 Douglas T. Beattie
Douglas T. Beattie Tim Lachnit
Tim Lachnit Elizabeth A. Dinsdale
Elizabeth A. Dinsdale Torsten Thomas
Torsten Thomas Peter D. Steinberg
Peter D. Steinberg- 1Centre for Marine Bio-Innovation and School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, NSW, Australia
- 2Zoological Institute, Christian-Albrechts-University Kiel, Kiel, Germany
- 3Department of Biology, San Diego State University, San Diego, CA, United States
- 4Sydney Institute of Marine Science, Mosman, NSW, Australia
Kelp forests provide essential habitats for organisms in temperate rocky shores. Loss of kelp forests has occurred over large areas in a number of temperate regions, including in Australia, where the dominant kelp Ecklonia radiata has been lost from substantial areas of the shoreline. Loss of E. radiata has been associated with environmental stressors, including increased temperature and anthropogenic contaminants, as well as biological factors, such as herbivory. Disease may also play a role, but there is little information on the role of disease in the loss of kelp from coastal ecosystems or on the potential role of pathogenic microorganisms, such as viruses. E. radiata across much of its distribution in Australia can develop a “bleached” phenotype, which may be a disease. To investigate whether the phenotype was associated with a potential viral agent, we shotgun sequenced viral particles that were isolated from kelp with normal (healthy) and bleached phenotypes. Each virome consisted of ~380,000 reads, of which ~25% were similar to known viruses. All samples were dominated by bacteriophages, but novel ssDNA virus sequences were detected that were almost exclusively in viromes from the bleached kelp phenotype. These ssDNA viruses are covered by 11 contigs that contained complete capsids and characteristic rep genes that were 30–60% similar to those of circular, Rep-encoding ssDNA viruses (CRESS-DNA viruses). CRESS-DNA viruses have not previously been described from macroalgae, and the rep genes were similar to CRESS-DNA viruses from marine water samples, snails, crabs, anemones, but also dragonflies. This raises the interesting possibility that the kelp could be a vector of the CRESS-DNA viruses to other organisms that are associated with the bleached state.
Introduction
Ecklonia radiata is a large, stipitate brown alga (class Phaeophyceae, order Laminariales, or commonly, kelp), and the dominant primary habitat-forming organism over 71,000 km2 of the temperate rocky coastlines of Australia (Bennett et al., 2016). Kelps, such as E. radiata, provide habitat and nutrients that are essential to coastal food webs (Steneck et al., 2002), and the importance of E. radiata habitats in temperate Australia has led to them being described as “The Great Southern Reef” (Bennett et al., 2016). Fisheries, tourism, and other industries taking place within these habitats are estimated to be worth AU$10 billion annually to the Australian economy (Bennett et al., 2016).
In recent years there has been a loss of kelp cover in temperate regions of Australia from locations across much of the range of E. radiata, from Perth on the West coast, along the Southern coast, up to the Solitary Islands on the East coast (Connell et al., 2008; Ling and Johnson, 2012; Vergés et al., 2016; Wernberg et al., 2016). Loss of kelp results in major losses in biodiversity, ecosystem function, and associated fisheries (Andrew and O'Neill, 2000; Bishop et al., 2010; Burge et al., 2014; Vergés et al., 2016; Wernberg et al., 2016), and in Australia loss of E. radiata has been associated with a number of environmental stressors. This includes stressors associated with urbanization, such as increased nutrients and sedimentation of the coastal region (Connell et al., 2008), and ocean warming, through both direct physiological effects of temperature as well as indirect effects through range extensions of herbivorous sea urchins or fishes, which consume large areas of kelp forests (Wernberg et al., 2011; Ling and Johnson, 2012; Vergés et al., 2016). Though relatively little studied in seaweed systems, microbial disease may also play a role in loss of habitat-forming seaweeds (Egan et al., 2014). Disease in marine systems appears to be increasing globally, including in communities that are dominated by primary habitat-forming organisms, such as coral reefs and kelp forests (Rosenberg et al., 2007; Vega Thurber et al., 2009; Krediet et al., 2013), and disease in seaweeds has been linked to anthropogenic stressors, such as ocean warming (Campbell et al., 2011; Case et al., 2011).
Microbial communities are integral to the health of all organisms, such that hosts and their associated microbial communities (microbiome) are increasingly being treated as a holistic system, or holobiont (Egan et al., 2013). This approach is now common for understanding the interaction between marine habitat-forming organisms, such as corals, seaweeds, or sponges (Bourne et al., 2009; Egan et al., 2013; Webster and Thomas, 2016). Disturbances to the microbiome of these organisms are associated with stress and disease of the host (Morris et al., 2016). For example, disruption of the microbiome of the Australian macroalga Delisea pulchra leads to bleaching and reduced reproductive capacity (Egan et al., 2013; Campbell et al., 2014; Wahl et al., 2015), and microbial pathogens cause bleaching of D. pulchra in both the field (Campbell et al., 2011) and in laboratory inoculation experiments (Kumar et al., 2016). Disruption of the holobiont and resultant disease is increasingly seen as critical for other marine habitat forming organisms.
We have recently identified a bleaching phenotype in E. radiata, which occurs widely across its biogeographic range and which is characterized by bleached areas of the thallus with reduced photosynthetic capacity (Marzinelli et al., 2015). The bacterial components of the microbiome of bleached vs. non-bleached E. radiata were distinct, suggesting that the microbiome of the algae undergoes a shift associated with the disruption of normal function (e.g., photosynthesis; Marzinelli et al., 2015). However, specific bacterial pathogens, which induce bleaching, have not yet been extensively characterized. Other components of the microbiome may also affect the health of hosts. In particular, viruses are likely ubiquitous components of marine holobionts and are now emerging as pathogens or potential pathogens in a diverse range of marine organisms, including archaea (Rohwer and Thurber, 2009), microalgae (Brussaard, 2004; Weynberg et al., 2017), sea stars (Hewson et al., 2014), and macroalgae (Lachnit et al., 2015).
Viruses consist of two major groups, bacteriophages that infect bacteria and viruses that infect eukaryotes. The tailed bacteriophages with dsDNA genomes, Caudovirales, include the families Siphoviridae, Myoviridae, and Podoviridae. Bacteriophages also include Inoviridae, which possess ssDNA genomes, and together these bacteriophages form a major component of marine viral assemblages (Mann, 2005; Dinsdale et al., 2008b; Roux et al., 2012; Xue et al., 2012). Abundant eukaryote viruses detected in algae are from the family Phycodnaviridae, which have a dsDNA genome of 100–550 kb and infect both unicellular and multicellular algae (Wilson et al., 2009). Phycodnaviridae have been investigated in the filamentous brown-alga Ectocarpus siliculosus, a model organism, and a few genomes have been sequenced (Van Etten et al., 1991; Müller and Frenzer, 1993; Easton et al., 1997; Kapp et al., 1997; Maier et al., 1998; Delaroque and Boland, 2008). However, few studies have been conducted on macroalgae from the field (e.g., viruses in Laminaria digitata McKeown et al., 2017) and none on E. radiata.
ssDNA viruses, which have small circular genomes (1–5 kb), are an emerging group of viruses that are implicated in marine disease and mass mortalities across a number of marine phyla, including fishes, molluscs, echinoderms, and crustaceans (Hasson et al., 1995; Friedman et al., 2005; McLoughlin and Graham, 2007; Lang et al., 2009). ssDNA viruses include a subgroup which possess circular genomes, and a particular replication initiation gene, rep. These are therefore called Circular Rep-Encoding ssDNA (CRESS-DNA) viruses (Rosario et al., 2009; Delwart and Li, 2012; Labonté and Suttle, 2013; Simmonds et al., 2017). These include the Geminiviridae, Nanoviridae, Circoviridae, and Genomoviridae, which employ rolling-circle replication (RCR), facilitated by the Rep protein (Rosario et al., 2012b). In addition to the Rep protein, CRESS-DNA viruses have a characteristic stem-and-loop secondary structure, which facilitates RCR. The loop region contains a conserved nonanucleotide motif, NANTATTAC (Labonté and Suttle, 2013). The Circoviridae and Nanoviridae are diverse in aquatic and marine environments, including ocean, freshwater, wastewater, deep-sea vents, Antarctic lakes and ponds, and hot springs (Angly et al., 2006; Rosario et al., 2009, 2012a; Cassman et al., 2012; Liu et al., 2012; Labonté and Suttle, 2013).
Here we investigate the viruses associated with bleached and non-bleached E. radiata from Sydney, Australia. First, virus particles were extracted from kelp tissue. DNA was extracted from these and amplified using whole-genome amplification (WGA), which is biased toward small and circular genomes (Sabina and Leamon, 2015), but thus aids in detection of potentially rare and small genomes in metagenomic samples (Kim et al., 2008; Cheval et al., 2011; Rosario et al., 2012a; Fahsbender et al., 2015). This was followed by shotgun sequencing and metagenomic analysis. We describe the general features of the kelp virome, and test whether components of the virome differ between the non-bleached and bleached phenotype, in order to explore whether viral pathogens may be a possible cause of the bleaching phenotype. Our results show that circoviruses were nearly absent in the non-bleached kelp, but in high abundance in the bleached kelp, and we describe these novel viruses in detail.
Methods
Sampling
Individual thalli of E. radiata were sampled from Long Bay in Sydney, Australia (−33.97, 151.26) during June 2014 at a depth of 3 m. The water temperature at the time of sampling was 16°C. Tissue samples of blades were taken from three bleached individuals and three non-bleached, healthy, thalli. These phentoypes are described in Marzinelli et al. (2015). Viral particles were isolated from kelp tissue as described in Lachnit et al. (2015).
DNA Extraction and Metagenomic Sequencing
Viral DNA was extracted from virus particles in SDS-extraction buffer (1% SDS, 200 mM Tris, 50 mM EDTA, pH 7.5) in the presence of 1% (w/v) polyvinylpyrrolidone (MW 40,000), 1% 2-mercaptoethanol and proteinase K (0.5 mg ml−1) at 37°C for 30 min, followed by 56°C for 15 min. Double the volume of DNA-extraction buffer (2.6 M NaCl, 150 mM Tris, 75 mM EDTA, 2.5% cetyltrimethylammonium bromide (CTAB), 1% 2-mercaptoethanol, pH 7.5) was added and samples were incubated for another 15 min at 56°C. An equal volume of chloroform:isoamyl alcohol (24:1) was added to the solution, mixed and centrifuged at 12,000 RCF at room temperature for 5 min. The chloroform:isoamyl alcohol extraction of the supernatant was repeated three times to remove contaminating polysaccharides and proteins. The supernatant was transferred into a new tube and the nucleic acids were precipitated by the addition of 0.7 volume of isopropanol. After an incubation step at 20°C for 2 h nucleic acids were pelleted by centrifugation at 14,000 RCF at 4°C for 20 min. The pellet was washed with 75% (v/v) ethanol, air dried and dissolved in 30 μl molecular grade water.
All samples underwent whole genome amplification steps using a REPLI-g Mini Kit (QIAGEN), where the reaction was conducted using 20 μL of denatured DNA, incubating the reaction mix at 30°C for 16 h. DNA was frozen and submitted to the Ramaciotti Centre at UNSW for library preparation using a TruSeq Low Input kit (Illumina). Shotgun metagenomic sequencing was conducted using an Illumina MiSeq sequencer with 2 × 250 bp chemistry. A total of six viral metagenomes (or viromes) were sequenced, three from kelp with the bleached phenotype and three from the non-bleached phenotype. The reads were processed using Prinseq (Schmieder and Edwards, 2011) by trimming the ends where the quality score was below 20, and removing exact duplicates. The paired-end reads were assessed using default settings on PEAR (Zhang et al., 2014) to determine if they could be joined to form longer sequences.
Virome Profile Analysis
Several bioinformatics approaches were used to identify the types of phage and eukaryotic viruses in the kelp viromes. First, the reads were aligned against the NCBI non-redundant protein database (NR), downloaded on May 05 2017. Alignment was performed using BLASTx (Altschul et al., 1990) with an e-value of 10−5. Alignment matches were screened to remove all non-viral similarities found. Alignment results were analyzed using MEGAN (Huson et al., 2016) to infer best taxonomic and/or functional assignments. The Least Common Ancestor (LCA) parameters was used with a minimum score of 50, max expected e-value 10−5, top 10%, LCA 100%, minimum complexity 0. To detect significant differences in the relative abundances of assignments to each virus family, STAMP (Parks et al., 2014) was used to perform two tailed Welch's t-test (α = 0.05) with Bonferroni correction to compare the bleached and non-bleached kelp viromes.
To assess possible contamination of the viromes by host genomic or bacterial DNA, the entire set of similarities found, including similarities to viruses and non-viruses, was considered. Sequences, which had higher alignment bitscores to non-viruses, were aligned against all the brown-alga sequences in NCBI (downloaded on May 05 2017), using BLASTn with an e-value of 10−5. The sequences with <95% similarity to Ecklonia spp. suggested that they are not part of the brown algae. This threshold was chosen as essential genes (e.g., rbcL) of different species of Eckonia, and even some distally related members of the brown algae are >95% similar (Rothman et al., 2015). Decontamination programs (e.g., DeconSeq Schmieder and Edwards, 2011) were not employed to automate the removal of potential bacterial contamination, as genes are often shared between bacteriophage and their hosts. Instead, more information on the origin of the reads was obtained by assembly and assessment of the genomic content (see below), and contamination was retrospectively removed from the results and analyses.
The individual metagenomes were assembled to provide longer contigs using SPAdes 3.9.0 (using default parameters; Bankevich et al., 2012). SPAdes was chosen as it performs better than other assemblers on virome data (Hesse et al., 2017; Papudeshi et al., 2017; Vollmers et al., 2017). Reads were mapped to the contigs using Bowtie 2.2.5 (Langmead and Salzberg, 2012) and visualized using Tablet (Milne et al., 2009) to assess the evenness of read coverage and to identify possible chimeric assemblies, which were removed from further analysis. To investigate the distribution of viruses between the bleached and non-bleached kelp, the contigs were aligned to the NR database as described above. The contigs and their component reads were considered contamination and removed from the analysis and results if (a) the alignment bitscore to a virus reference was less than to a non-virus sequence, and (b) the alignment matches did not contain core genes of the assigned virus taxon e.g., phage-tail genes of Caudovirales.
Viral Genome Reconstruction
All six metagenomes were pooled and cross-assembled with SPAdes 3.9.0 to obtain longer contigs and/or complete genomes. Assessment and alignments of the cross-assembled contigs were performed as described above. At this stage of the analysis, circoviruses were identified as the only part of the eukaryotic DNA virus community that varied in abundance between bleached and non-bleached kelp, and therefore were investigated in more detail. Open reading frames (ORFs) on the circoviral contigs were detected using Prodigal (Hyatt et al., 2010) with a length of > 50 amino acids. Predicted protein sequences were aligned against one another, and their reference sequences using the MUSCLE aligner with default settings (Edgar, 2004). To visualize the diversity of the ORFs a phylogenetic tree was produced using the Jukes-Cantor genetic distance model and the neighbor-joining algorithm with 100 bootstraps. Circoviridae contigs were submitted to Mfold (Zuker, 2003) to determine their stem-and-loop secondary structure. The loop motif was compared across the Circoviridae group through alignment using MUSCLE.
Results
Overall Composition of the Virome of Ecklonia radiata
The six E. radiata viromes contained an average of 386,000 reads with an average length of 149 ± 2SD bp. (Supplementary information S1). No reads were removed through quality control, although 14% of the reads were trimmed up to 10 bp where the read quality was <20. Most paired reads could not be joined, and so forward-reads were only used in alignment searches. Analysis of alignment results found 81,256 ± 22,106 virus-like reads per metagenome (~20% of the sequences were similar to the NR database). The majority (86–94%) of virus-like reads were assigned to bacteriophage in the order Caudovirales (Figure 1). In order of abundance, similarities were detected to the viral groups Siphoviridae (average of 48% of virus-assigned reads), Myoviridae (16%), unclassified bacteriophages (12%), Inoviridae (8%), Podoviridae (7%), followed by Circoviridae (7%). Within the Caudovirales, reads similar to Siphoviridae were more abundant (adjusted p-value = 0.003) in the non-bleached kelp phenotype, however the total number of bacteriophage reads within the Caudovirales did not significantly differ between non-bleached and bleached kelp viromes. In the eukaryotic viral fraction, reads similar to Phycodnaviridae and other large dsDNA viruses were lower in abundance (<2% of reads assigned) compared to the bacteriophages and did not differ in abundance between viromes of non-bleached and bleached kelp (adjusted p-value = 0.785). Sequences of ssDNA viruses belonging to the Circoviridae were rare overall (0.48% of total reads), but were almost exclusively in bleached kelp viromes. Six reads were assigned to Circoviridae from a single non-bleached kelp virome, whereas 6,403 reads were assigned from bleached kelp viromes.
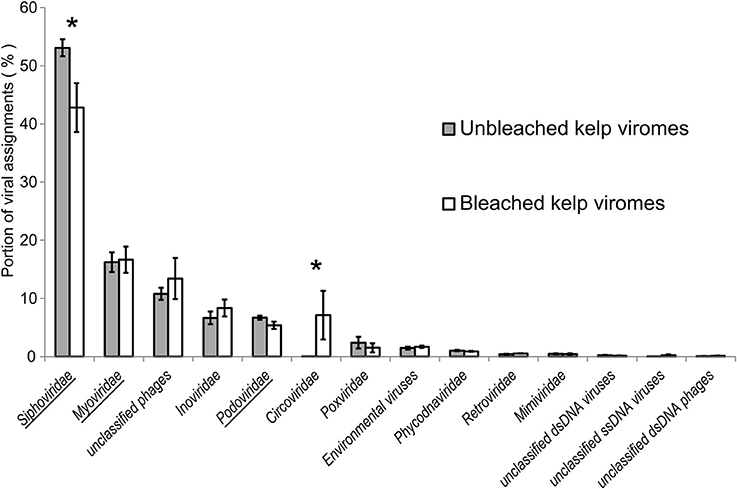
Figure 1. Reads from viromes of bleached and unbleached kelps that were assigned to virus references. Bars are ±1 standard error. Asterisk denotes an adjusted p-value < 0.005. Underlined taxa belong to the Caudovirales. Environmental viruses are annotated sequences from environmental metagenomes.
Assembly of all reads per individual metagenome provided an average of 4,011 ± 351 contigs of 38,709 ± 6,392 bp length. The taxonomic assignment of the contigs mirrored that of the reads (Supplementary information S2). Bacteriophage possessed the greatest amount of read coverage in the contigs (60%), followed by the Phycodnaviridae (8%) and Circoviridae (6%). The Circoviridae contigs (n = 42) were assembled only in the bleached kelp viromes, with the exception of a single contig from one non-bleached kelp virome. Similar to the read data, Circoviridae contigs were the only eukaryotic viral group that varied in coverage between the bleached and non-bleached kelp phenotypes (adjusted p-value = 0.002).
Cross-assembly of all six metagenomes yielded 7,561 contigs, the longest being 246,893 bp (N50 20,791 bp, N95 246 bp). When aligned against an NCBI virus-only protein database, 847 contigs showed similarity to viruses (Figure 2). In descending order of abundance these were assigned to Siphoviridae (97 contigs), environmental viruses from metagenomes (92), Myoviridae (59), Podoviridae (48), unclassified bacteriophages (47), Circoviridae (26), Phycodnaviridae (21), Mimiviridae (15), Inoviridae (9) and other virus families with very low abundances (total of 434).
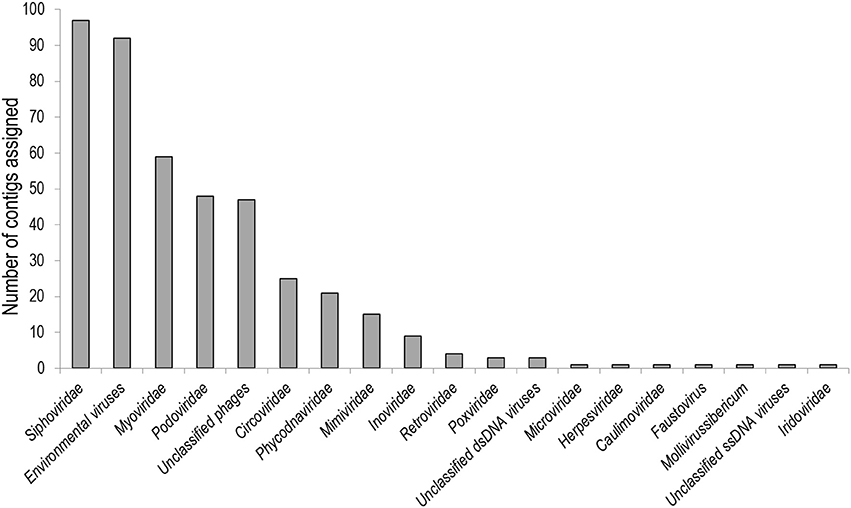
Figure 2. Contigs from the cross-assembly of kelp viromes that were assigned to virus references.
We assessed contamination by regarding the contig similarities to virus and non-viruses. Two thousand two hundred and sixty-seven contigs of the total 7,561 were given a taxonomic assignment through alignment to NR (Supplementary information S3). Eight hundred and forty-seven contigs had similarity to viruses. Of these, 146 contigs had no similarity to other references, 576 contigs were also similar to bacteria and 125 contigs were also similar to eukaryotes. The majority (36%) of contigs similar to both viruses and eukaryotes were similar to E. siliculosus Virus-1. Four contigs had both higher alignment bitscores to brown-alga sequences, and did not include characteristic virus genes or conserved domains and were thus assumed to be eukaryotic. The contigs with similarity to both viruses and bacteria (576) were examined further, finding 113 contigs with higher bitscores to bacterial references and no characteristic bacteriophage genes, and assumed to be from bacterial cells. Contigs and their constituent reads that were considered eukaryotic or bacterial contamination were removed from the analyses and results of virus abundances and gene content. Contigs that were assigned as circoviruses had no other similarities to references in NR, and as the circoviruses were the only eukaryotic virus that showed variation between kelp phenotypes, these were analysed in more detail.
Investigation of Circoviridae-Like Contigs
The Circoviridae contigs (n = 26) were 739–5,420 bp long, and contained up to two ORFs. The first ORF was 855 bp, and the only translated similarity between these and reference proteins were two ORFs with similarity to putative CRESS-DNA virus capsid genes from environmental metagenomes. These showed 30% pairwise identity to putative capsid genes of a circular virus associated with a sea anemone (Genbank accession: YP_009163900.1) and a circular ssDNA virus isolated from an Antarctic ice shelf pond (Genbank accession: YP_009047127.1). The second ORF was 305–336 amino acids and was the characteristic rep genes of CRESS-DNA viruses. The rep genes were found in three different lengths (915 bp, 924 bp, and 1,008 bp) suggesting three genotypes, which we refer to from hereon as A-C. There were three contig configurations of the circovirus sequences: Type-A (8 contigs), Type-B (2 contigs) and Type-C (single contig; Figure 3). Type-A contained both the capsid and repA, in reverse orientation with a conserved intergenic region (IR) between the two ORFs. The IR of the eight contigs shared a pairwise identity of 70%. Type-B contained repB and the IR. Type-C contained only repC and no capsid sequences or the IR. The origin of replication (ori), included the characteristic nonanucleotide stem and loop structures involved in rolling circle replication of CRESS-DNA viruses and was located upstream of the rep gene in all contigs (Figure 3). The nonanucleotide motif in each contig was TAGTATTAC. The other 15 of the 26 circovirus-like contigs that did not contain complete ORFs were found to contain fragments of rep and capsid genes of circoviruses, suggesting incomplete assembly.
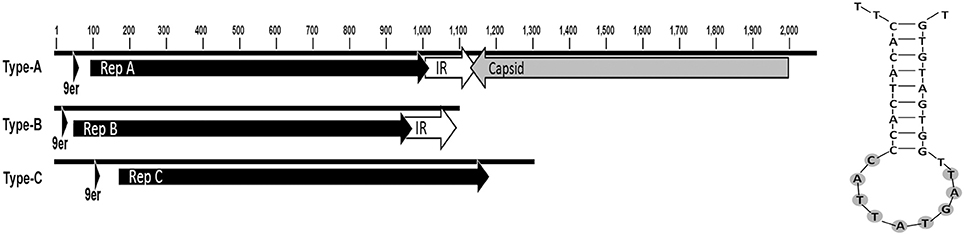
Figure 3. Circovirus-like contig ORFs Appeared in three different configurations (L) Rep types were present on three types of contig. (R) Predicted stem-loop structures associated with the RCR initiation nonanuclotide motif (9er) is highlighted.
The rep genes from the 11 contigs and the closest reference sequences in the database were aligned. The rep genes formed three clusters with each cluster showing >95% similarity. Between all rep genes in our dataset, and their most similar reference sequences, the rep sequences were 27–60% similar. The references with greatest similarity to the detected circoviruses were from viruses found in association with aquatic samples or invertebrates, including marine snails and hermit crabs, but also dragonflies (Figure 4).
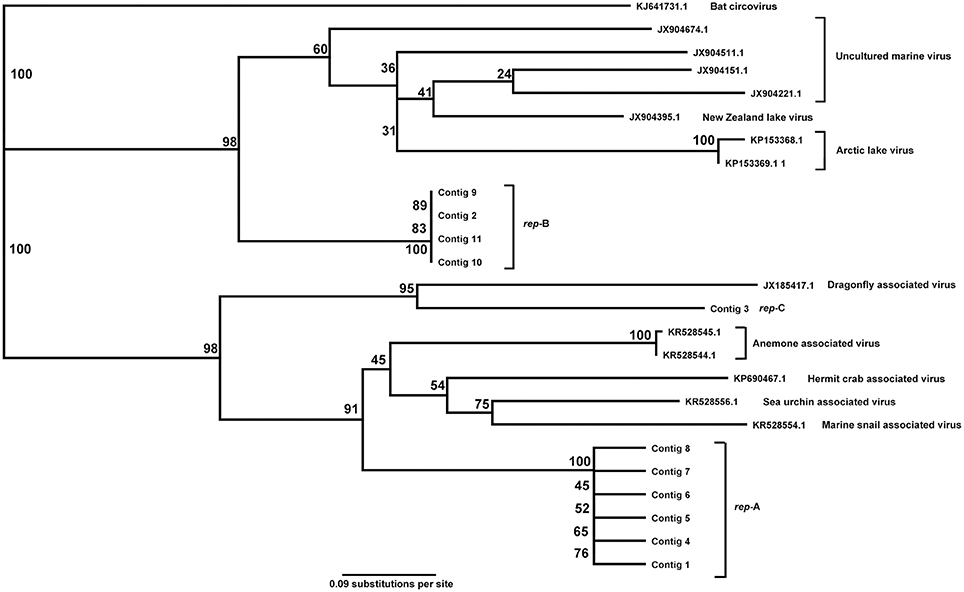
Figure 4. Alignment tree of rep ORFs in circovirus-like contigs.
Discussion
We characterized the virome of both bleached and non-bleached individuals of the kelp E. radiata. Sequences similar to bacteriophages in the Siphoviridae were detected in greatest abundance overall, followed by Myoviridae, unclassified phages, Inoviridae, and Podoviridae. For most viral taxa there was little difference in the relative abundance in reads between the two kelp phenotypes. However, eukaryote circovirus-like sequences differed significantly in abundance between bleached and non-bleached kelp, with bleached viromes containing the vast majority (>99%) of circovirus-like reads. The circoviruses sequences from E. radiata included three phylogenetic groups, which all contained the characteristic rep gene of CRESS-DNA viruses, and were most similar to recently described circoviruses from pelagic water samples and marine invertebrates.
Initial alignment of raw DNA reads of kelp-associated viromes against a non-redundant protein database showed that most sequences were unknown, which is often the case in environmental metagenomes, but especially in marine virome data (Dinsdale et al., 2008a,b; Willner et al., 2009; Cassman et al., 2012; Brussaard et al., 2016). The most abundant contigs with similarity to known viruses were the bacteriophage Caudovirales (Figure 2). While the overall abundance of bacteriophage-like contigs did not differ, contigs similar to Siphoviridae were more abundant in viromes of non-bleached kelp, perhaps as a cause or consequence of the differences in the bacterial community previously seen between bleached and non-bleached E. radiata (Marzinelli et al., 2015). Large dsDNA viruses, such as Phycodnaviridae and Mimiviridae, formed the second largest group of virus-like contigs detected, and found with similar relative read numbers in metagenomes of non-bleached and bleached kelp. The majority of similarities to Phycodnaviridae were to EsV-1, which only produces virus particles in the reproductive structures of its host. EsV-1 may share a similar life history to Phycodnaviridae present in E. radiata, in which case the virus particles likely originated from the sori on the sampled kelp thallus. This group of viruses may be part of a persistent and possibly stable viral assemblage of the kelp holobiont. In contrast, significantly more circovirus-like sequences were detected in bleached kelp viromes than in non-bleached ones.
The circovirus genomes assembled here were of 2–5 kb in length. The small size of ssDNA virus genomes means that their genetic material is predisposed to be relatively scarce when compared to other viruses that may have up to 100-fold larger genomes (e.g., Phycodnaviridae). Whole genome amplification using multiple displacement amplification (MDA) of circular genomes has been used to sequence these viruses, even from very small amounts of DNA (Binga et al., 2008). MDA is known to create biases toward viruses with small genomes. Alternative methods, such as random priming-mediated sequence-independent single-primer amplification (RP-SISPA), could have been used, but this method can cause an overrepresentation in the datasets of viruses with large genomes (Adriaenssens et al., 2017). This bias away from small genomes has been noted by other authors (Karlsson et al., 2013; Weynberg et al., 2014).
The genome structures of the CRESS-DNA virus sequences are similar to Type I and II ssDNA viruses, where the reading frames of the rep and capsid genes are positioned 3′–5′ away from the stem-loop replication initiation structure. These viral types include Circoviridae, Nanoviridae, Geminiviridae, and other unclassified CRESS-DNA viruses from aquatic and marine metagenomes (Rosario et al., 2012b).
ssDNA (including CRESS-DNA) viruses have been characterized in medical and agricultural fields, due to their significance as human and agricultural pathogens (Roossinck, 2013). Circoviridae, Anelloviridae, Genomoviridae, and Parvoviridae are pathogens of humans and other vertebrates, such as pigs, poultry, and other domestic animals, but to date none have been identified in macroalgae. The circoviruses detected in the bleached kelp viromes were similar to a diverse subsection of marine viruses isolated from hermit crabs, sea anemones, but also had similarity to CRESS-DNA viruses detected in dragonflies (Rosario et al., 2012a). CRESS-DNA viruses are found in association with diverse habitats and host phyla (Rosario et al., 2012a,b; Eaglesham and Hewson, 2013; Fahsbender et al., 2015; Reavy et al., 2015), and circovirus-like proteins have been detected in a wide range of organisms, including mammals, birds, fish, cnidarians, as well as in a diverse range of aquatic and marine environments (Rosario et al., 2012b). Many circoviruses are important pathogens, or putative pathogens, and given their high relative abundance on the bleached phenotype it may be that they play a role in bleaching of E. radiata. Their increase may also be a secondary effect of bleaching, with the primary causative agent(s) being other microbes, or an interaction among multiple viruses, bacteria, fungi and others that are present in the kelp microbiome. Microbial agents can also interact with eukaryotic natural enemies, such as grazers to damage seaweeds (Campbell et al., 2014), which adds complexity when attempting to distinguish pathogens which cause disease from other secondary organisms (e.g., saprophytes such as fungi or bacteria) that arrive after disease onset to feed on compromised or decaying hosts (Burge et al., 2013; Egan and Gardiner, 2016). A more definitive resolution of the role of circoviruses in kelp bleaching thus awaits a more experimental approach, e.g., inoculation experiments, such as those conducted for D. pulchra and its bacterial pathogens (Campbell et al., 2015; Kumar et al., 2016). The circoviruses were the only eukaryotic DNA virus in the viromes that was overrepresented in the bleached compared with the non-bleached phenotypes, suggesting it may be associated with the bleached phenotype. However, ssDNA viruses have a ubiquitous presence in a wide range of eukaryotes and natural environments. The contigs we identified showed similarities to circoviruses found in hosts as broad as sea stars and crustaceans, raising the question of whether the circovirus was infecting the kelp host, or one of the herbivores associated with the kelp. Therefore, kelp could be infected with the circovirus, directly causing the phenotype, or the kelp could be acting as a vector to one of the herbivores or symbionts.
Author Contributions
TL: performed isolation and sequencing of samples; DB: performed analysis onwards; Other authors are supervisors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding was provided by the University of New South Wales and the Centre for Marine Bio-Innovation, Sydney Australia and ARC Discovery Grant DP1401002776. TL acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG-La297 8/1-1).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2017.00441/full#supplementary-material
References
Adriaenssens, E. M., Kramer, R., Van Goethem, M. W., Makhalanyane, T. P., Hogg, I., and Cowan, D. A. (2017). Environmental drivers of viral community composition in Antarctic soils identified by viromics. Microbiome 5:83. doi: 10.1186/s40168-017-0301-7
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrew, N. L., and O'Neill, A. L. (2000). Large-scale patterns in habitat structure on subtidal rocky reefs in New South Wales. Mar. Freshw. Res. 51, 255–263. doi: 10.1071/MF99008
Angly, F. E., Felts, B., Breitbart, M., Salamon, P., Edwards, R. A., Carlson, C., et al. (2006). The marine viromes of four oceanic regions. PLoS Biol. 4:e368. doi: 10.1371/journal.pbio.0040368
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bennett, S., Wernberg, T., Connell, S. D., Hobday, A. J., Johnson, C. R., and Poloczanska, E. S. (2016). The ‘Great Southern Reef’: social, ecological and economic value of Australia's neglected kelp forests. Mar. Freshw. Res. 67, 47–56. doi: 10.1071/MF15232
Binga, E. K., Lasken, R. S., and Neufeld, J. D. (2008). Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J. 2, 233–241. doi: 10.1038/ismej.2008.10
Bishop, M. J., Coleman, M. A., and Kelaher, B. P. (2010). Cross-habitat impacts of species decline: response of estuarine sediment communities to changing detrital resources. Oecologia 163, 517–525. doi: 10.1007/s00442-009-1555-y
Bourne, D. G., Garren, M., Work, T. M., Rosenberg, E., Smith, G. W., and Harvell, C. D. (2009). Microbial disease and the coral holobiont. Trends Microbiol. 17, 554–562. doi: 10.1016/j.tim.2009.09.004
Brussaard, C. P. (2004). Viral control of phytoplankton populations—a review. J. Eukaryot. Microbiol. 51, 125–138. doi: 10.1111/j.1550-7408.2004.tb00537.x
Brussaard, C. P., Baudoux, A.-C., and Rodríguez-Valera, F. (2016). The Marine Microbiome. Cham: Springer.
Burge, C. A., Kim, C. J., Lyles, J. M., and Harvell, C. D. (2013). Special issue oceans and humans health: the ecology of marine opportunists. Microb. Ecol. 65, 869–879. doi: 10.1007/s00248-013-0190-7
Burge, C. A., Mark Eakin, C., Friedman, C. S., Froelich, B., Hershberger, P. K., Hofmann, E. E., et al. (2014). Climate change influences on marine infectious diseases: implications for management and society. Ann. Rev. Mar. Sci. 6, 249–277. doi: 10.1146/annurev-marine-010213-135029
Campbell, A. H., Harder, T., Nielsen, S., Kjelleberg, S., and Steinberg, P. D. (2011). Climate change and disease: bleaching of a chemically defended seaweed. Glob. Change Biol. 17, 2958–2970. doi: 10.1111/j.1365-2486.2011.02456.x
Campbell, A. H., Marzinelli, E. M., Gelber, J., and Steinberg, P. D. (2015). Spatial variability of microbial assemblages associated with a dominant habitat-forming seaweed. Front. Microbiol. 6:230. doi: 10.3389/fmicb.2015.00230
Campbell, A. H., Vergés, A., and Steinberg, P. D. (2014). Demographic consequences of disease in a habitat-forming seaweed and impacts on interactions between natural enemies. Ecology 95, 142–152. doi: 10.1890/13-0213.1
Case, R. J., Longford, S. R., Campbell, A. H., Low, A., Tujula, N., Steinberg, P. D., et al. (2011). Temperature induced bacterial virulence and bleaching disease in a chemically defended marine macroalga. Environ. Microbiol. 13, 529–537. doi: 10.1111/j.1462-2920.2010.02356.x
Cassman, N., Prieto-Davó, A., Walsh, K., Silva, G. G. Z., Angly, F., Akhter, S., et al. (2012). Oxygen minimum zones harbour novel viral communities with low diversity. Environ. Microbiol. 14, 3043–3065. doi: 10.1111/j.1462-2920.2012.02891.x
Cheval, J., Sauvage, V., Frangeul, L., Dacheux, L., Guigon, G., Dumey, N., et al. (2011). Evaluation of high-throughput sequencing for identifying known and unknown viruses in biological samples. J. Clin. Microbiol. 49, 3268–3275. doi: 10.1128/JCM.00850-11
Connell, S., Russell, B., Turner, D., Shepherd, A., Kildea, T., Miller, D., et al. (2008). Recovering a lost baseline: missing kelp forests from a metropolitan coast. Mar. Ecol. Prog. Ser. 360, 63–72. doi: 10.3354/meps07526
Delaroque, N., and Boland, W. (2008). The genome of the brown alga Ectocarpus siliculosus contains a series of viral DNA pieces, suggesting an ancient association with large dsDNA viruses. BMC Evol. Biol. 8:110. doi: 10.1186/1471-2148-8-110
Delwart, E., and Li, L. (2012). Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 164, 114–121. doi: 10.1016/j.virusres.2011.11.021
Dinsdale, E. A., Edwards, R. A., Hall, D., Angly, F., Breitbart, M., Brulc, J. M., et al. (2008a). Functional metagenomic profiling of nine biomes. Nature 452, 629–632. doi: 10.1038/nature06810
Dinsdale, E. A., Pantos, O., Smriga, S., Edwards, R. A., Angly, F., Wegley, L., et al. (2008b). Microbial ecology of four coral atolls in the Northern Line Islands. PLoS ONE 3:e1584. doi: 10.1371/journal.pone.0001584
Eaglesham, J. B., and Hewson, I. (2013). Widespread detection of circular replication initiator protein (rep)-encoding ssDNA viral genomes in estuarine, coastal and open ocean net plankton. Mar. Ecol. Prog. Ser. 494, 65–72. doi: 10.3354/meps10575
Easton, L. M., Lewis, G. D., and Pearson, M. N. (1997). Virus-like particles associated with dieback symptoms in the brown alga Ecklonia radiata. 30, 217–222.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Egan, S., and Gardiner, M. (2016). Microbial dysbiosis: rethinking disease in marine ecosystems. Front. Microbiol. 7:991. doi: 10.3389/fmicb.2016.00991
Egan, S., Fernandes, N. D., Kumar, V., Gardiner, M., and Thomas, T. (2014). Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environ. Microbiol. 16, 925–938. doi: 10.1111/1462-2920.12288
Egan, S., Harder, T., Burke, C., Steinberg, P., Kjelleberg, S., and Thomas, T. (2013). The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Fahsbender, E., Hewson, I., Rosario, K., Tuttle, A. D., Varsani, A., and Breitbart, M. (2015). Discovery of a novel circular DNA virus in the Forbes sea star, Asterias forbesi. Arch. Virol. 160, 2349–2351. doi: 10.1007/s00705-015-2503-2
Friedman, C. S., Estes, R. M., Stokes, N. A., Burge, C. A., Hargove, J. S., Barber, B. J., et al. (2005). Herpes virus in juvenile Pacific oysters Crassostrea gigas from Tomales Bay, California, coincides with summer mortality episodes. Dis. Aquat. Org. 63, 33–41. doi: 10.3354/dao063033
Hasson, K., Lightner, D. V., Poulos, B., Redman, R., White, B., Brock, J., et al. (1995). Taura syndrome in Penaeus vannamei: demonstration of a viral etiology. Dis. Aquat. Org. 23, 115–126. doi: 10.3354/dao023115
Hesse, U., van Heusden, P., Kirby, B. M., Olonade, I., van Zyl, L. J., and Trindade, M. (2017). Virome assembly and annotation: a surprise in the Namib Desert. Front. Microbiol. 8:13. doi: 10.3389/fmicb.2017.00013
Hewson, I., Button, J. B., Gudenkauf, B. M., Miner, B., Newton, A. L., Gaydos, J. K., et al. (2014). Densovirus associated with sea-star wasting disease and mass mortality. Proc. Natl. Acad. Sci. U.S.A. 111, 17278–17283. doi: 10.1073/pnas.1416625111
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Kapp, M., Knippers, R., and Müller, D. G. (1997). New members of a group of DNA viruses infecting brown algae. Phycological Res. 45, 85–90. doi: 10.1111/j.1440-1835.1997.tb00067.x
Karlsson, O. E., Belák, S., and Granberg, F. (2013). The effect of preprocessing by sequence-independent, single-primer amplification (SISPA) on metagenomic detection of viruses. Biosecur. Bioterror. 11, S227–S234. doi: 10.1089/bsp.2013.0008
Kim, K. H., Chang, H. W., Nam, Y. D., Roh, S. W., Kim, M. S., Sung, Y., et al. (2008). Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 74, 5975–5985. doi: 10.1128/AEM.01275-08
Krediet, C. J., Ritchie, K. B., Paul, V. J., and Teplitski, M. (2013). Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proc. R. Soc. Lond. B Biol. Sci. 280:20122328. doi: 10.1098/rspb.2012.2328
Kumar, V., Zozaya-Valdes, E., Kjelleberg, S., Thomas, T., and Egan, S. (2016). Multiple opportunistic pathogens can cause a bleaching disease in the red seaweed Delisea pulchra. Environ. Microbiol. 18, 3962–3975. doi: 10.1111/1462-2920.13403
Labonté, J. M., and Suttle, C. A. (2013). Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177. doi: 10.1038/ismej.2013.110
Lachnit, T., Thomas, T., and Steinberg, P. (2015). Expanding our understanding of the seaweed Holobiont: RNA viruses of the red alga Delisea pulchra. Front. Microbiol. 6:1489. doi: 10.3389/fmicb.2015.01489
Lang, A. S., Rise, M. L., Culley, A. I., and Steward, G. F. (2009). RNA viruses in the sea. FEMS Microbiol. Rev. 33, 295–323. doi: 10.1111/j.1574-6976.2008.00132.x
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Ling, S., and Johnson, C. R. (2012). Marine reserves reduce risk of climate-driven phase shift by reinstating size-and habitat-specific trophic interactions. Ecol. Appl. 22, 1232–1245. doi: 10.1890/11-1587.1
Liu, J., Wang, G., Wang, Q., Liu, J., Jin, J., and Liu, X. (2012). Phylogenetic diversity and assemblage of major capsid genes (g23) of T4-type bacteriophages in paddy field soils during rice growth season in Northeast China. Soil Sci. Plant Nutr. 58, 435–444. doi: 10.1080/00380768.2012.703610
Maier, I., Wolf, S., Delaroque, N., Müller, D., and Kawai, H. (1998). A DNA virus infecting the marine brown alga Pilayella littoralis (Ectocarpales, Phaeophyceae) in culture. Eur. J. Phycol. 33, 213–220. doi: 10.1080/09670269810001736713
Marzinelli, E. M., Campbell, A. H., Zozaya Valdes, E., Vergés, A., Nielsen, S., Wernberg, T., et al. (2015). Continental-scale variation in seaweed host-associated bacterial communities is a function of host condition, not geography. Environ. Microbiol. 17, 4078–4088. doi: 10.1111/1462-2920.12972
McKeown, D. A., Stevens, K., Peters, A. F., Bond, P., Harper, G. M., Brownlee, C., et al. (2017). Phaeoviruses discovered in kelp (Laminariales). ISME J. 11, 2869–2873. doi: 10.1038/ismej.2017.130
McLoughlin, M. E., and Graham, D. A. (2007). Alphavirus infections in salmonids–a review. J. Fish Dis. 30, 511–531. doi: 10.1111/j.1365-2761.2007.00848.x
Milne, I., Bayer, M., Cardle, L., Shaw, P., Stephen, G., Wright, F., et al. (2009). Tablet—next generation sequence assembly visualization. Bioinformatics 26, 401–402. doi: 10.1093/bioinformatics/btp666
Morris, M. M., Haggerty, J. M., Papudeshi, B. N., Vega, A. A., Edwards, M. S., and Dinsdale, E. A. (2016). Nearshore pelagic microbial community abundance affects recruitment success of giant kelp, Macrocystis pyrifera. Front. Microbiol. 7:1800. doi: 10.3389/fmicb.2016.01800
Müller, D., and Frenzer, K. (1993). Virus infections in three marine brown algae: Feldmannia irregularis, F. simplex, and Ectocarpus siliculosus. Hydrobiologia 260, 37–44.
Papudeshi, B., Haggerty, J., Doane, M., Morris, M., Walsh, K., Beattie, D., et al. (2017). Optimizing the reconstruction of microbial population genomes from metagenomes. BMC Genomics 18:915. doi: 10.1186/s12864-017-4294-1
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Reavy, B., Swanson, M. M., Cock, P. J. A., Dawson, L., Freitag, T. E., Singh, B. K., et al. (2015). Distinct circular single-stranded DNA viruses exist in different soil types. Appl. Environ. Microbiol. 81, 3934–3945. doi: 10.1128/AEM.03878-14
Rohwer, F., and Thurber, R. V. (2009). Viruses manipulate the marine environment. Nature 459, 207–212. doi: 10.1038/nature08060
Roossinck, M. J. (2013). Plant virus ecology. PLoS Pathog. 9:e1003304. doi: 10.1371/journal.ppat.1003304
Rosario, K., Dayaram, A., Marinov, M., Ware, J., Kraberger, S., Stainton, D., et al. (2012a). Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 93, 2668–2681. doi: 10.1099/vir.0.045948-0
Rosario, K., Duffy, S., and Breitbart, M. (2009). Diverse circovirus-like genome architectures revealed by environmental metagenomics. J. Gen. Virol. 90, 2418–2424. doi: 10.1099/vir.0.012955-0
Rosario, K., Duffy, S., and Breitbart, M. (2012b). A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Arch. Virol. 157, 1851–1871. doi: 10.1007/s00705-012-1391-y
Rosenberg, E., Koren, O., Reshef, L., Efrony, R., and Zilber-Rosenberg, I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362. doi: 10.1038/nrmicro1635
Rothman, M. D., Mattio, L., Wernberg, T., Anderson, R. J., Uwai, S., Mohring, M. B., et al. (2015). A molecular investigation of the genus Ecklonia (Phaeophyceae, Laminariales) with special focus on the Southern Hemisphere. J. Phycol. 51, 236–246. doi: 10.1111/jpy.12264
Roux, S., Krupovic, M., Poulet, A., Debroas, D., and Enault, F. (2012). Evolution and diversity of the microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 7:e40418. doi: 10.1371/journal.pone.0040418
Sabina, J., and Leamon, J. H. (2015). Bias in whole genome amplification: causes and considerations. Methods Mol. Biol. 1347, 15–41. doi: 10.1007/978-1-4939-2990-0_2
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Simmonds, P., Adams, M. J., Benko, M., Breitbart, M., Brister, J. R., Carstens, E. B., et al. (2017). Consensus statement: virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 15, 161–168. doi: 10.1038/nrmicro.2016.177
Steneck, R. S., Graham, M. H., Bourque, B. J., Corbett, D., Erlandson, J. M., Estes, J. A., et al. (2002). Kelp forest ecosystems: biodiversity, stability, resilience and future. Environ. Conserv. 29, 436–459. doi: 10.1017/S0376892902000322
Van Etten, J. L., Lane, L. C., and Meints, R. H. (1991). Viruses and viruslike particles of eukaryotic algae. Microbiol. Rev. 55, 586–620.
Vega Thurber, R., Willner-Hall, D., Rodriguez-Mueller, B., Desnues, C., Edwards, R. A., Angly, F., et al. (2009). Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11, 2148–2163. doi: 10.1111/j.1462-2920.2009.01935.x
Vergés, A., Doropoulos, C., Malcolm, H. A., Skye, M., Garcia-Pizá, M., Marzinelli, E. M., et al. (2016). Long-term empirical evidence of ocean warming leading to tropicalization of fish communities, increased herbivory, and loss of kelp. Proc. Natl. Acad. Sci. U.S.A. 113, 13791–13796. doi: 10.1073/pnas.1610725113
Vollmers, J., Wiegand, S., and Kaster, A.-K. (2017). Comparing and evaluating metagenome assembly tools from a microbiologist's perspective-not only size matters! PLoS ONE 12:e0169662. doi: 10.1371/journal.pone.0169662
Wahl, M., Molis, M., Hobday, A. J., Dudgeon, S., Neumann, R., Steinberg, P., et al. (2015). The responses of brown macroalgae to environmental change from local to global scales: direct versus ecologically mediated effects. Perspect. Phycol. 2, 11–29. doi: 10.1127/pip/2015/0019
Webster, N. S., and Thomas, T. (2016). The sponge hologenome. MBio 7, e00135–e00116. doi: 10.1128/mBio.00135-16
Wernberg, T., Bennett, S., Babcock, R. C., de Bettignies, T., Cure, K., Depczynski, M., et al. (2016). Climate-driven regime shift of a temperate marine ecosystem. Science 353, 169–172. doi: 10.1126/science.aad8745
Wernberg, T., Russell, B. D., Thomsen, M. S., Gurgel, C. F. D., Bradshaw, C. J., Poloczanska, E. S., et al. (2011). Seaweed communities in retreat from ocean warming. Current Biol. 21, 1828–1832. doi: 10.1016/j.cub.2011.09.028
Weynberg, K. D., Allen, M. J., and Wilson, W. H. (2017). Marine prasinoviruses and their tiny plankton hosts: a review. Viruses 9:E43. doi: 10.3390/v9030043
Weynberg, K. D., Wood-Charlson, E. M., Suttle, C. A., and van Oppen, M. J. (2014). Generating viral metagenomes from the coral holobiont. Front. Microbiol. 5:206. doi: 10.3389/fmicb.2014.00206
Willner, D., Thurber, R. V., and Rohwer, F. (2009). Metagenomic signatures of 86 microbial and viral metagenomes. Environ. Microbiol. 11, 1752–1766. doi: 10.1111/j.1462-2920.2009.01901.x
Wilson, W., Van Etten, J. L., and Allen, M. (2009). Lesser Known Large Dsdna Viruses. Cham: Springer.
Xue, H., Xu, Y., Boucher, Y., and Polz, M. F. (2012). High frequency of a novel filamentous phage, VCYφ, within an environmental Vibrio cholerae population. Appl. Environ. Microbiol. 78, 28–33. doi: 10.1128/AEM.06297-11
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Keywords: circovirus, microbial ecology, virome, kelp, marine disease, metagenome, CRESS-DNA, viral ecology
Citation: Beattie DT, Lachnit T, Dinsdale EA, Thomas T and Steinberg PD (2018) Novel ssDNA Viruses Detected in the Virome of Bleached, Habitat-Forming Kelp Ecklonia radiata. Front. Mar. Sci. 4:441. doi: 10.3389/fmars.2017.00441
Received: 20 November 2017; Accepted: 22 December 2017;
Published: 12 January 2018.
Edited by:
Matthias Wietz, University of Oldenburg, GermanyReviewed by:
Karen Dawn Weynberg, The University of Queensland, AustraliaSimon M. Dittami, UMR8227 Laboratoire De Biologie Intégrative Des Modèles Marins, France
Copyright © 2018 Beattie, Lachnit, Dinsdale, Thomas and Steinberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas T. Beattie, ZC5iZWF0dGllQHN0dWRlbnQudW5zdy5lZHUuYXU=