 Perrine Cruaud1,2,3*
Perrine Cruaud1,2,3* Adrien Vigneron1,2,3
Adrien Vigneron1,2,3 Patricia Pignet1,2,3
Patricia Pignet1,2,3 Jean-Claude Caprais4
Jean-Claude Caprais4 Françoise Lesongeur1,2,3
Françoise Lesongeur1,2,3 Laurent Toffin1,2,3Anne Godfroy1,2,3Marie-Anne Cambon-Bonavita1,2,3
Laurent Toffin1,2,3Anne Godfroy1,2,3Marie-Anne Cambon-Bonavita1,2,3- 1UMR6197, Laboratoire de Microbiologie des Environnements Extrêmes, IFREMER, Technopôle Brest Iroise, Plouzané, France
- 2Laboratoire de Microbiologie des Environnements Extrêmes, UMR6197, Université de Bretagne Occidentale, Plouzané, France
- 3Centre National de la Recherche Scientifique, Laboratoire de Microbiologie des Environnements Extrêmes, UMR6197, Technopôle Brest Iroise, Plouzané, France
- 4Laboratoire “Environnements Profonds,” EEP-LEP, IFREMER, Technopôle Brest Iroise, Plouzané, France
In the Guaymas Basin, the presence at a few tens of kilometers of cold seeps and hydrothermal vents coupled with comparable sedimentary settings and depths offer a unique opportunity to assess and compare the microbial community composition of these deep-sea ecosystems. The microbial diversity in sediments from three cold seep and two hydrothermal vent assemblages were investigated using high-throughput 16S rRNA-sequencing. Numerous bacterial and archaeal lineages were detected in both cold seep and hydrothermal vent sediments. Various potential organic matter degraders (e.g., Chloroflexi, Atribacteria, MBG-D) and methane and sulfur cycling related microorganisms (e.g., ANME and methanogenic lineages, sulfate-reducing lineages) were detected in both ecosystems. This suggests that analogous metabolic processes such as organic matter degradation and anaerobic methane oxidation coupled to sulfate reduction, were probably occurring in these two contrasted ecosystems. These highlighted “core microbiome” of the Guaymas Basin chemosynthetic ecosystems might therefore result from the combined presence of up-rising fluid emissions and high sedimentary rates of organic matter in the Basin. These results, coupled with the detailed ribotype analysis of major archaeal lineages (ANME-1, ANME-2, and MBG-D), also suggest a potential connectivity among deep-sea ecosystems of the Guaymas Basin likely due to the sedimentary context and the absence of physical border. However, thermophilic and hyperthermophilic lineages (e.g., Thermodesulfobacteria, Desulfurococcales, etc.) were exclusively identified in hydrothermally impacted sediments highlighting the strong influence of temperature gradients and other hydrothermally-related factors such as thermogenic sulfate reduction and sulfide formation on microbial community composition.
Introduction
Deep-sea hydrothermal vents and cold seeps are characterized by elevated microbial biomass and various faunal and microbial assemblages (Van Dover et al., 2002; Jørgensen and Boetius, 2007). The complex food webs of these ecosystems are mainly supported by microbial chemosynthetic primary production based on percolating or emitting fluids (Van Dover et al., 2002; Jørgensen and Boetius, 2007). Hydrothermal vents are characterized by the presence of geothermally heated fluids that spring through openings in the seafloor, especially along active mid-ocean ridges (Van Dover et al., 2002). By contrast, cold seeps are characterized by seafloor emissions of low-temperature hydrocarbon-rich fluids along continental margins (Sibuet and Olu, 1998). Diffusive and advective transports of potential substrates in sediments establish a succession of different redox zones allowing the presence of a large variety of metabolisms (Jørgensen, 1977; Froelich et al., 1979; Engelen and Cypionka, 2009; Orcutt et al., 2011). Indeed, chemosynthetic microorganisms can use available electron donors (e.g., H2S, H2, CH4, ) and electron acceptors (e.g., O2, , ), provided by the fluid seepage and the seawater, as energy sources to metabolize inorganic and organic carbon sources. Afterwards, by- and end-products of these activities can be metabolized in turn by secondary producers (Jørgensen and Boetius, 2007), leading to the establishment of rich benthic communities at the seafloor (Sibuet and Olu, 1998; Dubilier et al., 2008; Cambon-Bonavita et al., 2009; Lloyd et al., 2010; Grünke et al., 2011; McKay et al., 2012).
Biological assemblages exhibit a patchy distribution on the seafloor of these chemosynthetic environments and are mainly represented by symbiont-bearing invertebrates such as Vesicomyidae clams, Mytilidae mussels, and Siboglinidae tubeworms, but also by microbial mats usually formed by sulfur-oxidizing filamentous bacteria (Tunnicliffe, 1991; Sibuet and Olu, 1998; Dubilier et al., 2008). This patchy distribution was found to be correlated with the fluid composition and flow rate (Niemann et al., 2006), reflecting distinct life requirements (required substrates or sensibility toward toxic compounds present in the fluids; Barry et al., 1997; Sibuet and Olu, 1998; Sahling et al., 2002; Niemann et al., 2006; Lichtschlag et al., 2010; Pop Ristova et al., 2012; Ruff et al., 2013).
Comparative studies of faunal and microbial community structure between hydrothermal vent and cold seep ecosystems demonstrated functional and phylogenetic similarities but also numerous distinctive features (Sibuet and Olu, 1998; Tunnicliffe et al., 1998; Bernardino et al., 2012; Portail et al., 2016). These differences may arise from specific ecological niches related to the nature of fluids. However, they may also be explained by the existence of dispersal barriers between these usually distant ecosystems (e.g., biogeographic barrier). The Guaymas Basin in the Gulf of California (Mexico) has numerous unique features. Indeed the Basin is characterized by the presence of both active hydrothermal vents and hydrocarbon seeps in close proximity and without biogeographic barriers, allowing testing the influence of dispersal barriers on community compositions. Furthermore, this Basin harbors similar sedimentary setting (overlaid by 400–500 m of sediments, Simoneit et al., 1990), comparable water depths (ca 2,000 m), and visually similar surface biological assemblages (Simoneit et al., 1990; Otero et al., 2003; Vigneron et al., 2013) in both hydrothermal and cold seep areas. Therefore, the Guaymas Basin represents a unique opportunity to directly compare hydrothermal vent and cold seep ecosystems. Previous investigations on the sedimentary microbial community compositions of the Guaymas Basin, from either but not both of these ecosystems, suggested that analogous microbial processes might occur, despite contrasted temperatures (Teske et al., 2002; Dhillon et al., 2003; McKay et al., 2012; Vigneron et al., 2013; Cruaud et al., 2015; Lever and Teske, 2015). However, these results remain hardly comparable since different methodologies were used.
Therefore, the aim of this study is to compare Guaymas Basin cold seep and hydrothermal vent sedimentary ecosystems functioning, through the analysis of the microbial community composition. Diversity and spatial distribution of microbial communities were investigated to determine to what extent the microbial communities inhabiting seep and vent sediments are comparable in the unique context of the Guaymas Basin. In this light, we focused our work on the identification of the main drivers explaining the highlighted singularities and/or similarities between these two nearby ecosystems.
To answer these questions, sediment cores from three cold seep sites (Vasconcelos BIG13, Ayala BIG14, and Vasconcelos BIG18 sites) and two hydrothermal vent sites (Morelos and MegaMat sites) in the Guaymas Basin, which exhibited comparable surface assemblages (vesicomyid clams or microbial mat, Figure 1) were investigated. At each site, diversity and distribution of microbial communities from the seawater/sediment interface down to 12 cmbsf, were explored using 454-pyrosequencing coupled with geochemical analyses. For a more accurate comparison, all the samples were collected during the same oceanographic cruise and analyzed in identical conditions, by the same experimenters and with the same methodological approach.

Figure 1. Bathymetric map of the Sonora Margin (cold seep area) and Southern Trough (hydrothermal vent area) in the Guaymas Basin. Localization and pictures of the sampling sites.
Experimental Procedures
Sampling Sites and Methods
Five contrasted sediment areas of the Guaymas Basin were explored in this study. Three cold seep sites located at the Sonora Margin and two hydrothermal vent sites located at the Southern Trough were investigated. Sediment samples were collected using 20-cm long push cores operated by the manned submersible Nautile during the “BIG” cruise (June 2010). Sampling sites were selected according to visual observations of the seafloor and methane plumes in the water column.
Two cold seep sites (Ayala and Vasconcelos BIG13) and one hydrothermal vent site (Morelos) were colonized by chemosynthetic vesicomyid bivalves (Figure 1, Table S1). The Ayala site, located at 1,560 m water depth, was covered by abundant and scattered vesicomyid aggregates distributed on an estimated area within a 10-m radius. The Vasconcelos BIG13 site, located 1 km away from the Ayala site and at 1,570 m water depth, was characterized by vesicomyid populations surrounding a large and thin microbial mat. Finally, the Morelos site, located at 2,007 m water depth in the hydrothermal field, was colonized by vesicomyid populations surrounding a large area of sediments covered by a thin white layer, probably corresponding to a microbial mat.
Two sites hosted thick microbial mats at the sediment surface: the Vasconcelos BIG18 cold seep site and the MegaMat hydrothermal vent site (Figure 1, Table S1). The Vasconcelos BIG18 site, located 30 m away from the vesicomyid Vasconcelos BIG13 site, was colonized by a thick white Beggiatoa-like mat (called WM14 in Vigneron et al., 2013), whereas sediment surface at the MegaMat site, located about 240 m south from the Morelos vesicomyid site, was covered with white and yellow microbial mats near a hydrothermal mound overgrown by Riftia bushes.
At each location, sediment push cores were collected for microbiological and geochemical analyses. At each cold seep site, one core (10 or 12 cm length) was collected for microbiological analyses (Table S1) whereas two cores of 12 cm length were collected for microbiological analyses at the vent sites. At Morelos vesicomyid site, CT2 core was collected in a vesicomyid area close to a microbial mat whereas CT8 core was collected ~1 m away in the same vesicomyid area. At MegaMat site, sediment core CT3 was collected in sediments covered by a white microbial mat whereas GCT1 sediment core was collected in sediments covered by a yellow microbial mat (Table S1). Porewater analyses were performed as described in Vigneron et al. (2013). Temperature measurements were performed in situ using an independent thermal lance T-Rov (NKE Electronics, France) for cold seep habitats or using the temperature sensor of the submersible for hydrothermal vent sediments.
After recovering on board, sediment cores were immediately transferred in cold room (~8°C) for sub-sampling. Sediment cores were cut into 2-cm thick layers and then frozen at −80°C for further nucleic acid extractions.
DNA Extraction
In order to increase the diversity of extracted lineages, total nucleic acids were extracted with two different methods for each sample: 4 × 0.6 g of frozen sediments were extracted using FastDNA® SPIN Kit for Soil (Bio101 Systems, MP Biomedicals™) with some modifications (Webster et al., 2003; Roussel et al., 2009) and 3 × 2.5 g of frozen sediments were extracted using a modified method described in Zhou et al. (1996) and detailed in Cruaud et al. (2014). Replicate of crude DNA extracts were then pooled together whatever the extraction method used and purified using the Wizard DNA clean-up kit (Promega, Madison, WI) according to the manufacturer instructions. Purified DNA extracts were stored at −20°C.
PCR Amplifications and Pyrosequencing
The 16S rRNA genes were amplified by PCR using archaeal and bacterial targeted primers. Bacterial 16S rRNA genes were amplified using SSU536F (Dufresne et al., 1996) and 907R (Yu and Morrison, 2004) primers, targeting the V4–V5 hypervariable regions whereas the V1–V2–V3 hypervariable regions of the archaeal 16S rRNA genes were amplified with 27F (Fish et al., 2002) and Arc518R (Sørensen and Teske, 2006) primers (Table S2). The primers were fused to 5- to 10-nucleotide key tags and to the 454 GS-FLX sequencing adaptor using the Lib-A chemistry. To allow multiplex sequencing of the 22 different samples (five sampling locations with the different sediment sections), fusion primers were designed to minimize secondary structures and following Roche recommendations (Table S3). The 16S rRNA genes amplifications were performed under the following conditions: 10 min at 95°C for denaturation, then 30 cycles for Bacteria or 35 cycles for Archaea with 30 s at 95°C, 45 s at 58°C, and 45 s at 72°C followed by a final elongation step of 6 min at 72°C. PCR amplifications were performed in triplicate using Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (Agilent Technologies, Palo Alto, CA), 0.5 μM of each primer (Eurofins MWG Operon, Ebersberg, Germany), and 1 μL of purified DNA template. The final volume was adjusted to 25 μL with sterile water. Absence of contaminations was checked by negative controls. PCR products were pooled and purified on TAE agarose gel (1.2%) using PCR clean-up Gel extraction Nucleospin® Gel and PCR clean-up kit (Macherey-Nagel, Düren, Germany).
Microfluidic digital PCR (Fluidigm Corporation, San Francisco, CA) was used to quantify nucleic acids in purified amplicons. Amplicon products obtained for each PCR replicate were mixed equimolarly (108 molecules per microliter of each amplicon). Emulsion PCR and sequencing were then performed on a 454 Life Sciences Genome Sequencer GS-FLX (PicoTiterPlate divided in four regions; Roche Diagnostics, Indianapolis, IN). Quantification, emulsion PCR and sequencing were performed by the Biogenouest platform (Rennes, France).
Pyrosequencing Data Analyses
To minimize the effect of random sequencing errors (Huse et al., 2007; Kunin et al., 2010), pyrosequencing reads analyses and filtering were performed from the sff file using the Mothur pipeline (Schloss et al., 2009). Sequences (i) shorter than 200 bp, (ii) containing homopolymers longer than 8 bp, (iii) that aligned to the incorrect region within the 16S rRNA gene (Silva release 119, www.arb-silva.de), (iv) identified as chimeras using Uchime algorithm (Edgar et al., 2011; Schloss et al., 2011), and (v) affiliated to Archaea when Bacteria was targeted and inversely were removed from the dataset.
Using the tag combinations, sequences were assigned to their respective sample. Sequences were pre-clustered using the Mothur pipeline, as recommended to reduce potential pyrosequencing errors (Schloss et al., 2009). Then samples were normalized and all the following analyzes were performed on the same number of sequences per sample for the bacterial (1,096 sequences) and the archaeal (1,413 sequences) regions.
Taxonomic assignments of the reads were performed as previously described in detail in Cruaud et al. (2015) using the Mothur version of the Bayesian classifier (Schloss et al., 2009) on both SILVA database (release 119, 464,618 sequences of Bacteria and 18,797 sequences of Archaea, www.arb-silva.de) and an in-house database composed of deep-sea marine sediment sequences from reference publications updated and completed for this study using the SILVA database release 119, as described in Cruaud et al. (2015) (2,860 bacterial and 978 archaeal sequences, Table S4).
Statistical analyses were performed using the software environment R (v. 3.4.0) with the RStudio toolkit (v. 1.0.143). Venn diagrams were constructed using assigned sequences independently at the genus and phylum levels using the VennDiagram package (Chen and Boutros, 2011). Non-metric multidimensional scaling (NMDS) (Kruskal, 1964) based on the Bray-Curtis dissimilarity measure (Bray and Curtis, 1957) and Redundancy Analysis (RDA) were carried out using the Vegan package (Oksanen et al., 2007). RDA were performed using relative abundances of taxa at the taxonomical level 6 (genus level) transformed with the Hellinger transformation and standardized environmental data at zero mean and unit variance (z-scores). To select the significant explanatory variables, we performed a forward selection using the ordiR2step function of the Vegan package using adjusted R2 as the stopping criterion and 1,000 as the number of permutation in one step. Analyses of similarity (ANOSIM) based on Bray-Curtis dissimilarity measure were used to determine significant differences between sampling locations using the Vegan package. SIMPER (Similarity Percentages) analyses were used to determine the contribution of each microbial groups to the Bray-Curtis dissimilarity (Clarke, 1993) using the Vegan package. Ribotype (representing 16S rRNA sequences being identical at 99.6% similarity) distribution within ANME-1, ANME-2, and MBG-D lineages were illustrated by network analyses of the 16S rRNA gene sequences using the ape and pegas packages (Paradis et al., 2004; Paradis, 2010).
Sequences have been assigned to a same taxonomical level (phylum or genus) within an analysis. The sequences that could not be assigned to the taxonomical level used were affiliated to “Unclassified” followed by the lowest identified taxonomical level (e.g., Unclassified Desulfobacteraceae).
The raw sequencing data have been submitted to the NCBI database under BioProject accession numbers PRJNA278499 and PRJNA394915.
Results
Geochemical Characterization
Although methane concentrations might have been underestimated due to outgassing during core retrieval, methane was detected in all sediment samples (Figure 2). Highest methane concentrations were measured throughout the sediment cores of the microbial mat habitats, with up to 1 mM (min: 0.2 mM, max: 1 mM, Average: 0.7 mM) at Vasconcelos BIG18 cold seep site and up to 0.9 mM (min: 0.01 mM, max: 0.9 mM, average: 0.3 mM) at the MegaMat vent site (Figures 2D,E). Methane concentrations were lower in vesicomyid cold seep habitats with 0.013 mM and 0.003 mM at the Ayala and Vasconcelos BIG13 sites, respectively (Figures 2A,B), whereas up to 0.18 mM of methane were detected at 15 cmbsf in sediment core of Morelos vesicomyid vent site.
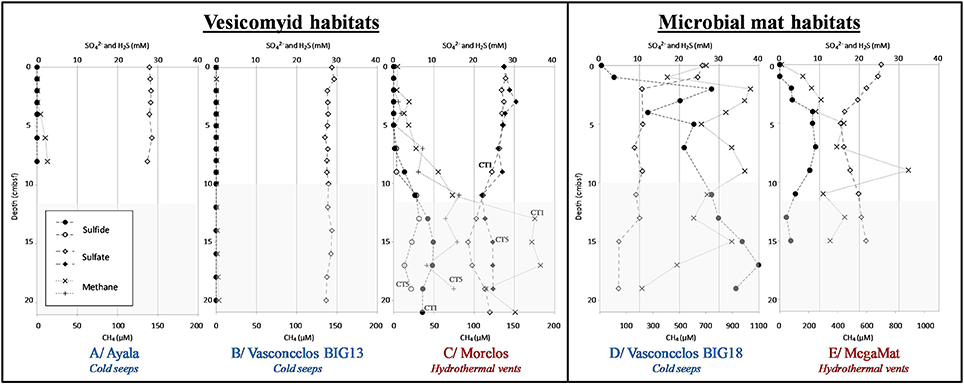
Figure 2. Geochemical profiles in sediments of the Guaymas Basin. Dissolved methane (cross), dissolved sulfate (full and empty diamond), and dissolved sulfide (full and empty circle) concentrations in porewater sediments. White areas correspond to depth of sediment cores used for microbial community analysis. Sediment cores were from vesicomyid habitats: (A) Ayala—cold seeps, (B) Vasconcelos BIG13—cold seeps, (C) Morelos—hydrothermal vent, and from microbial mat habitats: (D) Vasconcelos BIG18—cold seeps and (E) MegaMat M27—hydrothermal vent.
Hydrogen sulfide porewater concentrations were higher in sediments underlying cold seep microbial mats, increasing with depth and reaching up to 40 mM at the deepest layers of the Vasconcelos BIG18 seep site (Figure 2D). By contrast, a peak of hydrogen sulfide concentration was detected between 4 and 10 cmbsf in MegaMatvent site with up to 9.0 mM (Figure 2E). Peak of hydrogen sulfide was also detected at 15 cmbsf of Morelos vesicomyid vent site with concentration up to 9.9 mM (Figure 2C). Sulfide porewater concentrations were below the technical detection limit [10 μM] in cold seep sediments colonized by vesicomyids (Ayala and Vasconcelos BIG13, Figures 2A,B).
Dissolved sulfate concentrations in porewater mirrored the hydrogen sulfide concentrations. In Vasconcelos BIG18 microbial mat seep site, porewater sulfate concentrations decreased with depth from 26 mM in surface layer down to 5 mM at 15 cmbsf (Figure 2D). At the MegaMat vent sites, inverted peak of sulfate concentration was detected between 4 and 10 cmbsf with sulfate concentration down to 15 mM (Figure 2E). Sulfate depletion was also measured at 11 cmbsf for the Morelos vesicomyid vent sites with sulfate concentration down to 21.9 mM (Figure 2C). Sulfate concentrations at vesicomyid cold seep habitats remained constant with 28 mM throughout the sediment core (Ayala and Vasconcelos BIG13, Figures 2A,B).
In situ temperature measurements at the cold seep sites (Vasconcelos BIG18, Ayala and Vasconcelos BIG13) indicated a constant temperature around 3°C throughout the forty-first centimeters of sediments. By contrast in the hydrothermal vent area the temperature increased with depth. In Morelos vesicomyid vent sediments, temperature increased from 3.2°C at 5 cmbsf to 15.6°C at 55 cmbsf whereas the temperature strongly increased in MegaMat microbial mat vent sediments, reaching 55°C at 10 cmbsf, 92.5°C at 20 cmbsf, and up to 120.5°C at 50 cmsbf below the white microbial mat and 113°C at 10 cmbsf and up to 123°C at 50 cmbsf in sediment underlying the yellow microbial mat (Figure S1).
All sediment cores had a limited oxygen depth penetration ranging from 0.2 to 0.55 cmbsf.
Bacterial Community Composition
After sequence quality filtering, a total of 37,261 partial 16S rRNA gene sequences were used for the bacterial community composition analysis. Taxonomic affiliation of the sequences highlighted different bacterial community structures according to sampling sites (Figure 4 and Table S6).
Shared and unique bacterial phyla and genera were identified in the different habitats (Figure 3). A high proportion of bacterial phyla (68.8%) and genera (33.5%) was identified in all habitats [e.g., Chloroflexi, SEEP-SRB1, Candidate divisions JS1 (Atribacteria), OP3 (Omnitrophica), OP8 (Aminicenantes), MSBL8]. Around 80% of the detected phyla and 63.5% of the detected genera were shared between at least one seep site and one vent site. The large majority (up to 98% of the detected phyla and 92.5% of the detected genera) of bacterial lineages identified in cold seep sites were also detected in vent sites. By contrast, 7 phyla (16%; e.g., GBG/HotSeep-1) and 56 genera [28%; e.g., candidate div. OD1 (Parcubacteria)] were specific to vent sites and 3 phyla (Thermodesulfobacteria, Thermotoga, and candidate div. KB1) and 40 genera (e.g., Caldimicrobium) were only detected in hydrothermal vent microbial mat sediments. At the deepest sections (8–12 cmbsf) of the yellow mat, hydrothermal sediment core (MegaMat-GCT1, in situ temperature 110°C), sequences affiliated to betaprotebacterial lineages were identified. However, these lineages have been reported to be potential contaminants from DNA extraction kits and other laboratory reagents (Salter et al., 2014). Therefore, these betaproteobacterial lineages are likely to be contaminants, sequenced due to the potential low biomass present in these deep sections of the sediments with extremely high temperature (no amplifiable DNA in similar samples in McKay et al., 2016).
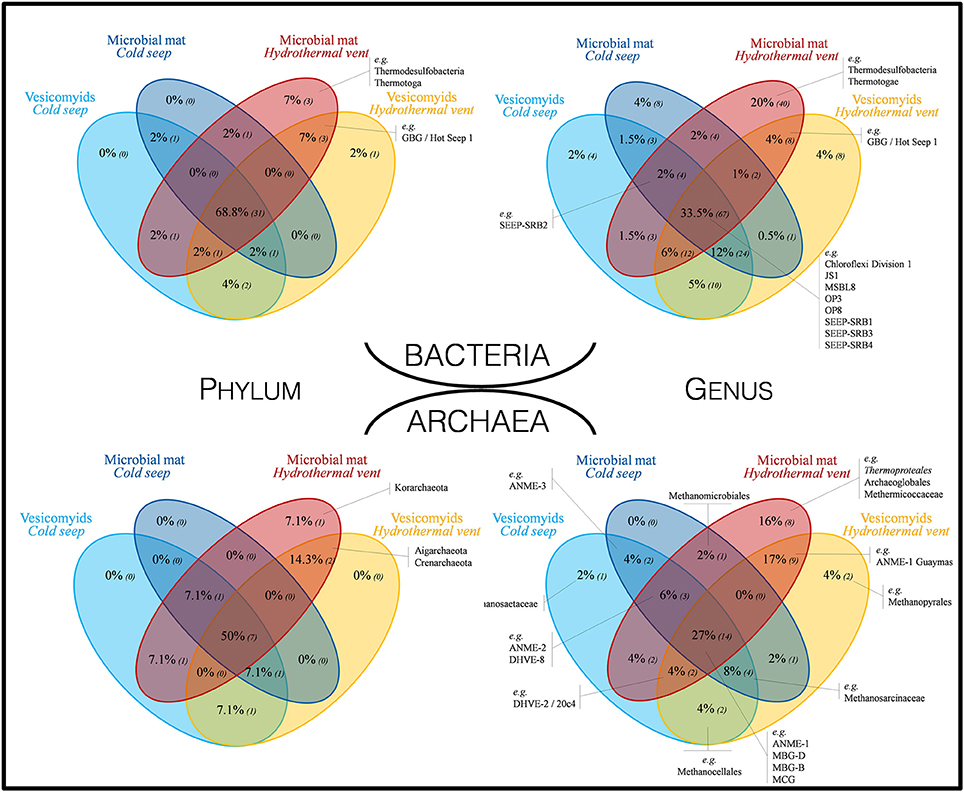
Figure 3. Venn diagrams representing the proportion of shared bacterial and archaeal taxa at phylum and genus levels, across all investigated habitats. Numbers in brackets correspond to the number of different lineages. Taxa indicated in black were selected as representative taxa of seep and vent sediment microbial communities.
Whatever the surface assemblage (vesicomyid or microbial mat), detected bacterial communities were significantly different between cold seep sediments and hydrothermal vent sediments (ANOSIM statistic R 0.777 and 0.421, p = 0.001 and 0.004 for vesicomyid and for microbial mat habitats, respectively). RDA analyses highlighted strong correlations between the bacterial community composition and temperature and dissolved sulfide porewater concentrations in the samples (p = 0.002 and 0.02, respectively, Figure 5). The difference between the bacterial community of hydrothermal vent and cold seep mat sediments was mainly due to the relative proportion of Thermodesulfobacteria (Caldimicrobium relatives) (explaining 14.1% of the dissimilarity; SIMPER) and GBG/HotSeep-1 (8.6% of the dissimilarity), both exclusively detected in hydrothermal vent sediments. The relative proportion of Sulfurovum relatives (Epsilonproteobacteria) (5.5% of the dissimilarity) and unclassified Desulfobacteraceae (4.6% of the dissimilarity), mainly identified in cold seeps, also contributed to the difference between hydrothermal vent and cold seep microbial mat communities. In sediments colonized by vesicomyids, various gammaproteobacterial groups frequently identified in cold seep sediments (MHGSII, JTB255 marine benthic group, BD7–8 and unclassified Gammaproteobacteria), were the main lineages responsible for the difference of bacterial community composition observed between vent and seep sites (30.7% contribution of the dissimilarity) (Figures 4,5, Figure S2, and Table S6).

Figure 4. Relative abundance of bacterial and archaeal taxa (16S rRNA gene sequences affiliated to taxonomical level 6) detected in vesicomyid and microbial mat habitats, both for cold seep and hydrothermal vent areas. Bacterial and archaeal groups represented <1% of the sequenced reads were clustered in “Other.”
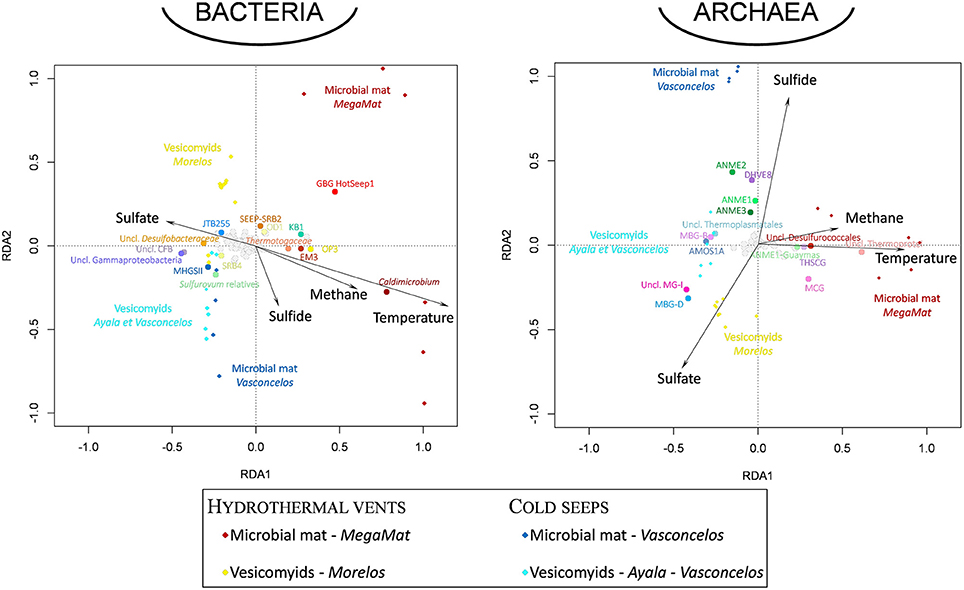
Figure 5. Visualization of the results of the redundancy analysis (RDA) on triplots identifying the relationships between the bacterial and archaeal taxonomical composition of the sedimentary microbial communities detected at each site and environmental geochemical parameters as explanatory variables. Diamond-shaped dots correspond to microbial community for each sampling site (dark red: MegaMat microbial mat vent site, yellow: Morelos vesicomyid vent sites, blue: Vasconcelos BIG18 microbial mat cold seep sites, light blue: Ayala and Vasconcelos BIG13 vesicomyid cold seep sites). Black dotted vectors correspond to the explanatory variables (temperature and sulfate, sulfide, methane porewater concentrations). Round-shaped dots correspond to bacterial and archaeal taxa (16S rRNA gene sequences affiliated to taxonomical level 6). Emblematic taxa of seep and vent sediments discussed in the text are highlighted in colored dots (dots color coded as in Figure 4). The environmental variables explained 57.4 and 62.9% of the variance in archaeal and bacterial community composition, respectively, and the RDA models were significant (p = 0.001).
In both hydrothermal and cold seep environments, bacterial community composition was significantly different between sediments colonized by vesicomyid assemblages and sediments underlying microbial mats (Figures 4, 5 and Figure S2) (ANOSIM statistic R 0.529, p = 0.001). SIMPER analyses highlighted that Thermodesulfobacteria (Caldimicrobiumrelatives) (13.6% contribution) and GBG/HotSeep-1 (Guaymas Basin Group) (9.5% contribution), predominantly identified in sediments underlying microbial mats, were the main lineages responsible for the differences between vesicomyid and microbial mat habitats in hydrothermal area (Figures 4, 5 and Figure S2). By contrast, gammaproteobacterial lineages (21% contribution), mainly detected in sediments colonized by vesicomyids, and Desulfobacteraceae (6.7% contribution) and Sulfurovum relatives (9.2% contribution), mainly detected in sediments underlying microbial mats, were the major lineages responsible for the difference between vesicomyid and microbial mat habitats in cold seep area (Figure 4, Figure S2, and Table S6).
Archaeal Community Composition
After sequence quality filtering, a total of 48 042 partial 16S rRNA gene archaeal sequences were analyzed to explore the archaeal community composition (Figure 4 and Table S5).
A high proportion of taxa were shared among sites, while unique taxa were detected primarily in hydrothermally active sites (Figure 3). Up to 50% of phyla and 27% of genera were shared between all sites; e.g., ANME-1, MBG-D, MBG-B, MCG (Bathyarchaea). Most of the taxa were found both in cold seep and hydrothermal vent sediments (78% of the taxa and 57% of the genera were shared between at least two different habitats). All the detected phyla and 94% of the detected genera in seep sites have also been detected in vent sites. By contrast, 3 phyla (21.4%; Korarchaeota, Aigarchaeota, and Crenarchaeota) and 19 genera (37%; e.g., ANME-1 Guaymas, Methanopyrales, Thermoproteales) were specific to vent sites and 1 phylum (Korarchaeota) and 8 genera (e.g., Thermoproteales, Archaeoglobales, Methermicoccaceae) were only detected in hydrothermal microbial mat site (Figure 3). Various potential methanogenic lineages (Unclassified Methanocellales, unclassified Methanomicrobiales, Methermicoccaceae, Methanosaetaceae, Methanopyraceae) were detected in small proportions in all sampled habitats (Figure 3 and Table S5).
Statistical analysis of community composition highlighted distinct archaeal community compositions between the different habitats (Figures 4 and Figure S2). RDA analyses indicated that temperature and dissolved sulfide porewater concentration were the main environmental parameters shaping the archaeal community composition in the samples (p = 0.002, Figure 5).
In both vesicomyid and microbial mat habitats, archaeal community compositions were significantly different between cold seep and hydrothermal vent habitats (ANOSIM statistic R 0.779 and 0.616, p = 0.001 for vesicomyid and for microbial mat habitats, respectively). In sediments associated with microbial mat habitat, ANME-2 (16.1% contribution) and DHVE-8 (9.5% contribution), mainly identified under the cold seep mats, and Thermoprotei (10.2% contribution) and ANME-1 Guaymas (9.7% contribution), mainly detected under hydrothermal vent mats (Figures 4 and Figure S2), were the major lineages responsible for the difference between hydrothermal vent and cold seep mats. In sediments colonized by vesicomyid, MBG-D (31% contribution), detected as predominant (up to 58%) Archaea in cold seep sediments, and MG-I (31.7% contribution), detected as predominant (up to 53%) in hydrothermal sediments, were the major lineages responsible for the difference between hydrothermal vent and cold seep vesicomyid habitats (Figure 4, Figure S2, and Table S5).
Archaeal communities observed in sediments colonized by vesicomyid assemblages were also significantly different from those detected in sediment underlying microbial mats (Figure S2) (ANOSIM statistic R 0.55, p = 0.001). SIMPER analyses indicated that relative proportion of MBG-D (15.8% contribution) and MG-I (14.5% contribution), largely represented in vesicomyid habitats (respectively, up to 58 and 53% of the sequences in vesicomyid sediment push cores, Figure 4, Figure S2, and Table S5) as well as Thermoprotei (8.3% contribution), ANME-1 Guaymas (7.8% contribution), and ANME-2 (5.2%), predominant in sulfide-rich microbial mat habitats (up to 54, 82, and 53%, respectively), were mainly responsible for the differences between the vesicomyid and microbial mat habitats (Figure 4, Figure S2, and Table S5).
As major archaeal lineages responsible for the difference between sites, ANME-1, ANME-2 and MBG-D lineages were selected for detailed ribotype analyses. A ribotype represented several 16S rRNA sequences being identical at 99.6% along the sequenced DNA fragment (sequences that are within 2 bp of a more abundant sequence have been merged to reduce sequencing errors). Network constructions revealed distinct identity, composition and diversification patterns depending on the analyzed lineage and the sampling sites (Figure 6). The network analysis performed on the ANME-1 related sequences showed a site-dependent distribution with a clear dichotomy between hydrothermal and cold seep sediments (Figure 6A). Analysis of ANME-2 16S rRNA gene sequences indicated a different pattern with a larger ribotype diversity and no clear dichotomy between hydrothermal and cold seep habitats (Figure 6B). Likewise, network analyses performed on the MBG-D sequences exhibited a high ribotype diversity without clear dichotomy between vent and seep sediments (Figure 6C). Up to 16% of the MBG-D ribotypes, representing 70% of the MBG-D 16S rRNA gene sequences, were detected in both cold seep and hydrothermally-influenced sediments.
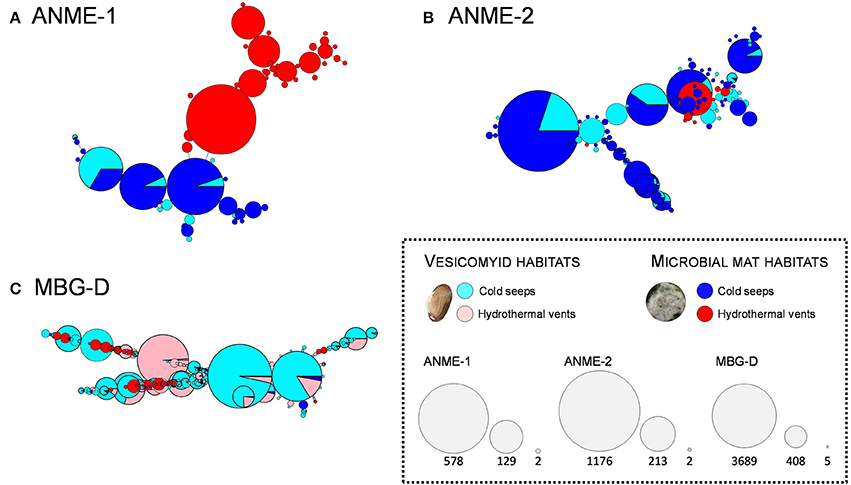
Figure 6. Network representing ribotype (16SrRNA sequences being identical at 99.6% similarity) distribution among the ANME-1 (A), ANME-2 (B), and MBG-D (C) lineages according to the sampling sites. Each circle represents a ribotype and diameter of the circles represents the number of sequences included in each ribotype (keys to the node size represent the highest number, the lowest number and an intermediate value, for each of the three lineages). Color of the circles illustrates the proportion of the sequences detected in each sampling site (blue and light blue: microbial mat and vesicomyid cold seep sites, red and light red: microbial mat and vesicomyid hydrothermal vent sites). Singletons were removed for this analyses to make the graphs more readable. The three lineages represented were selected as representative taxa of chemosynthetic ecosystems detected in hydrothermal vent sites as well as cold seep sites in our study.
Discussion
The Guaymas Basin is a unique environment which harbors not only hydrothermal chimneys, mounds (Callac et al., 2015) and hydrothermally impacted sediments (i.e., sediments percolated by hydrothermal fluid) (Teske et al., 2002) but also cold seep sediments (Vigneron et al., 2013; Cruaud et al., 2015), within few kilometers. In this study, area of hydrothermally impacted sediments and cold seep sediments were investigated to compare the microbial community compositions and potential functions present in these contrasted ecosystems in absence of physical borders and in a similar geochemical context characterized by the accumulation of organic-rich sediments.
Sedimentary Context Leads to Similar Microbial Communities
Comparable features were observed between hydrothermal and cold seep sediments. First, analogous surface assemblages were observed on the seafloor with either microbial mats or vesicomyids assemblages (Figure 1, Table S1). Then, similar geochemical features were measured in the sediments. Indeed, presence of sulfate to methane transition zones and high hydrogen sulfide concentrations were detected in sediments underlying microbial mats. Moreover, low methane concentration increasing with depth and steady sulfate concentrations were detected in sediments colonized by vesicomyids (Figure 2). Additionally, comparable temperature conditions were also detected at the seawater/sediment interface (Figure S1). These common features are likely due to the unusual sedimentary context of the Basin.
Bacterial and archaeal 16S rRNA gene pyrosequencing analyses revealed that a large proportion of the microbial taxa (69% of bacterial phylum, 50% of archaeal phyla and about 30% of bacterial and archaeal genera) was shared between all the sites (Figure 3), constituting the “core microbiome” of the Guaymas Basin chemosynthetic areas. This “core microbiome” was mainly represented by Thermoplasmatales relatives (e.g., MBG-D), previously detected in marine sediments and deep biosphere (Ruff et al., 2015), Miscellaneous Crenarchaeotic Group (MCG, Bathyarchaeota), which are widespread in marine environments (Kubo et al., 2012), Chloroflexi and candidate division JS-1 (Atribacteria), also frequently detected in hydrocarbon-rich and subsurface marine sediments (Blazejak and Schippers, 2010), and ANME and SEEP-SRB lineages, frequently detected in methane-rich sediments (Ruff et al., 2015) (Figures 3, 4, and Tables S5, S6). These lineages also align with the concept of “seep microbiome” identified in Ruff et al. (2015) that gather microbial lineages systematically identified in seep sediments. Moreover, up to 92% of the bacterial and archaeal lineages detected in cold seep sediments were also identified in hydrothermally altered sediments (Figure 3). Consistently, nearly similar ribotypes of ANME-2 and MBG-D lineages were detected between hydrothermal and cold seep sites (Figures 6B,C). This supports a potential continuity within these ecosystems and the possible occurrence of contemporary exchanges among neighboring seeps and vents within the Basin potentially through the hydrosphere dispersion of microorganisms, as previously suggested (Hoshino et al., 2017). However, these common lineages and ribotypes were mainly detected at the surface sediment layers of the hydrothermal sediments (0–2 cmbsf, Figure 4). This suggests that the presence of a comparable shallow microbial community in both cold seep and hydrothermal sediments was likely allowed by the seawater cooling of the hydrothermal sediments (<20°C at 5 cmbsf; Figure S1).
Although predicting functional profiles from 16S rRNA data and related known lineages can be misleading, some potential metabolic capacities can be hypothesized from the community composition identified in the sediments.
Candidate divisions JS-1 (Atribacteria), OP3 (Omnitrophica), and OP8 (Aminicentantes), Chloroflexi and MBG-D lineages were detected in all investigated habitats. Based on genome-centric metagenomic, members of these taxa have been proposed as potential degraders of organic matters in marine sediments, degrading aromatic compounds (Wasmund et al., 2016), scavenging dead cells (Lloyd et al., 2013; Robbins et al., 2016; Lazar et al., 2017) or fermenting various carbohydrates (Nobu et al., 2016). These heterotrophic microbial communities identified in both cold seep and hydrothermal sediments might be supported by the high sedimentary rates and the important microbial and meiofaunal biomass (Portail et al., 2015) likely generating the important organic carbon concentrations measured in the Guaymas Basin seafloor (Lin et al., 2017). Thus, these potential organic matter degraders might represent an important proportion of the “core microbiome” of the Guaymas Basin chemosynthetic areas.
ANME lineages were also detected in all investigated habitats (Figures 3, 4, and Tables S5, S6). These lineages have been previously associated with anaerobic oxidation of methane (AOM) process, forming large consortia with sulfate reducing SEEP-SRB1 and -SRB2 lineages (Kleindienst et al., 2012; Vigneron et al., 2014), or GBG/HotSeep-1 lineage for ANME-1 Guaymas (Holler et al., 2011). Consistently, these sulfate-reducing lineages were also detected in large proportion in the sediment samples (Figures 4, Figure S2, and Table S5). Occurrence of AOM associated with sulfate reduction in hydrothermally affected and cold seep sediments is also supported by the geochemical measurements of the porewater that highlighted a sulfate to methane transition zone in both ecosystems (Figure 2), suggesting concomitant sulfate reduction and methane oxidation processes. These results are consistent with previous molecular data and in vitro incubation studies (Holler et al., 2011) as well as FISH observations of Guaymas Basin sediment samples (Kleindienst et al., 2012; Vigneron et al., 2014) and support that AOM coupled to sulfate reduction can be an important process in both hydrothermal and cold seep sediments of the Guaymas Basin (Holler et al., 2011; Vigneron et al., 2013). Noteworthily, ANME community composition in the hydrothermal sediments changed with depth and therefore with temperature. At surface sediments, hydrothermal vent ANME community was comparable to the ANME community identified in cold seep sediments with ANME-2 and ANME-1 Archaea groups (Figures 4, Figure S2, and Table S5). However, with increasing sediments depth and temperature, ANME-1 Guaymas lineage became prevalent in hydrothermal vent sites (Figure 4, Figure S2, and Table S5), as previously observed (Holler et al., 2011; Biddle et al., 2012; Kellermann et al., 2012). This result aligns with previous activity measurements and cultivation efforts of ANME-1 Guaymas lineage that demonstrated an active anaerobic methane oxidation at higher temperature than any other ANME lineages and suggested an adaptation to extreme temperatures (Wegener et al., 2015).
Numerous potential sulfate reducers such as the SEEP-SRB3 and four lineages and Desulfatiglans relatives were also identified in the sediments (Figure 4, Figure S2, and Table S6), as previously observed by microscopy (Kleindienst et al., 2012) and sulfate reducing genes surveys (Dhillon et al., 2003). These microorganisms have been previously suspected to be associated with hydrocarbons (methane and/or short chain alkanes) oxidation (Kleindienst et al., 2012), suggesting that despite different origins and probably compositions (Portail et al., 2016), hydrocarbons of the seepages might sustain similar microbial functions in these two ecosystems.
Potential methanogenic related lineages were detected in all sampled habitats (Figure 3 and Table S5), which is consistent with previous functional gene survey (Dhillon et al., 2005) and enrichment cultures (Vigneron et al., 2015). However, taxonomic affiliation of the sequences suggests a strong site specificity of these potential methanogenic lineages with various taxa detected only in a single habitat (Figure 3 and Table S5). This indicates that, despite the specificity of each habitat that probably leaded to the selection of different lineages (e.g., temperature, composition of the fluid, presence of macrofauna, different level of competition with sulfate-reducers, etc.), potential methanogenic populations occur in shallow sediments and are likely to contribute to the methane production in the seafloor (Vigneron et al., 2015).
Together, these results suggest that a common “core microbiome” occurs in the seepage-influenced sediments and that major microbial functions associated with organic matter recycling, hydrocarbon degradation as well as methane and sulfur cycles are potentially sustained in both hydrothermally impacted and cold seep sediments. These processes probably generate the high concentrations of hydrogen sulfide measured in porewaters, and might fuel the surface assemblages such as mat-forming sulfur-oxidizing giant bacteria (Lloyd et al., 2010; Grünke et al., 2011; McKay et al., 2012) or the sulfur-oxidizing symbionts of the invertebrates living at the sediment surface (Sibuet and Olu, 1998; Dubilier et al., 2008; Cambon-Bonavita et al., 2009), resulting in visually similar landscape on the seafloor.
Hydrothermal Influence Leads to Contrasted Microbial Community
Despite these similarities, hydrothermally influenced sediments harbored numerous endemic lineages (seven bacterial and three archaeal phyla—Figure 3) with members of the ANME-1 Guaymas, Thermoprotei, Archaeoglobales, Thermotogales, GBG/HotSeep-1 and Thermodesulfobacteraceae, previously detected in hydrothermal chimneys (Callac et al., 2015) and sediments of the Guaymas Basin (Teske et al., 2002). Supporting the positive correlation between these lineages and the temperatures measured in the sediments (Figure 5 and Figure S1), cultivated representatives of these lineages are thermophiles, growing at temperatures from 50 to 90°C (Amend and Shock, 2001). A higher level of endemism was detected in the deepest microbial mat sediment layers where the highest in situ temperatures were measured, as previously reported (Mackay et al., 2011). Furthermore, analysis of ANME-1 sequences revealed specific ribotypes associated with hydrothermal sediments, suggesting a distinct evolutionary history. Since these ANME-1 sequences were detected in the higher temperature sediments of the microbial mat vent site (Figure 4, Figure S1, and Table S5), the detection of these hydrothermal ANME-1 lineages probably reflect the selective pressure exerted by the high temperatures measured in vent sediments rather than an absence of continuity between cold seep and vent areas. Altogether, this indicates that presence of endemic lineages in hydrothermal vent sediments is likely correlated with high temperatures (Figure 5).
Metabolic features of cultivated representatives of the microbial lineages exclusively identified in hydrothermally impacted sediments suggest that additional metabolisms might occur in these habitats. Indeed, cultivated representatives of the hyperthermophilic Caldimicrobium relatives (Thermodesulfobacteria), Thermotogales, Desulfurococcales, and Archaeoglobales identified in hydrothermal sediments (Figure 4 and Table S5) have metabolic potentials mainly based on sulfur compounds utilization. Various lineages affiliated to Thermotogales and Desulfurococcales can use elemental sulfur and polysulfide as electron acceptor (Erauso et al., 1993; González et al., 1998; Adams et al., 2001; Huber and Stetter, 2006; Gorlas et al., 2015) whereas Caldimicrobium, identified in large proportion in the deepest sediment layers (Figure 4 and Table S5), can disproportionate elemental sulfur and generate sulfate and hydrogen sulfide as described for Caldimicrobium thiodismutans sp. nov (Kojima et al., 2016). Polysulfide and elemental sulfur are likely to be abundant in the hydrothermal fluid where dissolved sulfide can abiotically react with iron or manganese, detected in the Guaymas Basin hydrothermal fluid (Von Damm et al., 1985). Potential elemental sulfur disproportionation by Caldimicrobium relatives in the deepest sediment layers is also consistent with the high sulfate porewater concentrations measured in the same sediment horizons (Figure 2). Presence of these potential metabolisms only in hydrothermal sediments was therefore likely due to the hydrothermal context where substrates for these activities were generated by abiotic reactions allowed in high temperature conditions (Von Damm et al., 1985; Boyd and Druschel, 2013). These metabolisms could therefore provide additional sulfate and sustain Archaeoglobales metabolism, mainly based on sulfate or iron reduction (Brileya and Reysenbach, 2014), as well as the other potential sulfate reducing lineages identified in these sediments (e.g., GBG/HotSeep-1, SEEP-SRB, Desulfatiglans relatives, etc.).
Cold seep and hydrothermal vent sediments colonized by vesicomyids harbored comparable in situ temperature at the time of sampling (5°C at 10 cmbsf). Sequences of microbial communities potentially involved in AOM (ANME and SEEP-SRB) were detected in minority in both hydrothermal vent and cold seep sediment samples colonized by vesicomyids (Figures 4 and Tables S5, S6). This feature has been previously observed in similar environments and might be due to deeper sulfate to methane transition zone probably due to a lower fluid flow and/or bioturbation activity of the animals settled in the sediments (Barry et al., 1997; Fischer et al., 2012). However, different microbial community compositions were detected between seep and vent sediments colonized by vesicomyids (Figure 4 and Figure S2). Indeed, the archaeal community in vesicomyid cold seep sediments was dominated by members of the Thermoplasmatales (MBG-D) (Figure 4, Figure S2, and Table S5). These Archaea have been previously proposed to be involved in organic matter degradation (Lloyd et al., 2013) and might thrive in cold seep sediments by degrading vesicomyid exudates (Joye et al., 2010; Pop Ristova et al., 2012; Cruaud et al., 2015). By contrast, thermophilic lineages (e.g., Methanopyrales, Thermoprotei) were detected in hydrothermal vent sediments colonized by vesicomyids (Table S5), confirming that this site was fueled by a hydrothermal fluid. Nonetheless, the archaeal community in hydrothermally influenced sediments was dominated by members of the Thaumarchaeota (MG-1) (Figure 4, Figure S2, and Table S5), previously identified in water column (DeLong, 1992) and detected in sediments outside seepage area of the Guaymas Basin (Vigneron et al., 2013; Cruaud et al., 2015). One hypothesis to explain the difference between microbial community of vent and seep vesicomyids habitats would be that the hydrothermal sampling site could not be continuously colonized by the bivalves but only be a transition zone for vesicomyids. Indeed, numerous vesicomyids movement paths were observed on the seafloor (Figure 1) and some vesicomyids were also observed within microbial mats. Furthermore, sulfur storage capacity have been observed for the vesicomyid species found in the Morelos site (Archivesica gigas) (Cruaud et al., unpublished), suggesting that hydrothermal vesicomyids could explore the seafloor and potentially exploit sulfide-rich areas, such as microbial mats, and then move away from those zones when/where sulfide concentrations and/or temperature conditions are not adapted to vesicomyid requirements (Vetter et al., 1991; Barry and Kochevar, 1998; Sahling et al., 2002). Indeed, substantial thermal fluctuations and fluid flow variations have been reported on the scale of hours or days in microbial mat areas (McKay et al., 2016), potentially allowing transient crossing of vesicomyids. Therefore, the sampled sediments in the Morelos site might be not sufficiently influenced by the metabolism of the bivalves (exudates, bioturbation) and the difference of microbial community composition between vesicomyid-colonized sediments might also be due to an influence of the hydrothermal fluid on vesicomyids behavior.
Conclusion
In this study, microbial community structure in sediment cores sampled in both cold seep and hydrothermally influenced sediments of the Guaymas Basin were explored and compared. Our results revealed that the sedimentary environment, by the presence of geochemical gradients and similar substrates (organic matter, sulfate, methane, hydrogen sulfide, and hydrocarbon), likely influences the microbial community composition and potential activities. This might explain the detection in both ecosystems of similar microbial lineages associated with analogous metabolic processes as well as comparable surface assemblages. Therefore, the “core microbiome” of the Guaymas Basin, identified in this study, might result from the influence of both deep-rising fluid emissions and organic matter inputs from the highly productive water of the Basin (Calvert et al., 1992).
Nonetheless, the proportion of endemic microorganisms was correlated with temperature, confirming that this parameter might be one of the key constraints on the spatial extent, physiological and phylogenetic diversity, and biogeochemical function in marine sediment microbial communities (Teske et al., 2014). Furthermore, more than high temperature conditions specific to the hydrothermal vent area, other hydrothermal activity-related features, such as abiotic reactions and vesicomyids behavior toward hydrothermal fluid emission, might shape the sedimentary microbial community composition and associated metabolisms and might also be likely responsible for the differences observed between these two environments.
Finally, the existence of similar microbial populations in the surface sediments of the two ecosystems and the potential continuity revealed by ribotype network analyses of archaeal lineages support the hypothesis of a potential connectivity among deep-sea ecosystems (Portail et al., 2016). In absence of physical borders, microorganisms might be dispersed globally across the seafloor or the hydrosphere and environmental conditions (temperature, specific compounds associated with hydrothermal fluids) might then select specific and highly adapted microorganisms.
Author Contributions
PC, AV, AG, and M-AC-B: designed the study; AV, J-CC, FL, LT, and AG: collected the samples; PC, PP, and J-CC: performed laboratory work; PC and AV: analyzed the data; PC, AV, LT, and M-AC-B: discussed and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to the crews of the research vessel l'Atalante and the submersible Nautile of the cruise “BIG” and the scientific team for their work on board. This cruise was funded by IFREMER (France) and has benefited from a work permit in Mexican waters (DAPA/2/281009/3803, 28 October 2009). We thank Maria-Cristina Ciobanu, Philippe Vandenkoornhuyse, Alexandra Dheilly, and Sophie Coudouel for helpful scientific discussions. This study was supported by a Carnot Institute funding and by an IFREMER/Brittany region Ph.D. grant for PC.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2017.00417/full#supplementary-material
References
Adams, M. W., Holden, J. F., Menon, A. L., Schut, G. J., Grunden, A. M., Hou, C., et al. (2001). Key role for sulfur in peptide metabolism and in regulation of three hydrogenases in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 183, 716–724. doi: 10.1128/JB.183.2.716-724.2001
Amend, J. P., and Shock, E. L. (2001). Energetics of overall metabolic reactions of thermophilic and hyperthermophilic Archaea and Bacteria. FEMS Microbiol. Rev. 5, 175–243. doi: 10.1111/j.1574-6976.2001.tb00576.x
Barry, J. P., Kochevar, R. E., and Baxter, C. H. (1997). The influence of pore-water chemistry and physiology on the distribution of vesicomyid clams at cold seeps in Monterey Bay: implications for patterns of chemosynthetic community organization. Limnol. Oceanogr. 42, 318–328. doi: 10.4319/lo.1997.42.2.0318
Barry, J. P., and Kochevar, R. E. (1998). A tale of two clams: differing chemosynthetic life styles among vesicomyids in Monterey Bay cold seeps. Cah. Biol. Mar. 39, 329–331.
Bernardino, A. F., Levin, L. A., Thurber, A. R., and Smith, C. R. (2012). Comparative composition, diversity and trophic ecology of sediment macrofauna at vents, seeps and organic falls. PLoS ONE 7:e33515. doi: 10.1371/journal.pone.0033515
Biddle, J. F., Cardman, Z., Mendlovitz, H., Albert, D. B., Lloyd, K. G., Boetius, A., et al. (2012). Anaerobic oxidation of methane at different temperature regimes in Guaymas Basin hydrothermal sediments. ISME J. 6, 1018–1031. doi: 10.1038/ismej.2011.164
Blazejak, A., and Schippers, A. (2010). High abundance of JS-1- and Chloroflexi-related Bacteria in deeply buried marine sediments revealed by quantitative, real-time PCR. FEMS Microbiol. Ecol. 72, 198–207. doi: 10.1111/j.1574-6941.2010.00838.x
Boyd, E. S., and Druschel, G. K. (2013). Involvement of intermediate sulfur species in biological reduction of elemental sulfur under acidic, hydrothermal conditions. Appl. Environ. Microbiol. 79, 2061–2068. doi: 10.1128/AEM.03160-12
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Brileya, K., and Reysenbach, A.-L. (2014). “The Class Archaeoglobi,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg: Springer), 15–23.
Callac, N., Rouxel, O., Lesongeur, F., Liorzou, C., Bollinger, C., Pignet, P., et al. (2015). Biogeochemical insights into microbe–mineral–fluid interactions in hydrothermal chimneys using enrichment culture. Extremophiles 19, 597–617. doi: 10.1007/s00792-015-0742-5
Calvert, S. E., Bustin, R. M., and Pedersen, T. F. (1992). Lack of evidence for enhanced preservation of sedimentary organic matter in the oxygen minimum of the Gulf of California. Geology 20, 757–760. doi: 10.1130/0091-7613(1992)020<0757:LOEFEP>2.3.CO;2
Cambon-Bonavita, M. A., Nadalig, T., Roussel, E., Delage, E., Duperron, S., Caprais, J. C., et al. (2009). Diversity and distribution of methane-oxidizing microbial communities associated with different faunal assemblages in a giant pockmark of the Gabon continental margin. Deep Sea Res. II Top. Stud. Oceanogr. 56, 2248–2258. doi: 10.1016/j.dsr2.2009.04.007
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Cruaud, P., Vigneron, A., Lucchetti-Miganeh, C., Ciron, P. E., Godfroy, A., and Cambon-Bonavita, M.-A. (2014). Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl. Environ. Microbiol. 80, 4626–4639. doi: 10.1128/AEM.00592-14
Cruaud, P., Vigneron, A., Pignet, P., Caprais, J.-C., Lesongeur, F., Toffin, L., et al. (2015). Microbial communities associated with benthic faunal assemblages at cold seep sediments of the Sonora Margin, Guaymas Basin. Aquat. Microbiol. 2:53. doi: 10.3389/fmars.2015.00053
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689. doi: 10.1073/pnas.89.12.5685
Dhillon, A., Lever, M., Lloyd, K. G., Albert, D. B., Sogin, M. L., and Teske, A. (2005). Methanogen diversity evidenced by molecular characterization of methyl coenzyme M reductase A (mcrA) genes in hydrothermal sediments of the Guaymas Basin. Appl. Environ. Microbiol. 71, 4592–4601. doi: 10.1128/AEM.71.8.4592-4601.2005
Dhillon, A., Teske, A., Dillon, J., Stahl, D. A., and Sogin, M. L. (2003). Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl. Environ. Microbiol. 69, 2765–2772. doi: 10.1128/AEM.69.5.2765-2772.2003
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Dufresne, S., Bousquet, J., Boissinot, M., and Guay, R. (1996). Sulfobacillus disulfidooxidans sp. nov., a new acidophilic, disulfide-oxidizing, gram-positive, spore-forming bacterium. Int. J. Syst. Evol. Microbiol. 46, 1056–1064. doi: 10.1099/00207713-46-4-1056
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Engelen, B., and Cypionka, H. (2009). The subsurface of tidal-flat sediments as a model for the deep biosphere. Ocean Dyn. 59:385. doi: 10.1007/s10236-008-0166-1
Erauso, G., Reysenbach, A.-L., Godfroy, A., Meunier, J.-R., Crump, B., Partensky, F., et al. (1993). Pyrococcus abyssi sp. nov., a new hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent. Arch. Microbiol. 160, 338–349. doi: 10.1007/BF00252219
Fischer, D., Sahling, H., Nöthen, K., Bohrmann, G., Zabel, M., and Kasten, S. (2012). Interaction between hydrocarbon seepage, chemosynthetic communities, and bottom water redox at cold seeps of the Makran accretionary prism: insights from habitat-specific pore water sampling and modeling. Biogeosciences 9, 2013–2031. doi: 10.5194/bg-9-2013-2012
Fish, S. A., Shepherd, T. J., McGenity, T. J., and Grant, W. D. (2002). Recovery of 16S ribosomal RNA gene fragments from ancient halite. Nature 417, 432–436. doi: 10.1038/417432a
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., et al. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim. Cosmochim. Acta 43, 1075–1090. doi: 10.1016/0016-7037(79)90095-4
González, J. M., Masuchi, Y., Robb, F. T., Ammerman, J. W., Maeder, D. L., Yanagibayashi, M., et al. (1998). Pyrococcus horikoshii sp. nov., a hyperthermophilic archaeon isolated from a hydrothermal vent at the Okinawa Trough. Extremophiles 2, 123–130. doi: 10.1007/s007920050051
Gorlas, A., Marguet, E., Gill, S., Geslin, C., Guigner, J.-M., Guyot, F., et al. (2015). Sulfur vesicles from thermococcales: a possible role in sulfur detoxifying mechanisms. Biochimie 118, 356–364. doi: 10.1016/j.biochi.2015.07.026
Grünke, S., Felden, J., Lichtschlag, A., Girnth, A.-C., De Beer, D., Wenzhöfer, F., et al. (2011). Niche differentiation among mat-forming, sulfide-oxidizing bacteria at cold seeps of the Nile Deep Sea Fan (Eastern Mediterranean Sea). Geobiology 9, 330–348. doi: 10.1111/j.1472-4669.2011.00281.x
Holler, T., Widdel, F., Knittel, K., Amann, R., Kellermann, M. Y., Hinrichs, K.-U., et al. (2011). Thermophilic anaerobic oxidation of methane by marine microbial consortia. ISME J. 5, 1946–1956. doi: 10.1038/ismej.2011.77
Hoshino, T., Toki, T., Ijiri, A., Morono, Y., Machiyama, H., Ashi, J., et al. (2017). Atribacteria from the subseafloor sedimentary biosphere disperse to the hydrosphere through submarine mud volcanoes. Front. Microbiol. 8:1135. doi: 10.3389/fmicb.2017.01135
Huber, H., and Stetter, K. O. (2006). “Desulfurococcales,” in The Prokaryotes, eds M. D. P. S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 52–68.
Huse, S. M., Huber, J. A., Morrison, H. G., Sogin, M. L., and Welch, D. M. (2007). Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 8:R143. doi: 10.1186/gb-2007-8-7-r143
Jørgensen, B. B. (1977). Bacterial sulfate reduction within reduced microniches of oxidized marine sediments. Mar. Biol. 41, 7–17. doi: 10.1007/BF00390576
Jørgensen, B. B., and Boetius, A. (2007). Feast and famine [mdash] microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781. doi: 10.1038/nrmicro1745
Joye, S. B., Bowles, M. W., Samarkin, V. A., Hunter, K. S., and Niemann, H. (2010). Biogeochemical signatures and microbial activity of different cold-seep habitats along the Gulf of Mexico deep slope. Deep Sea Res. II Top. Stud. Oceanogr. 57, 1990–2001. doi: 10.1016/j.dsr2.2010.06.001
Kellermann, M. Y., Wegener, G., Elvert, M., Yoshinaga, M. Y., Lin, Y.-S., Holler, T., et al. (2012). Autotrophy as a predominant mode of carbon fixation in anaerobic methane-oxidizing microbial communities. Proc. Natl. Acad. Sci. U.S.A. 109, 19321–19326. doi: 10.1073/pnas.1208795109
Kleindienst, S., Ramette, A., Amann, R., and Knittel, K. (2012). Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments. Environ. Microbiol. 14, 2689–2710. doi: 10.1111/j.1462-2920.2012.02832.x
Kojima, H., Umezawa, K., and Fukui, M. (2016). Caldimicrobium thiodismutans sp. nov., a sulfur-disproportionating bacterium isolated from a hot spring, and emended description of the genus Caldimicrobium. Int. J. Syst. Evol. Microbiol. 66, 1828–1831. doi: 10.1099/ijsem.0.000947
Kruskal, J. B. (1964). Nonmetric multidimensional scaling: a numerical method. Psychometrika 29, 115–129. doi: 10.1007/BF02289694
Kubo, K., Lloyd, K. G., F Biddle, J., Amann, R., Teske, A., and Knittel, K. (2012). Archaea of the miscellaneous crenarchaeotal group are abundant, diverse and widespread in marine sediments. ISME J. 6, 1949–1965. doi: 10.1038/ismej.2012.37
Kunin, V., Engelbrektson, A., Ochman, H., and Hugenholtz, P. (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123. doi: 10.1111/j.1462-2920.2009.02051.x
Lazar, C. S., Baker, B. J., Seitz, K. W., and Teske, A. P. (2017). Genomic reconstruction of multiple lineages of uncultured benthic archaea suggests distinct biogeochemical roles and ecological niches. ISME J. 11, 1118–1129. doi: 10.1038/ismej.2016.189
Lever, M. A., and Teske, A. P. (2015). Diversity of methane-cycling archaea in hydrothermal sediment investigated by general and group-specific PCR primers. Appl. Environ. Microbiol. 81, 1426–1441. doi: 10.1128/AEM.03588-14
Lichtschlag, A., Felden, J., Brüchert, V., Boetius, A., and de Beer, D. (2010). Geochemical processes and chemosynthetic primary production in different thiotrophic mats of the Håkon Mosby Mud Volcano (Barents Sea). Limnol. Oceanogr. 55, 931–949. doi: 10.4319/lo.2009.55.2.0931
Lin, Y.-S., Koch, B. P., Feseker, T., Ziervogel, K., Goldhammer, T., Schmidt, F., et al. (2017). Near-surface heating of young rift sediment causes mass production and discharge of reactive dissolved organic matter. Sci. Rep. 7:srep44864. doi: 10.1038/srep44864
Lloyd, K. G., Albert, D. B., Biddle, J. F., Chanton, J. P., Pizarro, O., and Teske, A. (2010). Spatial structure and activity of sedimentary microbial communities underlying a Beggiatoa spp. Mat in a gulf of mexico hydrocarbon seep. PLoS ONE 5:e8738. doi: 10.1371/journal.pone.0008738
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218. doi: 10.1038/nature12033
Mackay, R. J., McEntyre, C. J., Henderson, C., Lever, M., and George, P. M. (2011). Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin. Biochem. Rev. 32, 33–43.
McKay, L. J., MacGregor, B. J., Biddle, J. F., Albert, D. B., Mendlovitz, H. P., Hoer, D. R., et al. (2012). Spatial heterogeneity and underlying geochemistry of phylogenetically diverse orange and white Beggiatoa mats in Guaymas Basin hydrothermal sediments. Deep Sea Res. I Oceanogr. Res. Pap. 67, 21–31. doi: 10.1016/j.dsr.2012.04.011
McKay, L., Klokman, V. W., Mendlovitz, H. P., LaRowe, D. E., Hoer, D. R., Albert, D., et al. (2016). Thermal and geochemical influences on microbial biogeography in the hydrothermal sediments of Guaymas Basin, Gulf of California. Environ. Microbiol. Rep. 8, 150–161. doi: 10.1111/1758-2229.12365
Niemann, H., Lösekann, T., de Beer, D., Elvert, M., Nadalig, T., Knittel, K., et al. (2006). Novel microbial communities of the Haakon Mosby mud volcano and their role as a methane sink. Nature 443, 854–858. doi: 10.1038/nature05227
Nobu, M. K., Dodsworth, J. A., Murugapiran, S. K., Rinke, C., Gies, E. A., Webster, G., et al. (2016). Phylogeny and physiology of candidate phylum “Atribacteria” (OP9/JS1) inferred from cultivation-independent genomics. ISME J. 10, 273–286. doi: 10.1038/ismej.2015.97
Oksanen, J., Kindt, R., Legendre, P., O'Hara, B., Stevens, M. H. H., Oksanen, M. J., et al. (2007). The vegan package. Commun. Ecol. Package 10, 631–637.
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark Ocean above, at, and below the Seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422. doi: 10.1128/MMBR.00039-10
Otero, X. L., Huerta-Diaz, M. A., and Macias, F. (2003). Influence of a turbidite deposit on the extent of pyritization of iron, manganese and trace metals in sediments from the Guaymas Basin, Gulf of California (Mexico). Appl. Geochem. 18, 1149–1163. doi: 10.1016/S0883-2927(02)00190-7
Paradis, E. (2010). pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420. doi: 10.1093/bioinformatics/btp696
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pop Ristova, P., Wenzhöfer, F., Ramette, A., Zabel, M., Fischer, D., Kasten, S., et al. (2012). Bacterial Diversity and Biogeochemistry of Different Chemosynthetic Habitats of the REGAB Cold Seep (West African Margin, 3160 m Water Depth). Available online at: http://agris.fao.org/agris-search/search.do?recordID=AV2012099958 (Accessed March 30, 2017).
Portail, M., Olu, K., Dubois, S. F., Escobar-Briones, E., Gelinas, Y., Menot, L., et al. (2016). Food-web complexity in Guaymas Basin hydrothermal vents and cold seeps. PLoS ONE 11:e0162263. doi: 10.1371/journal.pone.0162263
Portail, M., Olu, K., Escobar-Briones, E., Caprais, J. C., Menot, L., Waeles, M., et al. (2015). Comparative study of vent and seep macrofaunal communities in the Guaymas Basin. Biogeosciences 12, 5455–5479. doi: 10.5194/bg-12-5455-2015
Robbins, S. J., Evans, P. N., Parks, D. H., Golding, S. D., and Tyson, G. W. (2016). Genome-centric analysis of microbial populations enriched by hydraulic fracture fluid additives in a coal bed methane production well. Front. Microbiol. 7:731. doi: 10.3389/fmicb.2016.00731
Roussel, E. G., Sauvadet, A.-L., Allard, J., Chaduteau, C., Richard, P., Bonavita, M.-A. C., et al. (2009). Archaeal methane cycling communities associated with gassy subsurface sediments of Marennes-Oléron Bay (France). Geomicrobiol. J. 26, 31–43. doi: 10.1080/01490450802599284
Ruff, S. E., Arnds, J., Knittel, K., Amann, R., Wegener, G., Ramette, A., et al. (2013). Microbial communities of deep-sea methane seeps at hikurangi continental margin (New Zealand). PLoS ONE 8:e72627. doi: 10.1371/journal.pone.0072627
Ruff, S. E., Biddle, J. F., Teske, A. P., Knittel, K., Boetius, A., and Ramette, A. (2015). Global dispersion and local diversification of the methane seep microbiome. Proc. Natl. Acad. Sci. U.S.A. 112, 4015–4020. doi: 10.1073/pnas.1421865112
Sahling, H., Rickert, D., Lee, R. W., Linke, P., and Suess, E. (2002). Macrofaunal community structure and sulfide flux at gas hydrate deposits from the Cascadia convergent margin, NE Pacific. Mar. Ecol. Prog. Ser. 231, 121–138. doi: 10.3354/meps231121
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310. doi: 10.1371/journal.pone.0027310
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sibuet, M., and Olu, K. (1998). Biogeography, biodiversity and fluid dependence of deep-sea cold-seep communities at active and passive margins. Deep Sea Res. II Top. Stud. Oceanogr. 45, 517–567. doi: 10.1016/S0967-0645(97)00074-X
Simoneit, B. R. T., Lonsdale, P. F., Edmond, J. M., and Shanks, W. C. (1990). Deep-water hydrocarbon seeps in Guaymas Basin, Gulf of California. Appl. Geochem. 5, 41–49. doi: 10.1016/0883-2927(90)90034-3
Sørensen, K. B., and Teske, A. (2006). Stratified communities of active archaea in deep marine subsurface sediments. Appl. Environ. Microbiol. 72, 4596–4603. doi: 10.1128/AEM.00562-06
Teske, A., Callaghan, A. V., and LaRowe, D. E. (2014). Biosphere frontiers of subsurface life in the sedimented hydrothermal system of Guaymas Basin. Front. Microbiol. 5:362. doi: 10.3389/fmicb.2014.00362
Teske, A., Hinrichs, K.-U., Edgcomb, V., de Vera Gomez, A., Kysela, D., Sylva, S. P., et al. (2002). Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl. Environ. Microbiol. 68, 1994–2007. doi: 10.1128/AEM.68.4.1994-2007.2002
Tunnicliffe, V. (1991). The biology of hydrothermal vents : ecology and evolution. Oceanogr. Mar. Biol. Ann. Rev. 29, 319–407.
Tunnicliffe, V., McArthur, A. G., and McHugh, D. (1998). A biogeographical perspective of the deep-sea hydrothermal Vent Fauna. Adv. Mar. Biol. 34, 353–442. doi: 10.1016/S0065-2881(08)60213-8
Van Dover, C. L., German, C. R., Speer, K. G., Parson, L. M., and Vrijenhoek, R. C. (2002). Evolution and biogeography of deep-sea vent and seep invertebrates. Science 295, 1253–1257. doi: 10.1126/science.1067361
Vetter, R. D., Powell, M. A., and Somero, G. N. (1991). Metazoan adaptations to hydrogen sulphide. Metazoan Life Oxyg. 109–128.
Vigneron, A., Cruaud, P., Pignet, P., Caprais, J.-C., Cambon-Bonavita, M.-A., Godfroy, A., et al. (2013). Archaeal and anaerobic methane oxidizer communities in the sonora margin cold seeps, Guaymas Basin (Gulf of California). ISME J. 7, 1595–1608. doi: 10.1038/ismej.2013.18
Vigneron, A., Cruaud, P., Pignet, P., Caprais, J.-C., Gayet, N., Cambon-Bonavita, M.-A., et al. (2014). Bacterial communities and syntrophic associations involved in anaerobic oxidation of methane process of the Sonora Margin cold seeps, Guaymas Basin. Environ. Microbiol. 16, 2777–2790. doi: 10.1111/1462-2920.12324
Vigneron, A., L'Haridon, S., Godfroy, A., Roussel, E. G., Cragg, B. A., Parkes, R. J., et al. (2015). Evidence of active methanogen communities in shallow sediments of the sonora margin cold seeps. Appl. Environ. Microbiol. 81, 3451–3459. doi: 10.1128/AEM.00147-15
Von Damm, K. L., Edmond, J. M., Measures, C. I., and Grant, B. (1985). Chemistry of submarine hydrothermal solutions at Guaymas Basin, Gulf of California. Geochim. Cosmochim. Acta 49, 2221–2237. doi: 10.1016/0016-7037(85)90223-6
Wasmund, K., Cooper, M., Schreiber, L., Lloyd, K. G., Baker, B. J., Petersen, D. G., et al. (2016). Single-cell genome and group-specific dsrAB sequencing implicate marine members of the class dehalococcoidia (Phylum Chloroflexi) in sulfur cycling. MBio 7, e00266–16. doi: 10.1128/mBio.00266-16
Webster, G., Newberry, C. J., Fry, J. C., and Weightman, A. J. (2003). Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55, 155–164. doi: 10.1016/S0167-7012(03)00140-4
Wegener, G., Krukenberg, V., Riedel, D., Tegetmeyer, H. E., and Boetius, A. (2015). Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature 526, 587–590. doi: 10.1038/nature15733
Yu, Z., and Morrison, M. (2004). Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 70, 4800–4806. doi: 10.1128/AEM.70.8.4800-4806.2004
Keywords: microbial diversity, hydrothermal vents, cold seeps, ribotypes, methane, extreme environments
Citation: Cruaud P, Vigneron A, Pignet P, Caprais J-C, Lesongeur F, Toffin L, Godfroy A and Cambon-Bonavita M-A (2017) Comparative Study of Guaymas Basin Microbiomes: Cold Seeps vs. Hydrothermal Vents Sediments. Front. Mar. Sci. 4:417. doi: 10.3389/fmars.2017.00417
Received: 28 July 2017; Accepted: 04 December 2017;
Published: 18 December 2017.
Edited by:
Télesphore Sime-Ngando, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Mustafa Yucel, Middle East Technical University, TurkeyJulie L. Meyer, University of Florida, United States
Copyright © 2017 Cruaud, Vigneron, Pignet, Caprais, Lesongeur, Toffin, Godfroy and Cambon-Bonavita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Perrine Cruaud, cGVycmluZS5jcnVhdWRAZ21haWwuY29t