 Yan Cong
Yan Cong Hongxing Jin2†
Hongxing Jin2† Ke Wu
Ke Wu- 1Rehabilitation Department, Yiwu Maternity and Child Health Care Hospital, Yiwu, China
- 2Pediatric Department, Yiwu Maternity and Child Health Care Hospital, Yiwu, China
- 3Prenatal Diganosis Center, Yiwu Maternity and Child Health Care Hospital, Yiwu, China
Background: Coffin–Lowry syndrome (CLS) [OMIM#303600] is a rare X-linked dominant syndrome. CLS is caused by highly heterogeneous loss-of-function mutations in the RPS6KA3 gene (OMIM*300,075). CLS is characterized by intellectual disability (ID), short stature, tapered fingers, characteristic facial features, and progressive skeletal changes. Distal 22q11.2 microdeletion syndrome (OMIM#611867) is an autosomal dominant and recurrent genomic disorder. It mainly includes three types [distal type I (D–E/F), type II (E–F), and type III (F–G)] and exhibits variable clinical phenotypes (mild, moderate, or even normal): preterm birth, pre- and/or postnatal growth restriction, development delay, ID, behavioral problems, cardiovascular defects, skeletal anomalies, and dysmorphic facial features. We investigated the genetic etiology of a Chinese pedigree with ID, short stature, digit abnormalities, facial dysmorphism, and menstrual disorder. A heterozygous RPS6KA3 gene variant c.898C>T (p.R300X) was identified in this familial case. Two female CLS patients with distal 22q11.2 microdeletion presented with more severe clinical phenotypes. We provided clinical characteristics of these Chinese female CLS patients.
Case presentation: We described a Chinese family with three affected females (the mother, the elder sister, and the proband). The mother and the elder sister had more severe clinical phenotypes (moderate facial dysmorphism, more severe cognitive impairment, and shorter stature). The common characteristic phenotypes are ID, short stature, facial dysmorphism, irregular menstruation, and cardiovascular disorders. Peripheral blood samples were collected from the pedigree. Whole-exome sequencing (WES) identified a heterozygous nonsense RPS6KA3 gene variant c.898C>T (p.R300X). It was verified by Sanger sequencing. Copy number variation sequencing (CNV-seq) showed that both the mother and the elder sister carried a CNVseq [hg19] del (22) (q11.22-q11.23) (22997582–23637176)×0.5. RNA from peripheral blood samples was used for measuring the relative quantification of mRNA (expressed by exon 14 of RPS6KA3). The levels of mRNA relative expressions were significantly lower in the mother’s and the elder sister’s blood samples. The levels of mRNA relative expressions were significantly higher in the proband’s blood sample. X-chromosome inactivation (XCI) studies demonstrated that the proband showed extremely skewed XCI, and the XCI pattern of the elder sister was random.
Conclusion: Herein, we reported three Chinese female patients with a heterozygous nonsense RPS6KA3 gene variant c.898C>T. Further genetic studies were performed. To our knowledge, Chinese patients with this variant have not been previously reported in the literature. The three female patients presented with variable degrees of severity. The clinical characteristics of these Chinese female CLS patients could expand the phenotypic spectrum of CLS. We helped physicians to understand the genotype–phenotype correlation further.
Background
CLS is an X-linked dominant disorder characterized by ID, craniofacial features, and skeletal abnormalities. CLS is caused by heterozygous loss-of-function mutations in the RPS6KA3 gene. A total of 70–80% of probands of CLS had no family history, and roughly 2/3 occurred de novo. A total of 20–30% of CLS patients had more than one affected family member (Pereira et al., 2010). With the use of WES, we reported a Chinese pedigree with a heterozygous pathogenic variant c.898C>T (p.R300X) in RPS6KA3 gene. Further genetic studies were performed. A CNV was found in two female CLS patients with more severe clinical phenotypes, they may be associated with distal 22q11.2 microdeletion syndrome. We described clinical characteristics of these Chinese female CLS patients. Also, we hoped to improve the comprehensive understanding of CLS for pediatricians.
Case presentation
The proband (a 17 year-old female patient) was found by us in the countryside. The proband had additional two affected family members (Figure 1). They had similar symptoms. The parents were residents of adjacent villages, and they did not have a consanguineous relation. The proband’s father was phenotypically normal. The proband and her elder sister (21 years old) were born at full-term by spontaneous vaginal delivery without complication in the perinatal and neonatal period. The weight and height of the proband were 56 kg and 161 cm, respectively. The weight and height of her elder sister were 72 kg and 142 cm, respectively. The weight and height of her mother (47 years old) were 65 kg and 140 cm, respectively. The cognitive impairment tests showed that MMSE scores of the proband, her elder sister, and mother were 13, 8, and 5, respectively. The proband had received education at a special school for 2 years. Now she can recognize some Chinese characters, and she has simple communication with people. She had a poor memory like always forgetting her own birthday. Her elder sister and mother could not recognize any Chinese characters. They had little communication with people. The three patients all had coarse craniofacial features including widely spaced downward-slanting palpebral fissures, low nasal bridge, blunt tip, broad nose, anteverted nares, and thick lips (Figure 2C). In addition, her elder sister had acne all over her face (Figure 2B), and her mother had a wide and open mouth, and thick lips with everted lower lip (Figure 2A). The elder sister’s and mother’s fingers tapered markedly from relatively wide proximally to narrow distally with small terminal phalanges and nails. The proband’s fingers did not show anything abnormal. During their adolescent period, they all had irregular menstruation. Echocardiography showed that they all had mild mitral and tricuspid valve regurgitation. X-rays of spines showed that only the mother had scoliosis (Figure 3A). X-rays of hands did not show any signs of abnormality.
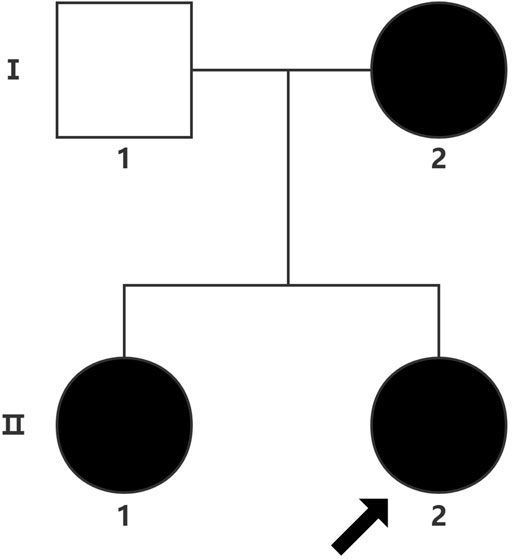
FIGURE 1. Proband (arrow).
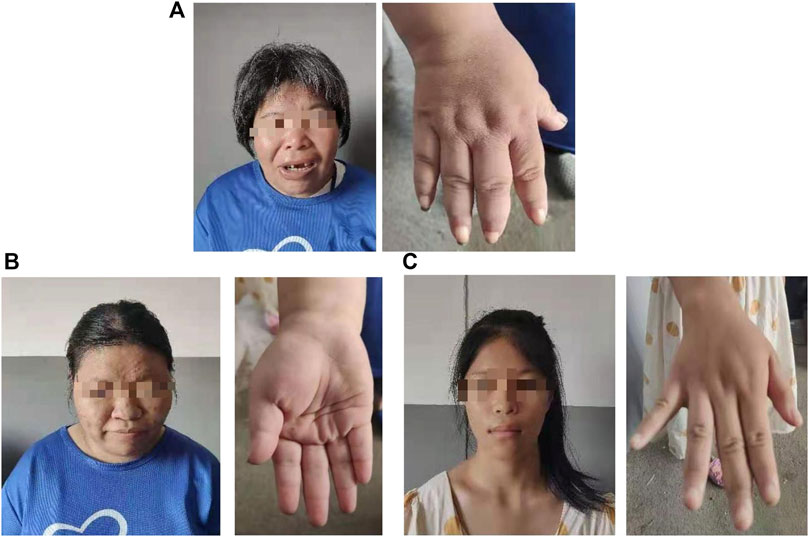
FIGURE 2. Hand and facial features of the mother (A). Hand and facial features of the elder sister (B). Hand and facial features of the proband (C).
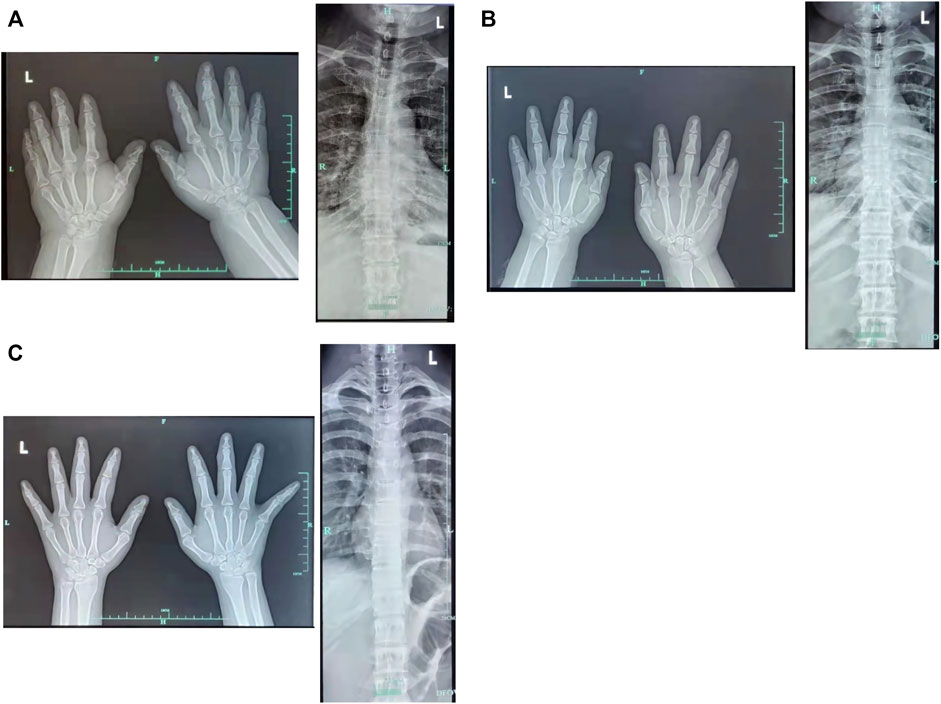
FIGURE 3. X-rays of hands and spines. (A) Mother (B) Elder sister (C) Proband.
Neuropsychological tests
The mini-mental state examination (MMSE) (Pangman et al., 2000) is a 30-point questionnaire. MMSE is one of the most widely used brief screening instruments for measuring cognitive impairment. It has been modified and translated into a Chinese version. The optimal cut-off points were determined according to the education level. The optimal cut-off points were 16/17 for illiterate individuals, 19/20 for individuals with 1–6 years of education, and 23/24 for individuals with seven or more years of education (Li et al., 2016).
Genetic tests
The guardian (the proband’s father) signed an informed consent for genetic analysis. Our legal ethics committee approved this genetic study. gDNAs were extracted from peripheral blood of the patients and phenotypically normal father for WES and CNVseq. Sanger sequencing was used for further verification. CNV-seq showed that both the mother and the elder sister carried a CNVseq [hg19] del (22) (q11.22-q11.23) (22997582–23637176) × 0.5 (Figure 4). WES identified a heterozygous nonsense RPS6KA3 gene variant c.898C>T (p.R300X), which was verified by Sanger sequencing (Figure 5).
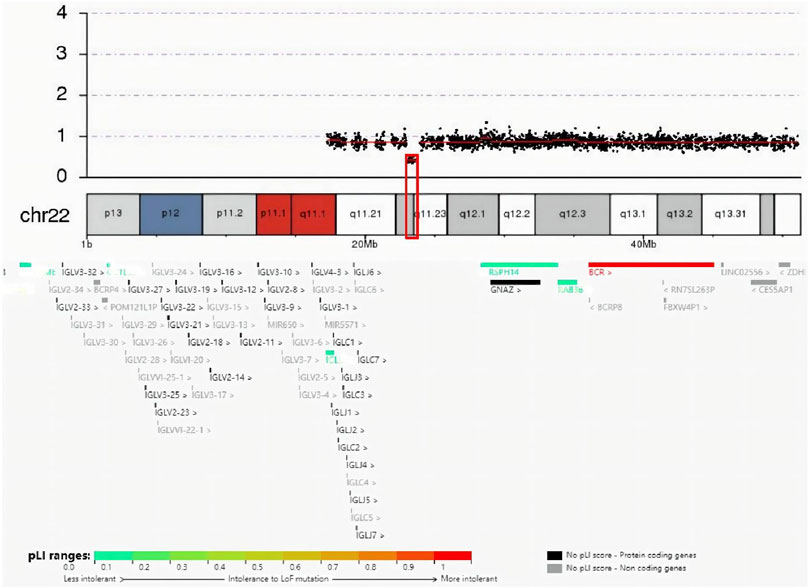
FIGURE 4. Result of CNV-seq was seq (hg19) del (22) (q11.22-q11.23) (22997582–23637176) × 0.5. In the deletion region (marked with a red box), genes were colored by the pLI score.
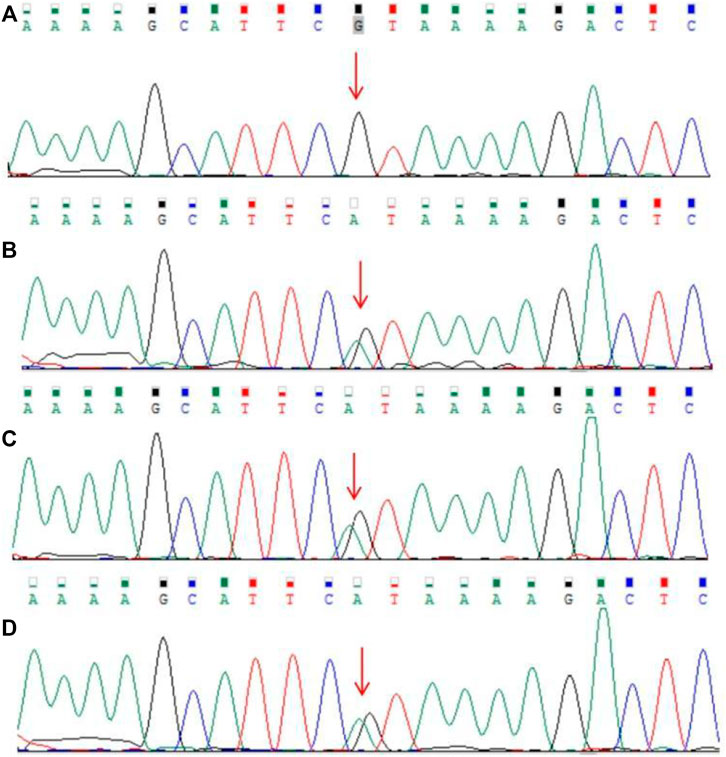
FIGURE 5. Results of Sanger sequencing. (A) Father (B–D). Heterozygous RPS6KA3 gene variant (C) 898C>T (NM_004,586.3) in the mother, the elder sister, and the proband (the variant was marked with a red arrow).
Levels of urine organic acids, plasma amino acids, lactate, and pyruvic acid were all normal in patients. Conventional G-banded chromosome analysis all showed a 46, XX karyotype. The results of genetic metabolic disease screening were all negative in patients.
As per the guidelines of the American College of Medical Genetics and Genomics (ACMG) for interpreting sequence variants (Richards et al., 2015), this variant was pathogenic (PVS1+PM2+PP1+PP3+PP4+PP5). Total cellular RNA was isolated from patients’ peripheral blood for measuring the relative quantification of mRNA (expressed by exon 14 of the RPS6KA3 gene). The quantitative PCR (qPCR) data demonstrated that the levels of mRNA relative expression were significantly lower in the mother’s and the elder sister’s blood samples, and the levels of the mRNA relative expression were significantly higher in the proband’s blood sample (Figure 6).
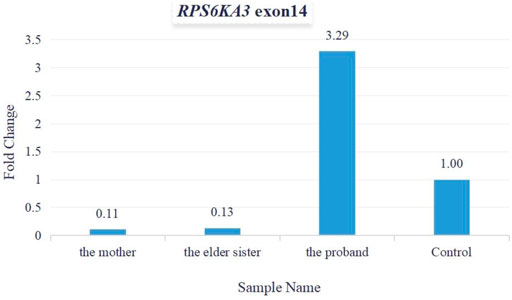
FIGURE 6. Fold change was used for calculating mRNA relative expression. The levels of mRNA relative expression were significantly lower in the mother’s and the elder sister’s blood samples, and the levels of mRNA relative expression were significantly higher in the proband’s blood sample.
gDNAs from peripheral blood samples of the two sisters and their mother were used for XCI analysis. The XCI studies demonstrated that the proband showed extremely skewed XCI and the XCI pattern of the elder sister was random.
Discussion
CLS is a well-described and rare X-linked dominant syndrome. CLS is caused by highly heterogeneous loss-of-function mutations in the RPS6KA3 gene. The ribosomal protein S6 kinase A3 (RPS6KA3) gene is located in Xp22.12, and it encodes ribosomal protein S6 kinase alpha-3 (UniProtKB-P51812). This transcript of the RPS6KA3 gene has 22 coding exons, a transcript length of 7987 base pairs, and a translation length of 740 amino acids (NM_004586.3). The RPS6KA3 is one of the serine/threonine-protein kinase. RPS6KA3 acts as one of many downstream effectors of the MAP-kinase ERK in the RAS-RAF-MEK-ERK signaling pathway. RPS6KA3 plays an important role in cell-cycle progression, differentiation, and cell survival (Fischer and Raabe, 2018). RPS6KA3-deficient mice displayed spatial learning impairment (Poirier et al., 2007). In RPS6KA3 knock-out mice, proliferation of adult-generated neurons was decreased and no pro-survival effect of learning was observed. Castillon et al. (2018) suggested that decreased young newborn neurons were associated with deficient long-term memory recall. Likewise, poor memory and learning impairment have been observed in our CLS patients.
Based on the authors’ experience, the prevalence of CLS was 1:40,000–1:100,000 (Pereira et al., 2010). CLS is usually characterized by severe-to-profound ID, characteristic craniofacial features, tapered fingers, and musculoskeletal features. Carrier females are less severely impaired. Approximately 14% of affected males and 5% of affected females had cardiovascular diseases (Hanauer and Young, 2002). A total of 13–20% of male CLS and 3–7% of female CLS presented with stimulus-induced drop episodes (SIDEs) (Rojnueangnit et al., 2014). In addition, a few CLS patients presented with diabetes type 2 (Boulos et al., 2021), epileptic seizures (Gschwind et al., 2015), growth retardation (Lv et al., 2019), and hearing or vision problems (Hunter, 2002). The risk of childhood-onset schizophrenia (COS) might be increased in CLS patients (Ambalavanan et al., 2019). Neuroimaging studies demonstrated some brain abnormalities, such as thinning and agenesis of the corpus callosum, mild dilatation of the ventricles, reduced gray- and white-matter volumes, and impacted cerebellum and hippocampus volumes (Wang et al., 2006; Kesler et al., 2007).
In this familial case, these three patients all had the ability to walk, no loss of hearing or vision. For now, there are no signs of SIDEs and epileptic seizures in them. They all had irregular menstruation at puberty. The length of their menstrual cycles kept changing, such as 45 days, 60 days, and 180 days. This symptom has not been previously reported in the literature. Ultrasonography of ovaries and uteruses all showed normal results. They refused to take brain magnetic resonance imaging (MRI) scans.
According to the ACMG standards and guidelines for the interpretation of sequence variants, we interpreted c.898C>T (p.R300X) as a pathogenic variant. This variant caused a premature termination of codon, resulting in strong protein truncation (more than 50% of protein length was missing) (PVS1). It was absent from normal population databases (Exome Sequencing Project, 1000 Genomes Project, the Genome Aggregation Database, and Exome Aggregation Consortium) (PM2). Silico predictive algorithms (SIFT, PolyPhen-2, and Mutation Taster) of pathogenicity all showed that this variant was damaging. This variant was segregated from the disease in multiple family members (PP1). The analysis of conserved sequences suggested that this variant was located in highly conserved sequences across several species (Figure 7) (PP3). Three female patients’ phenotypes and family histories were highly specific for CLS (PP4). Tos et al. (2015) reported that a familial CLS case was caused by the same variant (PP5).

FIGURE 7. In total, 30 amino acids surrounded the variant position (marked with a black box).
As shown in Table 1, the mother and the elder sister had the similar symptoms. They were more severely impaired than the proband. They presented with shorter stature, coarser facial features, tapered fingers, and poorer cognition. We speculated that more severe phenotypes may be connected with the CNV [ del (22) (q11.22-q11.23) (22997582–23637176)]. According to a proposed categorization system (Mikhail et al., 2014), they might be involved with distal 22q11.2 microdeletion syndrome. A cluster of low-copy repeats (LCRs) from A to H in chromosome 22q11.2 (chr22:17,900,001–25,900,000) mediated nonallelic homologous recombination and 22q11.2 chromosomal rearrangements. In the light of a systematic clinical overview of ClinGen curation (Xue et al., 2021), this CNV interval region contained the distal type II (E–F) region (chr22:23,119,414–23,649,111), and overlapped with the distal type I (D–E/F) region (chr22:21,917,117–23,649,111). Deletion of distal type I (D–E/F) or type II (E–F) regions may exhibit variable clinical phenotypes (mild, moderate, or even normal): prenatal growth restriction, short stature, ID, language delay, dysmorphic facial features, skeletal anomalies, cardiovascular defects, behavior problems, genitourinary anomalies, feeding problems, hypotonia, immune deficiency, and hypocalcemia (Burnside, 2015). Taken together, it was tempting to speculate that the CNV in the mother and elder sister made their phenotypes worse than the proband.
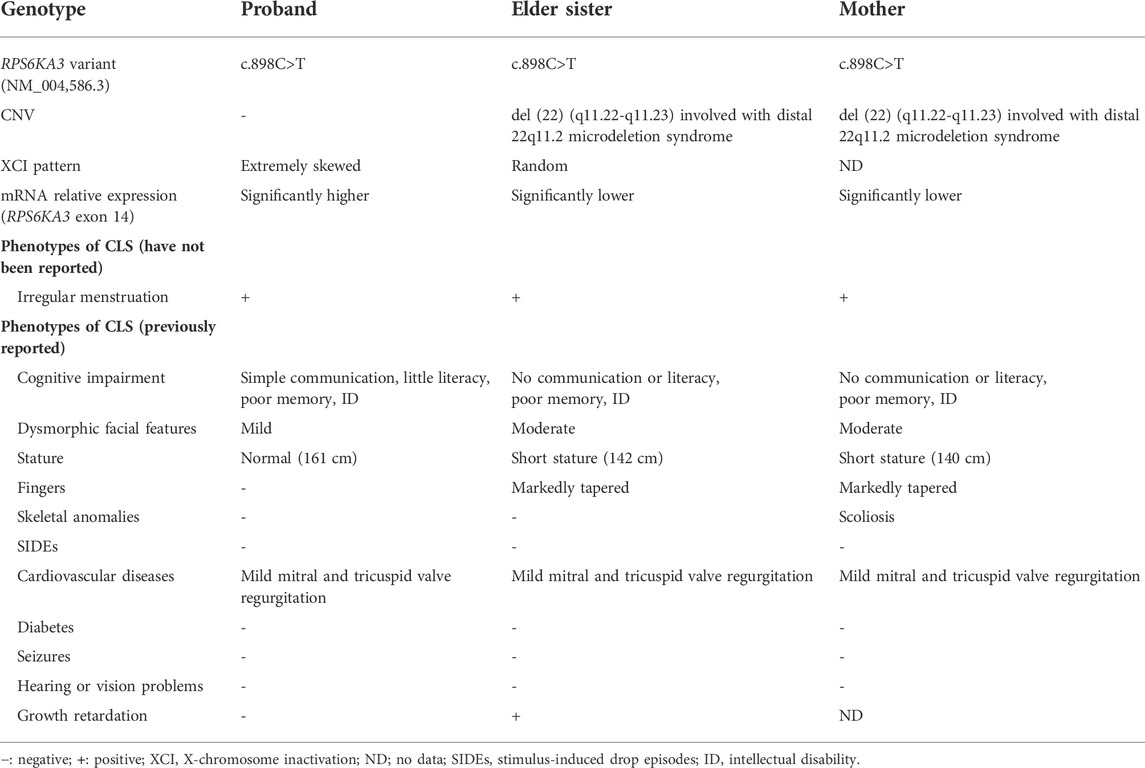
TABLE 1. Summary of genotype and phenotypes of the three female patients.
The two sisters showed different XCI patterns. We did not find a correlation between XCI patterns and phenotypic severity. It should be noted that the XCI pattern might be different in different tissues. Thus, XCI analysis in lymphocytes did not rule out this possibility. Also, the connection between the levels of mRNA (expressed by mutant RPS6KA3 in blood samples) and phenotypic severity should be studied further. As we know, mRNA of the RPS6KA3 gene is highly expressed in skeletal muscle. But we did not perform muscle biopsies, as we thought that it was kind of hard to get some tissues such as skeletal muscle, skin, or brain.
To sum up, severity of clinical features may be markedly variable. If possible, getting some tissues such as skeletal muscle or skin may be more useful for further genetic studies. These three female patients exhibited variable degrees of severity. Clinical characteristics of our CLS patients expanded the phenotypic spectrum of CLS. We hoped that our phenotypic and genetic studies could improve the comprehensive understanding of CLS for pediatricians.
Declarations
Ethics approval and consent to participate
All study procedures were approved by the ethical committee of Yiwu maternity and child health care hospital.
Consent for publication
Written informed consent of publication was obtained from the patients' guardian signed (the father). Also the guardian was informed and agreed to publish patients' photos.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethical Committee of Yiwu Maternity and Child Health Care Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YC and HJ contributed equally to the manuscript, YC and KW wrote the main manuscript text, HW carried out the molecular genetic experimental. HJ and DW prepared the clinical data and imaging data. KW contributed to the checking of revision, genetic evaluation, and genetic databases analysis. KW critically revised the manuscript. All authors reviewed the manuscript. All authors have read and approved the final manuscript.
Acknowledgments
We are grateful to the patients and their families for participation in this study, as well as the help of all the physicians in the course of the medical treatment. We wish to thank the staffs of Chigene (Beijing) Translational Medical Research Center Co. Ltd. for assisting with whole-exome sequencing and Sanger sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.900226/full#supplementary-material
Abbreviations
CLS, Coffin–Lowry syndrome; RPS6KA3, ribosomal protein S6 kinase A3; gDNA, genomic DNA; Trio-WES, trio-based whole-exome sequencing; NV, copy number variation; MMSE, the mini-mental state examination; MRI, magnetic resonance imaging; SIDAs, stimulus-induced drop attacks; XCI, X-chromosome inactivation; COS, childhood-onset schizophrenia (COS).
References
Ambalavanan, A., Chaumette, B., Zhou, S., Xie, P., He, Q., Spiegelman, D., et al. (2019). Exome sequencing of sporadic childhood-onset schizophrenia suggests the contribution of X-linked genes in males. Am. J. Med. Genet. B Neuropsychiatr. Genet. 180 (6), 335–340. doi:10.1002/ajmg.b.32683
Boulos, T., Moukarzel, Y., Yammine, T., Salem, N., Souaid, M., and Farra, C. (2021). Novel missense mutation c.1784A>G, p.Tyr595Cys in RPS6KA3 gene responsible for Coffin-Lowry syndrome in a family with variable features and diabetes 2. Clin. Dysmorphol. 30 (1), 32–35. doi:10.1097/MCD.0000000000000343
Burnside, R. D. (2015). 22q11.21 deletion syndromes: A review of proximal, central, and distal deletions and their associated features. Cytogenet. Genome Res. 146 (2), 89–99. doi:10.1159/000438708
Castillon, C., Lunion, S., Desvignes, N., Hanauer, A., Laroche, S., and Poirier, R. (2018). Selective alteration of adult hippocampal neurogenesis and impaired spatial pattern separation performance in the RSK2-deficient mouse model of Coffin-Lowry syndrome. Neurobiol. Dis. 115, 69–81. doi:10.1016/j.nbd.2018.04.007
Fischer, M., and Raabe, T. (2018). Animal models for coffin-lowry syndrome: RSK2 and nervous system dysfunction. Front. Behav. Neurosci. 12, 106. doi:10.3389/fnbeh.2018.00106
Gschwind, M., Foletti, G., Baumer, A., Bottani, A., and Novy, J. (2015). Recurrent nonconvulsive status epilepticus in a patient with Coffin-Lowry Syndrome. Mol. Syndromol. 6 (2), 91–95. doi:10.1159/000430429
Hanauer, A., and Young, I. D. (2002). Coffin-lowry syndrome: Clinical and molecular features. J. Med. Genet. 39 (10), 705–713. doi:10.1136/jmg.39.10.705
Hunter, A. G. (2002). Coffin-lowry syndrome: A 20-year follow-up and review of long-term outcomes. Am. J. Med. Genet. 111 (4), 345–355. doi:10.1002/ajmg.10574
Kesler, S. R., Simensen, R. J., Voeller, K., Abidi, F., Stevenson, R. E., Schwartz, C. E., et al. (2007). Altered neurodevelopment associated with mutations of RSK2: A morphometric MRI study of coffin-lowry syndrome. Neurogenetics 8 (2), 143–147. doi:10.1007/s10048-007-0080-6
Li, H., Jia, J., and Yang, Z. (2016). Mini-mental state examination in elderly Chinese: A population-based normative study. J. Alzheimers Dis. 53 (2), 487–496. doi:10.3233/JAD-160119
Lv, Y., Zhu, L., Zheng, J., Wu, D., and Shao, J. (2019). Growth concerns in coffin-lowry syndrome: A case report and literature review. Front. Pediatr. 6, 430. doi:10.3389/fped.2018.00430
Mikhail, F. M., Burnside, R. D., Rush, B., Ibrahim, J., Godshalk, R., Rutledge, S. L., et al. (2014). The recurrent distal 22q11.2 microdeletions are often de novo and do not represent a single clinical entity: A proposed categorization system. Genet. Med. 16 (1), 92–100. doi:10.1038/gim.2013.79
Pangman, V. C., Sloan, J., and Guse, L. (2000). An examination of psychometric properties of the mini-mental state examination and the standardized mini-mental state examination: Implications for clinical practice. Appl. Nurs. Res. 13 (4), 209–213. doi:10.1053/apnr.2000.9231
Pereira, P. M., Schneider, A., Pannetier, S., Heron, D., and Hanauer, A. (2010). Coffin-Lowry syndrome. Eur. J. Hum. Genet. 18 (6), 627–633. doi:10.1038/ejhg.2009.189
Poirier, R., Jacquot, S., Vaillend, C., Soutthiphong, A. A., LibbeyM., , DaviS, S., et al. (2007). Deletion of the Coffin-Lowry syndrome gene Rsk2 in mice is associated with impaired spatial learning and reduced control of exploratory behavior. Behav. Genet. 37 (1), 31–50. doi:10.1007/s10519-006-9116-1
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rojnueangnit, K., Jones, J. R., Basehore, M. J., and Robin, N. H. (2014). Classic phenotype of Coffin-Lowry syndrome in a female with stimulus-induced drop episodes and a genotype with preserved N-terminal kinase domain. Am. J. Med. Genet. A 164A (2), 516–521. doi:10.1002/ajmg.a.36299
Tos, T., Alp, M. Y., Aksoy, A., and HAnAuer, A. (2015). A familial case of Coffin-Lowry syndrome caused by RPS6KA3 C.898C>T mutation associated with multiple abnormal brain imaging findings. Genet. Couns. 26 (1), 47–52.
Wang, Y., Martinez, J. E., Wilson, G. L., He, X. Y., Tuck-Muller, C. M., Maertens, P., et al. (2006). A novel RSK2 (RPS6KA3) gene mutation associated with abnormal brain MRI findings in a family with Coffin-Lowry syndrome. Am. J. Med. Genet. A 140, 1274–1279. doi:10.1002/ajmg.a.31266
Xue, J., Shen, R., Xie, M., Liu, Y., Zhang, Y., Gong, L., et al. (2021). 22q11.2 recurrent copy number variation-related syndrome: A retrospective analysis of our own microarray cohort and a systematic clinical overview of ClinGen curation. Transl. Pediatr. 10 (12), 3273–3281. doi:10.21037/tp-21-560
Keywords: Coffin–Lowry syndrome, loss-of-function, intellectual disability, menstrual disorder, whole-exome sequencing, RPS6KA3 c.898C>T mutation
Citation: Cong Y, Jin H, Wu K, Wang H and Wang D (2022) Case Report: Chinese female patients with a heterozygous pathogenic RPS6KA3 gene variant c.898C>T and distal 22q11.2 microdeletion. Front. Genet. 13:900226. doi: 10.3389/fgene.2022.900226
Received: 20 March 2022; Accepted: 01 July 2022;
Published: 15 August 2022.
Edited by:
Dimitra Kiritsi, University of Freiburg Medical Center, GermanyReviewed by:
Hongbin Lv, Southwest Medical University, ChinaKitiwan Rojnueangnit, Thammasat University Hospital, Thailand
Copyright © 2022 Cong, Jin, Wu, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Wu, NzU0Mjk5MDU4QHFxLmNvbQ==
†These authors have contributed equally to this work