 Kristin M. Yoshimura
Kristin M. Yoshimura Joanna York
Joanna York Jennifer F. Biddle
Jennifer F. Biddle- School of Marine Science and Policy, University of Delaware, Lewes, DE, United States
Particulate matter in estuarine systems hosts microbial communities that can impact biogeochemical cycles. While the bacterial community composition on suspended particles has been previously investigated, especially with regards to how salinity may structure these communities, the archaeal fraction of the microbial community has not received the same attention. Here we investigate both the bacterial and archaeal community composition on two sizes of particles along a riverine discharge gradient in the Broadkill River, DE, USA, to determine whether the archaeal community is selected by similar environmental stressors as the bacteria. We measured salinity, nutrients, and diatom abundances, and use particle size as a proxy for oxygen concentrations. We show that salinity is a strong environmental factor that controls both bacterial and archaeal community composition and oxygen is an additional factor, impacting archaea more than bacteria.
Introduction
Particulate matter in aquatic and marine systems harbors diverse microbial communities that contribute to the cycling of nutrients such as carbon, nitrogen, and metals. Particle-associated bacterial communities have been studied in habitats around the world, including lakes (Allgaier and Grossart, 2006), rivers and estuaries (Crump et al., 1999; Selje and Simon, 2003; Campbell and Kirchman, 2013), coastal oceans (Bižic-Ionescu et al., 2014), and open oceans (Ganesh et al., 2014). These studies agree that like free-living bacterial communities (Troussellier et al., 2002; Kirchman et al., 2005; Herlemann et al., 2011), particle-associated bacterial communities are strongly influenced by the salinity of the water, which causes changes in the assemblage of core genes (Dupont et al., 2014) and gene expression (Smith et al., 2010). Understanding the community structure associated with these particles is a critical step to understanding the role of particle-associated microbes in the environment.
Particle-associated microbes differ from those in the free-living fraction of the community. The difference has been attributed to the concentration of nutrients associated with particulate matter, the need for cell-to-cell interactions and attachment, and particularly with the presence of anoxic microhabitats within these particles (Ganesh et al., 2014). These anoxic microhabitats have been well-documented and play a critical role in the assemblage of bacterial communities across studies of particle-associated bacteria. Oxygen gradients within marine snow have been measured with microelectrodes, and completely anoxic regions have been observed in large pieces of particulate matter such as fecal pellets (Alldredge and Cohen, 1987). Anaerobic and microaerophilic bacterial taxa including anaerobic green sulfur bacteria (Crump et al., 1999), sulfate reducers, manganese and iron reducers, and specific taxa including Anaerolineaceae and Chlorobiacaea (Smith et al., 2013) have all been found associated within large marine particles, but not in the free-living fraction of the water column. Additionally, genes involved with dissimilatory nitrate reduction, an anaerobic metabolic process, have been documented within particles (Smith et al., 2010) and particle-associated bacteria have been found to potentially produce reduced manganese (Klinkhammer and McManus, 2001). Findings of these specialist taxa and metabolisms associated with particulate matter solidify evidence that anaerobic and microaerophilic bacteria are able to take advantage of oxygen gradients within the interior of large particles. The effect of different size classes of particulate matter on these microbial communities, however, has yet to be addressed. Because particles of varying sizes have different surface area to volume ratios, oxygen penetration into the particle may differ and therefore have an effect on the attached microbial communities.
Though particle-associated bacteria have been extensively studied, the same attention has not been given to particle-associated archaea. Archaea attached to particulate matter have only been investigated in a handful of studies (Crump and Baross, 2000; Galand et al., 2008; Smith et al., 2013; Ganesh et al., 2014), which determined that salinity may play a role in determining archaeal community composition (Galand et al., 2008). Anaerobic methanogenic archaea have also been found associated with particulate matter (Crump and Baross, 2000; Smith et al., 2013), suggesting that the anaerobic microniches used by bacteria may also be used by anaerobic archaea. The previous studies utilized cloning and low-throughput amplicon sequencing methods or protein-coding genes from metagenomic analysis to provide taxonomic composition of the particle-associated archaeal communities, which have limited archaeal detection. Higher throughput sequencing studies specifically targeting the archaeal fraction of the microbial community would allow a more thorough understanding of particle-associated archaeal communities.
In this study, we examined the environmental factors that influence both the bacterial and archaeal communities in two size fractions of particulate matter along a riverine discharge gradient. We hypothesized that similar to bacteria, archaea would be influenced by salinity, and to a lesser degree, oxygen, which was determined by using particle size as a proxy. We show differences in both archaeal and bacterial community structure across the salinity gradient and between particle sizes, suggesting that both salinity and oxygen are forces acting on both microbial domains. Salinity was found to be a greater selecting factor for bacteria while oxygen was more important for archaea.
Materials and Methods
Study Site and Sampling
Samples were taken along a transect in the lower section of the Broadkill River in Southern Delaware, USA (Supplementary Figure 1). The Broadkill River, which is sourced from Wagamon's Pond in Milton, Delaware, is tidally influenced and ranges between 0 and 31 ppt. The depth of the river ranges from 2.8 to 4 m at mean low water along the upper and lower estuary. Historical measurements of dissolved oxygen (DO) show that tidal influence has more impact on DO concentrations than diel cycle in this river (Dewitt and Daiber, 1973). The river is surrounded by fresh and salt marshes, and runoff from residential and agricultural areas feed into the watershed. The Broadkill River empties into the Delaware Bay via the Roosevelt Inlet 21.4 km downriver (Dewitt and Daiber, 1973).
Samples were taken from 1 m below the water surface at 9 stations along the main channel of the Broadkill River in Delaware on July 10, 2014 (Table 1). This depth was chosen to ensure that a consistent depth could be sampled at all sites without the risk of sampling underlying sediment. Sites spanned the lower and upper estuary from the mouth of the river at Roosevelt Inlet (38° 47.419′N, 75° 9.743′W; salinity ≈ 30 ppt) to 12.7 km upstream (38° 47.469′N, 75° 15.054′W; salinity <1 ppt; Table 1). Sixty milliliters was filtered through 0.7 μm glass microfiber (Whatman GF/F) filters for chlorophyll analyses, 20 mL was filtered through 0.7 μm glass microfiber filters for nutrient analysis, and 1 L was sequentially vacuum filtered through individual 8.0, 0.8, 0.45, and 0.22 μm nitrocellulose filters to size fractionate the particulate matter in the sample for DNA analysis. After filtration the filters were immediately stored at −80°C. Sampling was conducted along a similar transect on July 21, 2014 (stations a–f; Table 1) to accomplish total carbon and nitrogen analysis and microscopy. This transect began and ended at the same coordinates as the previous transect and there were no major storms prior to either sampling trip.
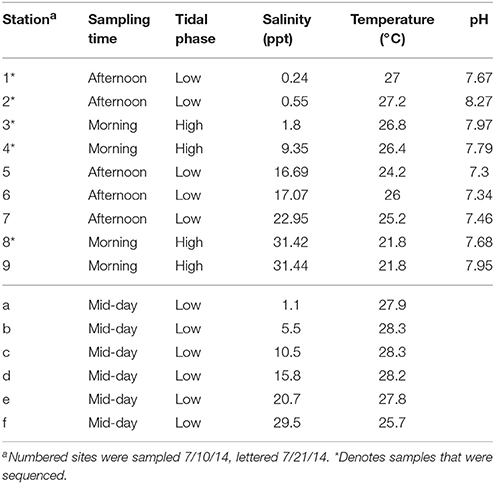
Table 1. Salinity of sampling sites.
Microscopy
Seawater was passed through an 8 μm nitrocellulose filter to concentrate the particulate matter in the sample. The particles were then transferred to a glass slide to be analyzed for apparent composition under an EVOS microscope (Life Technologies, Carlsbad, CA) at 40x and 100x magnifications. Apparent particle types and appearance were recorded and average particle size was determined for each station. A one-way ANOVA was performed on the data to test for statistical differences in particle sizes along the salinity gradient and a Tukey Honest Significant Difference (HSD) test was performed to determine differences between each individual sample.
Chlorophyll
Sixty milliliters of water from each sampling station was syringe filtered through a 0.7 μm glass microfiber filter, with duplicates from each station. Each filter was wrapped in foil to protect from light and frozen at −20°C. Pigments were extracted with 10 mL of acetone and quantified fluorometrically using a Turner 10-AU Fluorometer (Turner Designs, San Jose, CA).
Nutrients
Twenty milliliters of water from each station was syringe filtered through a 0.7 μm glass microfiber filter and the filtrate was immediately frozen at −20°C. Samples were defrosted prior to nutrient analyses and analyzed for nitrate, phosphate, silicate, and ammonium using a Seal Autoanalyzer 3 High Resolution Digital Colorimeter (Seal Analytical, Mequon, WI). Instrument error led to only four stations having valid ammonium measurements.
Total Carbon and Nitrogen
Carbon and nitrogen ratios were determined using a Costech Instruments Elemental Combustion System (Costech, Valencia, CA). Samples were prepared by syringe filtering 50 mL of seawater through a 0.7 μm glass microfiber filter previously baked at 450°C for 5 h. Filters were then dried overnight at 80°C before pelletizing and analyzing for total carbon and nitrogen mass relative to standard masses of EDTA and phenylalanine.
Nucleic Acids
DNA from each filter was extracted using a PowerSoil DNA Isolation kit (MoBio, Carlsbad, CA). Each sample filter was placed directly into a provided bead-beating tube and the manufacturer's protocol was followed. Yield was determined using a Qubit fluorometer (Quibit 2.0, Life Technologies, Carlsbad, CA) and DNA was stored at −20°C.
Quantitative PCR
Quantitative abundance of total archaea and bacteria, specific archaeal groups, and diatoms were determined using qPCR using ThermoScientific maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA) and measured on a MX3005P instrument (Stratagene, San Diego, CA). General 16S rRNA gene bacterial (B1114F and B1275R; Denman and McSweeney, 2006) and archaeal primers (A915F and A1059R; DeLong, 1992; Yu et al., 2005) were used to determine archaeal:bacterial abundance ratios in each sample. Methanogens were quantified using primers targeting the mcrA gene (qmcrA-F and qmcrA-R; Denman et al., 2007) and Miscellaneous Crenarchaeal Group archaea (MCG) were quantified using MCG410F and MCG528R (Kubo et al., 2012). Diatom 18S rRNA gene primers 1256F and 1536R (Lee, 2012; Tilney et al., 2014) were used to quantify diatoms in each sample. Standards were made using a dilution series of either cleaned PCR products from environmental samples known to contain the targeted group (bacteria and MCG) or from DNA from a cultured organism (Halobacterium salinarium for archaea, Methanosarcina acetivorans for methanogens, and Asterionellopsis glacialis for diatoms). One nanogram of template DNA was used in each reaction and optimizations were performed for each assay to determine optimal primer concentrations and thermal protocol (Supplementary Table 1).
Archaeal and Bacterial DNA Sequencing and Taxonomic Assignments
Two size fractions for 5 samples with the highest archaeal:bacterial DNA ratio as determined by qPCR were selected for amplicon sequencing of the 16S rRNA gene (Molecular Research, Clearwater TX) (Supplementary Table 2). These size fractions included 0.45–0.8 and >8.0 μm. The larger size fraction contained larger suspended particles, while the smaller size fraction contained small particles and free-living microbes. Using prior knowledge of oxygen trends on particulate matter, we worked under the assumption that the larger particles likely contained gradients of oxygen concentrations and possibly microaerophilic or anoxic microniches, while the greater surface area:volume ratio on smaller particles would result in less dramatic shifts in oxygen.
PCR and sequencing of 16S rRNA genes in each sample was performed using 349F and 806R archaeal primers (Swan et al., 2010) or 27F and 519R bacterial primers (Lane, 1991) and sequencing was performed by Molecular Research (Shallowater, TX) on an Illumina MiSeq (Ilumina, San Diego, CA). Forward sequences were run through the QIIME pipeline (Caporaso et al., 2010) to demultiplex and quality filter (minimum quality score of 25) the sequences before de novo OTU picking (Edgar, 2010) was performed at 97, 99, and 100% similarity. Taxonomy was assigned using the SILVA 111 database (Quast et al., 2013) as a reference. QIIME was further used to remove any non-archaeal or non-bacterial sequences from each respective dataset, remove singletons, and perform rarefaction of the samples. The resulting sequence depth was 5,664 archaeal sequences per sample and 17,718 bacterial sequences per sample. The 97% identity was used for all taxonomic analyses while all three OTU levels were used to create Bray Curtis similarity resemblance matrices. These matrices were analyzed via clustering of square root transformed or presence/absence data using SIMPROF in PRIMER (Clarke and Gorley, 2006). Comparison of our detected particle-associated archaeal groups with those from sediment samples from the same sites suggest our capture of suspended particulate matter rather than sedimentary particles (Chen, 2013). Sequences were submitted to the EMBL database under accession numbers PRJEB12826 and PRJEB12847.
Results
Microscopy and Nutrients
Particle composition changed along the transect downriver (Figure 1). Particles upriver contained pieces of organic matter, plant matter, and whole cells, while particles downriver were more lithogenous in origin. Average particle size also changed along the course of the transect (Figure 2), with the turbidity maximum occurring where the salinity = 15.8 ppt. Stations mid-river had small particle sizes and were not significantly different from each other (Supplementary Table 3). Nitrate concentrations were highest (~125 μM) upriver (Figure 3A), with concentrations decreasing dramatically as salinity increased downriver. C:N ratios of particles revealed that most locations in the river were nitrogen replete with ratios below 6.625:1, except for station E (Supplementary Figure 2). Ammonium concentrations increased with salinity in the upper estuary, but analysis error lead to a lack of ammonium data for the lower estuary. Silicate peaked mid-river, with concentrations up to 30 μM. Phosphate was present in concentrations <2 μM across the entire discharge gradient. Chlorophyll was also low along the discharge gradient (~5 mg/L), except for the first station where chlorophyll a showed a dramatic increase concurrent with high nitrate (Figure 3B) as well as diatom 18S rRNA gene qPCR measurement (Supplementary Figure 3).

Figure 1. Particle appearance across the salinity transect, from upriver (a) to downriver (e). Scale bars represent 200 μm. Panel letters correspond to station names from 7/21/14.
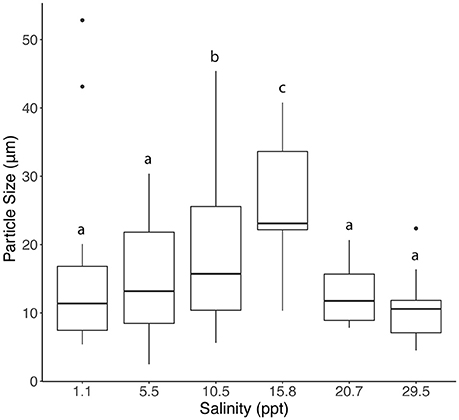
Figure 2. Average particle size, measured by microscopy, for each of six stations. Lowercase letters indicate statistically significant differences between stations. a is significantly different from c but not b, b is not significantly different from a or c. Significance was determined via a Tukey HSD test (Supplementary Table 3).
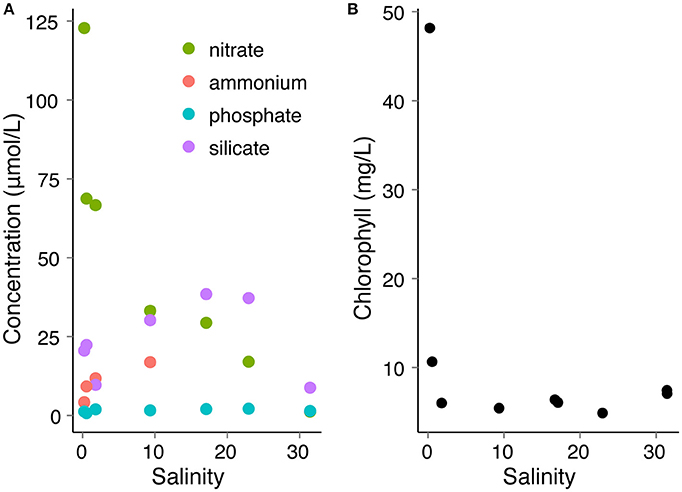
Figure 3. Nutrient (A) and chlorophyll (B) concentrations along the salinity gradient where samples were collected.
DNA Yields and Sample Choice
DNA yields were greatest from the >8 and 0.45–0.8 μm size fractions, with less in the 0.8–8 μm size fraction and virtually none in the 0.22–0.45 μm fraction (Supplementary Table 2). Quantitative PCR results show that in these samples, the percentage of archaea [archaea/(archaea + bacteria)] was up to 5.25% with an average of 2.23% among samples with the exception of station 4, which ranged from 8 to 10% in the 0.45 and 8 μm size fractions. Based on these results, we sequenced only the >8 and the 0.45–0.8 μm size fractions from stations 1, 2, 3, 4, and 8, as these represented a discharge gradient downstream, as well as had higher abundances of archaea, in order to have the highest chances of capturing the archaeal diversity present on particles.
Quantitative PCR
Diatom, methanogen, and MCG archaea abundances were determined using qPCR. Diatoms in the large fraction were more abundant than those from the smaller fraction (Supplementary Figure 3). In the large fraction, abundances of diatoms were highest at the beginning and end of the sampling transect, with lower abundances mid-transect. Results from both the mcrA (Figure 4A) and the MCG (Figure 4B) 16S rRNA gene qPCR assays showed that MCG and methanogenic archaea had the highest abundances on large particles and in freshwater.
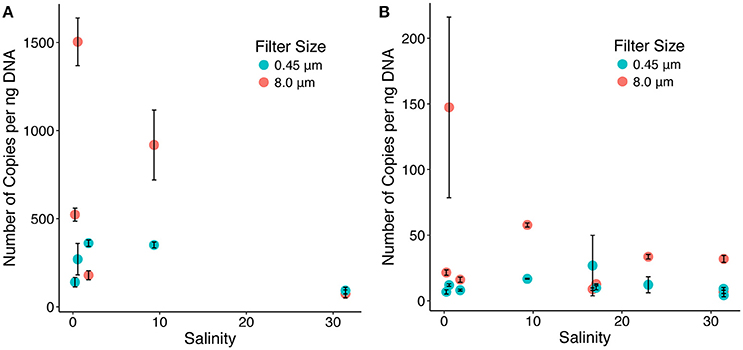
Figure 4. mcrA (A) and MGC archaea (B) copy numbers for small (blue) and large (red) size fractions.
Bacterial Community Composition
After non-bacterial sequences and chloroplasts were filtered from the bacterial dataset, the sequences were filtered to remove any singletons and rarefied, leaving a resulting dataset with 17,718 bacterial sequences. These sequences were analyzed for community composition at a 97% OTU level. The dataset consisted of a total of 12,387 OTUs in 15 major phyla (those that comprised at least 1% of the community in at least 1 sample) and at least 26 minor phyla (combined into “Other Bacteria”). Analyses at higher OTU levels resulted in greater diversity at the OTU level (16,438 OTUs at the 99% level and 23,834 OTUs at the 100% level), but yielded no change in taxonomic assignments at the phylum or class level.
The bacterial community exhibited several patterns that were consistent between the small and large particles. In both size fractions, several phyla including the Actinobacteria, Betaproteobacteria, and Verrucomicrobia were more abundant upriver and decreased in abundance as salinity increased downriver (Figures 5A,B). Specifically, the Actinobacteria comprised between 33 and 61% of the freshwater community on small particles and 21–28% on large particles, but in brackish and marine water, this phylum only comprised 18–22% of the community on small particles and 5–14% on large particles. The Betaproteobacteria comprised between 15 and 29% of the fresh and brackish communities, regardless of particle size, but only 1–2.4% on marine particles. The Verrucomicrobia on the other hand were abundant at Station 1, comprising 8.6% of the community on small particles and 17% on large particles, but were much less abundant (0.12–4.4% of the community) in all other samples. Conversely, phyla including the Alphaproteobacteria, Bacteroidetes, and Gammaproteobacteria were more prevalent downriver than upriver, regardless of particle size. The abundances of Alphaproteobacteria and Bacteroidetes both increased in brackish and marine water as the Alphaproteobacteria were 1.4–7.6% of the bacterial community in fresh water and between 13 and 31% in brackish and marine water while the Bacteroidetes comprised 5.9–18% in fresh water but 19–36% in brackish and marine water. The Gammaproteobacteria however steadily increased in abundance downriver, as 0.58–4.4% of the community in fresh water, 7.9–8.3% in brackish water, and 12–24% in marine water.
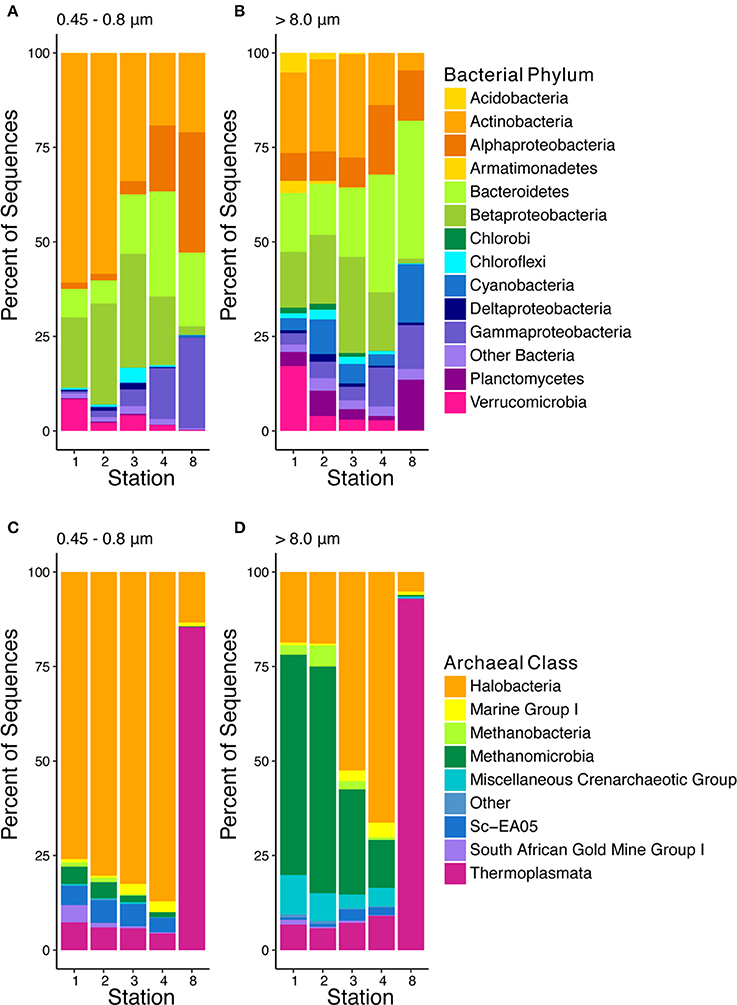
Figure 5. Community composition of bacteria (A,B) at the phylum level and archaea (C,D) at the class level, stations are listed by increasing salinity from left to right.
Other bacterial phyla, however, had differential relative abundances between particles of different sizes. Each of these phyla comprised less than half of 1% in any small particle sample, but was abundant to a greater extent in at least two samples on large particles. The Cyanobacteria comprised 2.8–15% on large particles and Planctomycetes composed up to 13% on large particles. Additionally, the Acidobacteria, Chlorobi, and Armatimonadetes were present only in freshwater and on large particles. The Acidobacteria composed 1.5–5.1% in Station 1 and 2, the Chlorobi comprised 1.7–1.8% of the community in Stations 1 and 2, and the Armatimonadetes comprised 2.9% of the bacterial community in Station 1.
Archaeal Community Composition
After the archaeal dataset was filtered for non-archaeal sequences and singletons and rarefied, each sample was left with 5,664 sequences. As with the bacteria, these sequences were analyzed for community composition at a 97% OTU level. In total, 4,230 OTUs spanning eight major archaeal classes and at least eight minor archaeal classes were present across the 10 samples. As with the bacterial samples, a different OTU diversity was seen when analyzing the sequences at a 99% (4,399 OTUs) or 100% (1,183 OTUs) OTU level, but no change in taxonomy was observed at the class level. The lower total OTUs across samples at the 100% level was due to a higher generation of singleton OTUs that were filtered out prior to subsequent analysis.
The archaeal community on the smaller particles was heavily dominated by members of the Halobacteria group in both the fresh and brackish stations (Figures 5C,D). The Halobacteria accounted for >77% of the archaeal community at each of these stations, with an increase in proportion to 88% occurring as salinity increased downstream. The Halobacteria mostly consisted of members of the Deep Sea Hydrothermal Vent Group, though the entire class exhibited a high level of diversity at the 97% OTU level, consisting of 3,310 OTUs. Halobacteria were also dominant on the large particles in the fresh and brackish stations. These, too, were mostly composed of members of the Deep Sea Hydrothermal Vent Group. The proportion of the overall archaeal community comprised by these archaea, however, were much lower than on the smaller particles, which accounted for 18% in the fresh samples and up to 69% in the brackish. In these samples, several groups of anaerobic archaeal groups including Methanobacteria, Methanomicrobia, and the MCG (also now referred to as Bathyarchaeota) were proportionally more abundant on the larger particles relative to the smaller particles. Methanogens, mostly comprised of Methanomicrobia, but also to a lesser extent by Methanobacteria, comprised up to 64% of the community in the fresh samples, whereas these groups comprised up to a total of 6% of the archaeal population on the smaller particles. Additionally members of the MCG were 5–9% of the fresh and brackish samples on large particles, but accounted for <1% of any sample on small particles. These relative abundances of methanogens and MCG archaea agree with qPCR measurements.
The archaeal community composition on both large and small particles in the marine station was markedly different from the fresh and brackish stations. On both size fractions, the community was heavily dominated by Thermoplasmata, which comprised 87% of the community in the smaller fraction and 93% in the larger fraction. Group II Euryarchaea accounted for a large percentage of this class. Eighty four percent of the small particle Marine Group II population and 79% of the large particle Marine Group II population were composed of just 2 OTUs, though a total of 357 OTUs composed the Thermoplasmata community across samples.
Sample Relatedness
Samples clustered differently based on the OTU level chosen (97, 99, and 100%), but the method of clustering (square root transform or presence/absence) did not change the clustering patterns. For all OTU levels and domains, Station 5 was the most different from other samples. The two particle sizes at this station were not significantly different from each other for either bacteria or archaea.
For the bacteria, clustering patterns at the 97% (Figure 6A) and 99% (Figure 6B) OTU levels were identical. Like the samples from Station 5, both size fractions of particles from Station 4 were significantly different from the fresh stations, but not significantly different from each other. The fresh samples were all more closely related to each other, but Stations 1 and 2 were more closely related to each other than to station 3. The large particles from Stations 1 and 2 were not significantly different from each other and the same was true for the small particles from these two stations. At the 100% OTU level, both the large and small particles from Station 4 were significantly different from other stations but not from each other, similarly to the 97 and 99% OTU clustering levels (Figure 6C). At this clustering level, however, all of the large particles from the fresh stations were closely related to each other, as were all of the small particles.
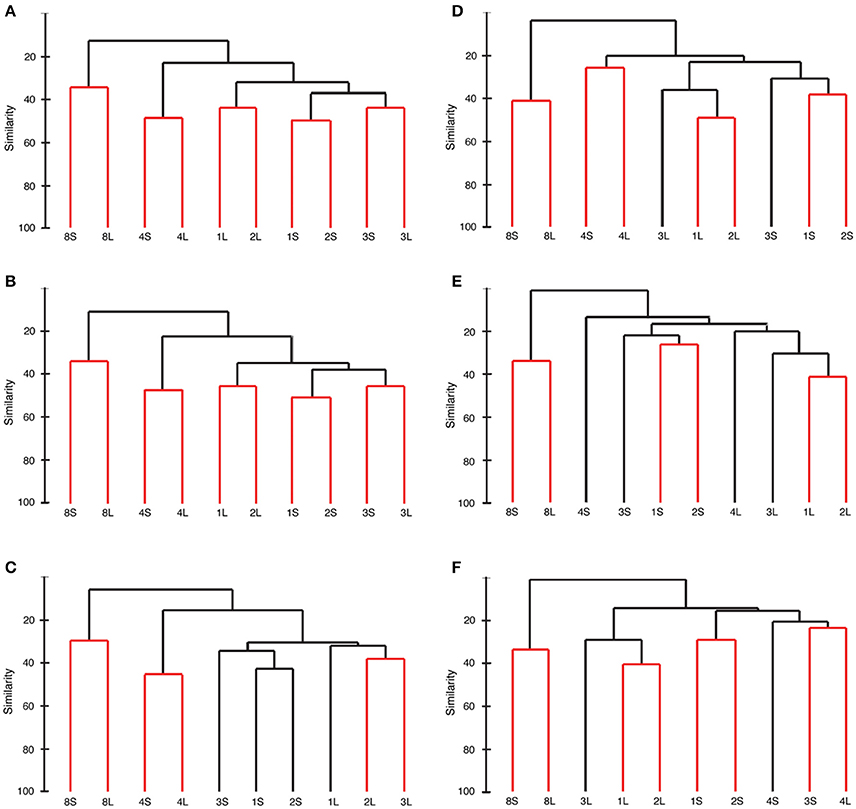
Figure 6. Sample relatedness of bacterial (A–C) and archaeal samples (D–F) when OTUs are picked at a 97% (A,D) 99% (B,E), and 100% (C,F) level. Black bars represent statistical significance, numbers represent the station, S denotes small particles between 0.45 and 0.8 μm and L denotes large particles >8.0 μm.
For the archaea clustered at 97% (Figure 6D), the fresh samples were all more closely related to each other than to the brackish samples, and the fresh samples were most closely related to other fresh samples of similar particle size. At more stringent OTU clustering, however, the brackish samples did not separate themselves as in the bacterial clustering or the 97% archaeal clustering. Instead, the stations predominantly broke into groups based on particle size rather than salinity (Figures 6E,F).
Discussion
Given the under-studied nature of particle-associated archaea, we investigated them alongside their bacterial counterparts in the Broadkill River, DE, along a riverine discharge gradient. Our aims were to determine if environmental factors have similar effects on the community structure between the two domains. We hypothesized that particle size, used as a proxy for oxygen concentrations, would have a greater effect on the archaeal population, but that salinity would also be important. Using high-throughput 16S rRNA amplicon sequencing methods, we determined the bacterial and archaeal community composition on two size fractions of particulate matter, the larger containing large particles 8 μm and above, and the smaller containing small particles and larger free-living microbes between 0.45 and 0.8 μm, and found both previously observed and new trends in both the bacterial and archaeal community structures.
Bacterial Community Composition
Salinity had a major impact on some bacterial groups despite differences in particle size (Figures 5A,B). Actinobacteria, Betaproteobacteria, and Verrucomicrobia were all present in higher abundances in freshwater across size fractions, while the Alphaproteobacteria, Gammaproteobacteria, Cyanobacteria, and Bacteroidetes were more abundant in higher salinities. These trends agree with those previously found in the Delaware Estuary, showing that Betaproteobacteria and Actinobacteria were abundant in the Delaware River, but not in the Delaware Bay, and Alphaproteobacteria were abundant in Delaware Bay, but not the Delaware River (Kirchman et al., 2005). In another study spanning fresh and marine gradients in the Bothnian Bay, Bothnian Sea, and the Baltic Proper sub-basins, Actinobacteria and Betaproteobacteria were associated with low-salinity habitats, whereas Gammaproteobacteria, Alphaproteobacteria, and Bacterioidetes all peaked in higher salinities (Dupont et al., 2014). Similar trends have also been documented in the Chesapeake Bay (Bouvier and del Giorgio, 2002) and the Pearl River Estuary (Zhang et al., 2006). Verrucomicrobia, however, have previously been shown to peak in brackish conditions in the Baltic Sea in several separate studies (Herlemann et al., 2011; Bergen et al., 2014), though different lineages of the Verrucomicrobia have been detected in diverse environments (Freitas et al., 2012). With the exception of the Verrucomicrobia, our data is in agreement with previous studies showing that salinity is a primary stressor for many phyla of particle-associated bacteria.
In contrast, bacterial members of the phyla Chlorobi, Armatimonadetes, Acidobacteria, and Planctomycetes showed differential abundances based on particle size rather than salinity. These groups were present in substantial abundances (>1%) on large particles, but not on small particles. Both the Chlorobi and Planctomycetes are generally considered anaerobes, and the greater presence of these phyla on large particles may be due to lower concentrations of oxygen, as found for large particles in previous studies (Alldredge and Cohen, 1987). While particle-associated bacteria have been studied in a wide array of environments, these trends have not been previously documented, likely due to the low-throughput analysis methods used by most other studies. The higher-throughput sequencing method employed here has allowed us to view the community at a finer resolution, revealing new trends in particle-associated bacterial communities.
Archaea
Similar to the trends exhibited by anaerobic bacteria, several classes of anaerobic archaea including Methanobacteria, Methanomicrobia, and Miscellaneous Crenarchaeotic Group archaea were found in higher abundances on large particles than small ones from the same stations (Figures 5C,D). These relative abundances were confirmed by qPCR, a method more quantitative than amplicon sequencing (Lloyd et al., 2013). These archaeal groups have all been previously documented in anaerobic sediments, which suggests that these classes of archaea may be taking advantage of low oxygen or oxygen-free microniches on larger particles. Given that these anaerobic groups comprise over half of the archaeal community in some stations, we conclude that oxygen has a greater effect on archaeal community composition than bacterial.
The Halobacteria archaea, on the other hand, were abundant on both particle sizes with a trend of increasing abundance downriver from fresh to brackish water, though this trend was more pronounced on larger particles. In this larger size fraction, these Halobacteria had an inverse relative abundance with the methanogens. Additionally, the Thermoplasmata, while present at abundances >1% in all size fractions and across the entire salinity gradient, was overwhelmingly dominant in the marine station on both particle sizes. The Thermoplasmata present in this station were mostly composed of Group II Euryarchaeota, an archaeal group that has been well documented in coastal marine waters (DeLong, 1992; Galand et al., 2008; Xie et al., 2018). A recent study from the Pearl River Estuary found that Group II Euryarchaeota are dominant in marine water, but not as common in brackish water (Xie et al., 2018). The increased relative abundance of the Halobacteria downstream and the dominance of Thermoplasmata in the marine station is likely related to increasing salinity, showing that in addition to anaerobic stress, salinity is an important factor in determining archaeal community composition.
Metagenomic studies have revealed that Group II Euryarchaeota have a photoheterotrophic metabolism that is enabled by a proteorhodopsin (Iverson et al., 2012). Water samples from Station 8/f had the lowest apparent turbidity (Supplementary Figure 4; Figure 2), suggesting that light penetration was high. While some light-dependent organisms such as the rhodopsin-containing Actinobacteria (Figure 3B) were highest upriver in Station 1, diatoms (Supplementary Figure 3) and Cyanobacteria (Figures 5A,B) both increase in relative abundance at Station 8. Diatoms and Group II Euryarchaeota are often found in association with each other (Lima-Mendez et al., 2015; Needham and Fuhrman, 2016) but we were unable to tell if this relationship has relevance to our study. While light may be an important resource for certain microbes, it is also clear that other factors including salinity and nutrients work together to determine the distribution of particle-associated microbes.
Salinity and Oxygen Impacts
We utilized a natural gradient to test salinity impacts on microbial communities and also used a particle size comparison to examine presumed oxygen impacts on community structure. Microbes in the marine station were significantly different than the fresh and brackish station and the small and large particles were not significantly different from each other (Figure 6). The brackish station, too, was significantly different from any of the fresh stations in all instances of bacterial OTU clustering, further showing the importance of salinity on the selection of bacterial communities. In most of these cases, the small fresh particles were not significantly different from each other, and neither were the large fresh particles. This shows that secondary to salinity stress, oxygen requirements and habitat space may also be important.
Unlike in the bacteria, the brackish station only distinguished itself entirely from the fresh stations at the 97% archaeal OTU clustering (Figure 6D). Small particles were most closely related to one another as were large particles showing that anoxic habitats are overall more important for the determination of archaeal community composition. Salinity was a secondary factor, as the marine station was always the most distantly related station.
Conclusion
While there are many environmental factors that can affect the abundance and distribution of particle-associated microbes, we found that salinity and oxygen are two stressors that play an important role in determining the community composition of both bacteria and archaea associated with particulate matter. Because most of the major trends in bacterial populations were between stations of different salinities rather than particle size, we determined that salinity is the greatest driving force behind bacterial diversity. Contrastingly, the greatest trends of archaeal communities were between particle sizes rather than stations. Therefore, we conclude that oxygen is a more important factor than salinity in driving archaeal community compositions, though salinity plays an important secondary role.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Veronique Oldham and Ian Rambo for sampling assistance and Rosa Leon Zayas, Dan Torre, Kathy Coyne, Kyra Kim, Tom Hanson, Patrick Gaffney, and Chris Main for assistance with sample analysis, project ideas, and data processing. We thank the Kerkhof lab at Rutgers for providing Halobacterium DNA and the Coyne lab at University of Delaware for providing Asterionellopsis DNA and diatom primers. This work was funded by the WM Keck Foundation, the University of Delaware and the Joanne Currier Daiber Fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00100/full#supplementary-material
References
Alldredge, E., and Cohen, Y. (1987). Can microscale chemical patches persist in the sea? Microelectrode study of marine snow, fecal pellets. Science 235, 689–691. doi: 10.1126/science.235.4789.689
Allgaier, M., and Grossart, H. P. (2006). Seasonal dynamics and phylogenetic diversity of free-living and particle-associated bacterial communities in four lakes in northeastern Germany. Aquat. Microb. Ecol. 45, 115–128. doi: 10.3354/ame045115
Bergen, B., Herlemann, D. P., Labrenz, M., and Jürgens, K. (2014). Distribution of the verrucomicrobial clade Spartobacteria along a salinity gradient in the Baltic Sea. Environ. Microbiol. Rep. 6, 625–630. doi: 10.1111/1758-2229.12178
Bižic-Ionescu, M., Zeder, M., Ionescu, D., Orlic, S., Fuchs, B. M., Grossart, H. P., et al. (2014). Comparison of bacterial communities on limnic versus coastal marine particles reveals profound differences in colonization. Environ. Microbiol. 17, 3500–3514. doi: 10.1111/1462-2920.12466
Bouvier, T. C., and del Giorgio, P. A. (2002). Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries. Limnol. Oceanogr. 47, 453–470. doi: 10.4319/lo.2002.47.2.0453
Campbell, B. J., and Kirchman, D. L. (2013). Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 7, 210–220. doi: 10.1038/ismej.2012.93
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, B. (2013). Variations of Archaeal Communities in Sediments of Coastal Delaware. Master's thesis, University of Delaware.
Crump, B. C., Armbrust, E. V., and Baross, J. A. (1999). Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 65, 3192–3204.
Crump, B. C., and Baross, J. A. (2000). Archaeaplankton in the Columbia River, its estuary and the adjacent coastal ocean, USA. FEMS Microbiol. Ecol. 31, 231–239. doi: 10.1111/j.1574-6941.2000.tb00688.x
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689. doi: 10.1073/pnas.89.12.5685
Denman, S. E., and McSweeney, C. S. (2006). Development of a real-time PCR assay for monitoring anaerobic fungal and cellulolytic bacterial populations within the rumen. FEMS Microbiol. Ecol. 58, 572–582. doi: 10.1111/j.1574-6941.2006.00190.x
Denman, S. E., Tomkins, N. W., and McSweeney, C. S. (2007). Quantitation and diversity analysis of ruminal methanogenic populations in response to the antimethanogenic compound bromochloromethane. FEMS Microbiol. Ecol. 62, 313–322. doi: 10.1111/j.1574-6941.2007.00394.x
Dewitt, P., and Daiber, F. C. (1973). The hydrography of the Broadkill River Estuary, Delaware. Chesapeake Sci. 14, 28–40. doi: 10.2307/1350700
Dupont, C. L., Larsson, J., Yooseph, S., Ininbergs, K., Goll, J., Asplund-Samuelsson, J., et al. (2014). Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS ONE 9:e89549. doi: 10.1371/journal.pone.0089549
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Freitas, S., Hatosy, S., Fuhrman, J. S., Huse, S. M., Welch, D. B. M., Sogin, M. L., et al. (2012). Global distribution and diversity of marine Verrucomicrobia. ISME J. 6, 1499–1505. doi: 10.1038/ismej.2012.3
Galand, P. E., Lovejoy, C., Pouliot, J., and Vincent, W. F. (2008). Heterogeneous archaeal communities in the particle-rich environment of an Arctic shelf ecosystem. J. Mar. Syst. 74, 774–782. doi: 10.1016/j.jmarsys.2007.12.001
Ganesh, S., Parris, D. J., DeLong, E. F., and Stewart, F. J. (2014). Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J. 8, 187–211. doi: 10.1038/ismej.2013.144
Herlemann, D. P., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579. doi: 10.1038/ismej.2011.41
Iverson, V., Morris, R. M., Frazar, C. D., Berthiaume, C. T., Morales, R. L., and Armbrust, E. V. (2012). Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335, 587–590. doi: 10.1126/science.1212665
Kirchman, D. L., Dittel, A. I., Malmstrom, R. R., and Cottrell, M. T. (2005). Biogeography of major bacterial groups in the Delaware Estuary. Limnol. Oceanogr. 50, 1697–1706. doi: 10.4319/lo.2005.50.5.1697
Klinkhammer, G. P., and McManus, J. (2001). Dissolved manganese in the Columbia River Estuary: production in the water column. Geochim. Cosmochim. Acta 65, 2835–2841. doi: 10.1016/S0016-7037(01)00650-0
Kubo, K., Lloyd, K. G., Biddle, J., Amann, R., Teske, A., and Knittel, K. (2012). Archaea of the miscellaneous crenarchaeotal group are abundant, diverse and widespread in marine sediments. ISME J. 6, 1949–1965. doi: 10.1038/ismej.2012.37
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Chichester: Wiley), 115–175.
Lee, K. (2012). Molecular Assessment of Benthic Diatom Assemblages in Delaware's Inland Bays. MS thesis, University of Delaware, Lewes.
Lima-Mendez, G., Faust, K., Henry, N., Decelle, J., Colin, S., Carcillo, F., et al. (2015). Determinants of community structure in the global plankton interactome. Science 348:1262073. doi: 10.1126/science.1262073
Lloyd, K. G., May, M. K., Kevorkian, R. T., and Steen, A. D. (2013). Meta-analysis of quantification methods shows that archaea and bacteria have similar abundances in the subseafloor. Appl. Environ. Microbiol. 79, 7790–7799. doi: 10.1128/AEM.02090-13
Needham, D. M., and Fuhrman, J. A. (2016). Pronounced daily succession of phytoplankton, archaea and bacteria folling a spring bloom. Nat. Microbiol. 1:16005. doi: 10.1038/nmicrobiol.2016.1035
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Selje, N., and Simon, M. (2003). Composition and dynamics of particle-associated and free-living bacterial communities in the Weser estuary, Germany. Aquat. Microb. Ecol. 30, 221–237. doi: 10.3354/ame030221
Smith, M. W., Herfort, L., Tyrol, K., Suciu, D., Campbell, V., Crump, B. C., et al. (2010). Seasonal changes in bacterial and archaeal gene expression patterns across salinity gradients in the Columbia River Coastal Margin. PLoS ONE 5:e13312. doi: 10.1371/journal.pone.0013312
Smith, M. W., Zeigler, A. L., Allen, A. E., Herfort, L., and Simon, H. M. (2013). Contrasting genomic properties of free-living and particle-attached microbial assemblages within a coastal ecosystem. Front. Microbiol. 4:120. doi: 10.3389/fmicb.2013.00120
Swan, B. K., Earhardt, C. J., Reifel, K. M., Moreno, L. I., and Valentine, D. L. (2010). Archaeal and bacterial communities respond differently to environmental gradients in anoxic sediments of a California hypersaline lake, The Salton Sea. Appl. Environ. Microbiol. 76, 757–768. doi: 10.1128/AEM.02409-09
Tilney, C. L., Pokrzywinski, K. L., Coyne, K. J., and Warner, M. E. (2014). Effects of a bacterial algicide, IRI-160AA, on dinoflagellates and the microbial community in microcosm experiments. Harmful Algae 39, 210–222. doi: 10.1016/j.hal.2014.08.001
Troussellier, M., Schäfer, H., Batailler, N., Bernard, L., Courties, C., Lebaron, P., et al. (2002). Bacterial activity and genetic richness along an estuarine gradient (Rhone River plume, France). Aquat. Microb. Ecol. 28, 13–24. doi: 10.3354/ame028013
Xie, W., Luo, H., Murugapiran, S. K., Dodsworth, J. A., Chen, S., Sun, Y., et al. (2018). Localized high abundance of Marine Group II archaea in the subtropical Pearl River Estuary: implications for their niche adaptation. Environ. Micriobiol. 20, 734–754. doi: 10.1111/1462-2920.14004
Yu, Y., Lee, C., and Hwang, S. (2005). Analysis of community structure in anaerobic processes using a quantitative real-time PCR method. Water Sci. Technol. 52, 85–91.
Keywords: particle-associated and free-living bacteria, salinity stress, oxygen stress, archaea, delaware
Citation: Yoshimura KM, York J and Biddle JF (2018) Impacts of Salinity and Oxygen on Particle-Associated Microbial Communities in the Broadkill River, Lewes DE. Front. Mar. Sci. 5:100. doi: 10.3389/fmars.2018.00100
Received: 17 October 2017; Accepted: 12 March 2018;
Published: 27 March 2018.
Edited by:
Hongyue Dang, Xiamen University, ChinaReviewed by:
Julian Damashek, University of Georgia, United StatesJennifer L. Bowen, Northeastern University, United States
Anton Hartmann, Helmholtz Zentrum München - Deutsches Forschungszentrum für Gesundheit und Umwelt, Germany
Copyright © 2018 Yoshimura, York and Biddle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer F. Biddle, amZiaWRkbGVAdWRlbC5lZHU=