 R. H. Ahmed
R. H. Ahmed C. Schmidtmann
C. Schmidtmann J. Mugambe
J. Mugambe G. Thaller1
G. Thaller1- 1Institute of Animal Breeding and Husbandry, Christian-Albrechts-University Kiel, Kiel, Germany
- 2IT Solutions for Animal Production (vit), Verden (Aller), Germany
Background: Beef on Dairy (BoD) calves are born from the crossing of dairy cows with beef breeds. The genetic architecture of these calves differs significantly from the parent breeds due to heterosis and other dominance effects. Identification of the genomic regions associated with traits in BoD calves and the inheritance pattern of these regions can assist in the selection process. We conducted a genome-wide association study (GWAS) for Belgian blue and Angus crossbreds born from a Holstein dam, incorporating additive and dominance effects to identify genomic regions associated with birth weight, calving difficulty, and gestation length. Additionally, a haplotype-based GWAS was performed to compare the effectiveness of these two different methodologies and to identify the parental origin of the haplotypes based on similar allelic patterns between crossbred and parental breeds.
Results: The heritability estimates for birth weight, calving difficulty, and gestation length were 0.29 (±0.03), 0.36 (±0.04), and 0.09 (±0.03), respectively. Using SNP-based GWAS for birth weight, a genomic region containing the GABRG1 gene on BTA 6 was identified. In addition, the haplotype-based analysis identified three more genes (CSER1, FAM13A, and LCORL) associated with birth weight. Incorporating dominance effects into the GWAS model led to the identification of an additional gene, SPP1, related to birth weight. For calving difficulty, SNP-based GWAS in Angus crossbreds revealed a genomic region containing the KCNIP4 gene. Most of the haplotypes associated with these traits originated from the three parental breeds, but six unique haplotypes for Angus and Belgian blue were identified.
Conclusion: Based on this study, Haplotype GWAS was found to have superior statistical power in the identification of associated genomic regions in BoD crossbreds. However, for traits such as calving difficulty, SNP-based GWAS proved to be more effective. Both approaches are essential for the identification of genomic regions associated with traits of interest in BoD calves.
Introduction
Mating dairy cows with beef bulls (Beef-on-Dairy, BoD) has become increasingly popular in recent years. This trend is aimed at producing calves with higher monetary value because such calves are expected to have better growth rates and superior carcass characteristics compared with purebred dairy calves (Bittante et al., 2023). However, at the same time, risk of calving difficulty is higher when applying BoD. The birth weight (BW) of the calf and the gestation length (GL) of the dam are well-known factors that influence calving difficulty (CD) (Kargo et al., 2014; Jenkins et al., 2016). In this regard, selection of beef sires to be used in dairy herds, which show a balanced genetic potential for optimal growth and calving ease are of high interest for farmers. To identify such bulls in view of negative genetic correlations between birth weight and calving ease requires a better understanding of the genetic architecture of relevant traits. This is especially important in crossbreeding systems where the performance of crossbred calves may vary from the purebred counterparts also due to heterosis and breed complementary effect (González-Diéguez et al., 2020; Khansefid et al., 2020). In the last 2 decades, genome-wide association studies (GWAS) have demonstrated their large potential to identify genes associated with different traits in cattle breeding, thereby pinpointing the selection process (Zhang et al., 2022). Traditionally, GWAS in purebred populations focuses on the additive effects associated with single-nucleotide polymorphisms (SNP), but for crossbred populations with differences in genetic architecture, the inclusion of dominance effects can help in identifying quantitative trait loci (QTL) influencing traits with low to moderate heritability (h2) (Zhang et al., 2008). Dominance described as non-additive interactions of different alleles at a specific locus is a major phenomenon to explain heterosis effect in animal breeding (Visscher et al., 2000). Especially in crosses of two different breeds interactions of differently selected alleles can account for a major fraction of the genetic variation (Cui et al., 2023). Historically, estimation of dominance based on pedigree data has been notoriously difficult due to the requirement of large numbers of full-sib families. But since the development and accessibility of large SNP panels, reliable estimation of dominance effects has become possible (Vitezica et al., 2013). Along with the SNP-based GWAS, GWAS based on haplotypes can also be of significant interest when analyzing crossbred populations due to the inheritance of such blocks with lower probability of recombination (Gabriel et al., 2002; Bovo et al., 2021). It has been shown that GWAS based on haplotypes can outperform SNP-based GWAS by exploiting aggregated effects of consecutive SNPs for quantitative traits, where individual loci usually have a small effect (Bickel et al., 2011). Additionally, haplotype blocks can be used to determine the parental origin of the regions with significant association for the traits of interest (Vandenplas et al., 2016). This can assist in making informed decisions about the selection of the sires with desired genetic architecture for the traits of economic interests. This study compares different approaches of GWAS based on SNP and haplotype blocks in BoD crossbreds for the traits BW, GL and CD and identifies genomic regions harbouring relevant genes associated with the traits. GWAS analyses are based on single SNPs as well as on haplotypes. In addition, haplotype blocks with significant association are traced to the respective parent breed to identify the origin of the gene variants in the crossbred population.
Materials and methods
Data and trait description
This study used 4,118 BoD crossbred calves sired by Belgian Blue (WBB) or Angus (ANG) and born to Holstein (HOL) dams. Between December 2021 to December 2023, the weight of calves was recorded once at the age of 0–40 days on 225 dairy farms in Schleswig-Holstein, Germany. Information regarding the insemination date and parity number of the dam, birth date and sex of the calf, type of birth (singleton or twin) and calving difficulty (CD) recorded on the official German scale (Arbeitsgemeinschaft Deutscher Rinderzüchter; organization of cattle production in Germany) and converted into binary scale (coded, 0 = no difficulty, 1 = difficult calving) was available. Gestation length (GL) was calculated as the duration (in days) between the date of insemination and the date of calving. Recorded values for weight and gestation length deviating ±4 SD from the mean were removed to filter for outliers. Additionally, twin calvings and farms with less than two observations were excluded from the analysis. After editing, 285 animals were excluded from the analysis and the final dataset contained 3,833 calves from 116 farms.
3,530 crossbred calves were genotyped using EuroG MD BeadChip (Illumina Inc). Quality control of genotypes was performed separately within ANG and WBB crossbreds, respectively, using PLINK 1.9 (Chang et al., 2015). SNPs with a minor allele frequency of <1%, a call rate lower than 90% and SNPs that deviate from the Hardy–Weinberg equilibrium at threshold of 1 × 10−6 along with variants located on sex chromosomes were excluded from the analysis (Supplementary Table S1).
Estimation of birth weight of calves
In order to approximate the birth weight (BW) of the calves recorded not at the day of birth, a correction of measured weight was performed within each breed using the following linear regression model (Equation 1):
where yg was the measured weight (kg) of a calf within the sire breed WBB or ANG, AGEg represented the regression of calf’s age at the day of measurement (g = 0, … ,40) and e.g., was the residual term. The model was fitted in R (Becker et al., 1988) using the package lme4 (Bates et al., 2015). BW of the calf was calculated by subtracting the average predicted weight gain over the period of time from the measured weight of the calf.
Analysis of population structure
Population stratification of the animals under study was examined using principal component analysis (PCA) in the individual breeds but also in the combined population (COM) via PLINK 1.9 (Chang et al., 2015). The first three components of PCA were used for visualization.
Haplotype phasing and block construction
Haplotype phasing was conducted using SHAPEIT v2 (Delaneau et al., 2013) for each autosomal chromosome with default parameters for MCMC iteration combined with pedigree information using–duoHMM option to apply pedigree based post hoc haplotypes correction (O’connell et al., 2014). Haplotypes for Bos taurus autosomes (BTA) 1 to 29 were combined and converted into the binary format using the R package GHap (Utsunomiya et al., 2016). For further analysis, haplotype blocks were generated with a sliding window of five consecutive SNPs as this approach has been found to have the highest power to detect associated QTLs (Braz et al., 2019).
Variance components and statistical model for GWAS
Genetic parameters for BW, GL and CD were estimated by using both univariate additive models and dominance models. SNP-based variance components were estimated using restricted estimation of maximum likelihood (REML) using the software GCTA (Yang et al., 2011) while narrow sense heritability for the trait was estimated as
GWAS were performed for each trait using univariate single SNP regression mixed linear models in GCTA (Yang et al., 2011). The additive model was (Equation 2):
where y is the vector of phenotypes (BW, GL or CD); b is a vector of fixed effects (herd-year-season, sex, parity number, first three principal components as covariates), a is the vector of polygenic effects with
For the dominance model (Equation 3), a modified version of Equation 2 was used:
where d is the vector of dominance effects with
where m is the number of SNPs, wD(ij) and wD(ik) dominance encoded genotypes,
Linkage disequilibrium and annotation of associated SNPs
Linkage disequilibrium (LD) for each crossbred population was measured as the correlation coefficient r2 (Hill and Robertson, 1968) by using PLINK v1.9 (Chang et al., 2015). LD decay between adjacent markers up to the distance of 500 kb was visualized in R using package ggplot2 (Wickham, 2011). For the functional annotation of SNPs with significant associations, base pair position was checked within 150 kb up- and downstream of the respective SNPs against the genome assembly Bos taurus UMD 3.1.1 to find relevant genes in the region. This distance was defined based on the LD decay (Qanbari, 2019), where the highest drop in first 150 kb of distance was observed in both crossbred populations (Additional file 1: F1). For haplotype blocks, screening for genes was limited to the significantly associated blocks in the analysis. The origin of significant haplotype blocks was determined by comparing them with parental haplotypes. This was achieved by matching the blocks with the corresponding dam and sire haplotypes with same block positions and allelic patterns to confirm their parental origins (Eiríksson et al., 2021a).
Results
Descriptive statistics
Descriptive statistics for the crossbred calves are depicted in Table 1. The distribution of the calves per parity number and age group at weight measurement is shown in Additional file 1: F2 & F3. The majority of calves belonged to WBB (Supplementary Table S2). These crossbreds had slightly higher daily weight gain (0.77 kg) compared to ANG crossbreds (0.66 kg). Moreover, calves from WBB had higher birth weight (48.5 kg ± 7.2) and longer gestation length (281.4d ± 4.7) compared to ANG sired calves (BW: 45.2 kg ± 7.2; GL: 280.1d ± 4.7). Incidences of calving difficulty were higher for ANG crossbreds, where 12.5% of calvings were labeled as difficult calving compared to WBB crossbreds with 11.0% difficult calvings.
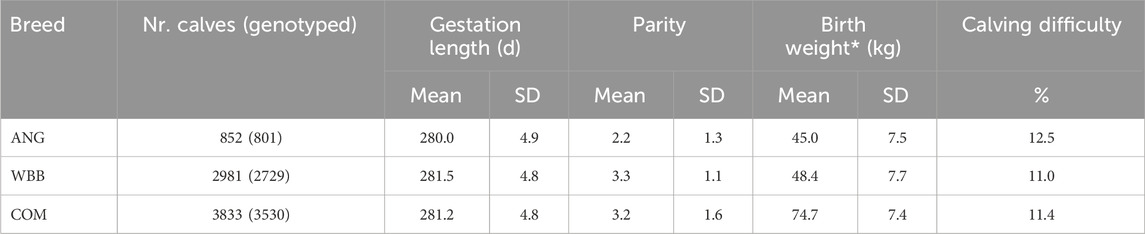
Table 1. Descriptive statistics of recorded traits of crossbred calves by sire breed.
Principal component analysis
Based on the PCA (Figure 1), clear clusters were identified. PC1 and PC2 collectively explained 48% of the variation in the combined data. Slight overlapping was observed for crossbred population based on PC1 and PC2 but with the addition of PC3 (58% variance), distinct clusters were observed across two studied populations (Additional file 1: F3, F4, F5)
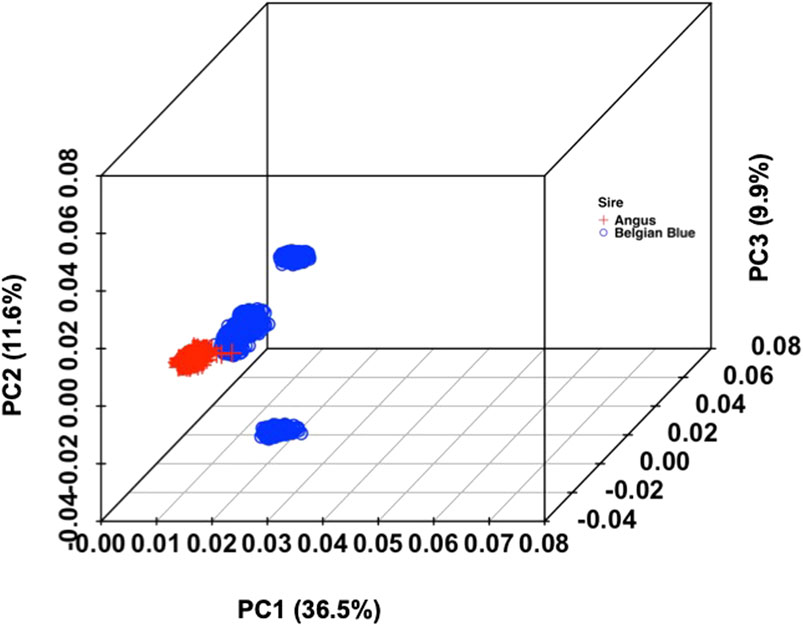
Figure 1. Population structure of the crossbred calves.
Haplotype blocks and variance components
Descriptive statistics for haplotype blocks are shown in Table 2. In total 11,160 haplotype blocks were created. In the combined population, highest number of haplotype blocks were observed on BTA1 (n = 726) while the lowest number of blocks were found on BTA28 (n = 210). Further details regarding haplotype blocks and number of markers per chromosome are presented in Additional file 1: F6, F7.
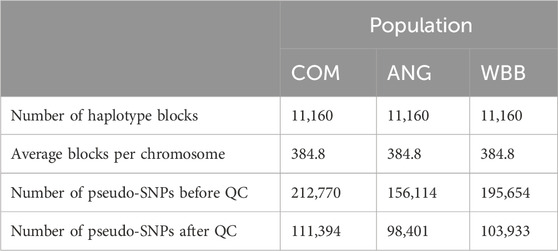
Table 2. Descriptive statistics of haplotype blocks in crossbred population.
Estimates of heritability (s.e.) for BW, GL and CD in the combined population using the additive model were 0.29 (0.03), 0.36 (0.04) and 0.09 (0.03), respectively. For WBB, Dominance variation at all SNPs
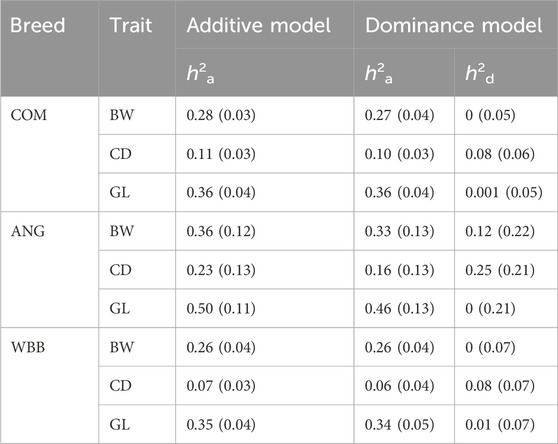
Table 3. Heritability estimates for birth weight, calving ease and gestation length.
Genome-wide associations of traits
Birth weight
In the GWAS with the combined crossbred population, one significantly associated SNP on BTA 6 around 6.26 Mb was found based on the additive model (Figure 2). This SNP falls in the region of gene GABRG1, which is responsible for the regulation of the ion channel and receptor activity along with the influence on puberty development (Tahir et al., 2021).
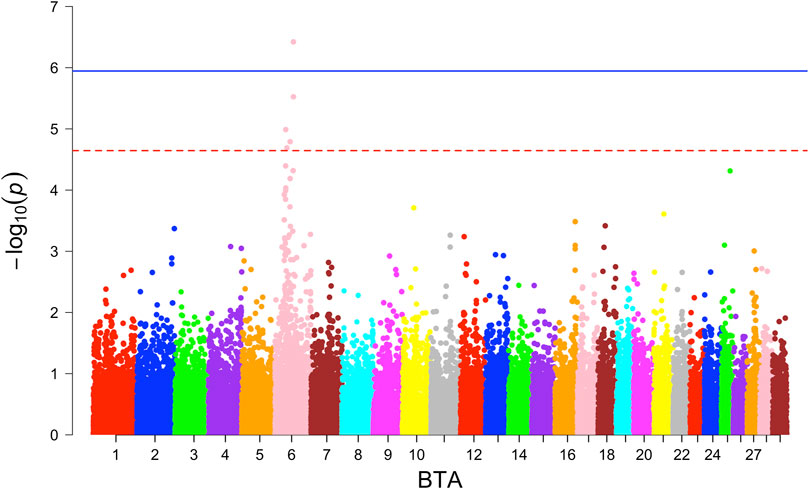
Figure 2. Manhattan plot for combined population GWAS for birth weight.
At the suggestive significance level, four SNPs were identified through the additive model, while an additional SNP on BTA 6 at 38.13 Mb was identified through the dominance model (Table 4).
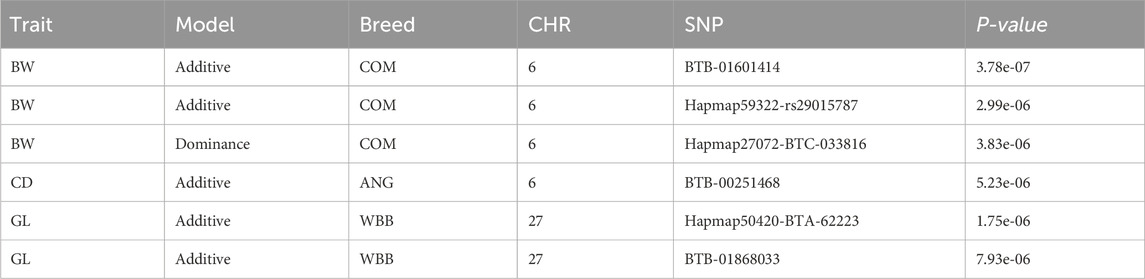
Table 4. Description of SNPs with significant association with traits.
For the BW, GWAS based on haplotypes revealed eight haplotype blocks on BTA 6, where three additional genes (CCSER1, FAM13A, LCORL) are located (Table 5). Furthermore, 14 haplotype blocks within 10 unique regions were identified (Supplementary Table S4) with suggestive association. Along with the previously mentioned three genes, seven more genes (SPP1, GABRG1, HERC6, LOC104972722, ADGRL3, SNCA, and PPARGC1A) were found in the associated genomic regions.
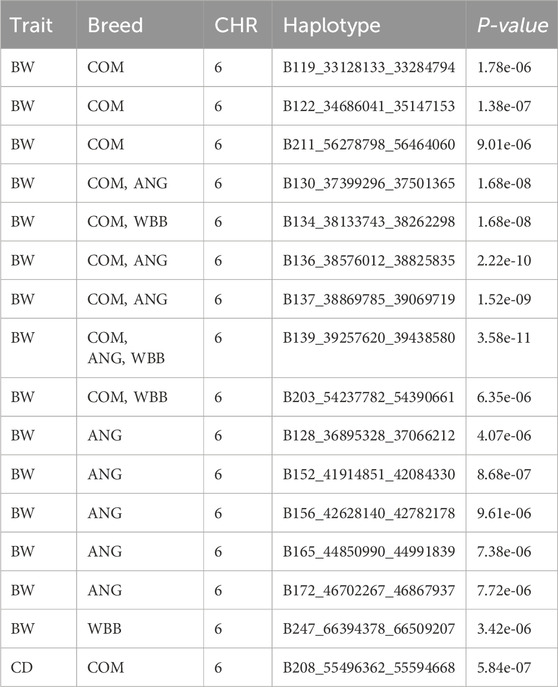
Table 5. Description of haplotypes with significant association with traits.
Calving difficulty
Neither additive nor dominance models identified SNPs with significant associations for the combined or individual WBB crossbred population. In the ANG crossbreds, two SNPs were identified on BTA 6 through the additive model (Figure 3) with the suggestive level of association, where SNP at 42.05 Mb harbouring the KCNIP4 gene in nearby region. Haplotype-based GWAS identified one haplotype block with suggestive association in the combined population (Supplementary Table S4).
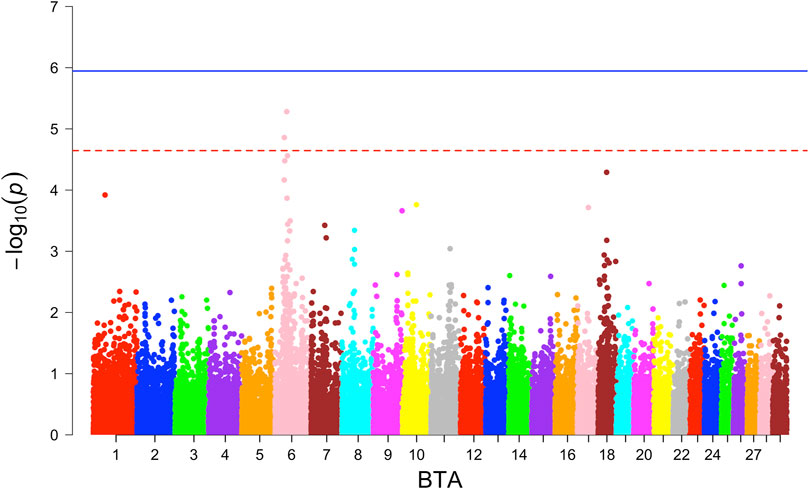
Figure 3. Manhattan plot for Angus crossbred population GWAS for calving difficulty.
Gestation length
For GL, two SNPs with suggestive associations were observed for WBB based on the additive model (Figure 4), while no associations were observed with either the additive or dominance model in the combined and ANG crossbreds. No haplotype block with significant association was identified across all crossbreds.
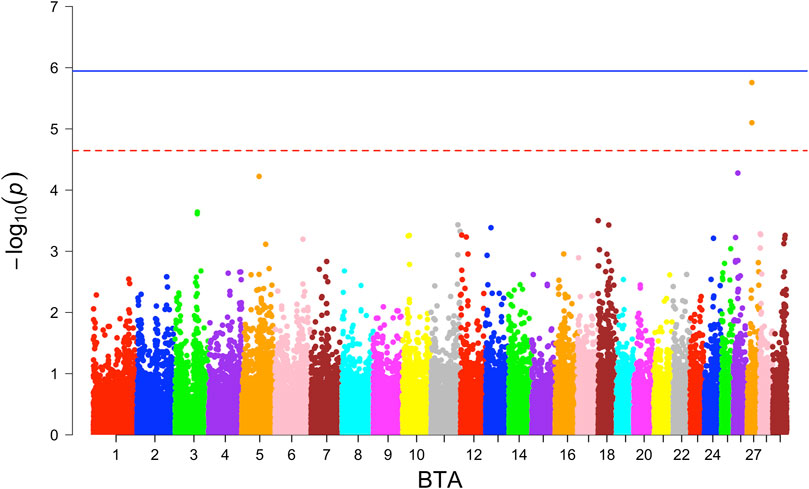
Figure 4. Manhattan plot for Belgian Blue crossbred population GWAS for gestation length.
Parental origin of haplotype blocks
For the haplotype blocks along with the presence of the gene in the genomic region, a comparison of the block with the respective allelic pattern was made to the maternal and paternal blocks. The majority of the blocks for the BW could be traced back to the HOL breed but for blocks with suggestive association, unique blocks were identified for the parental breeds (Table 6; Supplementary Table S5).
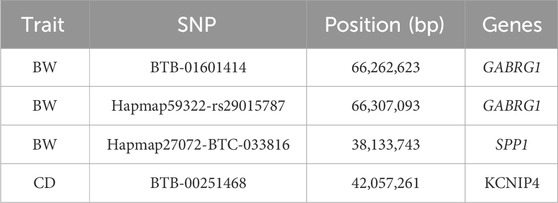
Table 6. Genes encoded with significantly associated SNPs.
Discussion
Heritability of traits
The study used two distinctive models to estimate heritability for BW, CD and GL. The effect of crossbreeding on heritability estimates depends upon the population and varies from trait to trait, but generally slightly lower heritability is expected for crossbred populations (Wientjes and Calus, 2017). This trend is also observed in the current study where BW heritability for WBB (0.26) crossbreds is lower compared to the purebred WBB (0.38) calves (Mota et al., 2017). However, heritability for ANG crossbred is slightly higher (0.36) compared to purebred ANG (0.33) (Torres-Vázquez et al., 2018). Non-additive genetic variance is expected to be higher in crossbred populations resulting in lower estimates for heritability (Fuerst and Solkneri, 1994). In the current study heritability estimates for CD varied from 0.11 (0.03) in the combined population to 0.07 for ANG and WBB BoD calves. These estimates are closer to the one reported in British herds, where heritability for CD in BoD crossbreds is reported to be 0.09 (0.01) (McGuirk et al., 1998), but lower to the once reported for purebred ANG (0.21) and WBB (0.25–0.34) breed (Cubas et al., 1991; Vanderick et al., 2017).
Heritability for GL varied from medium 0.36 (0.04) in combined crossbreds to high in ANG BoD 0.50 (0.11). The estimates for WBB and ANG BoD are closer to the purebred counterparts with 0.33 (0.04) for WBB and 0.59 (0.01) for ANG calves (Mota et al., 2017; Gilleland et al., 2021). A strong influence of the sire breed on gestation length is expected and can be the reason for the higher heritability estimates of ANG BoD calves (Haile-Mariam and Pryce, 2019). Heritability estimates can vary across the populations based on traits under observation, but generally traits with higher h2 in purebred animals will also have higher h2 in crossbred population (Wientjes and Calus, 2017). For the traits with dominant genes, crossbred animals may have higher additive genetic variance compared to the purebred animals (Wei et al., 1991). Comparison across purebred and crossbred populations is challenging due to the higher environmental variance in crossbred populations due to the scale effect (Habier et al., 2007), although no general trend is observed in this regard (Wientjes and Calus, 2017). Compared to purebred animals, the less controlled environmental conditions and greater variability in farming practices associated with crossbred animals may contribute to the observed differences in this regard (Wei and Van der Werf, 1995).
GWAS based on additive, dominance and haplotypes
Utilization of GWAS can help to understand the genetic architecture of relevant traits. In this study, GWAS was performed using three different models for BW, CD and GL in BoD crossbreds. GWAS based on haplotypes outperformed the SNP-based GWAS in terms of finding the associated genes for BW, but no significant differences were observed for GL and CD. The addition of dominance effects in the GWAS model was beneficial for BW only in terms of finding genomic regions with significant or suggestive associations. Based on SNP and haplotype GWAS, in total five genes with significant association and seven genes with suggestive association were identified for the BW trait. Role of these genes varied from ion-channels regulation to regulation of mineralization process of the bone. GABRG1 gene, identified through significant SNP association is responsible for the regulation of the ion channel and receptor activity along with the influence on puberty development (Tahir et al., 2021). The association of this gene with milk yield has also been previously reported in the Holstein population (Pedrosa et al., 2021). SPP1 gene identified based on the dominance model encodes for multifunctional cytokines (Matsumoto et al., 2019) and is vital for bone mineralization process (Rodriguez et al., 2014). Association between SPP1 gene and growth traits including yearling weight, live weight and average daily gain has been suggested in American beef crossbred herds (White et al., 2007). Moreover, the influence of this gene on the birth weight (Allan et al., 2007) and carcass weight has been established in beef cattle (Matsumoto et al., 2019). For the genes identified through haplotypes-based GWAS, the influence of CCSER1, KCNIP4, and LCORL genes has been reported on BW and growth traits in the Angus breed (Xia et al., 2017; Smith et al., 2022). The CCSER1 gene, controlling mitosis has association with milk yield in Holstein cows (Teng et al., 2023). Similarly, PPARGC1A gene, influencing fat deposition and energy metabolism has been associated with BW (Pasandideh, 2020). Previously FAM13A gene with regulatory role in metabolism has already been described in the Holstein breed for association with bone structure (Niu et al., 2021; Dominguez-Castaño et al., 2024) Moreover, associations between PPARGC1A, a gene with significant role in fat metabolism (Komisarek and Walendowska, 2012) and yearling weight along with carcass traits have been observed in beef cattle (Fonseca et al., 2015). In this study, higher statistical power was observed for Haplotype-based GWAS in identifying associations and candidate regions. Similar results have been previously described in various species, including cattle (Pryce et al., 2010; Barendse and Schneider, 2011) and pigs (Sato et al., 2016). To some extent these differences can be attributed to the window size for haplotypes and the number of markers per haplotype can potentially influence the accuracy of QTL mapping (Calus et al., 2009). In contrast to aforementioned studies, SNP-based GWAS was found to be more efficient for carcass traits in Simmental cattle (Wu et al., 2014).
Parental origin of haplotype blocks
Interest in the approaches in the partitioning of crossbred genome has gained significant interest in the recent years for the crossbred evaluation. Various methods with varying degree of success have been developed including breed base representation (BBR) and breed of origin of the allele (BOA) (VanRaden et al., 2020; Eiríksson et al., 2021b; Guillenea et al., 2023). Despite the advancements in the statistical models and the increased number of genotyped animals, 100% precise estimation of the share of the genome of each parent in the crossbred population is difficult due to the shared DNA content between breeds (VanRaden et al., 2020). In this study, despite the partial success of assigning haplotypes to the parental breeds, complete segregation of the haplotypes of the crossbred calves was not successful. For most of the haplotypes with significant associations, the origin of the haplotypes was traced back to both parental haplotypes, though six breed-specific haplotypes with similar pattern of alleles were identified for ANG (n = 3) and WBB (n = 3) breeds. All of these six haplotypes were also present in HOL population (Supplementary Table S5).
Limitations of the study
A key limitation of the current study was the insufficient number of genotyped dams as this may reduce the accuracy of haplotype phasing. Moreover, for calving difficulty and gestation length, maternal influence is significant, and unavailability of sufficient number of maternal genotypes and phenotype records can be a limiting factor in this regard. Furthermore, the unequal number of crossbreds from two beef breeds can have impact on the accuracy of genetic parameters and association studies.
Conclusion
Models based on combined populations resulted in the identification of more associated genes compared to the separate crossbred populations. Heritability estimates for BW and GL in BoD crossbred populations were similar to those found in purebred populations. However, the heritability estimates for CD were comparatively lower. Furthermore, combining crossbred populations with at least one common parent breed can be a viable strategy for estimation of SNP based genetic parameters and association studies. Incorporating dominance effects in both GWAS and heritability estimates resulted in modest improvements in model performance. Additionally, haplotype-based GWAS proved better in identifying genes associated with traits in crossbred animals compared to traditional SNP-based approaches. The origin of the majority of haplotype blocks with significant and suggestive associations can be traced back to the HOL breed, though some sire’s specific blocks were also identified in the current study.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: Ahmed (2025).
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because This data collection falls under regular evaluation and does not require any approval.
Author contributions
RA: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing. CS: Methodology, Supervision, Writing – original draft, Writing – review and editing. JM: Writing – original draft, Writing – review and editing. GT: Formal Analysis, Funding acquisition, Supervision, Validation, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study is part of the project GenoCross (funding code 28RZ3101) and was financially supported by the Federal Ministry of Food and Agriculture Germany (BMEL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1530310/full#supplementary-material
References
Ahmed, R. H. (2025). Data from: Genome-wide association studies for productive and reproductive traits in beef-on-dairy crossbreds. figshare. doi:10.6084/m9.figshare.29196206
Allan, M. F., Thallman, R. M., Cushman, R. A., Echternkamp, S. E., White, S. N., Kuehn, L. A., et al. (2007). Association of a single nucleotide polymorphism in SPP1 with growth traits and twinning in a cattle population selected for twinning rate. J. Anim. Sci. 85, 341–347. doi:10.2527/JAS.2006-460
Barendse, W., and Schneider, M. R. (2011). Haplotype analysis improved evidence for candidate genes for intramuscular fat percentage from a genome wide association study of cattle. PLoS One 6, 29601. doi:10.1371/journal.pone.0029601
Bates, D., Mächler, M., Bolker, B. M., and Walker, S. C. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. doi:10.18637/JSS.V067.I01
Becker, R. A., Chambers, J. M., and Wilks, A. R. (1988). The new S language: a programming environment for data analysis and graphics, 702.
Bickel, R. D., Kopp, A., and Nuzhdin, S. V. (2011). Composite effects of polymorphisms near multiple regulatory elements create a major-effect QTL. PLoS Genet. 7, e1001275. doi:10.1371/JOURNAL.PGEN.1001275
Bittante, G., Bergamaschi, M., Qianlin, N., Patel, N., Toledo-Alvarado, H., and Cecchinato, A. (2023). Veal and beef meat quality of crossbred calves from dairy herds using sexed semen and semen from double-muscled sires. Ital. J. Anim. Sci. 22, 169–180. doi:10.1080/1828051X.2023.2171919
Bovo, S., Ballan, M., Schiavo, G., Ribani, A., Tinarelli, S., Utzeri, V. J., et al. (2021). Single-marker and haplotype-based genome-wide association studies for the number of teats in two heavy pig breeds. Anim. Genet. 52, 440–450. doi:10.1111/AGE.13095
Braz, C. U., Taylor, J. F., Bresolin, T., Espigolan, R., Feitosa, F. L. B., Carvalheiro, R., et al. (2019). Sliding window haplotype approaches overcome single SNP analysis limitations in identifying genes for meat tenderness in Nelore cattle. BMC Genet. 20, 8–12. doi:10.1186/s12863-019-0713-4
Calus, M. P., Meuwissen, T. H., Windig, J. J., Knol, E. F., Schrooten, C., Vereijken, A. L., et al. (2009). Effects of the number of markers per haplotype and clustering of haplotypes on the accuracy of QTL mapping and prediction of genomic breeding values. Genet. Sel. Evol. 41, 11–10. doi:10.1186/1297-9686-41-11
Chang, C. C., Chow, C. C., Tellier, L. C. A. M., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
Cubas, A. C., Berger, P. J., and Healey, M. H. (1991). Genetic parameters for calving ease and survival at birth in Angus field data. J. Anim. Sci. 69, 3952–3958. doi:10.2527/1991.69103952X
Cui, L., Yang, B., Xiao, S., Gao, J., Baud, A., Graham, D., et al. (2023). Dominance is common in mammals and is associated with trans-acting gene expression and alternative splicing. Genome Biol. 24, 215–225. doi:10.1186/s13059-023-03060-2
Delaneau, O., Howie, B., Cox, A. J., Zagury, J. F., and Marchini, J. (2013). Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 93, 687–696. doi:10.1016/j.ajhg.2013.09.002
Dominguez-Castaño, P., Fortes, M., Tan, W. L. A., Toro-Ospina, A. M., and Silva, J. A. I. V. (2024). Genome-wide association study for milk yield, frame, and udder conformation traits of Gir dairy cattle. J. Dairy Sci. doi:10.3168/JDS.2024-24648
Eiríksson, J. H., Karaman, E., Su, G., and Christensen, O. F. (2021a). Breed of origin of alleles and genomic predictions for crossbred dairy cows. Genet. Sel. Evol. 53, 1–13. doi:10.1186/S12711-021-00678-3/TABLES/4
Eiríksson, J. H., Karaman, E., Su, G., and Christensen, O. F. (2021b). Breed of origin of alleles and genomic predictions for crossbred dairy cows. Genet. Sel. Evol. 53, 84. doi:10.1186/S12711-021-00678-3
Fonseca, P. D. da S., de Souza, F. R. P., De Camargo, G. M. F., Gil, F. M. M., Cardoso, D. F., Zetouni, L., et al. (2015). Association of ADIPOQ, OLR1 and PPARGC1A gene polymorphisms with growth and carcass traits in Nelore cattle. Meta Gene 4, 1–7. doi:10.1016/J.MGENE.2015.02.001
Fuerst, C., and Solkneri, J. (1994). Additive and nonadditive genetic variances for milk yield, fertility, and lifetime performance traits of dairy cattle. J. Dairy Sci. 77, 1114–1125. doi:10.3168/jds.S0022-0302(94)77047-8
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., et al. (2002). The structure of haplotype blocks in the human genome. Science 296 (296), 2225–2229. doi:10.1126/science.1069424
Gilleland, C., Retallick, K. J., Poole, D. H., Peppmeier, Z., and Knauer, M. (2021). Estimates of genetic variation for Angus gestation length. J. Anim. Sci. 99, 14–15. doi:10.1093/JAS/SKAB096.023
González-Diéguez, D., Tusell, L., Bouquet, A., Legarra, A., and Vitezica, Z. G. (2020). Purebred and crossbred genomic evaluation and mate allocation strategies to exploit dominance in pig crossbreeding schemes. G3 Genes|Genomes|Genetics 10, 2829–2841. doi:10.1534/G3.120.401376
Guillenea, A., Lund, M. S., Evans, R., Boerner, V., and Karaman, E. (2023). A breed-of-origin of alleles model that includes crossbred data improves predictive ability for crossbred animals in a multi-breed population. Genet. Sel. Evol. 55, 34–12. doi:10.1186/s12711-023-00806-1
Habier, D., Götz, K. U., and Dempfle, L. (2007). Estimation of genetic parameters on test stations using purebred and crossbred progeny of sires of the Bavarian Piétrain. Livest. Sci. 107, 142–151. doi:10.1016/J.LIVSCI.2006.09.012
Haile-Mariam, M., and Pryce, J. E. (2019). Genetic evaluation of gestation length and its use in managing calving patterns. J. Dairy Sci. 102, 476–487. doi:10.3168/JDS.2018-14981
Hill, W. G., and Robertson, A. (1968). Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38, 226–231. doi:10.1007/BF01245622
Jenkins, G. M., Amer, P., Stachowicz, K., and Meier, S. (2016). Phenotypic associations between gestation length and production, fertility, survival, and calf traits. J. Dairy Sci. 99, 418–426. doi:10.3168/jds.2015-9934
Kargo, M., Hjortø, L., Toivonen, M., Eriksson, J. A., Aamand, G. P., and Pedersen, J. (2014). Economic basis for the nordic total merit index. J. Dairy Sci. 97, 7879–7888. doi:10.3168/JDS.2013-7694
Khansefid, M., Goddard, M. E., Haile-Mariam, M., Konstantinov, K. V., Schrooten, C., de Jong, G., et al. (2020). Improving genomic prediction of crossbred and purebred dairy cattle. Front. Genet. 11, 598580. doi:10.3389/fgene.2020.598580
Komisarek, J., and Walendowska, A. (2012). Analysis of the PPARGC1A gene as a potential marker for productive and reproductive traits in cattle. Folia Biol. Czech Repub. 60, 171–174. doi:10.3409/FB60_3-4.171-174
Matsumoto, H., Kohara, R., Sugi, M., Usui, A., Oyama, K., Mannen, H., et al. (2019). The non-synonymous mutation in bovine SPP1 gene influences carcass weight. Heliyon 5, e03006. doi:10.1016/J.HELIYON.2019.E03006
McGuirk, B. J., Going, I., and Gilmour, A. R. (1998). The genetic evaluation of beef sires used for crossing with dairy cows in the UK 1. Sire breed and non-genetic effects on calving survey traits. Animal Sci. 66, 35–45. doi:10.1017/S135772980000881X
Mota, R. R., Mayeres, P., Bastin, C., Glorieux, G., Bertozzi, C., Vanderick, S., et al. (2017). Genetic evaluation for birth and conformation traits in dual-purpose Belgian Blue cattle using a mixed inheritance model. J. Anim. Sci. 95, 4288–4299. doi:10.2527/JAS2017.1748
Niu, Q., Zhang, T., Xu, L., Wang, T., Wang, Z., Zhu, B., et al. (2021). Identification of candidate variants associated with bone weight using whole genome sequence in beef cattle. Front. Genet. 12, 750746. doi:10.3389/fgene.2021.750746
O’connell, J., Gurdasani, J., Delaneau, D., Pirastu, O., Ulivi, N., Cocca, M., et al. (2014). A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, 1004234. doi:10.1371/journal.pgen.1004234
Pasandideh, M. (2020). Two SNPs in the bovine PPARGC1A gene are associated with the birth weight of Holstein calves. Meta Gene 25, 100732. doi:10.1016/J.MGENE.2020.100732
Pedrosa, V. B., Schenkel, F. S., Chen, S. Y., Oliveira, H. R., Casey, T. M., Melka, M. G., et al. (2021). Genomewide association analyses of lactation persistency and milk production traits in holstein cattle based on imputed whole-genome sequence data. Genes (Basel) 12, 1830. doi:10.3390/genes12111830
Pryce, J. E., Bolormaa, S., Chamberlain, A. J., Bowman, P. J., Savin, K., Goddard, M. E., et al. (2010). A validated genome-wide association study in 2 dairy cattle breeds for milk production and fertility traits using variable length haplotypes. J. Dairy Sci. 93, 3331–3345. doi:10.3168/jds.2009-2893
Qanbari, S. (2019). On the extent of linkage disequilibrium in the genome of farm animals. Front. Genet. 10, 1304. doi:10.3389/FGENE.2019.01304
Rodriguez, D. E., Thula-Mata, T., Toro, E. J., Yeh, Y. W., Holt, C., Holliday, L. S., et al. (2014). Multifunctional role of osteopontin in directing intrafibrillar mineralization of collagen and activation of osteoclasts. Acta Biomater. 10, 494–507. doi:10.1016/J.ACTBIO.2013.10.010
Sato, S., Uemoto, Y., Kikuchi, T., Egawa, S., Kohira, K., Saito, T., et al. (2016). SNP-and haplotype-based genome-wide association studies for growth, carcass, and meat quality traits in a Duroc multigenerational population. BMC Genet. 17, 60. doi:10.1186/s12863-016-0368-3
Smith, J. L., Wilson, M. L., Nilson, S. M., Rowan, T. N., Schnabel, R. D., Decker, J. E., et al. (2022). Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genomics 23 (1 23), 517–522. doi:10.1186/S12864-022-08667-6
Tahir, M. S., Porto-Neto, L. R., Gondro, C., Shittu, O. B., Wockner, K., Tan, A. W. L., et al. (2021). Meta-analysis of heifer traits identified reproductive pathways in bos indicus cattle. Genes (Basel) 12, 768. doi:10.3390/genes12050768
Teng, J., Wang, D., Zhao, C., Zhang, X., Chen, Z., Liu, J., et al. (2023). Longitudinal genome-wide association studies of milk production traits in Holstein cattle using whole-genome sequence data imputed from medium-density chip data. J. Dairy Sci. 106, 2535–2550. doi:10.3168/JDS.2022-22277
Torres-Vázquez, J. A., van der Werf, J. H. J., and Clark, S. A. (2018). Genetic and phenotypic associations of feed efficiency with growth and carcass traits in Australian Angus cattle. J. Anim. Sci. 96, 4521–4531. doi:10.1093/JAS/SKY325
Utsunomiya, Y. T., Milanesi, M., Utsunomiya, A. T. H., Ajmone-Marsan, P., Garcia, J. F., and Stegle, O. (2016). GHap: an R package for genome-wide haplotyping. 32, 2861, 2862. doi:10.1093/bioinformatics/btw356
Vandenplas, J., Calus, M. P. L., Sevillano, C. A., Windig, J. J., and Bastiaansen, J. W. M. (2016). Assigning breed origin to alleles in crossbred animals. Genet. Sel. Evol. 48, 61–22. doi:10.1186/s12711-016-0240-y
Vanderick, S., Gillon, A., Glorieux, G., Mayeres, P., Mota, R. R., and Gengler, N. (2017). Usefulness of multi-breed models in genetic evaluation of direct and maternal calving ease in Holstein and Belgian Blue Walloon purebreds and crossbreds. Livest. Sci. 198, 129–137. doi:10.1016/J.LIVSCI.2017.02.019
VanRaden, P. M., Tooker, M. E., Chud, T. C. S., Norman, H. D., Megonigal, J. H., Haagen, I. W., et al. (2020). Genomic predictions for crossbred dairy cattle. J. Dairy Sci. 103, 1620–1631. doi:10.3168/JDS.2019-16634
Visscher, P., Pong-Wong, R., Whittemore, C., and Haley, C. (2000). Impact of biotechnology on (cross)breeding programmes in pigs. Livest. Prod. Sci. 65, 57–70. doi:10.1016/S0301-6226(99)00180-3
Vitezica, Z. G., Varona, L., and Legarra, A. (2013). On the additive and dominant variance and covariance of individuals within the genomic selection scope. Genetics 195, 1223–1230. doi:10.1534/genetics.113.155176
Wei, M., Van der Steen, H. A. M., Van der Werf, J. H. J., and Brascamp, E. W. (1991). Relationship between purebred and crossbred parameters: I Variances and covariances under the one-locus model. J. Animal Breed. Genet. 108, 253–261. doi:10.1111/J.1439-0388.1991.TB00183.X
Wei, M., and Van der Werf, J. H. J. (1995). Genetic correlation and heritabilities for purebred and crossbred performance in poultry egg production traits. J. Anim. Sci. 73, 2220–2226. doi:10.2527/1995.7382220x
White, S. N., Casas, E., Allan, M. F., Keele, J. W., Snelling, W. M., Wheeler, T. L., et al. (2007). Evaluation in beef cattle of six deoxyribonucleic acid markers developed for dairy traits reveals an osteopontin polymorphism associated with postweaning growth. J. Anim. Sci. 85, 1–10. doi:10.2527/JAS.2006-314
Wientjes, Y. C. J., and Calus, M. P. L. (2017). Board invited review: the purebred-crossbred correlation in pigs: a review of theory, estimates, and implications. J. Anim. Sci. 95, 3467–3478. doi:10.2527/JAS.2017.1669
Wu, Y., Fan, H., Wang, Y., Zhang, L., Gao, X., Chen, Y., et al. (2014). Genome-wide association studies using haplotypes and individual SNPs in simmental cattle. PLoS One 9, e109330. doi:10.1371/journal.pone.0109330
Xia, J., Fan, H., Chang, T., Xu, L., Zhang, W., Song, Y., et al. (2017). Searching for new loci and candidate genes for economically important traits through gene-based association analysis of Simmental cattle. Sci. Rep. 2017 7 (1 7), 42048–42049. doi:10.1038/srep42048
Yang, J., Benyamin, B., McEvoy, B. P., Gordon, S., Henders, A. K., Nyholt, D. R., et al. (2010). Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569. doi:10.1038/NG.608
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi:10.1016/j.ajhg.2010.11.011
Zhang, L., Li, H., Li, Z., and Wang, J. (2008). Interactions between markers can Be caused by the dominance effect of quantitative trait loci. Genetics 180, 1177–1190. doi:10.1534/GENETICS.108.092122
Keywords: beef-on-dairy, haplotypes, birth weight, calving difficulty, gestation length
Citation: Ahmed RH, Schmidtmann C, Mugambe J and Thaller G (2025) Genomic-based genetic parameters and genome-wide association studies for productive and reproductive traits in Beef-on-Dairy crossbreds. Front. Genet. 16:1530310. doi: 10.3389/fgene.2025.1530310
Received: 18 November 2024; Accepted: 20 May 2025;
Published: 05 June 2025.
Edited by:
Arthur Francisco Araujo Fernandes, Cobb-Vantress, United StatesReviewed by:
Doreen Becker, Leibniz Institute for Farm Animal Biology (FBN), GermanyAli Esmailizadeh, Shahid Bahonar University of Kerman, Iran
Copyright © 2025 Ahmed, Schmidtmann, Mugambe and Thaller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. H. Ahmed, cmFobWVkQHRpZXJ6dWNodC51bmkta2llbC5kZQ==