordenskioldi to be paraphyletic with respect to E. tracta (Shekhovtsov et al., 2020), nuclear datasets unequivocally recover E. nordenskioldi as monophyletic. We also confirmed that neither E. n. nordenskioldi nor E. n. pallida are monophyletic.
E. nordenskioldi was found to be split into two large clades. One of those includes haplotypes belonging to the earlier recognized genetic lineages 6, 7, and 9 of E. n. nordenskioldi (Figure 3), and the other, to the rest of the specimens recognized as E. n. nordenskioldi, as well as those belonging to E. n. pallida. Comparison of the phylogenomic tree (Figure 3) and the COI tree (Figure 4) demonstrates that the phylogenomic datasets cover the majority of E. n. nordenskioldi diversity, and that most phylogenetic groupings coincide among those trees, although the COI tree provides far less resolution.
Current Delimitation of E. nordenskioldi
It was long known that E. nordenskioldi has extreme morphological, karyological, and ecological diversity (Malevich, 1956; Perel, 1979; Graphodatsky et al., 1982; Malinina and Perel, 1984; Viktorov, 1997; Shekhovtsov et al., 2017a). There were several attempts to split E. nordenskioldi into smaller taxa. The non-pigmented form was denoted as E. nordenskioldi forma pallida by Malevich (1956) and later on given the status of subspecies by Vsevolodova-Perel (1997). The obvious problem here is that these two forms are often sympatric (Vsevolodova-Perel and Leirikh, 2014), while subspecies have to be allopatric according to the conventional definition (Mayr, 1982). Vsevolodova-Perel probably felt that differences in pigmentation only without any other diagnostic characters did not justify the status of a separate species, so the recognition of E. n. pallida as a sympatric subspecies became established.
In this study, we found that E. n. pallida is unequivocally polyphyletic. Genetic lineages of E. n. pallida from different regions belong to different lineages and represent independent cases of pigmentation loss, probably as a consequence of the shift to the endogenic lifestyle.
Perel and Graphodatsky (1984) described two species, E. atlavinyteae and E. sibirica, earlier regarded as morphological forms slightly deviating from the typical diagnosis. E. atlavinyteae differed from E. nordenskioldi by the clitellum starting on the 26th segment, and by the tubercula pubertatis always present on the 28th segment. However, the authors noted that this arrangement was observed only in a portion of the type population, while in the rest, the position of these characters was close to those characteristic for E. nordenskioldi. E. sibirica had the clitellum spanning the segments 27–32, but again with significant intrapopulation variation overlapping the diagnosis of E. nordenskioldi (Perel and Graphodatsky, 1984).
These two newly described species were thus hard to distinguish from E. nordenskioldi in practice. In this study, we collected a set of individuals throughout the species' range identified as E. atlavinyteae from different locations. However, they did not form a separate clade and turned out to belong to lineages 1 and 3 of E. nordenskioldi. E. atlavinyteae, thus, does not represent a separate species and probably represents a morphological form that in some populations makes up the majority of individuals. Individuals identified as E. sibirica from a population from Western Siberia were found to belong to E. nordenskioldi lineage 2, along with those having the typical E. nordenskioldi diagnosis.
We can thus conclude that none of the major currently recognized taxa within E. nordenskioldi or related to it represent monophyletic clades: E. n. nordenskioldi is paraphyletic, E. n. pallida is polyphyletic, and E. atlavinyteae and E. sibirica probably represent morphological forms, not distinct species. The question on the latter two, however, cannot be considered as concluded without genetic investigation of type specimens.
Delimitation of the E. nordenskioldi Complex
As shown in the previous section, the current delimitation of the species complex is not consistent with its phylogeny. It is generally accepted that the newly described taxa should be monophyletic, however, criteria for species delimitation in earthworms are otherwise vague. One way to propose these putative species is to compare the extent of genetic variation among the detected clades and within them. Several algorithms of automated species delimitation exist. The two programs used in this study identifies from eight to as many as 77 species. One can see that the species' sampling is currently sufficiently dense (Figures 1, 2), so the observed deep split is not the result of sparse sampling of highly diverged haplotypes.
Although we could isolate these as new species or subspecies, splitting the species into so many new cryptic taxa would be highly impractical. We propose to divide E. nordenskioldi into two species. E. nordenskioldi was described by Eisen (1879) based on a set of populations from the Vaigach island and along the lower reaches of the Yenisei. Vaigach Island can be thus considered the type location. It is known to be inhabited by lineage 9, so we suggest that one of the detected clades including E. n. nordenskioldi lineages 6, 7, and 9 to be denoted as E. nordenskioldi.
The second clade on Figure 3 contained multiple clades corresponding to both pigmented and non-pigmented lineages. Phylogenetic relationships among them were only partially resolved, suggesting rapid diversification. Many of these clades have overlapping distributions and are found in sympatry (Figure 2) while being genetically distinct, and so might be viewed as distinct species. However, we suggest to isolate this whole clade as a putative new species, Eisenia sp. 1 (aff. E. nordenskioldi). Preliminary results of the morphological analysis suggest that E. nordenskioldi and Eisenia sp. 1 have some detectable differences (Golovanova et al., unpublished), which would allow a formal description of this species.
Conclusions
Phylogenomic data suggests that the E. nordenskioldi complex is split into two clades, one containing pigmented genetic lineages from the northern and western part of the distribution, and the second, both pigmented and non-pigmented ones mostly from the south and the southeast of the species' range. The existence of the two subspecies, E. n. nordenskioldi and E. n. pallida, as well as of the related species E. atlavinyteae and E. sibirica was not supported by the genetic analysis. We propose to split the E. nordenskioldi complex into two species corresponding to the abovementioned clades: the northern and western lineages as E. nordenskioldi, and the southern and southeastern ones, as Eisenia sp. 1 (aff. E. nordenskioldi).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA658219; https://www.ncbi.nlm.nih.gov/genbank/, MT863028–MT863089.
Author Contributions
SS designed the study. SS, GV, NB, and TS collected and identified the specimens. SS and TP extracted RNA. GV performed RNA sequencing. AS extracted sequence datasets. SS built phylogenetic trees, analyzed the data, and wrote the paper with input from AS, GV, and SP. All authors read and approved the final manuscript.
Funding
This study was financially supported by the Russian Fund for Basic Research (Grant No. 19-04-00661_a), ICG SB RAS Budget Project No. 0324-2019-0040-C-01 Genetic basis of biotechnology and bioinformatics, and Kurchatov Genomics Center of IC&G (075-15-2019-1662).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to D. I. Berman for his assistance in the collection of specimens and to N. I. Ershov for the help with bioinformatic analysis. Illumina sequencing was performed in the Center for Genome Studies of the Institute of Cytology and Genetics SB RAS.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.598196/full#supplementary-material
References
Blakemore, R. J. (2013). Earthworms newly from mongolia (Oligochaeta, Lumbricidae, Eisenia). Zookeys 285, 1–21. doi: 10.3897/zookeys.285.4502
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Ebersberger, I., Strauss, S., and von Haeseler, A. (2009). HaMStR: profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 9:157. doi: 10.1186/1471-2148-9-157
Eisen, G. (1879). On the Oligochaeta collected during the Swedish expeditions to the Arctic regions in the years 1870, 1875 and 1876. K. Sven. Vet. Akad. Forh. 15, 1–49.
Folmer, O., Hoeh, W. R., Black, M. B., and Vrijenhoek, R. C. (1994). Conserved primers for PCR amplification of mitochondrial DNA from different invertebrate phyla. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Graphodatsky, A. S., Perel, T. S., and Radzhabli, S. I. (1982). Chromosome sets of two forms of eisenia nordenskioldi (Eisen) (Oligochaeta, Lumbricidae). Dokl. AN SSSR 262, 1514–1516.
Hebert, P. D. N., Ratnasingham, S., and De Waard, J. R. (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. London. Ser. B Biol. Sci. 270, S96–S99. doi: 10.1098/rsbl.2003.0025
Hong, Y., and Csuzdi, C. (2016). New data to the earthworm fauna of the Korean Peninsula with redescription of Eisenia koreana (Zicsi) and remarks on the Eisenia nordenskioldi species group (Oligochaeta, Lumbricidae). Zool. Stud. 55:e12. doi: 10.6620/ZS.2016.55-12
Kapli, P., Lutteropp, S., Zhang, J., Kobert, K., Pavlidis, P., Stamatakis, A., et al. (2017). Multi-rate poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33, 1630–1638. doi: 10.1093/bioinformatics/btx025
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
King, R. A., Tibble, A. L., and Symondson, W. O. C. (2008). Opening a can of worms: unprecedented sympatric cryptic diversity within British lumbricid earthworms. Mol. Ecol. 17, 4684–4698. doi: 10.1111/j.1365-294X.2008.03931.x
Kocot, K. M., Citarella, M. R., Moroz, L. L., and Halanych, K. M. (2013). PhyloTreePruner: a phylogenetic tree-based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinform. 9:EBO-S12813. doi: 10.4137/EBO.S12813
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lechner, M., Findeiß, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics 12:124. doi: 10.1186/1471-2105-12-124
Malevich, I. I. (1956). On the study of earthworms in the Far East. Uch. Zap. Mosk. Gos. Ped. Inst. Im. Potemkina 61, 439–449.
Malinina, T. V., and Perel, T. S. (1984). Characteristic of chromosome race of Eisenia nordenskioldi (Oligochaeta, Lumbricidae) using biochemical markers. Dokl. Akad. Nauk SSSR 279:1265.
Marchán, D. F., Cosín, D. J. D., and Novo, M. (2018). Why are we blind to cryptic species? Lessons from the eyeless. Eur. J. Soil Biol. 86, 49–51. doi: 10.1016/j.ejsobi.2018.03.004
Martinsson, S., and Erséus, C. (2017). Cryptic speciation and limited hybridization within Lumbricus earthworms (Clitellata: Lumbricidae). Mol. Phylogenet. Evol. 106, 18–27. doi: 10.1016/j.ympev.2016.09.011
Perel, T. S. (1979). Range and Regularities in the Distribution of Earthworms of the USSR Fauna. Moscow: Nauka.
Perel, T. S., and Graphodatsky, A. S. (1984). New species of the genus Eisenia (Lumbricidae, Oligochaeta) and their chromosome sets. Zool. Zhurnal 63, 610–612.
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE. 5:e9490. doi: 10.1371/journal.pone.0009490
Puillandre, N., Lambert, A., Brouillet, S., and Achaz, G. (2012). ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Shekhovtsov, S. V., Bazarova, N. E., Berman, D. I., Bulakhova, N. A., Golovanova, E. V., Konyaev, S. V., et al. (2017b). DNA barcoding: how many earthworm species are there in the south of West Siberia? Russ. J. Genet. Appl. Res. 7, 57–62. doi: 10.1134/S2079059717010130
Shekhovtsov, S. V., Berman, D. I., Bazarova, N. E., Bulakhova, N. A., Porco, D., and Peltek, S. E. (2016a). Cryptic genetic lineages in Eisenia nordenskioldi pallida (Oligochaeta, Lumbricidae). Eur. J. Soil Biol. 75, 151–156. doi: 10.1016/j.ejsobi.2016.06.004
Shekhovtsov, S. V., Berman, D. I., Bulakhova, N. A., Makarova, O. L., and Peltek, S. E. (2018b). Phylogeography of earthworms from high latitudes of Eurasia. Acta Zool. Acad. Sci. Hungaricae 64, 369–382. doi: 10.17109/AZH.64.4.369.2018
Shekhovtsov, S. V., Berman, D. I., Bulakhova, N. A., Vinokurov, N. N., and Peltek, S. E. (2018a). Phylogeography of Eisenia nordenskioldi nordenskioldi (Lumbricidae, Oligochaeta) from the north of Asia. Polar Biol. 41, 237–247. doi: 10.1007/s00300-017-2184-2
Shekhovtsov, S. V., Berman, D. I., Golovanova, E. V., and Peltek, S. E. (2017a). Genetic diversity of the earthworm Eisenia nordenskioldi (Lumbricidae, Annelida). Vavilovskii Zhurnal Genet. Selektsii 21, 588–595. doi: 10.18699/VJ17.24-o
Shekhovtsov, S. V., Berman, D. I., and Peltek, S. E. (2015). Phylogeography of the earthworm Eisenia nordenskioldi nordenskioldi (Lumbricidae, Oligochaeta) in northeastern Eurasia. Dokl. Biol. Sci. 461, 85–88. doi: 10.1134/S0012496615020039
Shekhovtsov, S. V., Ershov, N. I., Vasiliev, G. V., and Peltek, S. E. (2019). Transcriptomic analysis confirms differences among nuclear genomes of cryptic earthworm lineages living in sympatry. BMC Evol. Biol. 19:50. doi: 10.1186/s12862-019-1370-y
Shekhovtsov, S. V., Golovanova, E. V., Ershov, N. I., Poluboyarova, T. V., Berman, D. I., Bulakhova, N. A., et al. (2020). Phylogeny of the Eisenia nordenskioldi complex based on mitochondrial genomes. Eur. J. Soil Biol. 96:103137. doi: 10.1016/j.ejsobi.2019.103137
Shekhovtsov, S. V., Golovanova, E. V., and Peltek, S. E. (2013). Cryptic diversity within the nordenskiold's earthworm, Eisenia nordenskioldi subsp. nordenskioldi (Lumbricidae, Annelida). Eur. J. Soil Biol. 58, 13–18. doi: 10.1016/j.ejsobi.2013.05.004
Shekhovtsov, S. V., Golovanova, E. V., and Peltek, S. E. (2016b). Mitochondrial DNA variation in Eisenia n. nordenskioldi (Lumbricidae) in Europe and southern urals. Mitochondrial DNA A 27, 4643–4645. doi: 10.3109/19401736.2015.1101594
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Viktorov, A. G. (1997). Diversity of polyploid races in the family lumbricidae. Soil Biol. Biochem. 29, 217–221. doi: 10.1016/S0038-0717(96)00086-7
Vsevolodova-Perel, T. S., and Bulatova, N. S. (2008). Polyploid races of earthworms (Lumbricidae, Oligochaeta) in the East European plain and Siberia. Biol. Bull. 35, 385–388. doi: 10.1134/S1062359008040092
Vsevolodova-Perel, T. S., and Leirikh, A. N. (2014). Distribution and ecology of the earthworm Eisenia nordenskioldi pallida (Oligochaeta, Lumbricidae) dominant in Southern Siberia and the Russian far east. Zool. Zhurnal 93, 45–52. doi: 10.1134/S0013873814040034
Yu, Y., Blair, C., and He, X. (2020). RASP 4: Ancestral state reconstruction tool for multiple genes and characters. Mol. Biol. Evol. 37, 604–606. doi: 10.1093/molbev/msz257
Keywords: phylogenomics, phylogeny, transcriptomes, Lumbricidae, Eisenia nordenskioldi, earthworms
Citation: Shekhovtsov SV, Shipova AA, Poluboyarova TV, Vasiliev GV, Golovanova EV, Geraskina AP, Bulakhova NA, Szederjesi T and Peltek SE (2020) Species Delimitation of the Eisenia nordenskioldi Complex (Oligochaeta, Lumbricidae) Using Transcriptomic Data. Front. Genet. 11:598196. doi: 10.3389/fgene.2020.598196
Received: 27 August 2020; Accepted: 09 November 2020;
Published: 07 December 2020.
Edited by:
Muniyandi Nagarajan, Central University of Kerala, IndiaReviewed by:
Svante Martinsson, University of Gothenburg, SwedenSamuel James, Maharishi University of Management, United States
Copyright © 2020 Shekhovtsov, Shipova, Poluboyarova, Vasiliev, Golovanova, Geraskina, Bulakhova, Szederjesi and Peltek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergei V. Shekhovtsov, c2hla2hvdnRzb3ZAYmlvbmV0Lm5zYy5ydQ==