 Neus Font-Porterias
Neus Font-Porterias Aaron Giménez2
Aaron Giménez2 Francesc Calafell
Francesc Calafell David Comas
David Comas- 1Departament de Ciències Experimentals i de la Salut, Institut de Biologia Evolutiva (UPF-CSIC), Universitat Pompeu Fabra, Barcelona, Spain
- 2Facultat de Sociologia, Universitat Autònoma de Barcelona, Barcelona, Spain
- 3Facultat de Geografia i Història, Universitat de Barcelona, Barcelona, Spain
Genetic patterns of inter-population variation are a result of different demographic and adaptive histories, which gradually shape the frequency distribution of the variants. However, the study of clinically relevant mutations has a Eurocentric bias. The Romani, the largest transnational minority ethnic group in Europe, originated in South Asia and received extensive gene flow from West Eurasia. Most medical genetic studies have only explored founder mutations related to Mendelian disorders in this population. Here we analyze exome sequences and genome-wide array data of 89 healthy Spanish Roma individuals to study complex traits and disease. We apply a different framework and focus on variants with both increased and decreased allele frequencies, taking into account their local ancestry. We report several OMIM traits enriched for genes with deleterious variants showing increased frequencies in Roma or in non-Roma (e.g., obesity is enriched in Roma, with an associated variant linked to South Asian ancestry; while non-insulin dependent diabetes is enriched in non-Roma Europeans). In addition, previously reported pathogenic variants also show differences among populations, where some variants segregating at low frequency in non-Roma are virtually absent in the Roma. Lastly, we describe frequency changes in drug-response variation, where many of the variants increased in Roma are clinically associated with metabolic and cardiovascular-related drugs. These results suggest that clinically relevant variation in Roma cannot only be characterized in terms of founder mutations. Instead, we observe frequency differences compared to non-Roma: some variants are absent, while other have drifted to higher frequencies. As a result of the admixture events, these clinically damaging variants can be traced back to both European and South Asian-related ancestries. This can be attributed to a different prevalence of some genetic disorders or to the fact that genetic susceptibility variants are mostly studied in populations of European descent, and can differ in individuals with different ancestries.
Introduction
Human genetic diversity is a continuum, which means that there are no fixed, immutable or discrete boundaries between populations. Linguistic, geographic and social factors can lead to different demographic histories and, in turn, to patterns of inter-population variability (Lao et al., 2008; Barbujani et al., 2013; Batai et al., 2020; Heyer and Reynaud-Paligot, 2020). It has been previously reported that this stratification has consequences in the genetics of complex traits and diseases [reviewed in Bentley et al. (2017); Sirugo et al. (2019)]. On the one hand, disease-associated variants present allele frequency differences across populations. For example, 70% of the cystic fibrosis cases in Europeans are due to ΔF508 mutation in CFTR gene, while the most common causal variant in South Africans with African ancestry is 3120 + 1G→A, and different mutations have different therapeutic targets (Padoa et al., 1999; Stewart and Pepper, 2017; Sirugo et al., 2019). On the other hand, genomic variation across populations is also observed for treatment response differences, especially in genes related to absorption, distribution, metabolism, and excretion (ADME) of drugs (Dopazo et al., 2016; Škarić-Jurić et al., 2018; Sirugo et al., 2019). For example, the metabolism of the anticoagulant warfarin can differ due to several genetic polymorphisms; however, their frequencies are different in European and African descent groups, which challenges the correct dosage prescription (Bress et al., 2012; Johnson et al., 2017; Sirugo et al., 2019).
Thus, an accurate clinical assessment relies on the study of clinically relevant genetic variants with different allele frequencies across groups. Yet, there is an underrepresentation of human populations in the screening of these variants. Particularly, genetic studies show a strong and systematic Eurocentric bias (Need and Goldstein, 2009; Popejoy and Fullerton, 2016; Bentley et al., 2017; Martin et al., 2019; Sirugo et al., 2019). As a consequence, this bias prevents to fully understand the genetic architecture of human disease and leads to an incomplete genetic assessment of complex traits, and to an inaccurate disease diagnosis and treatment in under-represented groups (Martin et al., 2019; Sirugo et al., 2019).
The Roma population, also known by the misnomer of “Gypsies,” has been under-represented in these genome-wide scans. They constitute the largest transnational minority ethnic group in Europe (Council of Europe, 2012). Linguistic and genetic evidence point to a South Asian origin and subsequent diaspora toward Europe, with extensive non-Roma gene flow and multiple founder effects shaping their demographic history (Hancock, 2002; Matras, 2002; Mendizabal et al., 2012; Moorjani et al., 2013; Morar et al., 2013; Font-Porterias et al., 2019). Most medical genetic studies on this population have been focused on targeting the genetic variants responsible for the increased prevalence of certain genetic diseases [reviewed in Kalaydjieva et al. (2001); Morar et al. (2013)]. In this sense, several founder mutations have been identified: e.g., the p.R299X mutation in the LTBP2 gene, which is responsible for congenital glaucoma (Azmanov et al., 2011). However, the distribution of disease-associated variants in this population has not been fully characterized. In addition, drug response-related genome-wide variation has only been deeply examined in Croatian Roma, where variants in ADME genes were found to have increased allele frequencies (Škarić-Jurić et al., 2018).
To fill this gap, we examine whole exome sequences (WES) and genome-wide array data of 89 healthy Spanish Roma individuals and characterize the functionally relevant genomic variants (i.e., associated to disease or to drug response) with either increased or decreased allele frequencies in the Roma. Beyond frequency distribution differences, and taking into account that Roma is an admixed population, we describe the ancestral origin of multiple variants by leveraging on the estimated local ancestry of their background haplotypes.
Materials and Methods
Data
We used WES (mean depth of 54X), and genome-wide autosomal SNP data (Affymetrix Axiom Genome-Wide Human Origins 1 array) for Spanish Roma individuals (89 and 62 samples, respectively) (Font-Porterias et al., 2021), deposited at EGA (EGAS00001004599). The Spanish Roma WES were merged with previously published non-Roma WES from 1000G (mean depth of 65.7X): Iberian Population in Spain (IBS), Toscani in Italia (TSI), Punjabi from Lahore (PJL), Indian Telugu from the United Kingdom (ITU), and Gujarati Indian from Houston (GIH) (Auton et al., 2015), resulting in a dataset with 512 individuals and 410,225 variants. The genome-wide SNP data was merged with IBS, TSI, PJL, ITU, and GIH from 1000G (1000 Genomes Project Consortium, 2012), with 474,632 genome-wide SNPs in 487 samples. Both datasets were then combined to increase the covered genomic variants, building a merged WES-array dataset with 487 individuals and 878,162 SNPs. Variant annotation was performed using the Variant Effect Predictor tool (VEP) from Ensembl (McLaren et al., 2016) focusing on three deleterious prediction scores: PolyPhen-2 (Adzhubei et al., 2010), GERP (Davydov et al., 2010) and CADD (Rentzsch et al., 2019), as previously explained (Font-Porterias et al., 2021). For each analysis, the corresponding dataset used is specified (WES dataset or merged WES-array dataset). In addition, we have included a glossary of terms that may have ambiguous meanings in genetic studies (Supplementary Note 1).
Local Ancestry Inference
The phasing of the merged WES-array dataset, with 405,814 variants with minor allele frequency (MAF) > 1%, was performed using SHAPEIT (O’Connell et al., 2014), using the population-averaged genetic map from the HapMap phase II (International HapMap Consortium, 2003) and the 1000G dataset as a reference panel (1000 Genomes Project Consortium, 2012). RFMix v1.5.4 (Maples et al., 2013) was run with one expectation-maximization (EM) iteration to infer the local ancestry of the phased haplotypes, using balanced reference panels representing European (IBS and TSI populations) and South Asian (PJL, GIH, and ITU populations) ancestries. As previously explained (Font-Porterias et al., 2021), the Roma individuals included in the present study show, on average, 68.4% and 31.6% of European and South Asian global ancestry proportions, with a standard deviation of 7%. Ancestry was assigned when RFMix posterior probability was higher than 0.9, resulting in 96.3% of the variants with assigned ancestry. In order to match the local ancestry inference in heterozygous variants and obtain the ancestry background of the allele, we adjusted the RFMix rephasing as previously performed (Browning et al., 2018), since RFMix partially rephases the data when assigning local ancestry. However, when the variant was filtered out (MAF < 1%) in the phasing, only genotype ancestries can be retrieved.
Genetic Portability
We computed the allele sharing ratio (proportion of variants at different frequency bins) from the Roma segregating in non-Roma populations from WES variants dataset. In addition, we compared the linkage disequilibrium (LD) decay patterns between Roma and non-Roma from the genome-wide array dataset using PopLDdecay (Zhang et al., 2018) with default parameters. We performed a two-sample Kolmogorov–Smirnov test to check whether the decay distributions of Roma and non-Roma groups were statistically different. For both analyses, we used the same number of individuals per population to avoid sample size biases (70 individuals per population for the allele sharing ratio and 62 individuals per population for the LD decay).
Gene Enrichment Analyses
Using the WES dataset, we performed two different gene enrichment analyses using WEB-based GEne SeT AnaLysis Toolkit (Liao et al., 2019) to identify categories (or classes) of genes that are over-represented in a particular set of genes, using a background gene set. First, we interrogated those genes with more deleterious mutations (i.e., deleterious Nalleles or Nhom per individual per gene). Although we expect that the most constrained genes (i.e., lower values of deleterious Nalleles or Nhom per individual per gene) will be shared across populations, we test whether there is a particular pathway enriched in the most mutated genes in the Roma samples and the non-Roma groups, independently. To do so, we normalized Nalleles or Nhom by the number of variants in each gene and we then examined the correlation between each pair of Roma to non-Roma populations per each gene. The over-representation analysis was performed with default parameters (Liao et al., 2019), Gene Ontology (GO) was selected as the functional database, and all the genes included in our variants set was used as background gene set. This analysis does not take into account the frequency of the variants, since the calculation is performed per individual. We also performed a gene enrichment analysis to test whether the genes with deleterious variants showing allele frequency increases in Roma to non-Roma (or non-Roma to Roma) belong to specific genetic disease clusters. We included in the analysis those genes with variants with a fold increase in minor allele frequency (MAF) > 5 or fold increase in minor allele count (MAC) > 5 for monomorphic variants. By using this restrictive threshold, we decrease the number of false positive results in the enrichment analyses, although we may lose some pathogenic variants found at low frequencies (and for that reason, we then focus on ClinVar pathogenic variants with less conservative thresholds). The same number of individuals were considered for this analysis (70 individuals per population) to avoid biases in MAC and MAF calculations. The over-representation analysis was performed with default parameters (Liao et al., 2019), OMIM was selected as the functional disease database and all the genes included in our variants set was used as background gene set. Once the enriched pathways were identified, we computed a chi-squared test to check whether the associated genetic variants described in OMIM present in our dataset have statistically different genotype frequencies between Roma and non-Roma groups.
Screening of Known Disease-Associated Variants
We first identified previously reported Mendelian mutations in the Roma, annotated in Bianco et al. (2020) and checked the ancestry of their haplotypes to trace their putative origin. However, this approach does not allow us to examine if there is a different frequency spectrum of disease-associated variants comparing Roma and non-Roma. To that end, we then annotated the set of WES variants using ClinVar database (Landrum et al., 2014) and compared the frequency of clinically validated variants among populations. We kept only variants with a clinical significance of “pathogenic,” which is the highest level of supported evidence. We selected those variants with a fold increase in risk allele frequency (RAF) > 1.5 between populations or 1.5% RAF for monomorphic variants. A chi-squared test was performed to test whether the genotype frequencies were significantly different across populations. The same number of individuals were considered for this analysis (70 individuals per population) to avoid biases in RAF calculations.
Screening Beyond Disease-Associated Variants
To examine pharmacogenetic variation in the WES dataset, we studied mutations that disrupt drug binding domains without being deleterious to the protein. These variants are based on function prediction: they might cause drug binding inhibition; however, not all variants have a reported association with drug response (Hopkins and Groom, 2002; Dopazo et al., 2016). We also examined variants found in 31 core ADME genes1 (Škarić-Jurić et al., 2018). We selected those variants with a fold increase in MAF > 1.5 between populations or 1.5% MAF for monomorphic variants. A chi-squared test was performed to test whether the genotype frequency was significantly different across populations. The same number of individuals were considered for this analysis (70 individuals per population) to avoid biases in MAF calculations. The selected variants in both analyses were searched in PharmGKB (Whirl-Carrillo et al., 2012) and the corresponding target drugs in DrugBank (Wishart et al., 2018) and PubChem (Kim et al., 2019).
Results
Initial Assessment of Functional Variants as a First Evidence of Inter-Population Variability
In order to assess the genetic portability from non-Roma to Roma, we examined the allele sharing and linkage disequilibrium patterns between populations. We have previously shown that Roma exhibit a considerable amount of private variants; however, their proportion is lower than in other populations: 15,287 population-specific variants in Roma; 25,060 in IBS; 24,158 in TSI; 21,160 in PJL; 22,040 in GIH; and 24,070 in ITU (Font-Porterias et al., 2021). In addition, allele sharing is high for common variants (MAF > 5%) (over 86% of Roma variants are present in non-Roma) (Figure 1). However, for rare variants (MAF < 5%), the allele sharing is around 30–40% (Figure 1). Regarding linkage disequilibrium, decay patterns are not statistically different between Roma and non-Roma (Supplementary Figure 1, p-value > 0.95). The total number of deleterious alleles per individual is similar among Roma and non-Roma groups (Font-Porterias et al., 2021), and here we further show that the genomic distribution of accumulation of deleterious mutations (i.e., number of deleterious alleles per gene per individual) has comparable patterns between populations (Supplementary Note 2, Supplementary Figures 2, 3, and Supplementary Tables 1, 2). Thus, the overall allele sharing and linkage disequilibrium patterns are comparable among populations. However, rare and private variants can present some challenges in the genetic characterization of Roma population, especially for those variants with a frequency lower than 5%.
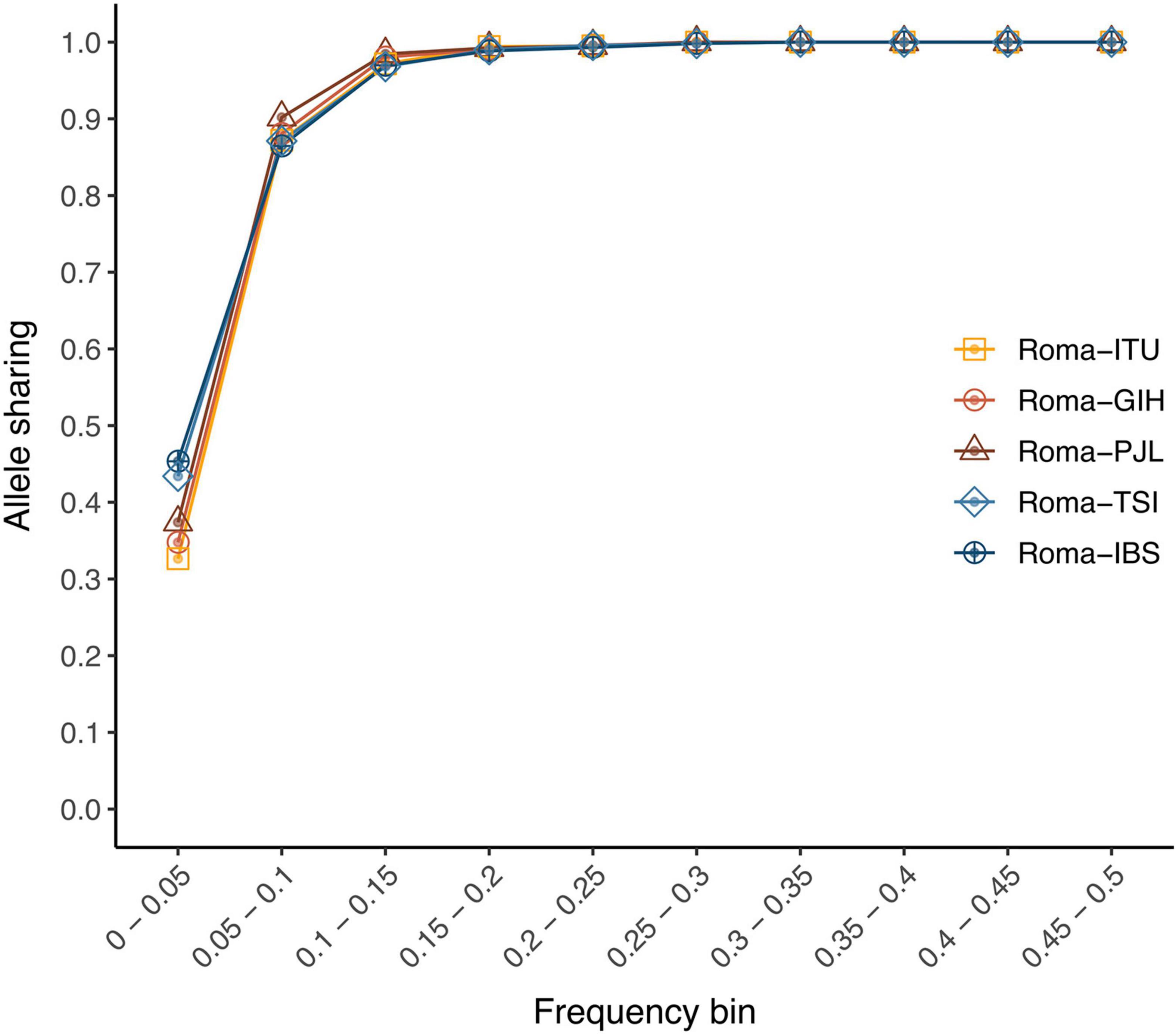
Figure 1. Allele-sharing ratios among Roma and non-Roma groups. Proportion of Roma variants from each minor allele frequency bin (from >0% to 50%) which are also segregating at each non-Roma population.
As shown in a previous study, Roma and non-Roma groups exhibit differences in the site frequency spectrum due to different demographic histories (Font-Porterias et al., 2021). An overrepresentation analysis including those genes with different allele frequency variants points to a differential OMIM trait enrichment in non-Roma and Roma (Supplementary Note 3 and Supplementary Tables 1, 3). Regarding rare conditions, the genes related to non-Herlitz type junctional epidermolysis bullosa are enriched when comparing deleterious variants with higher frequency in Roma than non-Roma. The prevalence of this disease in Roma is higher than non-Roma in Spain (Martinez-Frias and Bermejo, 1992), although none of the pathogenic variants described in OMIM are present in our dataset and only benign ones show increased frequencies (Supplementary Table 4). In addition, the tetralogy of Fallot is enriched in non-Roma (Table 1), with two pathogenic variants in GATA4 gene (rs56208331 and rs115099192) with increased frequencies in South Asian groups (Supplementary Table 4). This is consistent with the higher prevalence of this condition in Asia (Takkenberg and Roos-hesselink, 2011).
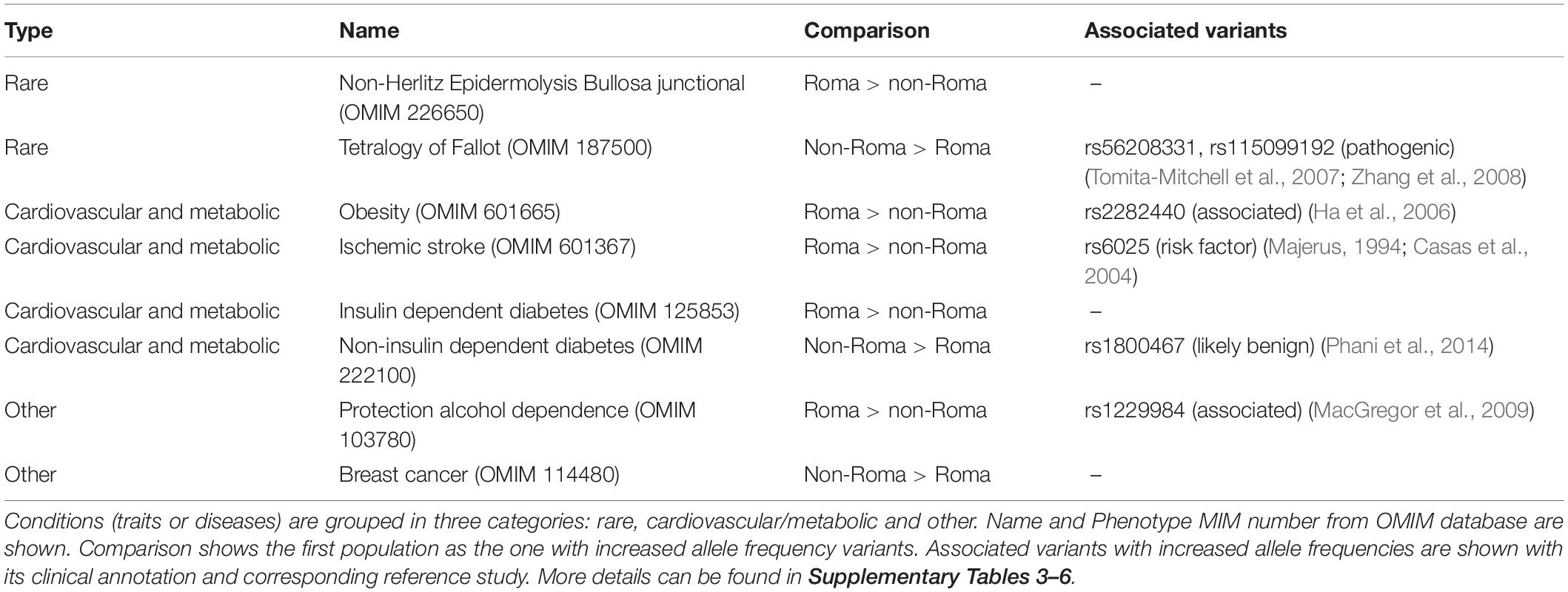
Table 1. Summary results of the overrepresentation analysis.
Multiple cardiovascular and metabolic disorders are also present in the gene overrepresentation analysis. Obesity is enriched comparing Roma with non-Roma, especially with Europeans. One variant in the SDC3 gene (rs2282440), which is associated with obesity in Asians (Table 1) has a significantly increased frequency in Roma (19%) and it is virtually absent in other European groups (Supplementary Tables 4, 5). All risk alleles of this variant in the Roma have South Asian ancestry (Supplementary Table 6), suggesting that this allele in the Roma has a South Asian origin. In addition, genes related to ischemic stroke are overrepresented in Roma (Table 1). One variant in the F5 gene, annotated as a risk factor in Europeans (rs6025), has significantly higher frequencies in Roma (8.6%) compared to all non-Roma (<1.5%) (Supplementary Tables 4, 5). This variant has a European ancestry background for 7 out of 8 risk alleles found (Supplementary Table 6), suggesting a European origin of this variant in the Roma. Lastly, both types of diabetes are enriched in our analysis: non-insulin dependent diabetes is enriched in non-Roma, while insulin dependent diabetes is overrepresented in Roma compared to non-Roma (Table 1). These results are consistent with previous literature (Mendizabal et al., 2013; Werissa et al., 2019), although our dataset does not include none of the pathogenic or risk-factor variants (more details in Supplementary Note 3).
Other conditions such as protection to alcohol dependence are enriched in Roma compared to non-Roma (Table 1). One variant in the ADH1B gene reported to be protective for alcohol dependence (rs1229984) shows a significantly higher allele frequency in Roma without a clear ancestry origin (Supplementary Tables 5, 6). The enrichment for breast cancer in non-Roma (Table 1) is due to variants with increased allele frequencies in ATM, BRCA2, and NQO2 genes. Roma show higher prevalence of triple negative (TN) breast tumors (Reckova et al., 2017), and mutations in the enriched genes are mostly linked to non-TN types (Peshkin et al., 2010; Lin et al., 2016; Decker et al., 2017; Slavin et al., 2017; Sirisena et al., 2018).
These results show multiple OMIM traits enriched for genes with a 5-fold increased allele frequency deleterious variants both in Roma and in non-Roma. Although this is an exploratory approach, it is consistent with previous literature and reports the first evidence that Roma do not show a systematically increased genetic susceptibility to disease.
A Genome-Wide Screening Does Not Support an Increased Susceptibility for Genetic Disorders
The presence of mutations responsible for particular Mendelian disorders in the Roma has been reported in several studies (see (Kalaydjieva et al., 2001; Álvarez et al., 2005; Bouwer et al., 2007; Gamella et al., 2013; Sevilla et al., 2013; Rocha et al., 2014; Cabrera-Serrano et al., 2018) among others). From these previously reported mutations, we found seven variants in our dataset (Table 2). Only in two out of seven variants, the risk allele is present in non-Roma populations: rs1801968 (chr9:132580901) and rs1126809 (chr11:89017961) (Table 2). The risk allele of the latter has a higher frequency in Europeans than South Asians (Table 2) and, in the Roma, it has a European assigned ancestry in 17 out of 19 alleles (Supplementary Table 7), suggesting that this variant (responsible for Oculocutaneous albinism) in the Roma has a European origin, as previously identified (Bianco et al., 2020). In addition, two variants responsible for Charcot–Marie–Tooth disease (rs119483085; chr8:134270617 and rs80338934; chr5:148389835) (Table 2) both have one risk allele with European ancestry (Supplementary Table 7). On the contrary, rs77931234 (chr1:76226846; Acetyl-coA dehydrogenase deficiency) and rs104894396 (chr13:20763650; Deafness) appear to have a South Asian origin (Supplementary Table 7). In fact, rs104894396 risk allele is also present in PJL population (Table 2), which is consistent with a South Asian origin. This variant, only present in Roma and Punjabi individuals in our dataset, is a non-synonymous mutation (W42X) responsible for autosomal recessive non-syndromic hearing loss. It has been previously reported in Spanish and Slovak Roma individuals and an Indian origin has been suggested (Minárik et al., 2003; Álvarez et al., 2005), which is congruent with the South Asian ancestry assignation of the risk allele.
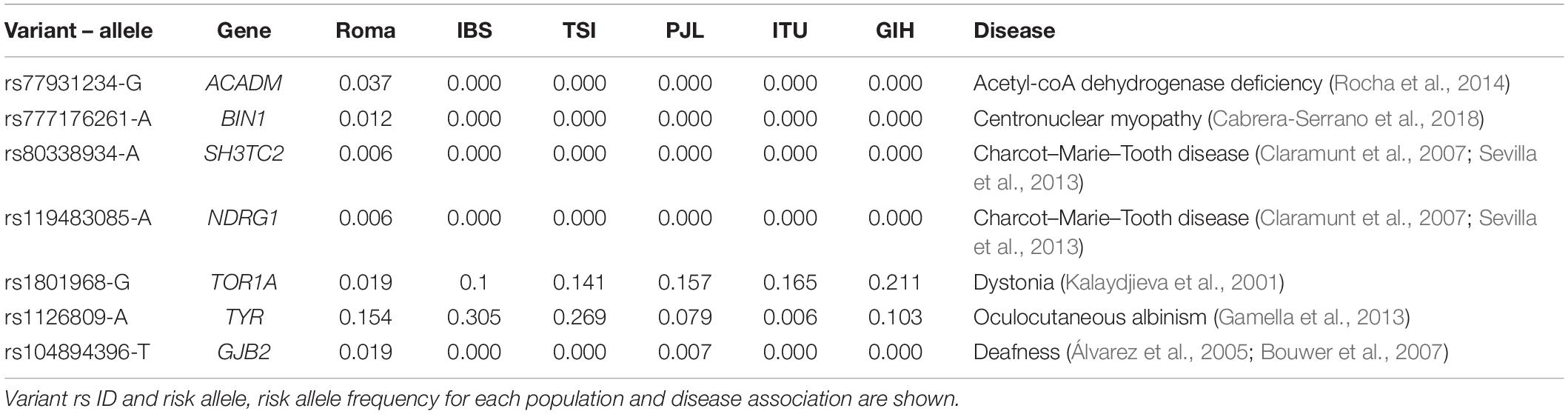
Table 2. List of previously reported Mendelian mutations in Roma present in this study.
We next investigated the 334 pathogenic variants described in the ClinVar database present in our dataset. In Roma, we found 60 out of 334 variants segregating at low RAF in the population. Only 27 variants have a RAF difference equal or higher than 1.5 comparing Roma and non-Roma (Supplementary Table 8). Although the RAF of these variants is low (below 5% in most cases), there are variants with increased frequency in Roma, but interestingly, we observe disease-associated variants with increased frequencies in European and South Asian non-Roma populations. For example, rs1799807 (chr 3:165548529) is a missense pathogenic variant causing the deficiency of butyrylcholine esterase and, consequently, postanesthetic apnea (McGuire et al., 1989; Jasiecki et al., 2019). This variant is only present in European populations and virtually absent in South Asia and Roma (Supplementary Table 8). On the contrary, rs137941190 (chr 11:126215441) is a missense pathogenic variant for Al-Raqad syndrome, described in Pakistani patients (Ahmed et al., 2014). The risk allele of this variant is absent in the European exomes, but it appears at low frequencies in the Roma and South Asian samples (Supplementary Table 8), although the genotype frequencies are not statistically different (Supplementary Table 9). Regarding the ancestry inference, European ancestry is assigned for the risk allele of most of these variants (Supplementary Table 10), except for rs104894396 (chr 13:20763650), as explained above.
The screening of known disease-associated variants together with the local ancestry inference has allowed to report both the presence and the absence of particular mutations in the Roma and to trace their most likely ancestral origin. However, most known clinically relevant variants have been discovered in European populations, which leads to an ascertainment bias that can weaken the results when studying the Roma population: out of the 334 pathogenic variants, less than 45 variants are segregating in South Asian populations (43 in PJL, 41 in GIH, and 31 in ITU), while there are 84 and 74 segregating in IBS and TSI, respectively.
Many Drug-Response Variants in Roma Are Related to Metabolic and Cardiovascular Disorders
Besides disease-associated variants, other functionally relevant mutations (e.g., pharmacogenomic variants) exhibit inter-population genetic variation in the human genome, as mentioned above. Regarding drug binding domains, we identified 101 variants in our dataset that disrupt the domains without being deleterious for the protein. This set is less biased toward European genetic variation, since it is based on impact prediction, rather than on previously discovered genetic associations (Dopazo et al., 2016). Only 26 variants were found to have a MAF fold increase ≥1.5 comparing Roma and non-Roma (Supplementary Table 11). Variants with known association drug phenotypes reported in PharmGKB (Whirl-Carrillo et al., 2012) have higher MAF in European populations, showing the European-centric bias in biomedical genetic studies. For example, the rs5918 variant, located in the ITGB3 gene (chr17:45360730), reduces the efficacy of aspirin and clopidogrel (Dropinski et al., 2007; Motovska et al., 2009) (indicated for coronary artery disease and myocardial infarction) and it shows a higher allele frequency in IBS and TSI than in Roma (Supplementary Table 11). A variant found in the GLP1R gene (rs6923761; chr6:39034072) reduces the treatment efficacy for obesity and type II diabetes (i.e., sitagliptin, vildagliptin, and liraglutide) (Javorský et al., 2016) and its frequency is significantly lower in Roma than in the tested European populations (Supplementary Table 11).
On the contrary, there are 13 variants with increased MAF in Roma (Supplementary Table 11) with significantly different genotype frequencies compared to non-Roma groups (Supplementary Table 12). For example, there is a variant in the CRYZ gene (chr1:75175886) with significantly higher MAF in Roma and South Asians than in Europeans (Supplementary Table 11). Its protein contains a drug binding domain for dicumarol, indicated for deep vein thrombosis (Supplementary Table 11) and 19 out of 27 minor alleles in Roma have South Asian ancestry (Supplementary Table 13). In the PTPRE gene drug binding domain for alendronate (indicated for osteoporosis) (Supplementary Table 11), we identify a variant (chr10:129868686) in Roma (3.7%), virtually absent in non-Roma, except GIH (0.5%) (Supplementary Table 11). Four Roma individuals were heterozygotes for this variant: one with both haplotypes assigned to South Asian ancestry and three with one European and one South Asian haplotype (Supplementary Table 13), which suggests that this variant in the Roma originated in South Asia. Regarding the ancestry inference and besides the mentioned examples, many of the variants have the minor allele with European ancestry: 72% of the minor alleles are assigned to a European-related ancestry; slightly above the mean genome-wide ancestry (68.4%), but within the first SD of the distribution. Although experimental evidence suggesting these variants affect the binding of these drugs is lacking, follow-up studies should be performed to validate the functional impact of these variants.
We next examined previously described variants in ADME genes. In our dataset, 14 out of 95 of them show increased MAF with a fold change equal or higher than 1.5 comparing Roma and non-Roma (Table 3). Some variants found in European groups are absent in the Roma exomes: e.g., rs34130495, which modifies the metabolism of tramadol (indicated for mild-to-moderate pain) (Table 3). However, many of the variants with increased frequencies in Spanish Roma are clinically associated with metabolic and cardiovascular-related drugs (Table 3) and some have significantly different genotype frequencies between Roma and non-Roma (Supplementary Table 14). For example, the rs4149056 variant shows a higher MAF in Roma than in IBS (18% and 11%, respectively) (Table 3) and it increases the risk of toxicity to simvastatin (indicated for hypercholesterolemia) (Table 3). A previous study reports a frequency of 17.2 and 18.9% of this variant in Roma and non-Roma groups from Hungary, respectively (Nagy et al., 2015), suggesting that this variant in the Spanish Roma was present before the arrival into the Iberian Peninsula. rs316019 variant also shows a significant MAF increase in Roma than in IBS and TSI populations (20%, 9%, and 11%, respectively) (Table 3) and it is reported to modify the metabolism of metformin, a drug used to treat type II diabetes (Table 3). Regarding ancestry inference, the minor alleles of these variants are almost exclusively of European ancestry (Supplementary Table 15). Lastly, three previously found variants with increased frequencies in Croatian Roma (Škarić-Jurić et al., 2018) do not show significantly higher MAFs in Spanish Roma (rs10509681, rs8192709, and rs34059508) (Table 3).
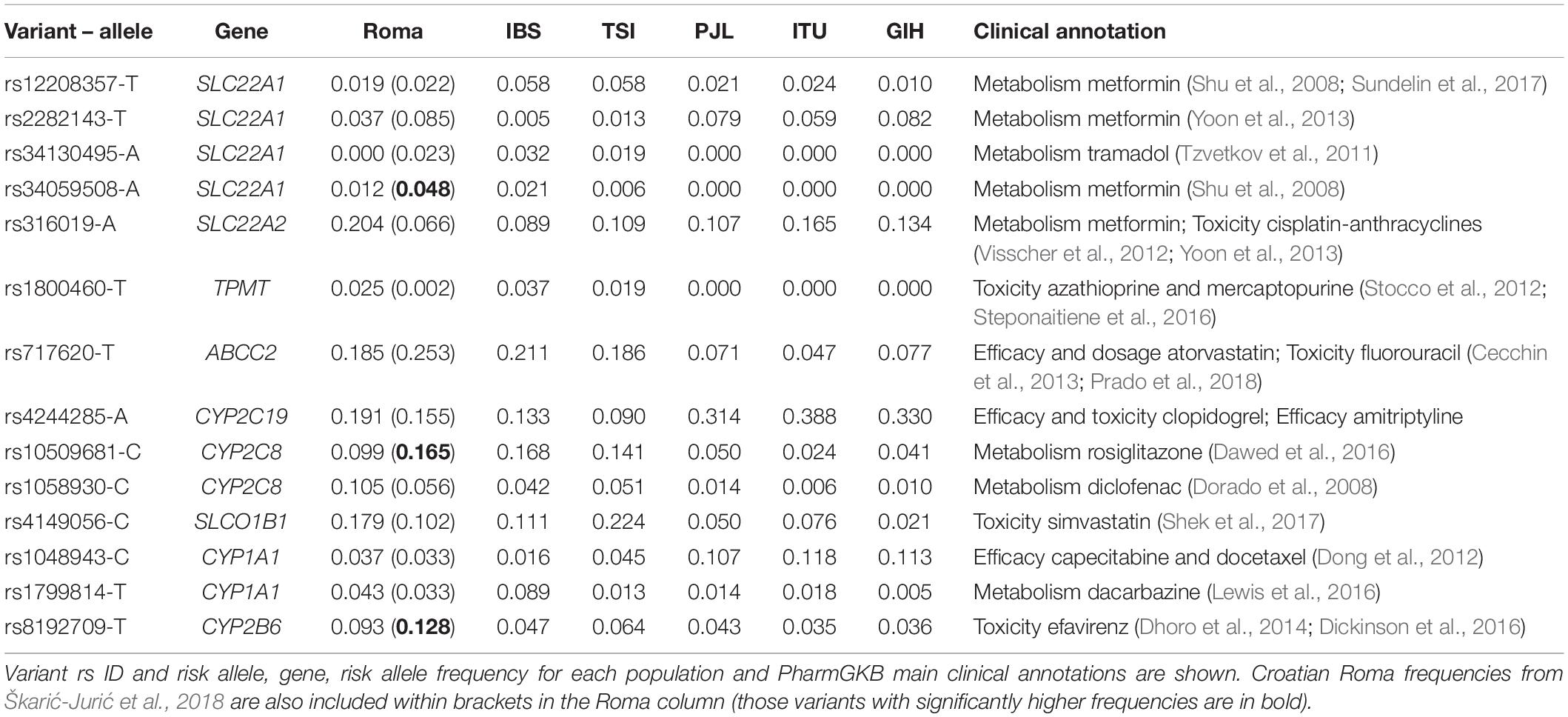
Table 3. List of variants in ADME genes found to have a fold increase in allele frequency equal or higher than 1.5 comparing Roma and non-Roma.
The screening beyond disease-associated variants in the Roma population reveals that most of them can change the response of drugs used for metabolic and cardiovascular disorders and that they might have a European origin. However, this analysis is based on the impact prediction and it is important to take into account that the phenotype is also influenced by the environment, non-coding variants and regulatory elements, among others.
Discussion
The underrepresentation of human populations in genetic studies impairs the understanding of genome architecture and exacerbates health differences. In order to overcome this limitation, the Eurocentric bias in the discovery of functional variants has to be taken into account (Need and Goldstein, 2009; Popejoy and Fullerton, 2016; Bentley et al., 2017; Martin et al., 2019; Sirugo et al., 2019). In the case of the Roma population, we found that the genetic portability with European populations is overall high: allele sharing and LD decay patterns are comparable among groups. However, low frequency variants (MAF < 5%) can present some challenges in the genetic characterization of this population, since only half of these variants in the Roma are also segregating in non-Roma populations. This is consistent with the fact that low frequency and rare variants are more population structured than common variants (Casals and Bertranpetit, 2012; Bomba et al., 2017).
The overrepresentation analysis shows enrichment of some gene sets with increased MAF deleterious variants for genetic disorders. Interestingly, this enrichment occurs both in Roma and in non-Roma, consistent with previous literature. For example, we identify an enrichment for non-triple negative breast cancer in non-Roma, in agreement with a lower incidence in Roma, which show more triple negative cases (Reckova et al., 2017). Triple negative refers to the overexpression of three common markers (i.e., estrogen receptor, progesterone receptor or HER2 oncogene) (Foulkes et al., 2010). However, the overexpression of these markers is common in breast cancer patients of European descent, but not in other groups, such as African-descent (Brewster et al., 2014) or Roma (Reckova et al., 2017) patients.
In the present study, we also examine previously defined disease-associated variants. Besides confirming the ancestry origin of some Mendelian mutations in the Roma, we provide new evidence: the risk allele responsible for Acetyl-coA dehydrogenase deficiency (rs77931234) is traced to a South Asian-like ancestry, while the risk alleles for Charcot–Marie–Tooth disease variants (rs119483085, rs80338934) show European-related ancestry. In addition, we perform a comprehensive study of the pathogenic variation reported in ClinVar database (Landrum et al., 2014). The results show a different frequency spectrum of previously identified variants when comparing Roma and non-Roma groups. However, most of the variants in the Roma are traced to a European origin, evidencing the Eurocentric bias in public databases (Kessler et al., 2016).
Regarding pharmacogenetic variation, we identify variants with increased and decreased MAF in Roma. Many of the variants in drug-binding domains that we found with increased frequencies in non-Roma European populations have been previously associated with a drug response trait, while those with increased frequencies in Roma or South Asian groups are still not specifically characterized. This is particularly relevant, since many of them are related to metabolic and cardiovascular drugs and previous studies suggest that Roma show higher prevalence of these diseases (Vozarova De Courten et al., 2003; Živković et al., 2010). Following the expectations of European ancestry proportions, most drug-response variants in Roma are traced to a European-related origin, which is not a signal of bias, because the list of variants that disrupt drug binding domains is not based on previously known associations discovered in populations of European descent (Dopazo et al., 2016). Thus, these variants show inter-population variability in the Roma, since a differential European gene flow among Roma groups has been previously described (Mendizabal et al., 2012; Font-Porterias et al., 2019). In this sense, the variants reported with increased frequency in Spanish Roma do not completely overlap with a similar study in Croatian Roma (Škarić-Jurić et al., 2018). Moreover, the significantly increased frequency of a CYP2C19 polymorphism (rs4244285) in Hungarian and Portuguese Roma groups (Sipeky et al., 2013; Teixeira et al., 2015) is not observed in Spanish Roma.
For complex diseases, polygenic risk scores (PRSs) are designed to predict the phenotype from genetic data, combining the effect sizes of multiple variants and their frequency (Wray et al., 2007). However, the pre-computed effect sizes usually derive from genome-wide association studies (GWAS), and given its Eurocentric bias, PRSs have a greater predictive accuracy in populations with European ancestry (Martin et al., 2017, 2019; De La Vega and Bustamante, 2018; Kim et al., 2018; Gurdasani et al., 2019). Particularly, they show a systematic bias when applied to other populations due to several factors: (i) GWAS are biased toward those variants segregating in the study population; (ii) when two populations have different LD patterns, tagSNPs and causal variants can differ; and (iii) environmental and genetic factors can be confounded when phenotypes are geographically stratified (Martin et al., 2019; Sirugo et al., 2019). Although new methods are emerging to overcome these limitations (Márquez-Luna et al., 2017; Marnetto et al., 2020), an in-depth analysis of PRS accuracy in non-European-descent populations is needed before implementing them to other under-represented populations (Sirugo et al., 2019).
Here, we provide new evidence of a different frequency spectrum of clinically relevant variants across populations: while some have increased allele frequencies in the Roma, others are virtually absent. This was possible due to the availability of a substantial number of whole-exome sequences at high coverage, which allows the study of clinically relevant genetic variation, enriched in low frequency variants (Bomba et al., 2017). However, this different frequency spectrum cannot be directly attributed to a lower prevalence of some diseases; instead, other unstudied variants might be responsible for disease susceptibility and drug response in this population. Although it is an exploratory approach without functional validation, this study aims to shift the traditional paradigm of focusing only on the increased genetic risk for some diseases in the so-called “isolated” populations. In fact, these results further confirm that Roma are not so genetically isolated. Gene flow with European groups accounts for 65% of their genetic ancestry (Font-Porterias et al., 2019); thus, clinically damaging variants are traced both to South Asian and European-related haplotypes. Lastly, we caution that these results are geographically limited to the Roma population from Spain, and further characterization should be performed in other groups with different demographic trajectories and with increasing sample sizes.
We would like to remark that this study does not aim to exacerbate the importance of inter-population variability, to justify health differences in minority ethnic groups, or to advocate for racialized medicine. In fact, genetic ancestry is not the only determinant of ethnicity or health, and social factors should be considered. Given that genetic diversity is a continuum, large scale genome-wide studies are needed to fully capture and represent human variation, without excluding any population while respecting their rights and interests and properly accounting for demographic differences. This would prevent the current overgeneralization of the results obtained from genetic studies on populations with only European ancestry in the assessment of disease risk testing and treatment response (Martin et al., 2019; Batai et al., 2020; Hudson et al., 2020).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the CEIC-Parc de Salut Mar 2019/8900/I. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
NF-P and DC contributed to the design and conception of the study. NF-P performed and implemented the data analysis. All authors contributed to the interpretation and discussion of the results and writing of the manuscript, and approved the submitted version.
Funding
This study was supported by the Spanish Ministry of Science, Innovation and Universities (MCIU), Agencia Estatal de Investigación (AEI) grant number PID2019-106485GB-I00/AEI/10.13039/501100011033, and “Unidad de Excelencia María de Maeztu” (AEI, CEX2018-000792-M). NF-P was supported by a FPU17/03501 fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the invaluable collaboration of the Federacioì d’Associacions Gitanes de Catalunya (FAGiC). We would also like to thank the members of the Human Genome Diversity Group and Genomics of Individuality Lab (Institut de Biologia Evolutiva, UPF-CSIC) for the insightful discussions about the results of this study and the computational support from the Scientific IT Core Facility from Universitat Pompeu Fabra.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.683880/full#supplementary-material
Footnotes
References
1000 Genomes Project Consortium, (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Ahmed, I., Buchert, R., Zhou, M., Jiao, X., Mittal, K., Sheikh, T. I., et al. (2014). Mutations in DCPS and EDC3 in autosomal recessive intellectual disability indicate a crucial role for mRNA decapping in neurodevelopment. Hum. Mol. Genet. 24, 3172–3180. doi: 10.1093/hmg/ddv069
Álvarez, A., Castillo, I., Villamar, M., Aguirre, L. A., González-Neira, A., López-Nevot, A., et al. (2005). High Prevalence of the W24X mutation in the gene encoding Connexin-26 (GJB2) in Spanish Romani (Gypsies) with autosomal recessive non-syndromic hearing loss. Am. J. Med. Genet. 137A, 255–258. doi: 10.1002/ajmg.a.30884
Auton, A., Abecasis, G. R., Altshuler, D. M., Durbin, R. M., Bentley, D. R., Chakravarti, A., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Azmanov, D. N., Dimitrova, S., Florez, L., Cherninkova, S., Draganov, D., Morar, B., et al. (2011). LTBP2 and CYP1B1 mutations and associated ocular phenotypes in the Roma/Gypsy founder population. Eur. J. Hum. Genet. 19, 326–333. doi: 10.1038/ejhg.2010.181
Barbujani, G., Ghirotto, S., and Tassi, F. (2013). Nine things to remember about human genome diversity. Tissue Antigens82, 155–164. doi: 10.1111/tan.12165
Batai, K., Hooker, S., and Kittles, R. A. (2020). Leveraging genetic ancestry to study health disparities. Am. J. Phys. Anthropol. 175, 1–13.
Bentley, A. R., Callier, S., and Rotimi, C. N. (2017). Diversity and inclusion in genomic research: why the uneven progress? J. Commun. Genet. 8, 255–266. doi: 10.1007/s12687-017-0316-6
Bianco, E., Laval, G., Font-Porterias, N., García-Fernandez, C., Dobon, B., Sabido-Vera, R., et al. (2020). Recent common origin, reduced population size, and marked admixture have shaped European Roma genomes. Mol. Biol. Evol. 37, 3175–3187. doi: 10.1093/molbev/msaa156
Bomba, L., Walter, K., and Soranzo, N. (2017). The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 18, 1–17.
Bouwer, S., Angelicheva, D., Chandler, D., Seeman, P., Tournev, I., and Kalaydjieva, L. (2007). Carrier rates of the ancestral Indian W24X mutation in GJB2 in the general Gypsy population and individual subisolates. Genet. Test. 11, 455–458. doi: 10.1089/gte.2007.0048
Bress, A., Patel, S. R., Perera, M. A., Campbell, R. T., Kittles, R. A., and Cavallari, L. H. (2012). Effect of NQO1 and CYP4F2 genotypes on warfarin dose requirements in Hispanic-Americans and African-Americans. Pharmacogenomics 13, 1925–1935. doi: 10.2217/pgs.12.164
Brewster, A. M., Chavez-MacGregor, M., and Brown, P. (2014). Epidemiology, biology, and treatment of triple-negative breast cancer in women of African ancestry. Lancet Oncol. 15, e625–e634.
Browning, S. R., Browning, B. L., Daviglus, M. L., Durazo, R. A., Schneiderman, N., Kaplan, R. C., et al. (2018). Ancestry-specific recent effective population size in the Americas. PLoS Genet. 14:e1007385. doi: 10.1371/journal.pgen.1007385
Cabrera-Serrano, M., Mavillard, F., Biancalana, V., Rivas, E., Morar, B., Hernández-Laín, A., et al. (2018). A Roma founder BIN1 mutation causes a novel phenotype of centronuclear myopathy with rigid spine. Neurology 91, e339–e348.
Casals, F., and Bertranpetit, J. (2012). Human genetic variation, shared and private. Science 336, 39–40. doi: 10.1126/science.1224528
Casas, J. P., Hingorani, A. D., Bautista, L. E., and Sharma, P. (2004). Meta-analysis of genetic studies in ischemic stroke: thirty-two genes involving approximately 18 000 cases and 58 000 controls. Arch. Neurol. 61, 1652–1661. doi: 10.1001/archneur.61.11.1652
Cecchin, E., D’Andrea, M., Lonardi, S., Zanusso, C., Pella, N., Errante, D., et al. (2013). A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharmacogenomics J. 13, 403–409. doi: 10.1038/tpj.2012.31
Claramunt, R., Sevilla, T., Lupo, V., Cuesta, A., Millán, J. M., Vílchez, J. J., et al. (2007). The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin. Genet. 71, 343–349. doi: 10.1111/j.1399-0004.2007.00774.x
Council of Europe, (2012). Human Rights of Roma and Travellers in Europe. Strasbourg: Council of Europe.
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
Dawed, A. Y., Donnelly, L., Tavendale, R., Carr, F., Leese, G., Palmer, C. N. A., et al. (2016). CYP2C8 and SLCO1B1 variants and therapeutic response to thiazolidinediones in patients with Type 2 diabetes. Diabetes Care 39, 1902–1908. doi: 10.2337/dc15-2464
De La Vega, F. M., and Bustamante, C. D. (2018). Polygenic risk scores: a biased prediction? Genome Med. 10:100.
Decker, B., Allen, J., Luccarini, C., Pooley, K. A., Shah, M., Bolla, M. K., et al. (2017). Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J. Med. Genet. 54, 732–741.
Dhoro, M., Ngara, B., Kadzirange, G., Nhachi, C., and Masimirembwa, C. (2014). Genetic variants of drug metabolizing enzymes and drug transporter (ABCB1) as possible biomarkers for adverse drug reactions in an HIV/AIDS Cohort in Zimbabwe. Curr. HIV Res. 11, 481–490. doi: 10.2174/1570162x113119990048
Dickinson, L., Amin, J., Else, L., Boffito, M., Egan, D., Owen, A., et al. (2016). Comprehensive pharmacokinetic, pharmacodynamic and pharmacogenetic evaluation of once-daily Efavirenz 400 and 600 mg in Treatment-Naïve HIV-Infected Patients at 96 weeks: results of the ENCORE1 study. Clin. Pharmacokinet. 55, 861–873. doi: 10.1007/s40262-015-0360-5
Dong, N., Yu, J., Wang, C., Zheng, X., Wang, Z., Di, L., et al. (2012). Pharmacogenetic assessment of clinical outcome in patients with metastatic breast cancer treated with docetaxel plus capecitabine. J. Cancer Res. Clin. Oncol. 138, 1197–1203. doi: 10.1007/s00432-012-1183-5
Dopazo, J., Amadoz, A., Bleda, M., Garcia-Alonso, L., Alemán, A., García-García, F., et al. (2016). 267 spanish exomes reveal population-specific differences in disease-related genetic variation. Mol. Biol. Evol. 33, 1205–1218. doi: 10.1093/molbev/msw005
Dorado, P., Cavaco, I., Cáceres, M. C., Piedade, R., Ribeiro, V., and LLerena, A. (2008). Relationship between CYP2C8 genotypes and diclofenac 5-hydroxylation in healthy Spanish volunteers. Eur. J. Clin. Pharmacol. 64, 967–970. doi: 10.1007/s00228-008-0508-4
Dropinski, J., Musial, J., Sanak, M., Wegrzyn, W., Nizankowski, R., and Szczeklik, A. (2007). Antithrombotic effects of aspirin based on PLA1/A2 glycoprotein IIIa polymorphism in patients with coronary artery disease. Thromb. Res. 119, 301–303. doi: 10.1016/j.thromres.2006.03.005
Font-Porterias, N., Caro-Consuegra, R., Lucas-Saìnchez, M., Lopez, M., Gimeìnez, A., Carballo, A., et al. (2021). The counteracting effects of demography on functional genomic variation: the Roma paradigm. Mol. Biol. Evol. msab070.
Font-Porterias, N., Arauna, L. R., Poveda, A., Bianco, E., Rebato, E., Prata, M. J., et al. (2019). European Roma groups show complex West Eurasian admixture footprints and a common South Asian genetic origin. PLoS Genet. 15:e1008417. doi: 10.1371/journal.pgen.1008417
Foulkes, W. D., Smith, I. E., and Reis-Filho, J. S. (2010). Triple-Negative Breast Cancer. N. Engl. J. Med. 363, 1938–1948.
Gamella, J. F., Carrasco-Muñoz, E. M., and Núñez Negrillo, A. M. (2013). Oculocutaneous albinism and consanguineous marriage among Spanish Gitanos or Calé-a study of 83 cases. Coll Antropol. 37, 723–734.
Gurdasani, D., Barroso, I., Zeggini, E., and Sandhu, M. S. (2019). Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 20, 520–535. doi: 10.1038/s41576-019-0144-0
Ha, E., Kim, M. J., Choi, B. K., Rho, J. J., Oh, D. J., Rho, T. H., et al. (2006). Positive association of obesity with single nucleotide polymorphisms of syndecan 3 in the Korean population. J. Clin. Endocrinol. Metab. 91, 5095–5099. doi: 10.1210/jc.2005-2086
Heyer, E., and Reynaud-Paligot, C. (2020). Us and them: from prejudice to racism. An original analysis of race and racism. Am. J. Phys. Anthropol. 175, 1–9. doi: 10.4135/9781446218877.n1
Hudson, M., Garrison, N. A., Sterling, R., Caron, N. R., Fox, K., Yracheta, J., et al. (2020). Rights, interests and expectations: indigenous. Nat. Rev. Genet. 21, 377–384.
International HapMap Consortium, (2003). The international HapMap project. Nature 426, 789–796. doi: 10.1038/nature02168
Jasiecki, J., Żuk, M., Krawczyńska, N., Jońca, J., Szczoczarz, A., Lewandowski, K., et al. (2019). Haplotypes of butyrylcholinesterase K-variant and their influence on the enzyme activity. Chem. Biol. Interact. 307, 154–157. doi: 10.1016/j.cbi.2019.05.007
Javorský, M., Gotthardová, I., Klimčáková, L., Kvapil, M., Židzik, J., Schroner, Z., et al. (2016). A missense variant in GLP1R gene is associated with the glycaemic response to treatment with gliptins. Diabetes Obes Metab. 18, 941–944. doi: 10.1111/dom.12682
Johnson, J., Caudle, K., Gong, L., Whirl-Carrillo, M., Stein, C., Scott, S., et al. (2017). Clinical pharmacogenetics implementation consortium (CPIC) guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin. Pharmacol. Ther. 102, 397–404. doi: 10.1002/cpt.668
Kalaydjieva, L., Gresham, D., and Calafell, F. (2001). Genetic studies of the Roma (Gypsies): a review. BMC Med. Genet. 2:5. doi: 10.1186/1471-2350-2-5
Kessler, M. D., Yerges-Armstrong, L., Taub, M. A., Shetty, A. C., Maloney, K., Jeng, L. J. B., et al. (2016). Challenges and disparities in the application of personalized genomic medicine to populations with African ancestry. Nat. Commun. 7, 1–8.
Kim, M. S., Patel, K. P., Teng, A. K., Berens, A. J., and Lachance, J. (2018). Genetic disease risks can be misestimated across global populations. Genome Biol. 19:179.
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2019). PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109.
Landrum, M. J., Lee, J. M., Riley, G. R., Jang, W., Rubinstein, W. S., Church, D. M., et al. (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, 980–985.
Lao, O., Lu, T. T., Nothnagel, M., Junge, O., Freitag-Wolf, S., Caliebe, A., et al. (2008). Correlation between genetic and geographic structure in Europe. Curr. Biol. 18, 1241–1248.
Lewis, B. C., Korprasertthaworn, P., and Miners, J. O. (2016). Impaired dacarbazine activation and 7-ethoxyresorufin deethylation in vitro by polymorphic variants of CYP1A1 and CYP1A2: implications for cancer therapy. Pharmacogenet. Genomics 26, 453–461. doi: 10.1097/fpc.0000000000000236
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z., and Zhang, B. (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205.
Lin, P. H., Kuo, W. H., Huang, A. C., Lu, Y. S., Lin, C. H., Kuo, S. H., et al. (2016). Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 7, 8310–8320.
MacGregor, S., Lind, P. A., Bucholz, K. K., Hansell, N. K., Madden, P. A. F., Richter, M. M., et al. (2009). Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: an integrated analysis. Hum. Mol. Genet. 18, 580–593. doi: 10.1093/hmg/ddn372
Maples, B. K., Gravel, S., Kenny, E. E., and Bustamante, C. D. (2013). RFMix?: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 93, 278–288. doi: 10.1016/j.ajhg.2013.06.020
Marnetto, D., Pärna, K., Läll, K., Molinaro, L., Montinaro, F., Haller, T., et al. (2020). Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat. Commun. 11, 1–9.
Márquez-Luna, C., Loh, P. R., Price, A. L., Kooner, J. S., Saleheen, D., Sim, X., et al. (2017). Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 41, 811–823. doi: 10.1002/gepi.22083
Martin, A. R., Gignoux, C. R., Walters, R. K., Wojcik, G. L., Neale, B. M., Gravel, S., et al. (2017). Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100, 635–649. doi: 10.1016/j.ajhg.2017.03.004
Martin, A. R., Kanai, M., Kamatani, Y., Okada, Y., Neale, B. M., and Daly, M. J. (2019). Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591. doi: 10.1038/s41588-019-0379-x
Martinez-Frias, M. L., and Bermejo, E. (1992). Prevalence of congenital anomaly syndromes in a Spanish gypsy population. J. Med. Genet. 29, 483–486.
McGuire, M. C., Nogueira, C. P., Bartels, C. F., Lightstone, H., Hajra, A., Van der Spek, A. F. L., et al. (1989). Identification of the structural mutation responsible for the dibucaine-resistant (atypical) variant form of human serum cholinesterase. Proc. Natl. Acad. Sci. U.S.A. 86, 953–957. doi: 10.1073/pnas.86.3.953
McLaren, W., Gil, L., Hunt, S.E., et al. (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. doi: 10.1186/s13059-016-0974-4
Mendizabal, I., Lao, O., Marigorta, U. M., Kayser, M., and Comas, D. (2013). Implications of population history of European Romani on genetic susceptibility to disease. Hum. Hered. 76, 194–200. doi: 10.1159/000360762
Mendizabal, I., Lao, O., Marigorta, U. M., Wollstein, A., Gusmão, L., Ferak, V., et al. (2012). Reconstructing the population history of European Romani from genome-wide data. Curr. Biol. 22, 2342–2349. doi: 10.1016/j.cub.2012.10.039
Minárik, G., Ferák, V., Feráková, E., Ficek, A., Poláková, H., and Kádasi, L. (2003). High frequency of GJB2 Mutation W24X among Slovak Romany (Gypsy) patients with non-syndromic hearing loss (NSHL). Gen. Physiol. Biophys. 22, 549–556.
Moorjani, P., Patterson, N., Loh, P.-R., Lipson, M., Kisfali, P., Melegh, B. I., et al. (2013). Reconstructing Roma history from genome-wide data. PLoS One 8:e58633. doi: 10.1371/journal.pone.0058633
Morar, B., Azmanov, D. N., and Kalaydjieva, L. (2013). “Roma (Gypsies): genetic Studies,” in eLS (Chichester: Wiley). Eds., John Wiley & Sons, Ltd
Motovska, Z., Widimsky, P., Kvasnicka, J., Petr, R., Bilkova, D., Hajkova, J., et al. (2009). High loading dose of clopidogrel is unable to satisfactorily inhibit platelet reactivity in patients with glycoprotein IMA gene polymorphism: a genetic substudy of PRAGUE-8 trial. Blood Coagul. Fibrinolysis 20, 257–262. doi: 10.1097/mbc.0b013e328325455b
Nagy, A., Sipeky, C., Szalai, R., Melegh, B. I., Matyas, P., Ganczer, A., et al. (2015). Marked differences in frequencies of statin therapy relevant SLCO1B1 variants and haplotypes between Roma and Hungarian populations. BMC Genet. 16:108. doi: 10.1186/s12863-015-0262-4
Need, A. C., and Goldstein, D. B. (2009). Next generation disparities in human genomics: concerns and remedies. Trends Genet. 25, 489–494. doi: 10.1016/j.tig.2009.09.012
O’Connell, J., Gurdasani, D., Delaneau, O., Pirastu, N., Ulivi, S., Cocca, M., et al. (2014). A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10:e1004234. doi: 10.1371/journal.pgen.1004234
Padoa, C., Goldman, A., Jenkins, T., and Ramsay, M. (1999). Cystic fibrosis carrier frequencies in populations of African origin. J. Med. Genet. 36, 41–44.
Peshkin, B. N., Alabek, M. L., and Isaacs, C. (2010). BRCA1/2 mutations and triple negative breast cancers. Breast Dis. 32, 25–33. doi: 10.3233/bd-2010-0306
Phani, N. M., Guddattu, V., Bellampalli, R., Seenappa, V., Adhikari, P., Nagri, S. K., et al. (2014). Population specific impact of genetic variants in KCNJ11 gene to type 2 diabetes: a case-control and meta-analysis study. PLoS One 9:e107021. doi: 10.1371/journal.pone.0107021
Popejoy, A. B., and Fullerton, S. M. (2016). Genomics is failing on diversity. Nature 538, 161–164. doi: 10.1038/538161a
Prado, Y., Arencibia, A., Zambrano, T., and Salazar, L. A. (2018). Gender-specific association between ABCC2 -24C>T SNP and reduction in triglycerides in chilean patients treated with atorvastatin. Basic Clin. Pharmacol. Toxicol. 122, 517–522. doi: 10.1111/bcpt.12943
Reckova, M., Mardiak, J., Plank, L., Vulevova, M., Cingelova, S., and Mego, M. (2017). Differences in incidence and biological characteristics of breast cancer between roma and non-roma patients in Slovakia. Klin Onkol. 30, 48–54. doi: 10.14735/amko201748
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894.
Rocha, H., Castiñeiras, D., Delgado, C., Egea, J., Yahyaoui, R., González, Y., et al. (2014). Birth prevalence of fatty acid β-Oxidation disorders in iberia. JIMD Rep. 16, 89–94. doi: 10.1007/8904_2014_324
Sevilla, T., Martínez-Rubio, D., Márquez, C., Paradas, C., Colomer, J., Jaijo, T., et al. (2013). Genetics of the charcot-marie-tooth disease in the spanish gypsy population: the hereditary motor and sensory neuropathy-Russe in depth. Clin. Genet. 83, 565–570. doi: 10.1111/cge.12015
Shek, A. B., Kurbanov, R. D., Abdullaeva, G. J., Nagay, A. V., Hoshimov, S. U., Nizamov, U. I., et al. (2017). Simvastatin intolerance genetic determinants: some features in ethnic Uzbek patients with coronary artery disease. Arch. Med. Sci. Atheroscler. Dis. 2, 68–75. doi: 10.5114/amsad.2017.70597
Shu, Y., Brown, C., Castro, R. A., Shi, R. J., Lin, E. T., Owen, R. P., et al. (2008). Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 83, 273–280. doi: 10.1038/sj.clpt.6100275
Sipeky, C., Weber, A., Szabo, M., Melegh, B. I., Janicsek, I., Tarlos, G., et al. (2013). High prevalence of CYP2C19∗2 allele in Roma samples: study on Roma and Hungarian population samples with review of the literature. Mol. Biol. Rep. 40, 4727–4735. doi: 10.1007/s11033-013-2569-4
Sirisena, N. D., Adeyemo, A., Kuruppu, A. I., Samaranayake, N., and Dissanayake, V. H. W. (2018). Genetic variants associated with clinicopathological profiles in sporadic breast cancer in Sri Lankan women. J. Breast Cancer 21, 165–172. doi: 10.4048/jbc.2018.21.2.165
Sirugo, G., Williams, S. M., and Tishkoff, S. A. (2019). The missing diversity in human genetic studies. Cell 177, 26–31. doi: 10.1016/j.cell.2019.02.048
Škarić-Jurić, T., Tomas, Ž, Petranović, M. Z., Božina, N., Naranćić, N. S., Janićijević, B., et al. (2018). Characterization of ADME genes variation in Roma and 20 populations worldwide. PLoS One 13:e0207671. doi: 10.1371/journal.pone.0207671
Slavin, T. P., Maxwell, K. N., Lilyquist, J., Vijai, J., Neuhausen, S. L., Hart, S. N., et al. (2017). The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. npj Breast Cancer 3:22.
Steponaitiene, R., Kupcinskas, J., Survilaite, S., Varkalaite, G., Jonaitis, L., Kiudelis, G., et al. (2016). TPMT and ITPA genetic variants in Lithuanian inflammatory bowel disease patients: prevalence and azathioprine-related side effects. Adv. Med. Sci. 61, 135–140. doi: 10.1016/j.advms.2015.09.008
Stewart, C., and Pepper, M. S. (2017). Cystic fibrosis in the African diaspora. Ann. Am. Thorac. Soc. 14, 1–7. doi: 10.1513/annalsats.201606-481fr
Stocco, G., Yang, W., Crews, K. R., Thierfelder, W. E., Decorti, G., Londero, M., et al. (2012). PACSIN2 polymorphism influences TPMT activity and mercaptopurine-related gastrointestinal toxicity. Hum. Mol. Genet. 21, 4793–4804. doi: 10.1093/hmg/dds302
Sundelin, E. I. O., Gormsen, L. C., Jensen, J. B., Vendelbo, M. H., Jakobsen, S., Munk, O. L., et al. (2017). Genetic polymorphisms in organic cation transporter 1 attenuates hepatic metformin exposure in humans. Clin. Pharmacol. Ther. 102, 841–848. doi: 10.1002/cpt.701
Takkenberg, J. J. M., and Roos-hesselink, J. W. (2011). Birth prevalence of congenital heart disease worldwide a systematic review and meta-analysis. JAC 58, 2241–2247.
Teixeira, J., Amorim, A., Prata, J. M., and Quental, S. (2015). Pharmacogenetic polymorphisms in a portuguese gypsy population. Curr. Pharmacogenomics Person. Med. 13, 36–40. doi: 10.2174/1875692113666150703180101
Tomita-Mitchell, A., Maslen, C. L., Morris, C. D., Garg, V., and Goldmuntz, E. (2007). GATA4 sequence variants in patients with congenital heart disease. J. Med. Genet. 44, 779–783. doi: 10.1136/jmg.2007.052183
Tzvetkov, M. V., Saadatmand, A. R., Lötsch, J., Tegeder, I., Stingl, J. C., and Brockmöller, J. (2011). Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin. Pharmacol. Ther. 90, 143–150. doi: 10.1038/clpt.2011.56
Visscher, H., Ross, C. J. D., Rassekh, S. R., Barhdadi, A., Dubé, M. P., Al-Saloos, H., et al. (2012). Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J. Clin. Oncol. 30, 1422–1428.
Vozarova De Courten, B., De Courten, M., Hanson, R. L., Zahorakova, A., Egyenes, H. P., Tataranni, P. A., et al. (2003). Higher prevalence of type 2 diabetes, metabolic syndrome and cardiovascular diseases in gypsies than in non-gypsies in Slovakia. Diabetes Res. Clin. Pract. 62, 95–103. doi: 10.1016/s0168-8227(03)00162-1
Werissa, N. A., Piko, P., Fiatal, S., Kosa, Z., Sandor, J., and Adany, R. (2019). SNP-based genetic risk score modeling suggests no increased genetic susceptibility of the roma population to Type 2 diabetes mellitus. Genes 10, 1–16.
Whirl-Carrillo, M., McDonagh, E. M., Hebert, J. M., Gong, L., Sangkuhl, K., Thorn, C. F., et al. (2012). Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417.
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082.
Wray, N. R., Goddard, M. E., and Visscher, P. M. (2007). Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 17, 1520–1528. doi: 10.1101/gr.6665407
Yoon, H., Cho, H. Y., Yoo, H. D., Kim, S. M., and Lee, Y. B. (2013). Influences of organic cation transporter polymorphisms on the population pharmacokinetics of metformin in healthy subjects. AAPS J. 15, 571–580. doi: 10.1208/s12248-013-9460-z
Zhang, C., Dong, S., Xu, J., He, W., and Yang, T. (2018). PopLDdecay?: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. doi: 10.1093/bioinformatics/bty875
Zhang, W., Li, X., Shen, A., Jiao, W., Guan, X., and Li, Z. (2008). GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur. J. Med. Genet. 51, 527–535. doi: 10.1016/j.ejmg.2008.06.005
Keywords: Romani, whole-exome sequences, clinically relevant variants, drug-response variants, local ancestry inference, Eurocentric bias
Citation: Font-Porterias N, Giménez A, Carballo-Mesa A, Calafell F and Comas D (2021) Admixture Has Shaped Romani Genetic Diversity in Clinically Relevant Variants. Front. Genet. 12:683880. doi: 10.3389/fgene.2021.683880
Received: 22 March 2021; Accepted: 13 May 2021;
Published: 16 June 2021.
Edited by:
Karoline Kuchenbaecker, University College London, United KingdomReviewed by:
Elizabeth Atkinson, Massachusetts General Hospital and Harvard Medical School, United StatesOlivia J. Veatch, University of Kansas Medical Center, United States
Copyright © 2021 Font-Porterias, Giménez, Carballo-Mesa, Calafell and Comas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Comas, ZGF2aWQuY29tYXNAdXBmLmVkdQ==