 Wirulda Pootakham1*
Wirulda Pootakham1* Chaiwat Naktang1Chutima Sonthirod1
Chaiwat Naktang1Chutima Sonthirod1 Thippawan Yoocha1Duangjai Sangsrakru1Nukoon Jomchai1Lalita Putchim2
Thippawan Yoocha1Duangjai Sangsrakru1Nukoon Jomchai1Lalita Putchim2 Sithichoke Tangphatsornruang1
Sithichoke Tangphatsornruang1- 1Genomic Research Laboratory, National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency, Pathum Thani, Thailand
- 2Phuket Marine Biological Center, Phuket, Thailand
Background
Elevation in seawater temperature associated with global climate change has caused coral bleaching problems and posed a significant threat to coral health and survival worldwide. Several studies have explored the effects of thermal stress on changes in gene expression levels of both coral hosts and their algal endosymbionts and provided evidences suggesting that corals could acclimatize to environmental stressors through differential regulation of their gene expression (Desalvo et al., 2008, 2010; Császár et al., 2009; Rodriguez-Lanetty et al., 2009; Polato et al., 2010; Meyer et al., 2011; Kenkel et al., 2013). Such information is crucial for understanding the adaptive capacity of the coral holobionts (Hughes et al., 2003). The availability of transcriptome data from a number of coral species and their associated Symbiodinium allows us to probe the molecular stress response of the organisms to heat stress (Traylor-Knowles et al., 2011; Moya et al., 2012; Kenkel et al., 2013; Shinzato et al., 2014; Kitchen et al., 2015; Anderson et al., 2016; Davies et al., 2016). Here, we report the first reference transcriptome for a scleractinian coral Porites lutea, one of the dominant reef-builders in the Indo-West Pacific (Yeemin et al., 2009). We applied both short-read Ion S5 RNA sequencing and long-read Pacific Biosciences (PacBio) isoform sequencing (Iso-seq) to generate transcriptome sequences of P. lutea under normal and heat stress conditions. The key advantage of PacBio's Iso-seq technology lies within its ability to capture full-length mRNA sequences. These full-length transcripts enable the identification of novel genes/isoforms and the detection of alternative splice variants, which have been shown to be overrepresented in stress responses (Iida et al., 2004; Reddy et al., 2013; Liu and Guo, 2017). We envision that this reference transcriptome will provide a coral research community a valuable resource for investigating changes in gene expression under various biotic/abiotic stress conditions.
Data Description
Sample Collection
Six P. lutea colonies were collected from the Maiton Island in the Andaman Sea (7°45′42.5″N 98°28′51.3″E) at the depth of 7–10 m and immediately placed in containers with aerated seawater (Supplementary Figure 1). Coral samples were transported back to shore (Phuket Marine Biological Center) within an hour of collection and transferred to flow-through aquaria, which circulated seawater pumped from the reef to the Phuket Marine Biological Center Research Station. Each colony was fragmented into 24 nubbins using a hammer and a chisel and acclimated for 14 days across four flow-through aquaria at ambient temperature (29°C). The position of the nubbins from each colony in the aquarium was randomized. During the experiment, two aquaria were maintained at ambient water temperature while the other two aquaria were exposed to a heat treatment, which involved an incremental ramping of seawater temperature at a rate of 0.5°C per day for six consecutive days. From day 7 to 13, the temperature was maintained at 34°C in the “heat treatment” aquaria. Coral nubbins were removed from the aquaria for sampling at the beginning of the experiment (day 0) and 6 and 13 days after the onset of the heat stress treatments. Coral tissues were collected using scalpel blades, placed in sterile 2-mL screw-capped tubes and immediately frozen in liquid nitrogen.
RNA Extraction and Transcriptome Sequencing
Frozen samples were pulverized in liquid nitrogen, and the CTAB buffer (2% CTAB, 1.4 M NaCl, 2% PVP, 20 mM EDTA pH 8.0, 100 mM Tris-HCl pH 8.0, 0.4% SDS) was added. RNA was extracted from the aqueous phase twice using 24:1 chloroform:isoamylalcohol and precipitated in 1/3 volume of 8 M LiCl overnight. RNA pellets were washed with 70% ethanol, air-dried, and resuspended in RNase-free water. Poly(A) mRNAs were enriched from total RNA samples using the Dynabeads mRNA purification kit (Thermo Fisher Scientific, Waltham, USA). RNA integrity was assessed with a BioAnalyzer (Agilent Technologies, Santa Clara, USA) prior to the construction of RNA sequencing libraries. We prepared the Iso-seq libraries according to Pacific Bioscience's Iso-seq protocol using the SMARTer PCR cDNA Synthesis Kit (Clontech, Mountain View, USA). The cDNA libraries were partitioned into 1–2, 2–3, and 3–6 kb ranges using the BluePippin Size Selection System (Sage Science, Beverly, USA) prior to being sequenced on a PacBio RS II instrument using P4-C6 polymerase and chemistry with 360 min movie times (Pacific Biosciences, Menlo Park, USA). To obtain short-read RNA sequences, 200 ng of poly(A) mRNA was used to construct a sequencing library using the Ion Total RNA Sequencing Kit (Thermo Fisher Scientific, Waltham, USA). The library was sequenced using the Ion 540TM chip on the Ion S5 XL sequencing system (Thermo Fisher Scientific, Waltham, USA).
Transcriptome Assembly and Annotation
Raw read data from the PacBio RS II were processed into error-corrected reads of insert using the PacBio SMRT Analysis Package (version 2.3) with default parameters. We obtained a total of 520,052 reads of insert totaling 47.82 Gb from 20 SMRT cells (Table 1). Using the Iso-seq protocol (the Classify module with default parameters), adapter sequences, poly-A tails, artificial concatemers and 3′ truncated transcript sequences were removed. We performed an additional round of error correction using PacBio ICE software without the Quiver step on the full-length non-chimeric transcripts (Gordon et al., 2015). The polished consensus isoforms generated were classified as “high-quality” if the consensus accuracy was no less than a cut-off value of 0.99 (default parameter; https://github.com/ben-lerch/IsoSeq-3.0/blob/master/README.md). Of the 520,052 reads of insert, 92,058 (18.5%) were high-quality full-length non-chimeric transcripts, and the remaining 427,967 were non-full-length reads of insert. The sizes of the reads of insert ranged from 356 to 4,657 nucleotides with an average read length of 1,950 and an N50 of 2,337 nucleotides.
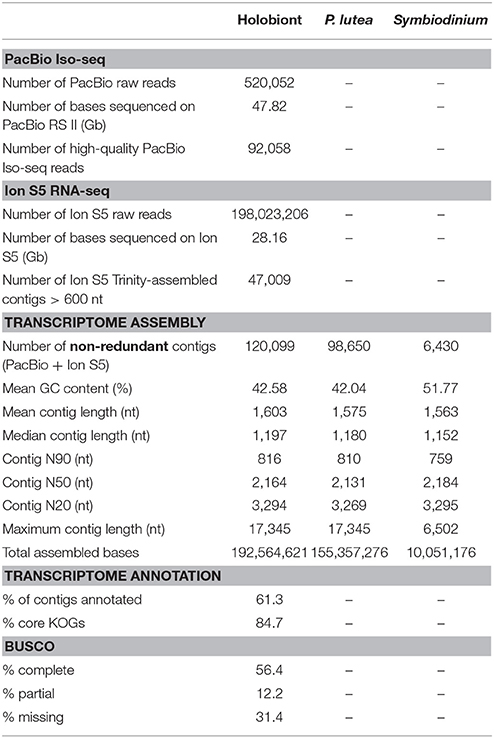
Table 1. Assembly statistics for the holobiont transcriptome.
We also carried out short-read RNA sequencing on the Ion S5 XL system and obtained 198,023,206 raw reads, totaling 28.16 Gb. After the error trimming and the removal of low-quality reads, we performed a de novo transcriptome assembly using default settings in Trinity v2.2.0 (Grabherr et al., 2011). We obtained 839,150 assembled transcripts ranging from 201 to 17,345 nucleotides. Of which, 47,009 sequences had a length of 600 nucleotides or longer (Table 1). To enhance the quality of the reference transcriptome assembly, we combined high-quality full-length transcripts from Iso-seq experiments with Trinity-assembled transcripts that were longer than 600 nucleotides. After clustering these transcript sequences at 100% identity using UCLUST (Edgar, 2010), we obtained a set of 120,160 non-redundant sequences in the metatranscriptome. These transcripts were further screened during an NCBI sequence submission process to remove sequences with similarity to known vectors/primers (https://www.ncbi.nlm.nih.gov/tools/vecscreen/contam/). We obtained a final set of 120,099 non-redundant transcript sequences with an average contig length of 1,603 nucleotides and an N50 length of 2,164 nucleotides (Table 1). Reference assembly annotations were determined by subsequent BLASTP queries against SwissProt and TrEMBL databases. EuKaryotic Orthologous Groups (KOG) annotations were assigned using an alignment search against the core eukaryotic gene set from the core eukaryotic genes mapping approach (CEGMA) pipeline (Parra et al., 2007). The completeness of the transcriptome was assessed through the coverage of the benchmarking universal single-copy orthologs (BUCSO) (Simão et al., 2015). Gene Ontology (GO) annotations describing biological processes, molecular functions, and cellular components were retrieved by mapping BLASTP hit results with their associated GO terms using Blast2Go suite (version 2.8) (Szklarczyk et al., 2011).
Specimens used in this study are expected to include RNA from both coral host and dinoflagellate symbionts. To identify the taxonomic origin of each transcript in the holobiont transcriptome, we conducted a hierarchical series of sequence comparisons against multiple databases similar to the protocol described in Kitchen et al. (2015). To remove ribosomal RNA contaminants, the assembly was aligned to eukaryotic rRNA database from SILVA release 128 (Quast et al., 2013) using BLASTN, and sequence matches with E < 10−5 and bit-scores higher than 45 were discarded. The transcriptome was subsequently compared with the complete mitochondrial genome sequences from Acropora tenuis (Van Oppen et al., 2002) and Porites lobata (Tisthammer et al., 2016) using BLASTN, and again contigs with E < 10−5 and bit-scores higher than 45 were removed. To identify the taxonomic origin of the remaining contigs, transcript sequences were queried against a custom database of publicly available Cnidarian transcriptomes and genomic sequences using BLASTN. The Cnidarian database contains transcriptome sequences from Acropora millepora (Moya et al., 2012), Acropora hyacinthus (Barshis et al., 2013), A. tenuis (Kenkel and Bay, 2017), Montastraea cavernosa (Kitchen et al., 2015), Seriatopora hystrix (Kitchen et al., 2015), Fungia scutaria (Kitchen et al., 2015), Porites astreoides (Kenkel et al., 2013), P. lobata (Kenkel and Bay, 2017), Montipora aequituberculata (Kenkel and Bay, 2017), Galaxea archelia (Kenkel and Bay, 2017), Galaxea astreata (Kenkel and Bay, 2017), Goniopora columna (Kenkel and Bay, 2017), Siderastrea siderea (Davies et al., 2016), Pocillopora damicornis (Traylor-Knowles et al., 2011), and genomic sequences from Acropora digitifera (Shinzato et al., 2011). The assembly was also screened against Symbiodinium transcriptome and genome sequences using BLASTN. The Symbiodinium database comprises transcriptome sequences from Symbiodinium clade A and B (Bayer et al., 2012), Symbiodinium kawagutii, Symbiodinium spp. Clades C1, C15, CCMP2430 and Mp (Lin et al., 2015) and genome sequences from Symbiodinium minutum (Shoguchi et al., 2013). For each transcript, the best hit (E < 10−5) was compared between coral and Symbiodinium database searches and assigned to the taxonomic origin with the smaller E-value. Transcripts matching Symbiodinium database more closely than coral database that did not return a metazoan hit as their best match in Blast nr database were assigned to the Symbiodinium category. The remaining transcript sequences that did not match either coral host or Symbiodinium (with E < 10−5) were classified as “others,” which represented taxa other than corals or symbionts that were members of the coral holobiont (e.g., fungi and benthic algae).
The initial holobiont transcriptome assembly contained 120,099 non-redundant contigs over 600 nucleotides in length, with 77,426 full-length Iso-seq contigs and 42,673 Trinity-assembled contigs. When we searched for potential non-mRNA sequence contaminants, 2,601 contigs matched sequences in the ribosomal database and 2,884 contigs matched the mitochondrial genomes. A total of 98,650 and 6,430 transcript sequences belonged to the coral and Symbiodinium transcriptomes, respectively, while 15,019 sequences matched neither database and were classified as “others.” The coral-specific assembly contained 62,907 full-length isoform sequences (PacBio Iso-seq) and 35,743 Trinity-assembled contigs (Ion S5 RNA-seq) while the Symbiodinium-specific assembly contained 3,839 full-length isoform sequences and 2,591 Trinity-assembled contigs.
Mean GC content of the coral-specific assembly was 42.04%, which was comparable to the numbers reported for other anthozoan transcriptomes (Figure 1) (Shinzato et al., 2014; Anderson et al., 2016; Mansour et al., 2016; Frazier et al., 2017; Kenkel and Bay, 2017). Mean GC content of the Symbiodinium transcripts (51.77%) was also consistent with previous reports (Bayer et al., 2012; Shinzato et al., 2014; Mansour et al., 2016). Interestingly, the GC content distribution of transcripts assigned to Symbiodinium transcriptome had a smaller peak at ~42% (Figure 1), suggesting that a small fraction of the “Symbiodinium” transcripts share the GC content characteristic with coral transcripts. These “Symbiodinium” transcripts with unusually low GC content could have been mistakenly assigned to the Symbiodinium transcriptome. Alternatively, they could be transcribed from genes that had been horizontally transferred into Symbiodinium genomes (although this is highly speculative).
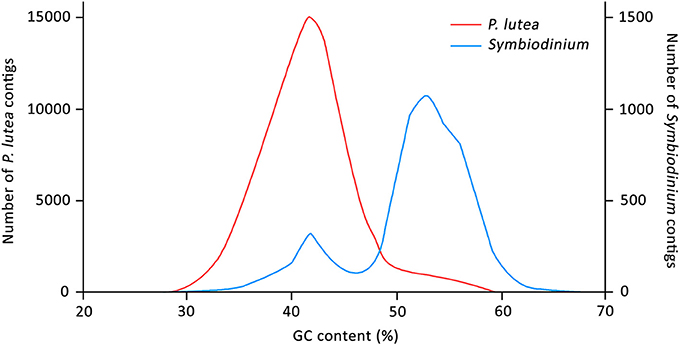
Figure 1. Distribution of GC content (in percentage) of the assembled transcripts. Red and blue lines represent coral- and Symbiodinium-specific transcripts, respectively.
Based on sequence comparison to SwissProt and TrEMBL databases, 73,565 out of 120,099 transcript sequences (61.3%) were annotated at an E-value of <10−6 (Table 1, Supplementary Table 1). Of 429 conserved eukaryotic genes, 68.6% were identified in the assembly, of which 56.4% were considered “complete” while an additional 12.2% were partially assembled. Our assembly contained 84.7% of KOG comprising the core eukaryotic gene set. GO terms were assigned to transcripts matching GO-annotated records in SwissProt and TrEMBL databases. The distribution of functional categories for coral and Symbiodinium transcriptomes is shown in Supplementary Figure 2.
Re-use Potential
The resources developed in this study provide the first reference transcriptome for P. lutea. This is also the first report that applied PacBio long-read single molecule real-time (SMRT) sequencing technology to generate full-length transcript sequences in corals. Future gene expression studies and genome sequencing projects in coral species will greatly benefit from the annotated transcriptome assembly contributed in this study.
Data Availability
This project has been deposited at DDBJ/EMBL/GenBank under the BioProject accession number PRJNA427745, with the PacBio and Ion S5 raw sequencing reads (FASTQ format) deposited under the accession numbers SRX3517969 and SRX3517968 and the transcriptome assembly (FASTA format) deposited under the accession numbers GGER00000000 and GGES00000000. Transcriptome annotations are provided in Supplementary Table 1.
Author Contributions
WP and ST: Conceived and designed the experiment; LP: Collected coral samples and performed the heat-stress experiments; TY, NJ and DS: Carried out the RNA extraction and sequencing; CS and CN: Performed bioinformatics analyses; WP: Wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Science and Technology Development Agency (NSTDA), Thailand (Grant number P-16-50720) and the Department of Marine and Coastal Resources, Ministry of Natural Resources and Environment, Thailand.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00122/full#supplementary-material
References
Anderson, D. A., Walz, M. E., Weil, E., Tonellato, P., and Smith, M. C. (2016). RNA-Seq of the Caribbean reef-building coral Orbicella faveolata (Scleractinia-Merulinidae) under bleaching and disease stress expands models of coral innate immunity. PeerJ 4:e1616. doi: 10.7717/peerj.1616
Barshis, D. J., Ladner, J. T., Oliver, T. A., Seneca, F. O., Traylor-Knowles, N., and Palumbi, S. R. (2013). Genomic basis for coral resilience to climate change. Proc. Natl. Acad. Sci. U.S.A. 110, 1387–1392. doi: 10.1073/pnas.1210224110
Bayer, T., Aranda, M., Sunagawa, S., Yum, L. K., Desalvo, M. K., Lindquist, E., et al. (2012). Symbiodinium transcriptomes: genome insights into the dinoflagellate symbionts of reef-building corals. PLoS ONE 7:e35269. doi: 10.1371/journal.pone.0035269
Császár, N. B. M., Seneca, F. O., and Van Oppen, M. J. H. (2009). Variation in antioxidant gene expression in the scleractinian coral Acropora millepora under laboratory thermal stress. Mar. Ecol. Prog. Ser. 392, 93–102. doi: 10.3354/meps08194
Davies, S. W., Marchetti, A., Ries, J. B., and Castillo, K. D. (2016). Thermal and pCO2 stress elicit divergent transcriptomic responses in a resilient coral. Front. Mar. Sci. 3:112. doi: 10.3389/fmars.2016.00112
Desalvo, M. K., Sunagawa, S., Voolstra, C. R., and Medina, M. (2010). Transcriptomic responses to heat stress and bleaching in the elkhorn coral Acropora palmata. Mar. Ecol. Prog. Ser. 402, 97–113. doi: 10.3354/meps08372
Desalvo, M. K., Voolstra, C. R., Sunagawa, S., Schwarz, J. A., Stillman, J. H., Coffroth, M. A., et al. (2008). Differential gene expression during thermal stress and bleaching in the Caribbean coral Montastraea faveolata. Mol. Ecol. 17, 3952–3971. doi: 10.1111/j.1365-294X.2008.03879.x
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Frazier, M., Helmkampf, M., Bellinger, M. R., Geib, S. M., and Takabayashi, M. (2017). De novo metatranscriptome assembly and coral gene expression profile of Montipora capitata with growth anomaly. BMC Genomics 18:710. doi: 10.1186/s12864-017-4090-y
Gordon, S. P., Tseng, E., Salamov, A., Zhang, J., Meng, X., Zhao, Z., et al. (2015). Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 10:e0132628. doi: 10.1371/journal.pone.0132628
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Hughes, T. P., Baird, A. H., Bellwood, D. R., Card, M., Connolly, S. R., Folke, C., et al. (2003). Climate change, human impacts, and the resilience of coral reefs. Science 301, 929–933. doi: 10.1126/science.1085046
Iida, K., Seki, M., Sakurai, T., Satou, M., Akiyama, K., Toyoda, T., et al. (2004). Genome-wide analysis of alternative pre-mRNA splicing in Arabidopsis thaliana based on full-length cDNA sequences. Nucleic Acids Res. 32, 5096–5103. doi: 10.1093/nar/gkh845
Kenkel, C. D., and Bay, L. K. (2017). Novel transcriptome resources for three scleractinian coral species from the Indo-Pacific. Gigascience 6, 1–4. doi: 10.1093/gigascience/gix074
Kenkel, C. D., Meyer, E., and Matz, M. V. (2013). Gene expression under chronic heat stress in populations of the mustard hill coral (Porites astreoides) from different thermal environments. Mol. Ecol. 22, 4322–4334. doi: 10.1111/mec.12390
Kitchen, S. A., Crowder, C. M., Poole, A. Z., Weis, V. M., and Meyer, E. (2015). De Novo assembly and characterization of four Anthozoan (Phylum Cnidaria) transcriptomes. G3 5, 2441–2452. doi: 10.1534/g3.115.020164
Lin, S., Cheng, S., Song, B., Zhong, X., Lin, X., Li, W., et al. (2015). The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350, 691–694. doi: 10.1126/science.aad0408
Liu, M., and Guo, X. (2017). A novel and stress adaptive alternative oxidase derived from alternative splicing of duplicated exon in oyster Crassostrea virginica. Sci. Rep. 7:10785. doi: 10.1038/s41598-017-10976-w
Mansour, T. A., Rosenthal, J. J., Brown, C. T., and Roberson, L. M. (2016). Transcriptome of the Caribbean stony coral Porites astreoides from three developmental stages. Gigascience 5:33. doi: 10.1186/s13742-016-0138-1
Meyer, E., Aglyamova, G. V., and Matz, M. V. (2011). Profiling gene expression responses of coral larvae (Acropora millepora) to elevated temperature and settlement inducers using a novel RNA-Seq procedure. Mol. Ecol. 20, 3599–3616. doi: 10.1111/j.1365-294X.2011.05205.x
Moya, A., Huisman, L., Ball, E. E., Hayward, D. C., Grasso, L. C., Chua, C. M., et al. (2012). Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO(2)-driven acidification during the initiation of calcification. Mol. Ecol. 21, 2440–2454. doi: 10.1111/j.1365-294X.2012.05554.x
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Polato, N. R., Voolstra, C. R., Schnetzer, J., Desalvo, M. K., Randall, C. J., Szmant, A. M., et al. (2010). Location-specific responses to thermal stress in larvae of the reef-building coral Montastraea faveolata. PLoS ONE 5:e11221. doi: 10.1371/journal.pone.0011221
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Reddy, A. S., Marquez, Y., Kalyna, M., and Barta, A. (2013). Complexity of the alternative splicing landscape in plants. Plant Cell 25, 3657–3683. doi: 10.1105/tpc.113.117523
Rodriguez-Lanetty, M., Harii, S., and Hoegh-Guldberg, O. (2009). Early molecular responses of coral larvae to hyperthermal stress. Mol. Ecol. 18, 5101–5114. doi: 10.1111/j.1365-294X.2009.04419.x
Shinzato, C., Inoue, M., and Kusakabe, M. (2014). A snapshot of a coral “holobiont”: a transcriptome assembly of the scleractinian coral, porites, captures a wide variety of genes from both the host and symbiotic zooxanthellae. PLoS ONE 9:e85182. doi: 10.1371/journal.pone.0085182
Shinzato, C., Shoguchi, E., Kawashima, T., Hamada, M., Hisata, K., Tanaka, M., et al. (2011). Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323. doi: 10.1038/nature10249
Shoguchi, E., Shinzato, C., Kawashima, T., Gyoja, F., Mungpakdee, S., Koyanagi, R., et al. (2013). Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408. doi: 10.1016/j.cub.2013.05.062
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A., Minguez, P., et al. (2011). The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568. doi: 10.1093/nar/gkq973
Tisthammer, K. H., Forsman, Z. H., Sindorf, V. L., Massey, T. L., Bielecki, C. R., and Toonen, R. J. (2016). The complete mitochondrial genome of the lobe coral Porites lobata (Anthozoa: Scleractinia) sequenced using ezRAD. Mitochondrial DNA Part B 1, 247–249. doi: 10.1080/23802359.2016.1157770
Traylor-Knowles, N., Granger, B. R., Lubinski, T. J., Parikh, J. R., Garamszegi, S., Xia, Y., et al. (2011). Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 12:585. doi: 10.1186/1471-2164-12-585
Van Oppen, M. J., Catmull, J., Mcdonald, B. J., Hislop, N. R., Hagerman, P. J., and Miller, D. J. (2002). The mitochondrial genome of Acropora tenuis (Cnidaria; Scleractinia) contains a large group I intron and a candidate control region. J. Mol. Evol. 55, 1–13. doi: 10.1007/s00239-001-0075-0
Keywords: coral, transcriptome, Porites lutea, coral bleaching, heat stress, Iso-seq, RNA-seq, PacBio sequencing
Citation: Pootakham W, Naktang C, Sonthirod C, Yoocha T, Sangsrakru D, Jomchai N, Putchim L and Tangphatsornruang S (2018) Development of a Novel Reference Transcriptome for Scleractinian Coral Porites lutea Using Single-Molecule Long-Read Isoform Sequencing (Iso-Seq). Front. Mar. Sci. 5:122. doi: 10.3389/fmars.2018.00122
Received: 23 January 2018; Accepted: 21 March 2018;
Published: 04 April 2018.
Edited by:
Noga Stambler, Bar-Ilan University, IsraelReviewed by:
Zac H. Forsman, University of Hawaii at Manoa, United StatesTamer Ahmed Mansour, University of California, Davis, United States
Copyright © 2018 Pootakham, Naktang, Sonthirod, Yoocha, Sangsrakru, Jomchai, Putchim and Tangphatsornruang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wirulda Pootakham, d2lydWxkYUBhbHVtbmkuc3RhbmZvcmQuZWR1