 Mark Meekan1
Mark Meekan1 Christopher M. Austin2,3,4,5Mun H. Tan2Nu-Wei V. Wei5Adam Miller2
Christopher M. Austin2,3,4,5Mun H. Tan2Nu-Wei V. Wei5Adam Miller2 Simon J. Pierce6David Rowat7Guy Stevens8Tim K. Davies9Alessandro Ponzo10
Simon J. Pierce6David Rowat7Guy Stevens8Tim K. Davies9Alessandro Ponzo10 Han Ming Gan2,3,4*
Han Ming Gan2,3,4*- 1Australian Institute of Marine Science, Indian Ocean Marine Research Centre (MO96), Crawley, WA, Australia
- 2Centre for Integrative Ecology, School of Life and Environmental Sciences, Deakin University, Geelong, VIC, Australia
- 3School of Science, Monash University Malaysia, Petaling Jaya, Malaysia
- 4Genomics Facility, Tropical Medicine and Biology Platform, Monash University Malaysia, Petaling Jaya, Malaysia
- 5Research Institute for the Environment and Livelihoods, Charles Darwin University, Darwin, NT, Australia
- 6Marine Megafauna Foundation, Truckee, CA, United States
- 7Marine Conservation Society, Seychelles, Seychelles
- 8The Manta Trust, Dorchester, United Kingdom
- 9MRAG Ltd., London, United Kingdom
- 10Large Marine Vertebrates Research Institute Philippines, Bohol, Philippines
The whale shark (Rhincodon typus) is an iconic and endangered species with a broad distribution spanning warm-temperate and tropical oceans. Effective conservation management of the species requires an understanding of the degree of genetic connectivity among populations, which is hampered by the need for sampling that involves invasive techniques. Here, the feasibility of minimally-invasive sampling was explored by isolating and sequencing whale shark DNA from a commensal or possibly parasitic copepod, Pandarus rhincodonicus that occurs on the skin of the host. We successfully recovered mitochondrial control region DNA sequences (~1,000 bp) of the host via DNA extraction and polymerase chain reaction from whole copepod specimens. DNA sequences obtained from multiple copepods collected from the same shark exhibited 100% sequence similarity, suggesting a persistent association of copepods with individual hosts. Newly-generated mitochondrial haplotypes of whale shark hosts derived from the copepods were included in an analysis of the genetic structure of the global population of whale sharks (644 sequences; 136 haplotypes). Our results supported those of previous studies and suggested limited genetic structuring across most of the species range, but the presence of a genetically unique and potentially isolated population in the Atlantic Ocean. Furthermore, we recovered the mitogenome and nuclear ribosomal genes of a whale shark using a shotgun sequencing approach on copepod tissue. The recovered mitogenome is the third mitogenome reported for the species and the first from the Mozambique population. Our invertebrate DNA (iDNA) approach could be used to better understand the population structure of whale sharks, particularly in the Atlantic Ocean, and also for genetic analyses of other elasmobranchs parasitized by pandarid copepods.
Introduction
The term environmental DNA (eDNA) refers to DNA extracted from cells that are not collected directly from a target organism, but are obtained from the environments they inhabit such as oceans, river water, soil, and air (Ficetola et al., 2008; Fonseca et al., 2010; Andersen et al., 2012; Sigsgaard et al., 2016). Invertebrate-derived DNA (iDNA) is an offshoot of this approach that involves the extraction of genetic material of animals via the flesh-eating or haematophagous invertebrates that parasitise them (Schnell et al., 2015; Schubert et al., 2015; Lee et al., 2016). To date, most iDNA studies have focused on terrestrial vertebrates and have extracted host DNA from insects, ticks, or leeches. There have been no analogous studies in marine environments, despite the potential usefulness of the approach for sampling large marine megafauna such as cetaceans, sirenians, pinnipeds, marine reptiles (sea turtles), elasmobranchs, and teleosts. For such taxa, iDNA sampling offers both ethical and practical advantages as it is minimally-invasive and thus preferable to the direct collection of blood or tissue, particularly where target species are rare or endangered.
The whale shark (Rhincodon typus), similar to many large marine vertebrates, poses significant challenges for researchers trying to understand their biology and ecology and for managers attempting to develop conservation strategies (Graham and Roberts, 2007; Rowat et al., 2009). Tagging, genetic and modeling studies suggest that individuals can disperse widely (Sequeira et al., 2013), although there is evidence of isolation between populations in the Indo-Pacific and Atlantic oceans (Vignaud et al., 2014). However, the scale at which population structure can be discerned is dependent on sample sizes, with low numbers reducing analytical power to reject the null hypothesis of a panmictic population (Castro et al., 2007). For whale sharks, low sample size tends to be a result of the rarity of the species and the difficulties in obtaining biopsies (usually of skin tissue) for genetic analyses, which requires appropriate permits and ethical approvals for invasive sampling of a species that is categorized as “Endangered” by the International Union for the Conservation of Nature and the use of trained divers and technicians. In this situation there are many benefits to the use of an iDNA approach, provided that external invertebrate parasites or commensals that harbor the intact DNA of the whale shark host can be successfully identified. Although eDNA techniques have recently been used to obtain whale shark DNA (Sigsgaard et al., 2016), iDNA still offers a major advantage because it enables haplotypes to be linked to individual whale sharks for the analysis of the genetic structures of populations.
Copepods (Crustacea, Maxillopoda, Copepoda) are an excellent candidate species for iDNA studies on marine vertebrates. Free-living forms are a dominant element of marine zooplankton and may even exceed insects in terrestrial environments in terms of sheer abundance of individuals. Less recognized is that approximately half of the nearly 30,000 described species live in parasitic or commensal associations with a diverse range of taxa, including fish and mammals (Boxshall, 2005). The order Siphonostomatoida contains largely parasitic copepods that feed on the blood, epidermal tissue, or mucus of many marine teleost fishes, sharks, and rays. Approximately 550 genera, representing nearly 40 families, are placed in the order and include economically important species such as sea lice (Brachiura) that parasitise farmed fish (Gunn and Pitt, 2012). Eleven siphonostomatoid families have been reported as symbionts of a diverse range of elasmobranchs (Dippenaar, 2009) and one family, the Pandaridae, is composed of 23 genera that include species that are ectoparasites or commensals of large sharks (Cressey and Boyle, 1978; Walter and Boxshall, 2017). Within the Pandaridae, the genus Pandarus currently has 17 recognized species of which P. rhincodonicus (Norman et al., 2000) is noteworthy as it is appears to be associated exclusively with the whale shark, where it is predominately found on the leading and trailing edges of fins and on the lips. Although it is thought to be a commensal that feeds off bacteria and other microorganisms on the skin of the shark (Norman et al., 2000), dietary studies of the species are incomplete.
Here, we demonstrate that P. rhincodonicus sampled from sites across the Indian Ocean contain sufficient DNA from their whale shark host to routinely recover mitochondrial sequences that allow analyses of the genetic structure of host populations, and the recovery of the complete whale shark mitogenome and ribosomal nuclear genes. The implications of these findings are discussed in relation to the use of copepods for iDNA studies of marine teleosts and sharks and the ecological status of Pandarus spp. as commensals.
Materials and Methods
Sampling and Sequencing
A total of 45 copepods were collected from 31 individual whale sharks sampled in Baa Atoll in the Maldives, at Ningaloo Reef, Western Australia, off Tofu Beach, Mozambique and off the coast of Mahe in the Seychelles as part of an earlier study using approved procedures under the Western Australian Department of Parks and Wildlife (WADPW) and University of Tasmania (UTAS) ethics permits (WADPW: SF009814 and SF009227; UTAS: 2255 and 2307) (Vignaud et al., 2014) (Supplementary Table 1). Copepods were scraped off the edges of the fins or lips using a plastic knife by a snorkeler who swam alongside an unrestrained animal, collected in an aquarium net and brought back to the vessel where they were preserved in 100% ethanol. DNA extraction was performed on whole copepod specimens using the DNeasy Blood and Tissue kit (Qiagen, Halden, Germany). Of the 45 copepods we collected, 44 were selected for the amplification of whale shark the mtDNA control region, using primers WSCR1-F and WSCR2-R, and following the PCR protocol as described by Castro et al. (2007). PCR products were purified using the Viogene Gel/PCR DNA Isolation System Kit (Viogene Biotek Corp, Taitung, Taiwan) and sequenced on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA) located at Charles Darwin University.
The remaining copepod, isolate 366.1, was used for partial genome sequencing. Briefly, 1 μg of genomic DNA (gDNA) was sheared to 300 bp using a Covaris Focused Ultrasonicator (Covaris, Woburn, MA), and subsequently processed with Truseq DNA library prep kit (Illumina, San Diego, CA) according to the manufacturer's instructions. This was followed by next-generation sequencing on a fraction of a HiSeq 2000 lane (Illumina, San Diego, CA), with a run setting of 2 × 100 bp, for the initial goal of recovering the copepod mitogenome (Austin et al., 2016) and microsatellite loci (unpublished).
Mitochondrial Control Region Analysis
Authenticity of the DNA sequences of the mitochondrial control region of whale sharks were validated by BLAST searches, and sequences were aligned with previously generated sequences (Castro et al., 2007; Vignaud et al., 2014) using MAFFT version 7.310 with the option “–adjustdirection” activated (Yamada et al., 2016). The 5′ and 3′ ends of the alignment were manually trimmed using MEGA 6 (Tamura et al., 2013) so that each aligned control region had the same flanking sequence (Supplementary Figure 1). The trimmed alignment was subsequently de-gapped using Seqret (Rice et al., 2000) and clustered with cd-hit-est at a 100% sequence identity and 100% sequence length coverage cut-off as implemented using the –c 1.0 and –a 1.0 setting (Li and Godzik, 2006).
Analysis of Population Differentiation
The mitochondrial control regions of whale sharks generated using iDNA were combined with 613 sequences published by previous studies of whale shark populations (Castro et al., 2007; Ramírez-Macías et al., 2007; Schmidt et al., 2010; Vignaud et al., 2014). Identical iDNA sequences from the same whale shark host were removed prior to genetic analysis, to eliminate the chance of biasing estimates of population genetic differentiation (Supplementary Table 1). Arlequin suite version 3.5.2.2 (Excoffier and Lischer, 2010) was used to estimate global and pairwise ϕST. A median-joining haplotype network was constructed using PopArt version 1.7 (Leigh and Bryant, 2015) and to simplify network representation, only the common haplotypes (a haplotype with more than one sequence representation) were used to generate the network graph.
Recovery of Whale Shark Whole Mitochondrial Genome and Nuclear Ribosomal Genes
Assembly used a baiting and iterative mapping approach as implemented in MITObim version 1.8 (Hahn et al., 2013) from the complete mitogenome of a whale shark sampled off Taiwan (Accession Number: NC_023455) as the reference sequence (Alam et al., 2014). In-silico circularization and annotation of the assembled mitogenome followed (Iwasaki et al., 2013; Gan et al., 2014). To improve alignment accuracy and specificity, mitogenome coverage was calculated by mapping the raw paired-end reads to the assembled mitogenome sequence using Bowtie2 version 2.3.2 (Langmead and Salzberg, 2012) with the setting “—score-min L,0.2,−0.2.” BRIG was used to visualize the mitogenome annotation and read mapping coverage (Alikhan et al., 2011). Similar Bowtie2 mapping setting was used to map the raw reads to a whale shark assembly contig containing the nuclear ribosomal genes (18S rRNA and 28S rRNA) and the read alignment was subsequently visualized using Integrative Genomics Viewer (Thorvaldsdottir et al., 2013).
Results
Mitochondrial Control Region Analysis
A ~1,000 bp DNA sequence fragment of the whale shark mitochondrial control region was successfully generated from the extracted gDNA from 44 copepods representing 31 sharks. Haplotype clustering confirmed a 100% sequence match for DNA sequences amplified from copepods collected from the same whale shark host (Supplementary Data 1). An initial alignment of the 31 newly-generated iDNA control region sequences with the 613 sequences provided by previous studies (Castro et al., 2007; Schmidt et al., 2010; Vignaud et al., 2014; Walter and Boxshall, 2017) yielded a 1,910 bp product (Supplementary Data 2). Trimming using 5′- and 3′-ends produced a final alignment length of 750 bp (Supplementary Data 3), from which a total of 163 unique haplotypes were evident (Supplementary Data 4). The Ningaloo Reef population had the highest number of sampled control region sequences (n = 163) and also had the greatest number of haplotypes (h = 36) (Figure 1A). Interestingly, the observed haplotype diversity for the Isla Holbox population was 50% lower (h = 11) than that of Djibouti population (Vignaud et al., 2014), despite similar sample sizes (Figures 1A,B).
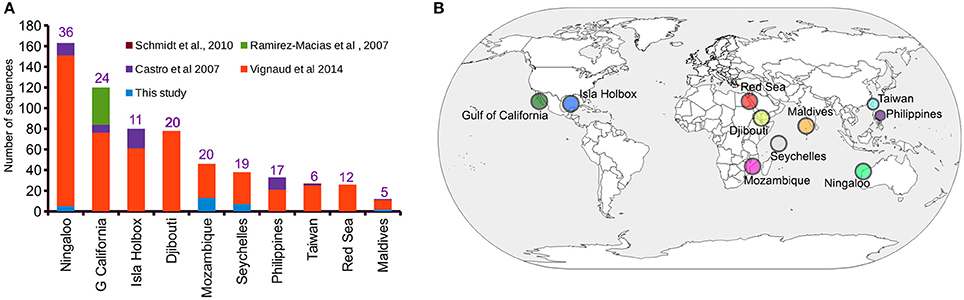
Figure 1. Sampling distribution of Rhincodon typus based on the mitochondrial control region sequences. (A) A summary of the number of publicly available mitochondrial control region sequences of R. typus from different populations and studies. Numbers on each bar indicate the number of haplotypes that are present in each population. (B) An annotated world map showing the locations of R. typus sampled by all studies.
Of the 31 deduplicated iDNA sequences reported in this study, five were found to represent replicate individuals from the Ningaloo population previously sequenced by Vignaud et al. (2014) using skin samples (Supplementary Table 1 and Supplementary Data 4). Despite the relatively low number of sequences reported by our study that represented sequences from new whale sharks (N = 27), our study contributed a 39 and 22% increase in the number of individuals sampled from the Mozambique and Seychelles populations, respectively (Figure 1A). In addition, four novel haplotypes for control region sequences of whale sharks were identified from copepod samples.
Population Structure of the Whale Shark
Global ϕST was weak but significantly different from zero, indicating limited gene flow among some sampling locations. This pattern was driven by a single population, with all pairwise ϕST calculations associated with the Isla Holbox population differing significantly from zero (average ϕST of 0.2), whereas all estimates of ϕST did not (Figure 2). This implied that the population at Isla Holbox was genetically isolated from an otherwise panmictic population spread across the Indo-Pacific Ocean. Three common haplotypes occurred at similar frequency across the sampling range (Figure 3). A few private haplotypes occurred in the Ningaloo Reef, Philippines and Seychelles populations. Consistent with ϕST estimates, the population at Isla Holbox was the most genetically differentiated, as it lacked one of the three common ancestral haplotypes (H108, 110, 125, 150, and 152), and had a high frequency of others that were rare (H67, H68, and H76).
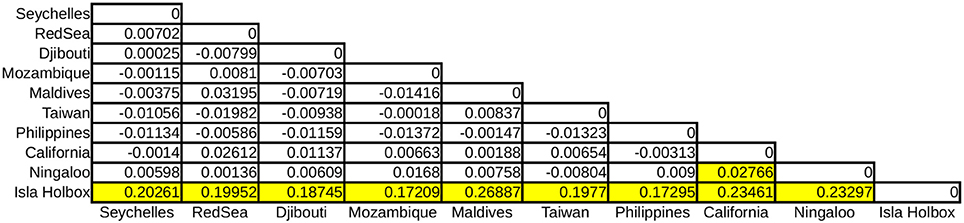
Figure 2. Pairwise ϕST calculations of R. typus populations with yellow boxes indicating significant values (p < 0.001).
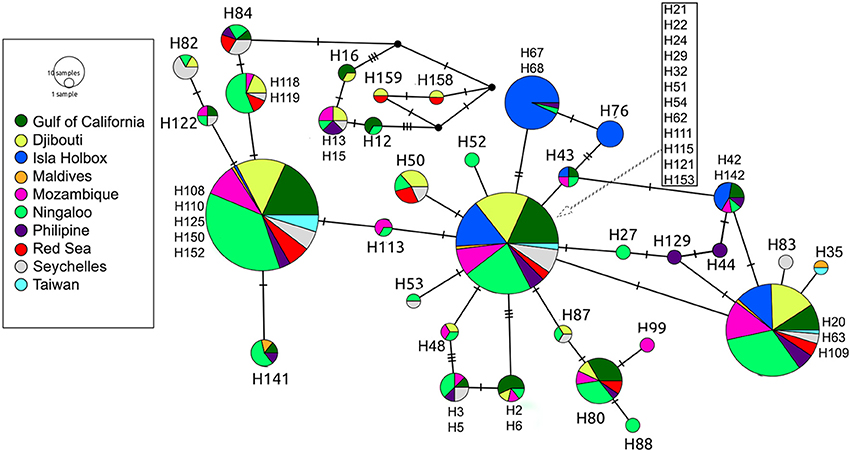
Figure 3. Median-joining population network based on control region sequences of R. typus. Each circle represents a unique haplotype, with colors depicting the proportion of individuals from the various sample sites sharing the haplotype. Ticks on connecting lines indicate mutational steps between haplotypes.
Recovery of the Whale Shark Mitogenome and Nuclear Genes from Shotgun Sequencing of Copepod Tissue
The complete mitochondrial genome of the whale shark was successfully assembled from partial genome sequencing of an iDNA sample from a copepod (isolate 366.1; Figure 4). Mapping of the 14 million paired-end reads to the reconstructed whale shark mitogenome gave a mapping rate of 0.006% (900 reads) representing approximately 5× mitogenome coverage (Figure 4). The mitogenomic composition of the whale shark were extracted was very similar (>99% identity) to those reconstructed from sharks from Taiwanese waters (Accession codes: KC633221 and NC_023445). A comparison of the 13 genes coding for mitochondrial proteins indicated one or two nucleotide mismatches, mostly associated with non-synonymous mutations found in the atp6, cox1, cytb, nad2, nad4, and nad5 genes. In addition, we also observed reads mapping to the whale shark 18S and 28S rRNA genes (Supplementary Figure 2 and Supplementary Data 5), indicating the presence of whale shark nuclear DNA in the copepod extracted gDNA, a phenomenon that will be useful for future population genetic studies of these sharks based on nuclear markers.
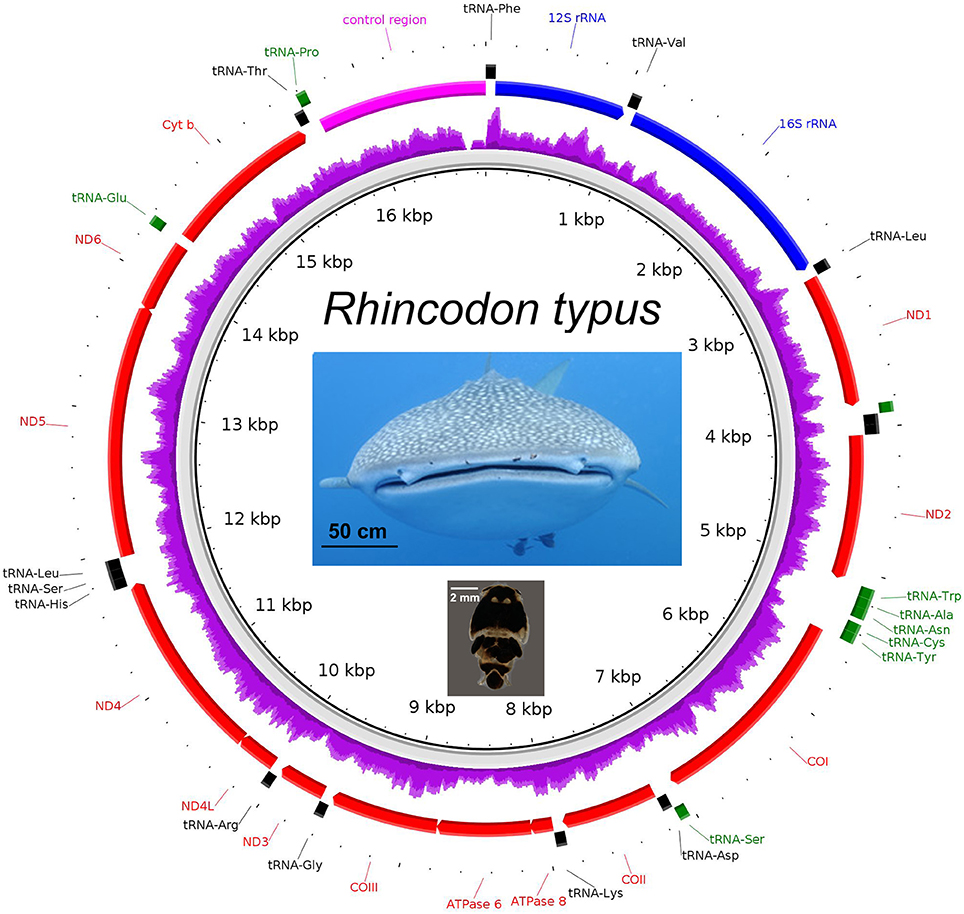
Figure 4. The mitogenome of Rhincodon typus (upper photo—note copepod parasites Pandarus rhincodonicus visible as black dots near the center of the upper lip) recovered from the shotgun sequencing of the parasitic copepod, P. rhincodonicus (lower photo). The purple ring indicates relative mapping coverage, and the transcriptional orientation of each protein coding gene is indicated by the red arrows.
Data Deposition
Mitochondrial control regions were submitted to NCBI under the accession numbers MF872682-MF872725. Raw reads from the shotgun sequencing of the copepod were submitted to Sequence Read Archive under the run number SRR4111090 and the reconstructed whale shark mitogenome has been assigned the accession number MF872726.
Discussion
Our study is the first demonstration of the use of iDNA sampling of a marine invertebrate in order to obtain mitochondrial genetic information from an elasmobranch host. We successfully amplified mitochondrial DNA fragments of whale sharks in the size range of ~1,000 bp from a copepod, one of the largest host DNA fragments to be recovered by an iDNA study (Schnell et al., 2012, 2015; Lee et al., 2016; Pérez-Flores et al., 2016; Rodgers et al., 2017). This suggests the presence of largely intact whale shark DNA in or on copepods, although the exact source could not be identified as copepods were analyzed whole. Targeted efforts to amplify DNA fragments of whale sharks from the intestine and epidermal layers of P. rhincodonicus will be useful to resolve this issue and clarify the role of the copepod as a commensal or parasite. We found iDNA sequences from multiple copepods sampled from the same whale shark host to be identical, suggesting copepods have a persistent, long-term association with their host shark, which contrasts with the more generalist and mobile host associations of other invertebrate ectoparasites in terrestrial and aquatic ecosystems, such as leeches, blow flies, and mosquitoes (Calvignac-Spencer et al., 2013). However, these findings should be treated with some degree of caution and a multi-locus approach using microsatellite markers (Olson et al., 2012) might offer a more powerful means to validate these findings. As reported by Vignaud et al. (2014), our estimates of genetic structure across the Indian, Pacific, and Atlantic oceans indicated the presence of two distinct populations, one in the Indo-Pacific and the other in the Atlantic Ocean. Isla Holbox was the only location sampled in the Atlantic Ocean and the minimally-invasive iDNA approach could potentially be used to improve the coverage of sampling in this region.
We also demonstrated that complete mitochondrial genome and nuclear ribosomal RNA sequences of whale sharks could be obtained using iDNA sampling. Although not the first for the species (Alam et al., 2014; Chen et al., 2016), the sequences generated by our study represent the first for the Mozambique population of whale sharks. The successful recovery of the mitogenome and nuclear ribosomal RNA from the shotgun sequencing of P. rhincodonicus was probably due to the high copy number of mitochondrial and nuclear ribosomal genes in somatic cells, coupled with high sequencing depth (~14 million paired-end reads). Efficiency of the recovery of whale shark mitogenomes from copepod tissue samples might be improved by sequencing gDNA extracted from the part of the copepod body that contains higher concentrations of whale shark tissue. In addition, given the intactness of whale shark gDNA (>1,000 bp) that was present in P. rhincodonicus, long range PCR of additional mitogenome regions followed by high throughput amplicon sequencing could also be explored to dramatically increase the coverage of the whale shark mitogenome for population genetic and phylogeographic applications (Deiner et al., 2017; Pavlova et al., 2017). Given that mtDNA only represents a fraction of the evolutionary history of a species due to its strict maternal inheritance and small genome size (16 kb), studies of the population genetics of whale sharks may benefit from the combination of both mtDNA and nuclear data (Godinho et al., 2008; Schmidt et al., 2009; Vignaud et al., 2014). The presence of nuclear ribosomal RNA reads in the copepod shotgun sequencing library suggests that it is possible to amplify both nuclear DNA and mtDNA of whale sharks from the copepod and the recent publication of the first draft genome for the whale shark (Read et al., 2017) will greatly facilitate the design of a targeted next-generation sequencing panel to enhance future studies of whale shark population genetics.
The Pandaridae comprises 23 genera and more than 100 species, many of which are ectoparasites or symbionts of large sharks (Walter and Boxshall, 2017), and there are nearly 30 other families of copepods that include species that are parasites of fishes (Boxshall, 2005). These offer an opportunity to extend iDNA methods to other taxa of large marine vertebrates. Conversely, the presence of gDNA of whale sharks in the extracted DNA of P. rhincodonicus will need to be considered as a possible contaminant in future whole genome sequencing or population genomic projects that target this copepod.
Author Contributions
MM and CA: Original concept, sample collection, analyses, interpretation of results, writing, proof-reading. MT: Bioinformatics analysis. N-WW: Control region sequencing. AM: interpretation of results, writing, proof-reading. SP, DR, GS, TD, and AP: sample collection proof-reading. HG: analyses, writing, interpretation of results, proof-reading.
Funding
Funding for this study was provided by the SeaWorld Research and Rescue Foundation, the Save our Seas Foundation, the Monash University Malaysia Tropical Medicine and Biology Platform, Monash University Malaysia School of Science, Quadrant Energy, Australian Institute of Marine Science, The Western Australian Department of Environment and Conservation, The Four Seasons Resorts Maldives. Funding for SP came from Aqua-Firma, the Shark Foundation, the Save Our Seas Foundation and two private trusts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GS declared a shared affiliation, with no collaboration,with several of the authors, CA, MT, AM, and HG, to the handling Editor.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2017.00420/full#supplementary-material
Supplementary Table 1. Sample collection information.
Supplementary Figure 1. Visualization of aligned control region sequences (one representative from each study). Arrows indicate regions upstream and downstream of the 5′ and 3′ ends, respectively, that will be trimmed off for subsequent haplotype clustering.
Supplementary Figure 2. Bowtie2 alignment of Illumina reads generated from copepod gDNA to the whale shark genomic region containing 18S and 28S nuclear rRNA genes. Red and blue arrow bars indicate forward and reverse pair-end reads, respectively.
Supplementary Data 1. Cd-hit clustering of trimmed whale shark control region sequences that were amplified from copepods.
Supplementary Data 2. Alignment of whale shark control region sequences.
Supplementary Data 3. Trimmed alignment of whale shark control region sequences based on the conserved region depicted in Supplementary Figure 1.
Supplementary Data 4. Cd-hit Clustering of trimmed whale shark control region sequences.
Supplementary Data 5. Paired-end reads mapped to the whale shark genomic region containing the 18S and 28S ribosomal RNA genes.
References
Alam, M. T., Petit, R. A. III., Read, T. D., and Dove, A. D. (2014). The complete mitochondrial genome sequence of the world's largest fish, the whale shark (Rhincodon typus), and its comparison with those of related shark species. Gene 539, 44–49. doi: 10.1016/j.gene.2014.01.064
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Andersen, K., Bird, K. L., Rasmussen, M., Haile, J., Breuning-Madsen, H., Kjaer, K. H., et al. (2012). Meta-barcoding of 'dirt' DNA from soil reflects vertebrate biodiversity. Mol. Ecol. 21, 1966–1979. doi: 10.1111/j.1365-294X.2011.05261.x
Austin, C. M., Tan, M. H., Lee, Y. P., Croft, L. J., Meekan, M. G., Pierce, S. J., et al. (2016). The complete mitogenome of the whale shark parasitic copepod Pandarus rhincodonicus norman, Newbound and Knott (Crustacea; Siphonostomatoida; Pandaridae)–a new gene order for the copepoda. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 694–695. doi: 10.3109/19401736.2014.913147
Boxshall, G. (2005). “Curstacean parasites: Branchiura (fish lice),” in Marine Parasitology, ed K. Robde (Collingwood, VIC: CSIRO publishing), 145–147.
Calvignac-Spencer, S., Leendertz, F. H., Gilbert, M. T., and Schubert, G. (2013). An invertebrate stomach's view on vertebrate ecology: certain invertebrates could be used as “vertebrate samplers” and deliver DNA-based information on many aspects of vertebrate ecology. Bioessays 35, 1004–1013. doi: 10.1002/bies.201300060
Castro, A. L., Stewart, B. S., Wilson, S. G., Hueter, R. E., Meekan, M. G., Motta, P. J., et al. (2007). Population genetic structure of Earth's largest fish, the whale shark (Rhincodon typus). Mol. Ecol. 16, 5183–5192. doi: 10.1111/j.1365-294X.2007.03597.x
Chen, X., Ai, W., Pan, L., and Shi, X. (2016). Complete mitochondrial genome of the largest living fish: whale shark Rhincodon typus (Orectolobiformes: Rhincodontidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 425–426. doi: 10.3109/19401736.2014.898288
Cressey, R., and Boyle, H. (1978). A new genus and speices of parasitic copepod (Pandaridae) from a unique new shark. Pac. Sci. 32, 25–30.
Deiner, K., Renshaw, M. A., Li, Y., Olds, B. P., Lodge, D. M., and Pfrender, M. E. (2017). Long-range PCR allows sequencing of mitochondrial genomes from environmental DNA. Methods Ecol. Evol. 8, 1888–1898. doi: 10.1111/2041-210X.12836
Dippenaar, S. (2009). Estimated molecular phylogenetic relationships of six siphonostomatoid families (Copepoda) symbiotic on elasmobranchs. Crustaceana 82, 1547–1567. doi: 10.1163/001121609X12511103974538
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under linux and windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Ficetola, G. F., Miaud, C., Pompanon, F., and Taberlet, P. (2008). Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425. doi: 10.1098/rsbl.2008.0118
Fonseca, V. G., Carvalho, G. R., Sung, W., Johnson, H. F., Power, D. M., Neill, S. P., et al. (2010). Second-generation environmental sequencing unmasks marine metazoan Biodiversity 1:98. doi: 10.1038/ncomms1095
Gan, H. M., Schultz, M. B., and Austin, C. M. (2014). Integrated shotgun sequencing and bioinformatics pipeline allows ultra-fast mitogenome recovery and confirms substantial gene rearrangements in Australian freshwater crayfishes. BMC Evol. Biol. 14:19. doi: 10.1186/1471-2148-14-19
Godinho, R., Crespo, E. G., and Ferrand, N. (2008). The limits of mtDNA phylogeography: complex patterns of population history in a highly structured Iberian lizard are only revealed by the use of nuclear markers. Mol. Ecol. 17, 4670–4683. doi: 10.1111/j.1365-294X.2008.03929.x
Graham, R. T., and Roberts, C. M. (2007). Assessing the size, growth rate and structure of a seasonal population of whale sharks (Rhincodon typus Smith 1828) using conventional tagging and photo identification. Fish. Res. 84, 71–80. doi: 10.1016/j.fishres.2006.11.026
Gunn, A., and Pitt, S. J. (eds.). (2012). “Arthropod Parasites,” in Parasitology: An Integrated Approach (Chichester: John Wiley and Sons, Ltd), 137–179.
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129. doi: 10.1093/nar/gkt371
Iwasaki, W., Fukunaga, T., Isagozawa, R., Yamada, K., Maeda, Y., Satoh, T. P., et al. (2013). MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 30, 2531–2540. doi: 10.1093/molbev/mst141
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, P.-S., Gan, H. M., Clements, G. R., and Wilson, J.-J. (2016). Field calibration of blowfly-derived DNA against traditional methods for assessing mammal diversity in tropical forests. Genome 59, 1008–1022. doi: 10.1139/gen-2015-0193
Leigh, J. W., and Bryant, D. (2015). Popart: full-feature software for haplotype network construction. Meth. Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Norman, B. M., Newbound, D. R., and Knott, B. (2000). A new species of Pandaridae (Copepoda), from the whale shark Rhincodon typus (Smith). J. Nat. Hist. 34, 355–366. doi: 10.1080/002229300299534
Olson, Z. H., Briggler, J. T., and Williams, R. N. (2012). An eDNA approach to detect eastern hellbenders (Cryptobranchus a. alleganiensis) using samples of water. Wildlife Res. 39, 629–636. doi: 10.1071/WR12114
Pavlova, A., Gan, H. M., Lee, Y. P., Austin, C. M., Gilligan, D. M., Lintermans, M., et al. (2017). Purifying selection and genetic drift shaped Pleistocene evolution of the mitochondrial genome in an endangered Australian freshwater fish. Heredity 118, 466–476. doi: 10.1038/hdy.2016.120
Pérez-Flores, J., Rueda-Calderon, H., Kvist, S., Siddall, M. E., and Oceguera-Figueroa, A. (2016). From the worm in a bottle of mezcal: iDNA confirmation of a leech parasitizing the Antillean Manatee. J. Parasitol. 102, 553–555. doi: 10.1645/16-46
Ramírez-Macías, D., Vázquez-Juárez, R., Galván-Magaña, F., and Munguía-Vega, A. (2007). Variations of the mitochondrial control region sequence in whale sharks (Rhincodon typus) from the Gulf of California, Mexico. Fish. Res. 84, 87–95. doi: 10.1016/j.fishres.2006.11.038
Read, T., Petit, R. A., Joseph, S. J., Alam, M. T., Weil, M. R., Ahmad, M., et al. (2017). Draft sequencing and assembly of the genome of the world's largest fish, the whale shark: Rhincodon typus Smith 1828. BMC Genomics 18:532. doi: 10.1186/s12864-017-3926-9
Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Rodgers, T. W., Xu, C. C. Y., Giacalone, J., Kapheim, K. M., Saltonstall, K., Vargas, M., et al. (2017). Carrion fly-derived DNA metabarcoding is an effective tool for mammal surveys: evidence from a known tropical mammal community. Mol. Ecol. Resour. 17, e133–e145. doi: 10.1111/1755-0998.12701
Rowat, D., Speed, C. W., Meekan, M. G., Gore, M. A., and Bradshaw, C. J. (2009). Population abundance and apparent survival of the vulnerable whale shark Rhincodon typus in the Seychelles aggregation. Oryx 43, 591–598. doi: 10.1017/S0030605309990408
Schmidt, J., Chien-Chi, C., Sheikh, S., Meekan, M., Norman, B., and Joung, S.-J. (2010). Paternity analysis in a litter of whale shark embryos. Endanger. Species Res. 12, 117–124. doi: 10.3354/esr00300
Schmidt, J. V., Schmidt, C. L., Ozer, F., Ernst, R. E., Feldheim, K. A., Ashley, M. V., et al. (2009). Low genetic differentiation across three three major ocean populations of the whale shark, Rhincodon typus. PLoS ONE 4:e4988. doi: 10.1371/journal.pone.0004988
Schnell, I. B., Sollmann, R., Calvignac-Spencer, S., Siddall, M. E., Douglas, W. Y., Wilting, A., et al. (2015). iDNA from terrestrial haematophagous leeches as a wildlife surveying and monitoring tool–prospects, pitfalls and avenues to be developed. Front. Zool. 12:24. doi: 10.1186/s12983-015-0115-z
Schnell, I. B., Thomsen, P. F., Wilkinson, N., Rasmussen, M., Jensen, L. R. D., Willerslev, E., et al. (2012). Screening mammal biodiversity using DNA from leeches. Curr. Biol. 22, R262–R263. doi: 10.1016/j.cub.2012.10.014
Schubert, G., Stockhausen, M., Hoffmann, C., Merkel, K., Vigilant, L., Leendertz, F. H., et al. (2015). Targeted detection of mammalian species using carrion fly-derived DNA. Mol. Ecol. Resour. 15, 285–294. doi: 10.1111/1755-0998.12306
Sequeira, A. M., Mellin, C., Meekan, M. G., Sims, D. W., and Bradshaw, C. J. (2013). Inferred global connectivity of whale shark Rhincodon typus populations. J. Fish Biol. 82, 367–389. doi: 10.1111/jfb.12017
Sigsgaard, E. E., Nielsen, I. B., Bach, S. S., Lorenzen, E. D., Robinson, D. P., Knudsen, S. W., et al. (2016). Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nat. Ecol. Evol. 1:4. doi: 10.1038/s41559-016-0004
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Vignaud, T. M., Maynard, J. A., Leblois, R., Meekan, M. G., Vázquez-Juárez, R., Ramírez-Macías, D., et al. (2014). Genetic structure of populations of whale sharks among ocean basins and evidence for their historic rise and recent decline. Mol. Ecol. 23, 2590–2601. doi: 10.1111/mec.12754
Walter, T. C., and Boxshall, G. (2017). World of Copepods Database [Online]. Available online at: http://www.marinespecies.org/copepoda (Accessed August 21, 2017).
Keywords: eDNA, sharks, minimally-invasive sampling, copepod, control region, population genetics, parasite, commensal
Citation: Meekan M, Austin CM, Tan MH, Wei N-WV, Miller A, Pierce SJ, Rowat D, Stevens G, Davies TK, Ponzo A and Gan HM (2017) iDNA at Sea: Recovery of Whale Shark (Rhincodon typus) Mitochondrial DNA Sequences from the Whale Shark Copepod (Pandarus rhincodonicus) Confirms Global Population Structure. Front. Mar. Sci. 4:420. doi: 10.3389/fmars.2017.00420
Received: 08 October 2017; Accepted: 07 December 2017;
Published: 18 December 2017.
Edited by:
Rob Harcourt, Macquarie University, AustraliaReviewed by:
Celine Frere, University of the Sunshine Coast, AustraliaGail Schofield, Deakin University, Australia
Copyright © 2017 Meekan, Austin, Tan, Wei, Miller, Pierce, Rowat, Stevens, Davies, Ponzo and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Han Ming Gan, aGFuLmdhbkBkZWFraW4uZWR1LmF1