 Laura Hernández Javier1
Laura Hernández Javier1 Hicham Benzekri2Marta Gut3,4
Hicham Benzekri2Marta Gut3,4 M. Gonzalo Claros2
M. Gonzalo Claros2 Stefanie van Bergeijk5
Stefanie van Bergeijk5 José Pedro Cañavate5
José Pedro Cañavate5 Manuel Manchado5*
Manuel Manchado5*- 1Programa de Genómica Evolutiva, Centro de Ciencias Genómicas, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
- 2Departamento de Biología Molecular y Bioquímica, Universidad de Málaga, Málaga, Spain
- 3CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology, Barcelona, Spain
- 4Universitat Pompeu Fabra, Barcelona, Spain
- 5IFAPA Centro El Toruño, Junta de Andalucía, El Puerto de Santa María, Spain
Iodine metabolism is essential for the antioxidant defense of marine algae and in the biogeochemical cycle of iodine. Moreover, some microalgae can synthetize thyroid hormone-like compounds that are essential to sustain food webs. However, knowledge regarding iodine-related molecular processes in microalgae is still scarce. In this study, a de novo transcriptome of Tisochrysis lutea cultured under high iodide concentrations (5 mM) was assembled using both long and short reads. A database termed IsochrysisDB was established to host all genomic information. Gene expression analyses during microalgal growth showed that most of the antioxidant- (aryl, ccp, perox, sod1, sod2, sod3, apx3, ahp1) and iodide-specific deiodinase (dio) genes increased their mRNA abundance progressively until the stationary phase to cope with oxidative stress. Moreover, the increase of dio mRNA abundance in aging cultures indicated that this enzyme was also involved in senescence. Cell treatments with iodide modified the expression of perox whereas treatments with iodate changed the transcript levels of gpx1 and ccp. To test the dependence of perox on iodide, microalgae cells were treated with hydrogen peroxide (H2O2) either in presence or absence of iodide observing that several genes related to reactive oxygen species (ROS) deactivation (perox, gpx1, apx2, apx3, ahp1, ahp2, sod1, sod3, and aryl) were transcriptionally activated although with some temporal differences. However, only the expression of perox was dependent on iodide levels indicating this enzyme, acquired by horizontal gene transfer (HGT), could act as a haloperoxidase. All these data indicate that T. lutea activates coordinately the expression of antioxidant genes to cope with oxidative stress. The identification of a phase-regulated deiodinase and a novel haloperoxidase provide new clues about the origin and evolution of thyroid signaling and the antioxidant role of iodine in the marine environment.
Introduction
Phytoplankton is responsible for approximately half of the atmospheric oxygen through oxygenic photosynthesis and provides food resources to sustain the vast majority of marine life (Falkowski, 2012; Chapman, 2013). Along evolution, oxygenic photosynthesis created an oxygen-rich environment that generated high toxic reactive oxygen species (ROS) which are prone to damage functional macromolecules including DNA, proteins and structural lipids (Yilancioglu et al., 2014). Aside of exogenous ROS sources, several endogenous metabolic pathways in microalgae produce ROS including hydrogen peroxide (H2O2) as a by-product of glycolate recycling and fatty acid oxidation in the peroxisome and superoxide () during oxidative phosphorylation in the mitochondria and photosynthesis via the Mehler reaction in the chloroplast (Cirulis et al., 2013). To detoxify ROS and preserve cellular homeostasis, microalgae have developed a wide range of protective mechanisms including ROS-specific antioxidant enzymes and low molecular weight ROS scavengers. With respect the enzymes, the most important are superoxide dismutases (SODs) which remove by dismutation into H2O2, and peroxidases that catalyze the reduction of H2O2 to water. Among scavenging molecules, polyphenols, pigments, ascorbate, glutathione, and some halogenated metabolites including iodide appear as the most important chemical ROS scavengers to prevent oxidative injury (Cirulis et al., 2013; Gribble, 2015).
In marine environments, Iodide is considered as the most ancient and powerful mechanism against ROS (Venturi and Venturi, 1999; Venturi, 2011). Küpper et al. (2008) demonstrated that macroalgae accumulate iodide as an antioxidant mechanism that can rapidly and non-enzymatically scavenge ROS (i.e., O3, O2, 1O2, and OH·), or can act as an electron donor for haloperoxidase enzymes to degrade H2O2. Moreover, this enzyme has been revealed as critical for extracellular iodide uptake thanks to iodine oxidation to hypoiodous acid as a previous step to cell membrane diffusion. One part of the iodine is transformed to volatile halocarbons responsible for the maintenance of the geochemical functioning of the global iodine. Another iodine fraction is intracellularly reduced to iodide or iodinated to organic substrates that act as antioxidant defense reservoirs to scavenge ROS or against excessive predation (Küpper et al., 1998; Gribble, 2003; Iwamoto and Shiraiwa, 2012). The most common organic forms of iodine in algae (that can comprise up to 1% iodine by weight) are iodomethane (CH3I) and its derivatives. In this regard, it is worth noting that some algae are able to accumulate iodotyrosine-derived compounds including monoiodotyrosine (MIT), diiodotyrosine (DIT), triiodothyronine (T3), and thyroxine (T4) (Chino et al., 1994; Heyland and Moroz, 2005; La Barre et al., 2010; Taylor and Heyland, 2017). The spontaneous reaction between tyrosine and iodine to synthetize MIT and DIT reported in some eukaryotic phyla is believed to occur spontaneously (Crockford, 2009; La Barre et al., 2010). This reaction is involved in cell-to-cell endocrine communication and it is also critical for consumers to sustain developmental processes such as the larval metamorphosis of marine invertebrates. Metabolic functions associated to oxidative stress can thus be involved in the origin and evolution of thyroid regulatory pathways in deusterostomes (Laudet, 2011; Taylor and Heyland, 2017). Despite the important antioxidant function of iodide in the algae and its qualitative role to sustain development of key organisms in main food webs, mechanisms related with iodide metabolism are poorly understood in microalgae.
Tisochrysis lutea, previously named Isochrysis aff. galbana (Clone Tahiti) (Bendif et al., 2013) is a small haptophyte within the class Coccolithophyceae. This microalga is widely used in aquaculture due to its high content in polyunsaturated fatty acids, particularly in docosahexaenoic acid (DHA, 22:6n-3) that can reach 8–10% of total fatty acids (Guedes and Malcata, 2012; Rasdi and Qin, 2015). Hence, this microalga has become very popular in most hatcheries for fish and invertebrate larval production as well as for the extensive production of bivalves (Guedes and Malcata, 2012). In addition to its nutritional value, T. lutea is a microalga that accumulates thyroid hormone (TH)-like compounds (Heyland and Moroz, 2005), indicating a particular iodide-related metabolism, what confers novel functional characteristics. Previous data of our group demonstrated this microalga is able to incorporate iodide from the environment after oxidation by enzymatic (haloperoxidase-mediated) or non-enzymatic ways (catalyzed by iron) (van Bergeijk et al., 2013, 2016). Moreover, a fraction of intracellular iodine is organified as TH-like hormones (Heyland and Moroz, 2005). However, Iodine is not essential for growth in T. lutea (Iwamoto and Shiraiwa, 2012; van Bergeijk et al., 2016) suggesting this element could exert other functions such as that of antioxidant defense. The identification of a putative haloperoxidase involved in iodine organification as well as putative deiodinases controlling iodine mobilization will be important to better understand iodine metabolism and its function in this microalga.
Four different types of haloperoxidases are currently considered in a wide range of organisms including brown algae, red algae, gram-negative bacteria, fungi, and some invertebrates (Gwon et al., 2014). In Laminaria digitata, the haloperoxidases play a key role in the specific uptake of iodide from seawater and nucleotide sequences encoding for vanadium iodoperoxidases were reported (Colin et al., 2005; Leblanc et al., 2006; Verhaeghe et al., 2008). In microalgae, some studies reported haloperoxidase activity (Moore et al., 1996; Murphy et al., 2000; Hill and Manley, 2009; Hughes and Sun, 2016), however, putative genes encoding putative iodoperoxidases have not been identified to date. Moreover, iodotyrosine deiodinases were only described in metazoan and bacteria (Phatarphekar et al., 2014; Taylor and Heyland, 2017) and iodothyronine deiodinases were restricted to metazoa and between social amoebae (Lobanov et al., 2007; Orozco et al., 2012; Singh et al., 2014). To increase the knowledge about iodine metabolism and oxidative defenses in T. lutea, the aims of this study were: (i) de novo assembly and characterization of a transcriptome for T. lutea cultured at high iodide concentrations; (ii) To quantify the expression patterns of antioxidant- and iodine-related genes during growth; (iii) To quantify the expression patterns of selected genes in response to iodide and iodate treatments (iv) To quantify the expression patterns of selected genes in response to H2O2 and iodide treatments. Results represent new important clues about the emergence of deiodinases and TH signaling in the marine environment as well as the identification of a novel functional haloperoxidase acquired by horizontal gene transfer (HGT).
Materials and Methods
Microalgal Strain
The marine microalga T. lutea, was purchased from the Culture Collection of Algae and Protozoa in Oban, Scotland (CCAP 927/14). Stock cultures were treated with a mix of streptomycin (50 mg l−1 final concentration) and ampicillin (250 mg l−1 final concentration) or with chloramphenicol (35 mg l−1 final concentration) to remove any bacterial contamination. The lack of bacteria was checked microscopically and by plating on Marine Agar 2216 (Difco). Cultures were grown and maintained in an illuminated incubator (Sanyo MLR-351) at 20°C and an irradiance of 50 μmol photons m−2 s−1. Standard stock cultures (vol 0.35 L) were carried out in 0.5 L by weekly transferring sterile artificial seawater (ASW; salinity 35 and pH 8.0) with f/2 nutrients as previously described (van Bergeijk et al., 2016).
Experimental Set-Up
Growth Conditions and Culture Media
Microalga cultures were carried out in a temperature-controlled room (20°C) under continuous illuminating conditions using a cool white fluorescent light (100 μmol photons m−2 s−1). These conditions are considered optimal to ascertain less variable redox conditions with an optimal quantum yield (Fv/Fm) kept at maximum values (van Bergeijk et al., 2016). Cultures were bubbled with filter-sterilized air (Acro 50 Vent devices, 0.2 μm PTFE, Pall Corporation) enriched with 1% CO2. All cultures were carried out using autoclaved ASW enriched with filter-sterilized f/2 nutrients (0.5 mM N) as previously described (van Bergeijk et al., 2016). All manipulations of the cultures were aseptically carried out in a laminar flow hood.
To evaluate expression patterns during microalga growth, three replicate cultures in 5L-flasks containing 5 L of medium were grown for 17 days under the conditions described above. Replicate flasks were inoculated with starting cultures to obtain an initial cell density of 0.5 × 106 cells ml−1. Daily, each flask was sampled for cell density and gene expression analyses. Cell density was estimated in samples fixed with Lugol's fixative. For gene expression, cells (~2 × 108) were harvested by centrifugation at 4,083 × g for 10 min and 4°C. Pellets were washed in autoclaved ASW and then centrifuged at 5,900 × g for 5 min at 4°C. The cell pellets were stored at −80°C until analysis.
To evaluate the effect of iodide (I−) and iodate () on gene expression, nine 2-L flasks containing 1.5 L of medium were inoculated with T. lutea and cultivated for 4 days (exponential phase) under the above conditions. Three flasks were added potassium iodide (final concentration 5 mM; Sigma-Aldrich) and other three flasks were added potassium iodate (5 mM; Sigma-Aldrich), whereas the remaining three flasks were kept as the untreated control group. Microalgae were harvested at 3 and 6 h after the treatments. The samples were collected as indicated above and kept at −80°C until analysis.
To evaluate the effect of H2O2 and iodide on gene expression, one 2-L flask containing 1.5 L medium was inoculated with T. lutea (initial density 0.5 × 106 cells ml−1) and grown for 4 days (in exponential phase) under the same conditions reported above. Then, this culture was split into twenty-four 50-mL flasks containing 40 mL of culture. Half of flasks were firstly added iodide (0.5 mM final concentration). Then, half of I−-added and half of non-I− added flasks were treated with H2O2 (200 μM; Sigma-Aldrich). Microalgae were harvested at 5 and 45 min after H2O2 addition by centrifuging at 6,000 × g for 5 min and 5°C. H2O2 was immediately measured in the supernatant. The pellets were kept at −80°C until analysis.
Biological Culture Parameters
Cell density
Microalgae cell densities determined with the hemocytometer were used to calculate the specific growth rate was according to the equation: μ = (lnNt-lnN0) (t–t0)−1, where N0 is the cell number at the start of the exponential phase (t0), and Nt is the cell number at time t of the exponential phase.
Hydrogen peroxide quantification
The concentration of extracellular H2O2 was measured colorimetrically using the Quantitative Peroxide Assay Kit (Prod No. 23285; Thermo Scientific). In this assay hydrogen peroxide converts Fe2+ to Fe3+ at acidic pH, which complexes with a xylenol orange dye to yield a purple product with maximum absorbance at 560 nm. Samples were measured in a spectrophotometer against a calibration curve of H2O2 in ASW, which was linear up to 100 μM. When needed, samples were diluted with ASW before analysis.
Transcriptome Sequencing and Characterization
Total RNA was isolated using the RNeasy Plant Mini Kit (Qiagen) according to the manufacturer's instructions. Before sequencing, RNA integrity was checked using the Bioanalyzer 2100 (Agilent Technologies). To achieve a good transcript representation and assembly, both long (454 Roche) and short (Illumina) reads were generated using different microalgal samples and methodological approaches. In the case of long-reads, six 0.5-L flasks containing 0.4 L of ASW enriched with f/2 nutrients were inoculated with T. lutea (initial cell density of 0.5 × 106 cells ml−1) and cultivated for 4 days. Three flasks were added chloramphenicol (35 mg l−1 final concentration) and all of them were added potassium iodide (5 mM final concentration). At the fourth day, microalgae (~2 × 108 cells) were harvested by centrifugation at 4,083 × g for 10 min at 4°C. Pellets were washed in autoclaved ASW, transferred to 1.5-mL tubes and then centrifuged at 5,900 × g for 5 min at 4°C. The cell pellets were stored at −80°C until analysis. Total RNA from T. lutea samples grown without chloramphenicol was isolated as indicated below. For sequencing, the three biological replicates were pooled in the same proportion and cDNA normalized using the SMART technology. In the case of microalgae cultivated with chloramphenicol, total RNA from the three biological replicates was also pooled and the cDNA was not normalized but the most abundant bands visible by electrophoreses corresponding to chloroplast sequences were cut and removed before 454 libraries preparation. These samples were sequenced by 454/Roche technology as previously described (Benzekri et al., 2014).
For the generation of the transcriptomic short-reads, two types of Illumina libraries were prepared. Firstly, a pool of total RNA from microalgae cultivated with chloramphenicol and potassium iodide, as indicated above for long-read sequencing, was normalized using the Mint Universal (Evrogen) and Trimmer (Evrogen) kits and libraries were constructed using the TruRNA-Seq kitv2. Secondly, seven samples of the experiment described above to evaluate the effect of H2O2 and iodide on gene expression were selected: untreated control at 5 and 45 min, I−-added (0.5 mM) samples at 5 and 45 min, I−- and H2O2-added (200 μM) samples at 5 and 45 min and sample H2O2-added at 5 min. The three biological replicates of each condition (~2 × 108 cells at 4 day of culture) were equally pooled and seven cDNA libraries were prepared using the mRNA-Seq sample preparation kit as previously described (Benzekri et al., 2014).
Bioinformatic Analysis
Raw data were processed and assembled at the Plataforma Andaluza de Bioinformática (PAB). The strategy for transcriptome assembly is indicated in Supplementary data sheet 1. Roche/454 long-reads and Illumina short-reads were pre-processed using the SeqTrimNext pipeline (http://www.scbi.uma.es/seqtrimmex; Falgueras et al., 2010), available at the PAB using the specific NGS technology configuration parameters. Long-reads were assembled using MIRA3 (Chevreux et al., 2004) and EULER-SR (Pevzner et al., 2001) with the default parameters and a k-mer = 29 (maximum length allowed). Contigs obtained were mapped onto the original reads using Bowtie (Langmead and Salzberg, 2012) allowing two mismatches to confirm the goodness of the final consensus. Unmapped contigs were considered a sign of misassembling and were submitted to Full-LengtherNext (Seoane et al, in preparation) analysis to recover putative coding sequences.
The Illumina short-reads were assembled with SOAP de novo (Luo et al., 2012) using two k-mers (23 and 47) since the use of multiple k-mers is reported to improve the quality and good performance of de novo assembly (Surget-Groba and Montoya-Burgos, 2010). Processed single reads and paired reads were assembled independently. Thereafter, a script was used to discard misassembled contigs based on the presence of exact, internal, direct or inverse repetitions. Preassemblies were finally reconciled using CAP3 with default parameters to provide the maximal set of transcripts that resulted in T. luteav1 transcriptome. To avoid contaminant sequences due to the sequencing depth used, a BLAST comparison was carried out with the T. lutea transcriptome previously described by Carrier et al. (2014). Those transcripts only represented in T. luteav1 transcriptome with a low number of reads mapped (<10 reads by transcript) with annotation hits for other organisms like H. sapiens or fish species were considered as sequence contamination and removed to generate the T. luteav2 transcriptome.
Transcripts were annotated using Sma3s (Muñoz-Mérida et al., 2014), AutoFact and Full-LengtherNext filtering for E < 10−10 and a minimal identity of 30% as reported in Benzekri et al. (2014). The homology identification analysis was performed using Blastx at NCBI database. All proteins with E < 10−6 and an identity ≥25% were considered.
For phylogenetic analysis, a set of sequences encoding for deiodinases in metazoans was retrieved from GenBank/EMBL/DDBJ and SoleaDB (Benzekri et al., 2014) databases (Accession numbers are shown in Supplementary data sheet 2). Sequences were aligned using ClustalV methods implemented in Megalign (Dnastar). Maximum likelihood (ML) phylogenetic analysis was carried out using ProtTestv3.2 and the PHYLIP package. The best-fit model for sequence evolution was JTT+I+G+F. At tree was drawn using Figtree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). Sequence names used in the phylogeny are indicated in Supplementary data sheet 2.
RNA Isolation and Gene Expression Analysis
Homogenization of microalgal cells was carried out using Lysing Matrix D (MP Biomedicals) for 40 s at speed setting 6 in the Fastprep FG120 instrument (Bio 101). Total RNA was extracted from frozen samples using the RNeasy Plant Mini Kit (Qiagen) according to the manufacturer's instructions. In all cases, total RNA was treated twice with DNase I using the RNase-Free DNase Kit (Qiagen) to avoid amplification of contaminated genomic DNA. RNA sample quality was assessed by electrophoresis and quantification was performed spectrophotometrically (NanoDrop 8000). Total RNA (1 μg) from each sample was reverse-transcribed using the iScript™ cDNA Synthesis kit (Bio-Rad).
Real-time analysis was carried out on an iCycler (Bio-Rad). Reactions were performed in a 10 μl volume containing cDNA generated from 10 ng of original RNA template, 300 nM each of specific forward and reverse primers, and 5 μl of Mix SYBR Premix Ex TaqTM (Takara). Matching oligonucleotide primers were designed using Oligo ν6.89 software (Medprobe) (Table 1). The amplification protocol used was as follows: initial 15 min denaturation and enzyme activation at 95°C, 40 cycles of 95°C for 30 s, 68°C for 15 s, and 72°C for 1 min. Each assay was performed in duplicate. For normalization of cDNA loading, all samples were run in parallel using the elongation factor (eef1a) and ubiquitin (ubi) as reference genes but only the latter was used due to its higher stability. Relative mRNA expression was determined using the 2−(ΔΔCt) method. Data were calibrated to the first day of culture in microalgal growth experiment, to the untreated control in the iodide and iodate experiment and to the 5 min control samples added iodide but not hydrogen peroxide.
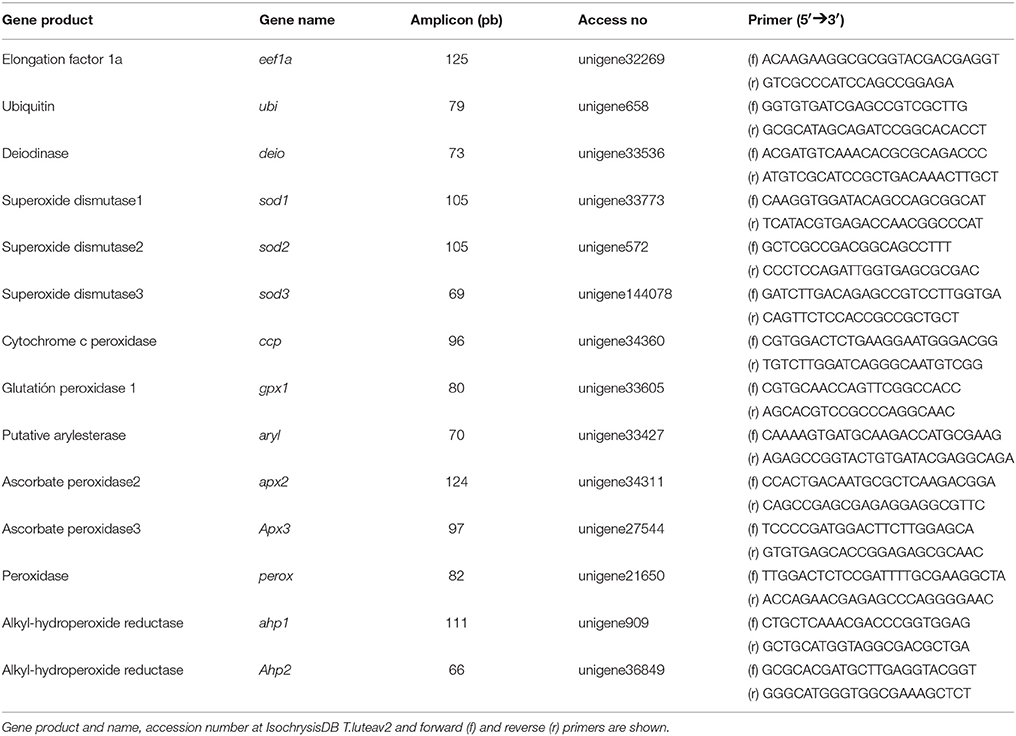
Table 1. Primers used for qPCR analyses.
Statistical Analysis
All data were checked for normal distribution with the Kolmogorov–Smirnov test as well as for homogeneity of the variances with the Levene's test and when necessary, log transformation was applied. A simple ANOVA was used to test significant differences along the growth curves. Moreover, a General Linear Model (GLM) analysis using time, H2O2 and iodide levels as fixed factors was carried out to test significant differences associated with these treatments.
Results
De Novo Sequencing of T. lutea Transcriptome
The transcriptome of T. lutea cultivated under different iodide and hydrogen peroxide conditions was sequenced using long- (454) and short-reads (Illumina). A total of 1.6 × 106 and 1,539 × 106 of raw sequence reads, were generated respectively (Supplementary data sheet 1) and deposited in the BioProject PRJNA418294. Intriguingly, data pre-processing showed that normalized libraries resulted in the highest levels of redundant sequences (66.2–68.3%) mainly originating from chloroplasts. The main characteristics of the assembled transcriptomes are depicted in Table 2 and Supplementary data sheet 1. Assembly of long reads (0.72 × 106 of clean sequences) generated 23,405 transcripts with 52.2% longer than 500 bp (Table 2). When both long and short reads were jointly processed (T. luteav2 assembly), the total number of transcripts increased to 69,800 (50.9% longer than 500 bp), with 23.2% representing different complete open reading frames (ORFs) and 47.6% different gene orthologs. To reduce transcript redundancy (splicing forms and partial sequences encoding the same gene) in the full transcriptome, Full-LengtherNext was used to select a subset of unique sequences (23,671) to build a reference transcriptome. In silico Blastx comparisons between the reference transcriptome and the transcriptomic information previously reported by Carrier et al. (2014) for this microalga demonstrated a matching of 98.7% (sequence alignment >95% and cut-off E-value <1.0E−10; Supplementary Table 1). A total of 16,363 sequences perfectly matched (100%) between both transcriptomes. For the full transcriptome, the matching correspondence with Carrier's transcriptome ranged between 89.3 and 92.0% (Supplementary Table 1).
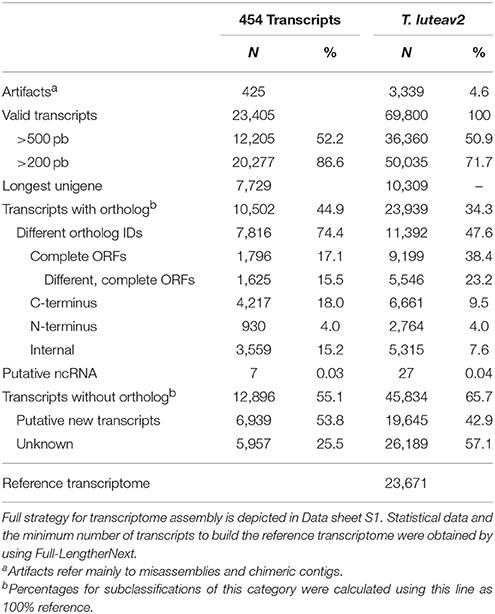
Table 2. Overview of quality parameters for assembled long-reads (454 transcripts) and final T.luteav2 transcriptome.
Annotation percentages were 42.4 and 57.0% for the full and reference transcriptomes, respectively. Gene Ontology analysis showed that categories were equally represented in both transcriptomes with catalytic activity (51.1%), cellular (38.4%), and cellular process (28.6%) as the most represented ones in the molecular function, cellular component and biological process domains, respectively (Figure S1). KEGG analysis indicated that all major metabolic pathways were represented and a total of 1,757 transcripts could be assigned to at least one known universal KEGG pathway.
All transcriptomic information generated in this study was hosted at IsochrysisDB (http://www.scbi.uma.es/isochrysisdb/). This database was built, structured and designed in a user-friendly manner showing all information regarding experimental conditions, information about NGS libraries and processing bioinformatics pipelines to clean, assemble and annotate these transcriptomes. In the “All Assemblies” tab, the different assembly versions can be browsed. In the “Unigenes” section, users can search for specific transcripts and browse specific information about annotation, ORF prediction, and putative markers (Figure 1). Moreover, transcripts can be browsed for SNPs, SSRs, Descriptions, GOs, EC, KEGGs, and InterPros annotations.
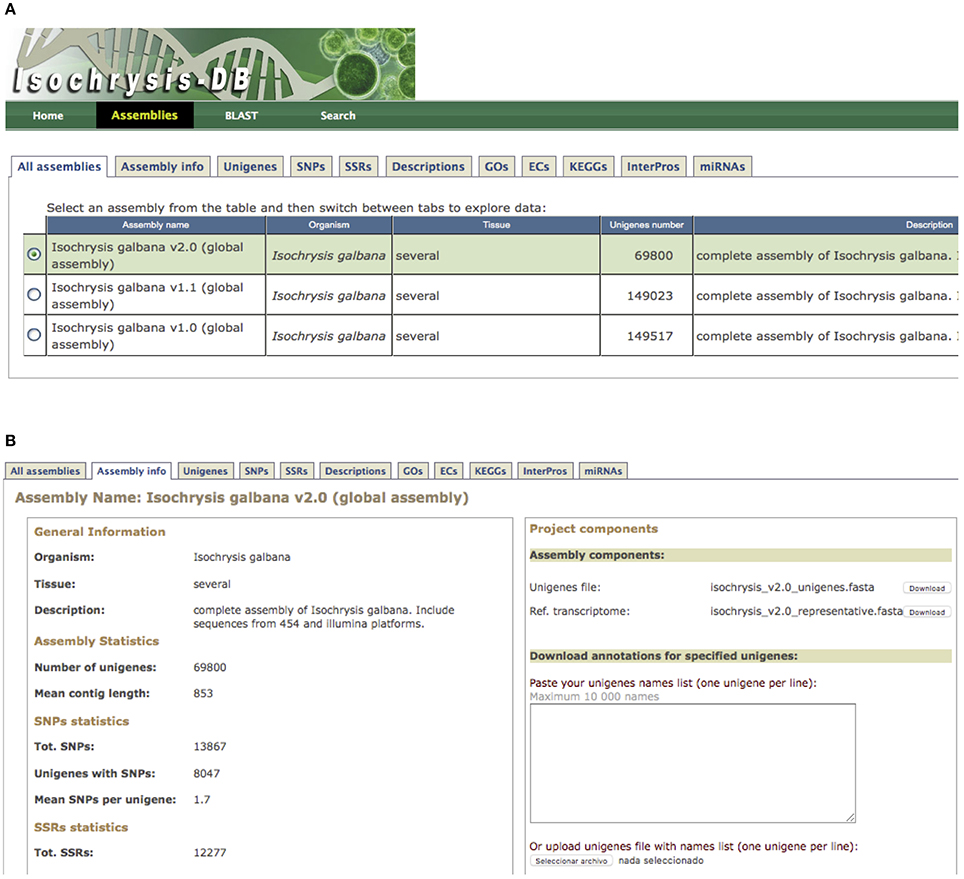
Figure 1. IsochrysisDB interface. (A) “Assemblies” tab containing all information about transcriptomes; (B) Navigation window for T. luteav2.0 that includes the different tabs (“Assembly info,” “Unigenes,” “SNPs,” “SSRs,” “Descriptions,” “GO,” “ECs,” “KEGGs,” “InterPros,” and “miRNAs”) is shown. Moreover, the fasta files for full and reference transcripomes are available in the assembly components section.
Identification of Transcripts Related to Antioxidant Defenses and Iodide Metabolism
Several searches using key terms were carried out at IsochrysisDB and three SODs (sod1, sod2, and sod3), some peroxidases including the cytochrome c peroxidase (ccp), one glutathione peroxidase (gpx1), one putative peroxidase (perox), and two ascorbate peroxidases (apx2 and apx3), two alkyl-hydroperoxide reductases (ahp1 and ahp2), one arylesterase (aryl), and one iodothyronine deiodinase (dio) were identified (Supplementary Table 2; E-value <1.0E-06). The annotation was further confirmed by BLASTx at NCBI (Supplementary Table 2). Analysis of homologous transcripts in bacteria, fungi, plants and vertebrates showed a high number of conserved domains in all of them except in perox (Table 3 and Supplementary data sheet 3) with only 18 similar sequences in ciliates, amoeba, bacteria, and dinoflagellates. The best hit with annotation showed an E-value < 10−17 and a putative peroxidase function of Oxytricha trifallax (EJY77502) (Table 3).
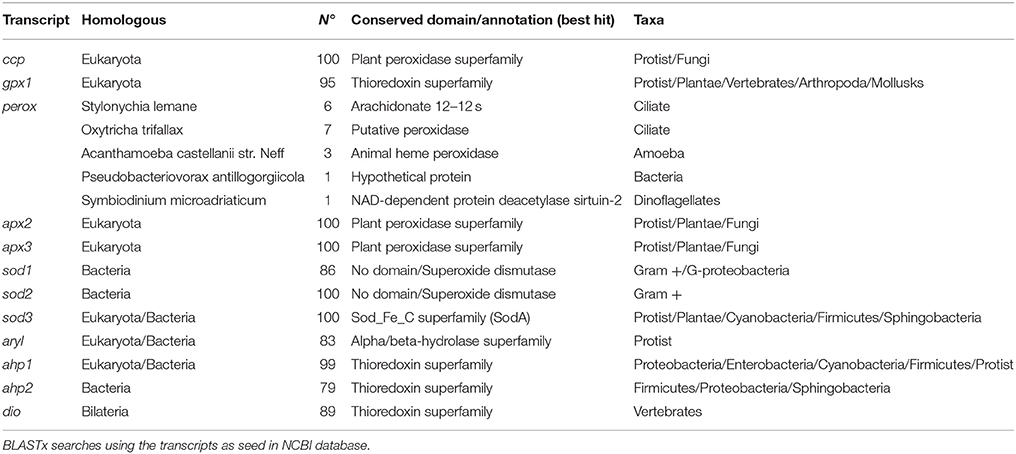
Table 3. Homologous sequences to transcripts selected for PCR studies.
About conserved domains, sod1 and sod2 appeared as Cu,Zn SOD sod3 as Mn-Fe SOD (Table 3). The peroxidase domain was identified in ccp, apx2, apx3, and perox genes and the aryl appeared as alpha/beta-hydrolase. Moreover, a thioredoxin domain was found in transcripts such as gpx1, ahp1, ahp2, and dio. This latter was highly homologous of type I iodothyronine deiodinase in vertebrates (Supplementary data sheet 2). A search for SeCIS elements confirmed the presence in the putative sequence gpx1 but failed in the dio encoding sequence one. A phylogenetic analysis of dio with homologous in invertebrates and vertebrates indicated that this deiodinase clustered separately for their counterparts indicating its ancestral origin (Figure 2). Moreover, a Cys residue instead of SeCys highly conserved in vertebrates was found in the active site (Supplementary data sheet 2).
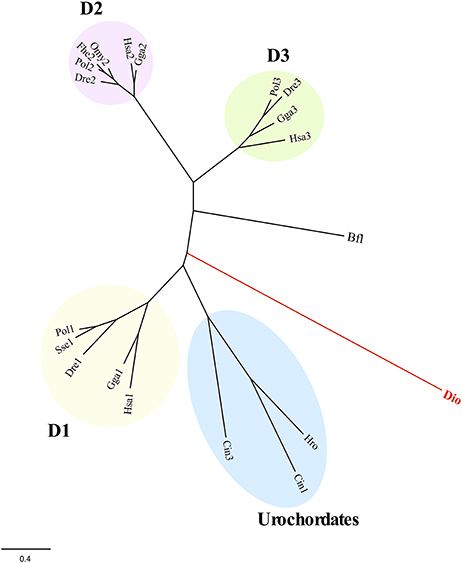
Figure 2. Maximum likelihood phylogeny of T. lutea deiodinase (dio) and counterpart from vertebrates and invertebrates (see Supplementary data sheet 2). The D1-, D2-, and D3-type deiodinases in vertebrates and the urochordates branch are indicated. The B. floridae (Bfl) was used as outgroup to root tree. The scale for branch length (0.4 substitutions/site) is shown below the tree. Bfl, Branchiostoma floridae; Cio, Ciona intestinalis; Dre, Danio rerio: Fhe, Fundulus heteroclitus; Gga, Gallus gallus; Hro, Halocynthia roretzi; Hsa, Homo sapiens; Omy, Oncorhynchus mykiss; Pol, Paralichthys olivaceus; Sse, Solea senegalensis.
Expression Patterns During Microalgal Growth
Growth of T. lutea under controlled conditions of temperature and illumination was monitored for 17 days and cell density was estimated daily. The exponential phase lasted until the day 6, the phase of declining relative growth from days 6 to 9, the stationary phase from days 9 to 15 and the death phase from days 15 to 17 (Figure 3, left upper panel). Expression profiles indicated a significant increase of mRNA levels for perox, apx3, ahp1, sod2, aryl, and dio transcripts along the growth curve (Figures 3, 4). A wide set of transcripts (including aryl, ccp, perox, sod1, sod2, sod3, apx3, ahp1, and dio) peaked their mRNA levels coinciding with declining growth rate and the beginning of stationary phase, with small gene-specific differences in the peaking time (Figures 3, 4). Thereafter, most of these transcripts reduced expression except ahp1 and dio, which increased their expression at the end of the stationary phase. Unlike these transcripts, the gpx1 transcript decreased its expression progressively during the growth of T. lutea, and no significant differences were observed in apx2 and ahp2 mRNA levels (Figures 3, 4).
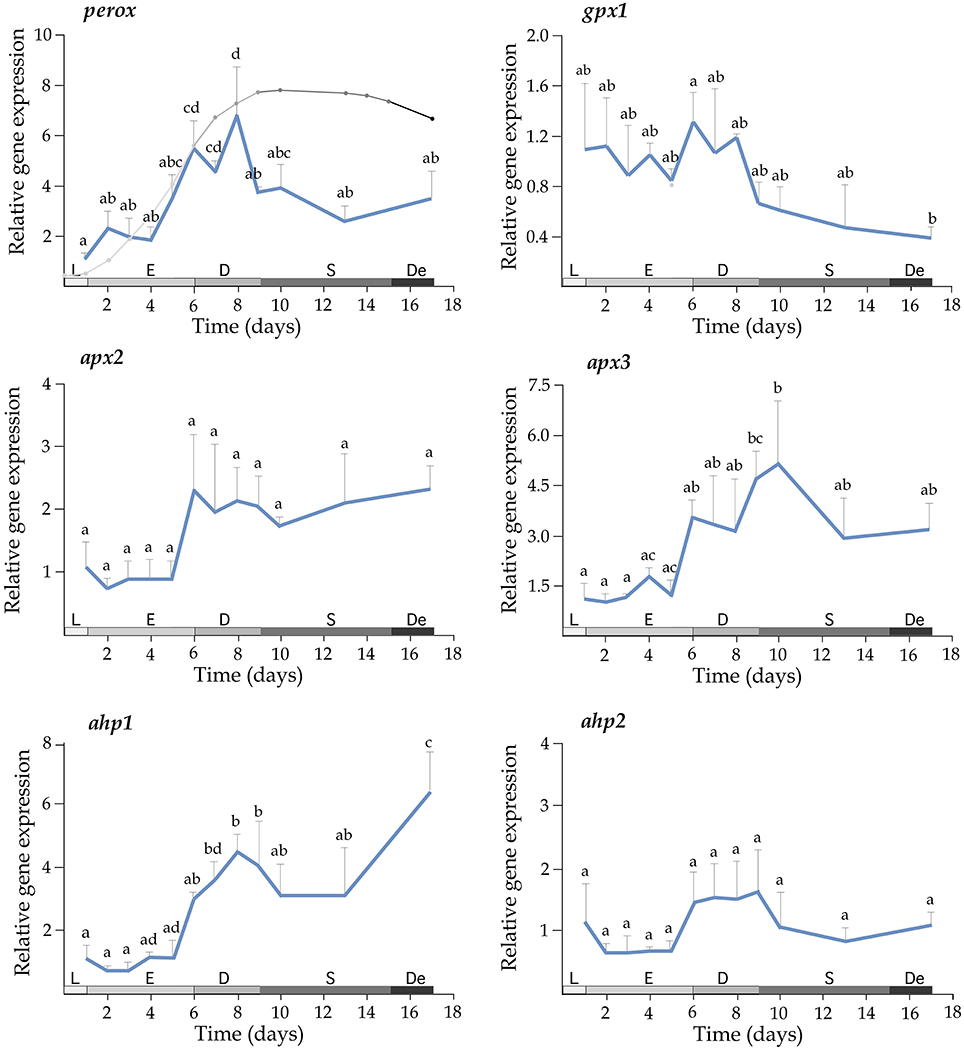
Figure 3. Expression profiles of perox, gpx1, apx2, apx3, ahp1, and ahp2 during the growth of T. lutea. Data represent the mean ± SEM. Data were normalized using ubi and calibrated to 1 day of culture. Different letters denote statistically significant differences between sampling times after the post-hoc test. The growth stages including lag phase (L), exponential phase (E), retardation phase (D), stationary phase (S), and declining phase (De) are shown below.
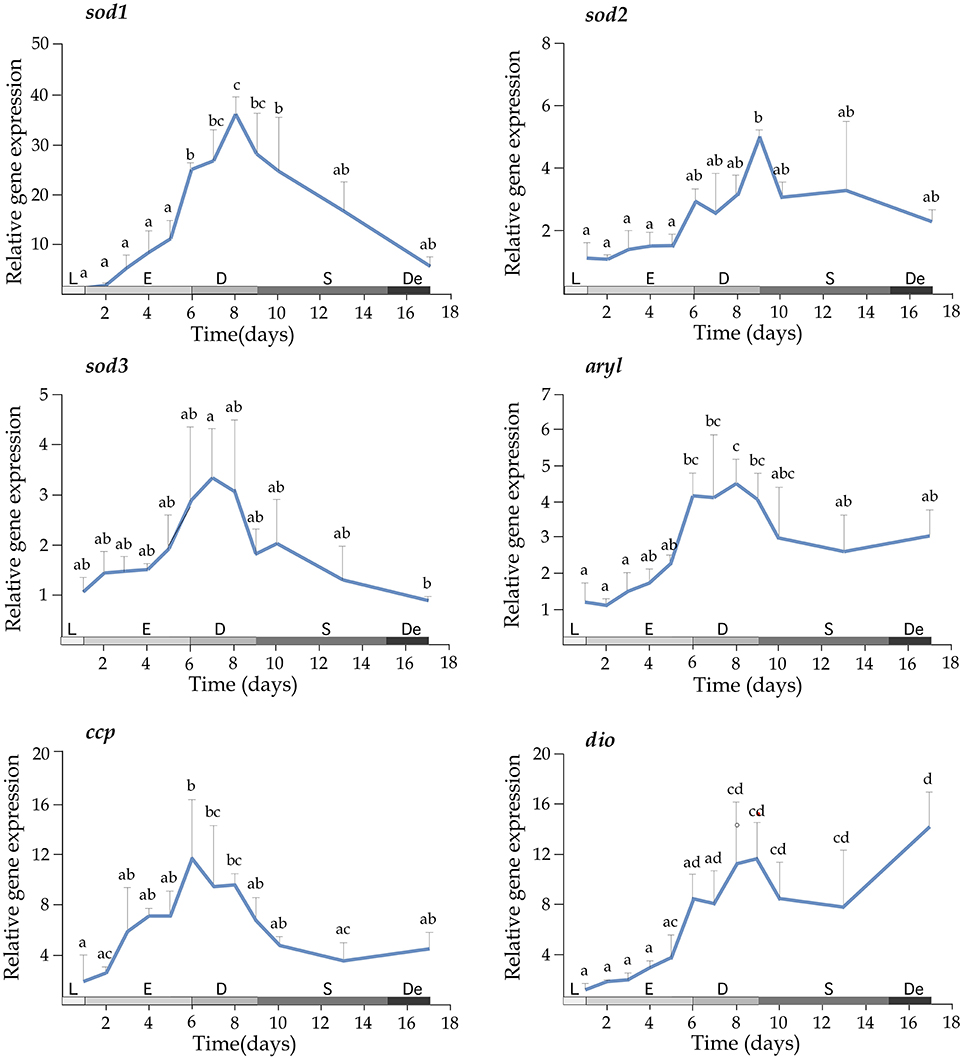
Figure 4. Expression profiles of sod1, sod2, sod3, aryl, ccp, and dio during the growth of T. lutea. Data represent the mean ± SEM. Data were normalized using ubi and calibrated to 1 day of culture. Different letters denote statistically significant differences between sampling times after the post-hoc test. The growth stages including lag phase (L), exponential phase (E), declining of growth phase (D), stationary phase (S), and death phase (De) are shown below.
Effects of Iodide and Iodate on Gene Expression
In order to determine the effects of iodide and iodate on gene expression, mRNA levels of selected transcripts were quantified in T. lutea cultures exposed to 5 mM iodide and 5 mM iodate for 3 h. Iodide significantly up-regulated perox gene expression (2.25-fold with respect to the untreated control). In contrast, iodate increased gpx1 mRNA levels (3.12-fold) and down-regulated ccp gene expression (1.96-fold) (Table 4). A further analysis of activated genes (perox and gpx1) 6 h after the treatment confirmed the expression profiles (Figure S2).
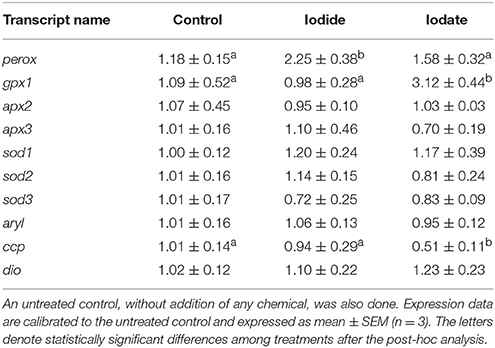
Table 4. Expression levels of perox, gpx1, apx2, apx3, sod1, sod2, sod3, aryl, ccp, and dio in T. lutea cultures at 3 h after iodide (5 mM) or iodate (5 mM).
Effects of H2O2 and Iodide on Gene Expression
In order to evaluate a possible interaction between iodide levels and H2O2 to modulate gene expression, a preliminary trial was carried out to evaluate the removal rate of H2O2 either in the presence or absence of iodide using ASW and T. lutea cells. In ASW free of T. lutea cells and without any added iodide, the H2O2 reduction was ~7% of initial H2O2 concentration after 6 h. The addition of iodide to the ASW clearly accelerated the removal of H2O2 (p < 0.01; Figure 5). On the other hand, when H2O2 was added to the microalgae cultures, 90% was rapidly removed in 45 min independently of iodide levels. Following these results, a new trial using microalgae in exponential phase and exposed to both H2O2 and iodide was carried out and gene expression of a subset of genes involved in oxidative stress and iodide regulation quantified at 5 and 45 min after treatments.
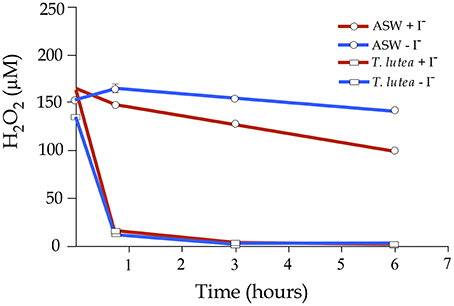
Figure 5. Hydrogen peroxide decomposition in artificial seawater (ASW) and in an exponential culture of T. lutea. Half of the samples were added iodide (500 μM) and then all samples were treated with H2O2 (200 μM). H2O2 levels were monitored for 6 h. Bars represent mean values ± SEM (n = 4). Red lines indicated iodide-added samples and blue lines the controls without iodide.
H2O2 treatments significantly activated the expression of perox, gpx1, apx2, apx3, ahp1, ahp2, sod1, sod3, and aryl (p < 0.05) although with temporal differences, confirming their participation in antioxidant defensive response to detoxify H2O2 (Figures 6 and Figure S3). However, only apx2 and perox showed a significant response to iodide treatments. The apx2 mRNA levels were lower in the iodide-treated samples but the activation response to H2O2 was not dependent on iodide levels. On the contrary, mRNA perox abundance was higher in iodide treated cells. More interestingly, the transcriptional induction triggered by H2O2 in this gene was highly dependent on the presence of iodide to the medium. This observation was consistent in a different experimental replicate in which iodide treatments were done during separate experiments (Figure S3).
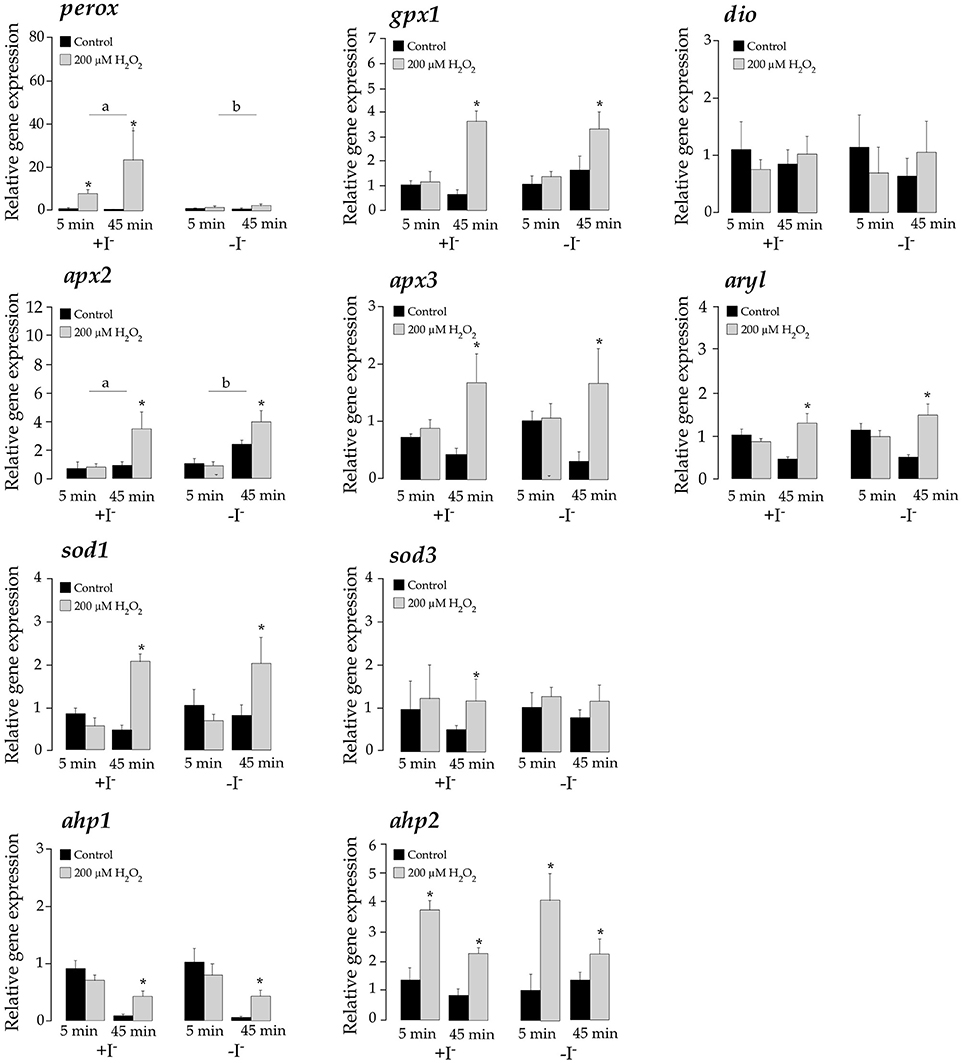
Figure 6. Expression levels of perox, gpx1, dio, apx2, apx3, aryl, sod1, sod3, ahp1, and ahp2 in T. lutea cultures at 5 and 45 min after adding iodide (+I−) or not (–I−) followed by a H2O2 treatment (200 μM). Negative controls without H2O2 treatments were also done for each iodide concentration. Data represent the mean ± SEM. The asterisk denotes statistically significant differences between non-treated and H2O2-treated at each sampling point and iodide condition. The letters denote significant differences between iodide-added and non-iodide added samples. All data were calibrated to the 5 min control samples added iodide without H2O2.
Discussion
The metabolism of iodine in microalgae is far to be properly understood. In this study, the transcriptome of T. lutea, one microalga that accumulates THs, using both long (Roche/454) and short (Illumina) reads was assembled. A previous transcriptome of T. lutea using cells in the retardation phase cultivated standard culture conditions in batch was reported (Carrier et al., 2014), however, our experimental conditions using cells in exponential phase cultivated under high iodide and peroxide treatments enriched in transcripts specifically related with iodine metabolism. Although normalized libraries were originally intended to minimize transcript redundancy, the cleaning results indicated a high number of rejected sequences, most of them from plastids (66.2% in 454 and 68.3% in Illumina). These data agree with previous data from our group in the flatfish Solea senegalensis, in which 454-normalized libraries resulted in a high rate of rejected redundant sequences and more fragmented assemblies (Benzekri et al., 2014). Our data confirm that normalization is not a good strategy when a maximal number of correctly predicted full-length cDNAs is intended unless a deep coverage is expected (Cahais et al., 2012).
The full transcriptome T. luteav2 contained 69,800 valid transcripts that resulted in a reference transcriptome of 23,671 transcripts with similar GO categories representation. This reference transcriptome is considered a useful tool for deep sequencing studies to minimize data processing times without losing any significant information about regulatory pathways in the functional analysis (Hachero-Cruzado et al., 2014; Fatsini et al., 2016). Sequence comparisons of the reference transcriptome with the previously published transcriptome in T. lutea (Carrier et al., 2014) indicated an almost complete matching (98.7%). Nevertheless, comparison of full transcriptomes reduced similarities between 89.3 and 92.0% indicating a small subset of assembly-specific transcripts that could be due to different experimental conditions, strains or assembly artifacts. In any case, both transcriptomes represent a valuable genomic information for the annotation and characterization of the upcoming genome of this microalga of high importance in aquaculture and biotechnology.
It is noteworthy that all transcriptomes assembled in this study were hosted in a specific database named IsochrysisDB. The information was structured in a similar way to SoleaDB (Benzekri et al., 2014), EuroPineDB (Fernández-Pozo et al., 2011), EuroPineDB (Fernández-Pozo et al., 2011), and ReprOlive (Carmona et al., 2015). All transcriptomic information can be browsed by assemblies (with history versions) that contains all relevant information about bioinformatic pipeline, experimental conditions and main statistical features. Within each assembly, all information about the transcript sequences, ORF prediction, annotations (including GO, KEGGs, InterPros) and molecular markers (SNPs and SSRs) can be browsed. Moreover, custom searches by keywords and Blast can be carried out to rapidly identify target sequences. This database represents a powerful tool for research focused on microalgae genomics, specifically in T. lutea that can be progressively updated to improve the current information and provide new services for transcriptomics.
Our experimental design used a permanent and non-saturating illumination conditions that ensured stable oxidative stress conditions. The irradiance used was well below photoinhibition values as demonstrated by optimal quantum yield achieved (van Bergeijk et al., 2016). Under these conditions, a set of transcripts related to iodine and/or oxidative metabolism were identified in the IsochrysisDB and were further characterized by in silico and gene expression analyses. Of particular interest are the peroxidases since they play a central role in H2O2 detoxification and iodine uptake and organification. All peroxidases showed a high number of homologous sequences in other microalgae and bacteria except perox that had low identities with respect to protist homologs (ciliates and amoeboids). The best hit indicated that the closest peroxidase is in Acanthanieba castellanii homolog, an animal-like heme-dependent peroxidase (Clarke et al., 2013), that belongs to the peroxidase-cyclooxygenase family, which includes among others the myeloperoxidase or thyroid peroxidase (Zámocký and Obinger, 2010; Zámocký et al., 2014). We hypothesize that T. lutea could have retained this peroxidase by HGT. Extensive bacterium-derived HGT not related to plastid functions has been described in red algal genomes (Qiu et al., 2013, 2015). More importantly, these non-endosymbiont gene transfers can be propagated through additional rounds of secondary and tertiary endosymbiosis to several other protist hosts such as stramenopiles, alveolates, haptophytes, cryptophytes, and amoeba (Qiu et al., 2013), supporting the HGT as the most plausible hypothesis with fixation in some specific protist lineages.
A striking result was the identification of a type I iodothyronine deiodinase-like sequence in T. lutea. In vertebrates, this kind of enzyme plays a key role in the peripheral regulation of THs actions catalyzing the conversion of T4 to the active form T3 (Alves et al., 2017). Although the thyroid system appeared at the beginning of vertebrate evolution, some studies reported thyroxine deiodinases in invertebrates and social amoebas (Heyland et al., 2006; Lobanov et al., 2007; Wu et al., 2012; Singh et al., 2014). Our study is the first report identifying a thyroxine deiodinase in a microalga species with capacity to synthetize TH-like compounds (Heyland and Moroz, 2005) representing one of the most ancestral deiodinase that later propagated in eukaryote lineages. The molecular analysis identified a Cys instead of SeCys in the active site as occurs in some dictyostelids and the cephalochordate amphioxus Branchiostoma floridae (Holland et al., 2008; Klootwijk et al., 2011; Singh et al., 2014) indicating that ancestral deiodinases lacked the selenocysteine. The residue in the active site not only modulates catalytic efficiency but also molecule affinity as observed B. floridae biasing preferentially the activity toward the 3,3′5-triiodothyroacetic acid (TA3) and tetraiodothyroacetic acid (TA4) instead of triiodothyronine (T3) and tetraiodothyronine (T4) (Paris et al., 2008). Some authors have suggested the TA3 as the ancient bioactive hormone whereas T3 acquired a predominant role later during evolution probably reflecting a co-evolution enzyme and substrate (Klootwijk et al., 2011). In any case, the identification of TH-like compounds in some microalgae including T. lutea (Heyland and Moroz, 2005) suggests that this novel deiodinase should participate in TH metabolism. The increase of dio mRNA levels during the exponential growth phase, in a similar way to most antioxidant genes, suggests this enzyme acts modulating the bulk of intracellular iodide to scavenge ROS (Venturi, 2011). It is thus likely that iodide mobilization might contribute to the pool of antioxidant mechanisms acting in response to elevated ROS production due to high photosynthetic activity during exponential growth. The nature of the response of marine microalgae to irradiance is highly species-specific (Janknegt et al., 2009) and iodide could be playing a yet uncovered function at this regard. The later increase of dio in the declining phase suggests an additional role for iodide in other cellular processes not directly linked to photosynthesis. In mammals, molecular iodine induces cell death through the activation of a mitochondria-mediated apoptotic pathway (Shrivastava et al., 2006) and in the amoeba D. discoideum, the deiodinase is essential for the formation of multiple signaling centers regulating growth and development (Singh et al., 2014) indicating that deiodinase could also regulate gene life cycle in the microalga.
In batch-cultures, the stationary growth phase coincides with a decrease in cell viability, depletion of pigments and essential nutrients and deterioration of thylakoid membranes. Under these conditions, photosynthetic rates and CO2 fixation drop resulting in different metabolic processes becoming more relevant at generating oxidative stress (Sigaud-Kutner et al., 2002, 2005; Liu et al., 2012). Cells then need to rearrange mobilization of oxidative defense mechanisms including peroxidases, SODs, and other antioxidant defenses such as carotenoids (Blokhina et al., 2003). Although previous studies in diverse microalgae demonstrated absence of correlation between irradiance intensity and SOD activity (Janknegt et al., 2009), an increased SOD activity in the stationary growth phase associated to oxidative stress has been reported (Sigaud-Kutner et al., 2002). Moreover, SOD, catalase and APx activities are also activated under nitrogen depletion to cope with the physiological stress induced by such condition (Yilancioglu et al., 2014). In this study, we demonstrated a coordinated activation of genes involved in ROS detoxification, including SOD (sod1, sod2, sod3) and peroxidases (apx3, aryl, ccp, perox, ahp1) that peaked at the beginning of the stationary phase, when nutrients started to be depleted (Liu et al., 2012) indicating that cells activated antioxidant enzymes transcripts coinciding with ROS production likely derived from nutrient scarcity.
External addition of H2O2 rapidly activated the expression of some peroxidases (perox, gpx1, apx2, apx3, aryl) and even of two SODs (sod1 and sod3) to quickly remove toxic levels from the T. lutea cultures (Figure 6). The perox and ahp2 rapidly expressed after 5 min of the H2O2 insult, whereas gpx1, sod1, sod3, apx2, apx3, aryl, and ahp1 increased later, after 45 min. These temporal expression patterns might be associated with the nature of the detoxifying activity and its localization in the cell. The activation of SOD-encoding genes by H2O2 could be an indirect effect resulting of a change cell redox state that can activate a global defensive response in the cell. A similar effect was detected in E. coli in which an external H2O2 insult could activate the regulator SoxRS and trigger the expression of several genes related with detoxification of (Manchado et al., 2000). However, the most striking result is the iodide-dependent response of perox gene expression after the H2O2 treatment. A search in the IsochrysisDB by annotations or blast failed to identify any haloperoxidase-encoding sequence in T. lutea. However, the dependence of perox expression on environmental iodide (and not iodate), as revealed by threshold mRNA abundance and the response to H2O2, indicates that this gene could be a putative haloperoxidase. A previous study of our group indicated that iodide uptake in T. lutea could possibly be promoted by an enzymatic oxidation using H2O2 (van Bergeijk et al., 2013). The similarity of perox with peroxidase-cyclooxygenases in metazoans and its origin by HGT, as previously discussed, makes this gene a good candidate to participate in iodide metabolism processes such as uptake and organification and it could thus also have an impact on the biosynthesis of TH-like-compounds. Further studies will be necessary to elucidate the precise role of this enzyme on THs metabolism in T. lutea.
In conclusion, the transcriptomic analysis and the database IsochrysisDB provided new genomic resources to boost research in T. lutea improving the management of information and our understanding about the molecular mechanisms that control iodide and oxidative metabolism in microalgae. Moreover, some genes involved in ROS detoxification were identified and characterized. Of particular interest is the identification of one iodothyronine deiodinase and one putative haloperoxidase that could represent the ancestral origin of TH-signaling and hence be essential to understand the evolution of TH in vertebrates.
Author Contributions
JC, MM, and SvB conceived the overall study. LH and SvB carried out the microalgae trials and sample preparation. MM carried out the qPCR analyses and wrote the manuscript with contributions from all coauthors. HB and MC carried out the bioinformatic analysis (assembly, annotation and database development). MG prepared RNA-seq libraries and sequenced iodide- and peroxide-treated samples.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was carried out in the framework of research project RTA2009-00054-00-00, funded by the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA) and European Regional Development Funds and the FEDER project Nuevas herramientas genómicas para el análisis genético y evaluación transcriptómica de compuestos funcionales basados en microalgas para impulsar la acuicultura del lenguado (SOLEALGAE) PP.AVA.AVA201601.9 cofunded 80% by Fondo Europeo de Desarrollo Regional, dentro del Programa Operativo FEDER de Andalucía 2014-2020. We wish to thank to Dr. Miguel Ángel Cevallos and Dr. Santiago Castillo Ramírez from Center for Genomic Sciences at Mexico for their comments and support during the preparation of this manuscript. Additionally, LH is thankful to the Center for Genomic Sciences and the Mexican Secretary of Foreign Affairs, which supported her by a fellowship (AMEXCID number HERLAU8104116) during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00134/full#supplementary-material
References
Alves, R. N., Cardoso, J. C., Harboe, T., Martins, R. S., Manchado, M., Norberg, B., et al. (2017). Duplication of Dio3 genes in teleost fish and their divergent expression in skin during flatfish metamorphosis. Gen. Comp. Endocrinol. 246, 279–293. doi: 10.1016/j.ygcen.2017.01.002
Bendif, E. M., Probert, I., Schroeder, D. C., and De Vargas, C. (2013). On the description of Tisochrysis lutea gen. nov. sp. nov. and Isochrysis nuda sp. nov. in the Isochrysidales, and the transfer of Dicrateria to the Prymnesiales (Haptophyta). J. Appl. Phycol. 25, 1763–1776. doi: 10.1007/s10811-013-0037-0
Benzekri, H., Armesto, P., Cousin, X., Rovira, M., Crespo, D., Merlo, M. A., et al. (2014). De novo assembly, characterization and functional annotation of Senegalese sole (Solea senegalensis) and common sole (Solea solea) transcriptomes: integration in a database and design of a microarray. BMC Genomics 15:952. doi: 10.1186/1471-2164-15-952
Blokhina, O., Virolainen, E., and Fagerstedt, K. V. (2003). Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann. Bot. 91, 179–194. doi: 10.1093/aob/mcf118
Cahais, V., Gayral, P., Tsagkogeorga, G., Melo-Ferreira, J., Ballenghien, M., Weinert, L., et al. (2012). Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol. Ecol. Resour. 12, 834–845. doi: 10.1111/j.1755-0998.2012.03148.x
Carmona, R., Zafra, A., Seoane, P., Castro, A. J., Guerrero-Fernandez, D., Castillo-Castillo, T., et al. (2015). ReprOlive: a database with linked data for the olive tree (Olea europaea L.) reproductive transcriptome. Front. Plant Sci. 6:625. doi: 10.3389/fpls.2015.00625
Carrier, G., Garnier, M., Le Cunff, L., Bougaran, G., Probert, I., De Vargas, C., et al. (2014). Comparative transcriptome of wild type and selected strains of the microalgae Tisochrysis lutea provides insights into the genetic basis, lipid metabolism and the life cycle. PLoS ONE 9:e86889. doi: 10.1371/journal.pone.0086889
Chapman, R. L. (2013). Algae: the world's most important “plants”—an introduction. Mitig. Adapt. Strateg. Glob. Change 18, 5–12. doi: 10.1007/s11027-010-9255-9
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A. J., Muller, W. E., Wetter, T., et al. (2004). Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14, 1147–1159. doi: 10.1101/gr.1917404
Chino, Y., Saito, M., Yamasu, K., Suyemitsu, T., and Ishihara, K. (1994). Formation of the adult rudiment of sea urchins is influenced by thyroid hormones. Dev. Biol. 161, 1–11. doi: 10.1006/dbio.1994.1001
Cirulis, J. T., Scott, J. A., and Ross, G. M. (2013). Management of oxidative stress by microalgae. Can. J. Physiol. Pharmacol. 91, 15–21. doi: 10.1139/cjpp-2012-0249
Clarke, M., Lohan, A. J., Liu, B., Lagkouvardos, I., Roy, S., Zafar, N., et al. (2013). Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol. 14:R11. doi: 10.1186/gb-2013-14-2-r11
Colin, C., Leblanc, C., Michel, G., Wagner, E., Leize-Wagner, E., Van Dorsselaer, A., et al. (2005). Vanadium-dependent iodoperoxidases in Laminaria digitata, a novel biochemical function diverging from brown algal bromoperoxidases. J. Biol. Inorg. Chem. 10, 156–166. doi: 10.1007/s00775-005-0626-8
Crockford, S. J. (2009). Evolutionary roots of iodine and thyroid hormones in cell-cell signaling. Integr. Comp. Biol. 49, 155–166. doi: 10.1093/icb/icp053
Falgueras, J., Lara, A. J., Fernandez-Pozo, N., Canton, F. R., Perez-Trabado, G., and Claros, M. G. (2010). SeqTrim: a high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinformatics 11:38. doi: 10.1186/1471-2105-11-38
Falkowski, P. (2012). Ocean science: the power of plankton. Nature 483, S17–S20. doi: 10.1038/483S17a
Fatsini, E., Bautista, R., Manchado, M., and Duncan, N. J. (2016). Transcriptomic profiles of the upper olfactory rosette in cultured and wild Senegalese sole (Solea senegalensis) males. Comp. Biochem. Physiol. Part D Genomics Proteomics 20, 125–135. doi: 10.1016/j.cbd.2016.09.001
Fernández-Pozo, N., Canales, J., Guerrero-Fernandez, D., Villalobos, D. P., Diaz-Moreno, S. M., Bautista, R., et al. (2011). EuroPineDB: a high-coverage web database for maritime pine transcriptome. BMC Genomics 12:366. doi: 10.1186/1471-2164-12-366
Gribble, G. W. (2003). The diversity of naturally produced organohalogens. Chemosphere 52, 289–297. doi: 10.1016/S0045-6535(03)00207-8
Gribble, G. W. (2015). Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 13, 4044–4136. doi: 10.3390/md13074044
Guedes, A. C., and Malcata, F. X. (2012). “Nutritional value and uses of microalgae in aquaculture,” in Aquaculture, ed Z. Muchlisin (Rijeka: InTech), 59–78.
Gwon, H. J., Teruhiko, I., Shigeaki, H., and Baik, S. H. (2014). Identification of novel non-metal haloperoxidases from the marine metagenome. J. Microbiol. Biotechnol. 24, 835–842. doi: 10.4014/jmb.1310.10070
Hachero-Cruzado, I., Rodriguez-Rua, A., Roman-Padilla, J., Ponce, M., Fernandez-Diaz, C., and Manchado, M. (2014). Characterization of the genomic responses in early Senegalese sole larvae fed diets with different dietary triacylglycerol and total lipids levels. Comp. Biochem. Physiol. Part D Genomics Proteomics 12, 61–73. doi: 10.1016/j.cbd.2014.09.005
Heyland, A., and Moroz, L. L. (2005). Cross-kingdom hormonal signaling: an insight from thyroid hormone functions in marine larvae. J. Exp. Biol. 208, 4355–4361. doi: 10.1242/jeb.01877
Heyland, A., Price, D. A., Bodnarova-Buganova, M., and Moroz, L. L. (2006). Thyroid hormone metabolism and peroxidase function in two non-chordate animals. J. Exp. Zool. B Mol. Dev. Evol. 306, 551–566. doi: 10.1002/jez.b.21113
Hill, V., and Manley, S. (2009). Release of reactive bromine and iodine from diatoms and its possible role in halogen transfer in polar and tropical oceans. Limnol. Oceanogr. 54, 812–822. doi: 10.4319/lo.2009.54.3.0812
Holland, L. Z., Albalat, R., Azumi, K., Benito-Gutierrez, E., Blow, M. J., Bronner-Fraser, M., et al. (2008). The amphioxus genome illuminates vertebrate origins and cephalochordate biology. Genome Res. 18, 1100–1111. doi: 10.1101/gr.073676.107
Hughes, C., and Sun, S. (2016). Light and brominating activity in two species of marine diatom. Mar. Chem. 181, 1–9. doi: 10.1016/j.marchem.2016.02.003
Iwamoto, K., and Shiraiwa, Y. (2012). Characterization of intracellular iodine accumulation by iodine-tolerant microalgae. Proced. Environ. Sci. 15, 34–42. doi: 10.1016/j.proenv.2012.05.007
Janknegt, P. J., De Graaff, C. M., Van De Poll, W. H., Visser, R. J. W., Rijstenbil, J. W., and Buma, A. G. J. (2009). Short-term antioxidative responses of 15 microalgae exposed to excessive irradiance including ultraviolet radiation. Eur. J. Phycol. 44, 525. doi: 10.1080/09670260902943273
Klootwijk, W., Friesema, E. C., and Visser, T. J. (2011). A nonselenoprotein from amphioxus deiodinates triac but not T3: is triac the primordial bioactive thyroid hormone? Endocrinology 152, 3259–3267. doi: 10.1210/en.2010-1408
Küpper, F. C., Carpenter, L. J., Mcfiggans, G. B., Palmer, C. J., Waite, T. J., Boneberg, E. M., et al. (2008). Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proc. Natl. Acad. Sci. U.S.A. 105, 6954–6958. doi: 10.1073/pnas.0709959105
Küpper, F. C., Schweigert, N., Ar Gall, E., Legendre, J.-M., Vilter, H., and Kloareg, B. (1998). Iodine uptake in Laminariales involves extracellular, haloperoxidase-mediated oxidation of iodide. Planta 207, 163–171. doi: 10.1007/s004250050469
La Barre, S., Potin, P., Leblanc, C., and Delage, L. (2010). The halogenated metabolism of brown algae (Phaeophyta), its biological importance and its environmental significance. Mar. Drugs 8, 988–1010. doi: 10.3390/md8040988
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Laudet, V. (2011). The origins and evolution of vertebrate metamorphosis. Curr. Biol. 21, R726–R737. doi: 10.1016/j.cub.2011.07.030
Leblanc, C., Colin, C., Cosse, A., Delage, L., La Barre, S., Morin, P., et al. (2006). Iodine transfers in the coastal marine environment: the key role of brown algae and of their vanadium-dependent haloperoxidases. Biochimie 88, 1773–1785. doi: 10.1016/j.biochi.2006.09.001
Liu, W. H., Huang, Z. W., Li, P., Xia, J. F., and Chen, B. (2012). Formation of triacylglycerol in Nitzschia closterium f. minutissima under nitrogen limitation and possible physiological and biochemical mechanisms. J. Exp. Mar. Biol. Ecol. 418, 24–29. doi: 10.1016/j.jembe.2012.03.005
Lobanov, A. V., Fomenko, D. E., Zhang, Y., Sengupta, A., Hatfield, D. L., and Gladyshev, V. N. (2007). Evolutionary dynamics of eukaryotic selenoproteomes: large selenoproteomes may associate with aquatic life and small with terrestrial life. Genome Biol. 8:R198. doi: 10.1186/gb-2007-8-9-r198
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18
Manchado, M., Michan, C., and Pueyo, C. (2000). Hydrogen peroxide activates the SoxRS regulon in vivo. J. Bacteriol. 182, 6842–6844. doi: 10.1128/JB.182.23.6842-6844.2000
Moore, R. M., Webb, M., Tokarczyk, T., and Wever, R. (1996). Bromoperoxidase and iodoperoxidase enzymes and production of halogenated methanes in marine diatom cultures. J. Geophys. Res. 101, 20899–20908. doi: 10.1029/96JC01248
Muñoz-Mérida, A., Viguera, E., Claros, M. G., Trelles, O., and Perez-Pulido, A. J. (2014). Sma3s: a three-step modular annotator for large sequence datasets. DNA Res. 21, 341–353. doi: 10.1093/dnares/dsu001
Murphy, C. D., Moore, R. M., and White, R. L. (2000). Peroxidases from marine microalgae. J. Appl. Phycol. 12, 507–513. doi: 10.1023/A:1008154231462
Orozco, A., Valverde, R. C., Olvera, A., and Garcia, G. C. (2012). Iodothyronine deiodinases: a functional and evolutionary perspective. J. Endocrinol. 215, 207–219. doi: 10.1530/JOE-12-0258
Paris, M., Escriva, H., Schubert, M., Brunet, F., Brtko, J., Ciesielski, F., et al. (2008). Amphioxus postembryonic development reveals the homology of chordate metamorphosis. Current Biol. 18, 825–830. doi: 10.1016/j.cub.2008.04.078
Pevzner, P. A., Tang, H., and Waterman, M. S. (2001). An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci, U.S.A. 98, 9748–9753. doi: 10.1073/pnas.171285098
Phatarphekar, A., Buss, J. M., and Rokita, S. E. (2014). Iodotyrosine deiodinase: a unique flavoprotein present in organisms of diverse phyla. Mol. Biosyst. 10, 86–92. doi: 10.1039/C3MB70398C
Qiu, H., Price, D. C., Yang, E. C., Yoon, H. S., and Bhattacharya, D. (2015). Evidence of ancient genome reduction in red algae (Rhodophyta). J. Phycol. 51, 624–636. doi: 10.1111/jpy.12294
Qiu, H., Yoon, H. S., and Bhattacharya, D. (2013). Algal endosymbionts as vectors of horizontal gene transfer in photosynthetic eukaryotes. Front. Plant Sci. 4:366. doi: 10.3389/fpls.2013.00366
Rasdi, N. W., and Qin, J. G. (2015). Effect of N:P ratio on growth and chemical composition of Nannochloropsis oculata and Tisochrysis lutea. J. Appl. Phycol. 27, 2221–2230. doi: 10.1007/s10811-014-0495-z
Shrivastava, A., Tiwari, M., Sinha, R. A., Kumar, A., Balapure, A. K., Bajpai, V. K., et al. (2006). Molecular iodine induces caspase-independent apoptosis in human breast carcinoma cells involving the mitochondria-mediated pathway. J. Biol. Chem. 281, 19762–19771. doi: 10.1074/jbc.M600746200
Sigaud-Kutner, T. C. S., Neto, A. M. P., Pinto, E., and Colepicolo, P. (2005). Diel activities of antioxidant enzymes, photosynthetic pigments and malondialdehyde content in stationary-phase cells of Tetraselmis gracilis (Prasinophyceae). Aquat. Bot. 82, 239–249. doi: 10.1016/j.aquabot.2005.02.011
Sigaud-Kutner, T. C., Pinto, E., Okamoto, O. K., Latorre, L. R., and Colepicolo, P. (2002). Changes in superoxide dismutase activity and photosynthetic pigment content during growth of marine phytoplankters in batch-cultures. Physiol. Plant. 114, 566–571. doi: 10.1034/j.1399-3054.2002.1140409.x
Singh, S. P., Dhakshinamoorthy, R., Jaiswal, P., Schmidt, S., Thewes, S., and Baskar, R. (2014). The thyroxine inactivating gene, type III deiodinase, suppresses multiple signaling centers in Dictyostelium discoideum. Dev. Biol. 396, 256–268. doi: 10.1016/j.ydbio.2014.10.012
Surget-Groba, Y., and Montoya-Burgos, J. I. (2010). Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 20, 1432–1440. doi: 10.1101/gr.103846.109
Taylor, E., and Heyland, A. (2017). Evolution of thyroid hormone signaling in animals: non-genomic and genomic modes of action. Mol. Cell. Endocrinol. 459, 14–20. doi: 10.1016/j.mce.2017.05.019
van Bergeijk, S. A., Hernández Javier, L., Heyland, A., Manchado, M., and Cañavate, J. P. (2013). Uptake of iodide in the marine haptophyte Isochrysis sp. (T.ISO) driven by iodide oxidation. J. Phycol. 49, 640–647. doi: 10.1111/jpy.12073
van Bergeijk, S. A., Hernandez, L., Zubía, E., and Cañavate, J. P. (2016). Iodine balance, growth and biochemical composition of three marine microalgae cultured under various inorganic iodine concentrations. Mar. Biol. 163:107. doi: 10.1007/s00227-016-2884-0
Venturi, S. (2011). Evolutionary significance of iodine. Curr. Chem. Biol. 5, 155–162. doi: 10.2174/187231311796765012
Venturi, S., and Venturi, M. (1999). Iodide, thyroid and stomach carcinogenesis: evolutionary story of a primitive antioxidant? Eur. J. Endocrinol. 140, 371–372. doi: 10.1530/eje.0.1400371
Verhaeghe, E. F., Fraysse, A., Guerquin-Kern, J. L., Wu, T. D., Devés, G., Mioskowski, C., et al. (2008). Microchemical imaging of iodine distribution in the brown alga Laminaria digitata suggests a new mechanisms for its accumulation. J. Biol. Inorg. Chem. 13, 257–269. doi: 10.1007/s00775-007-0319-6
Wu, T., Shi, X., Zhou, Z., Wang, L., Wang, M., Wang, L., et al. (2012). An iodothyronine deiodinase from Chlamys farreri and its induced mRNA expression after LPS stimulation. Fish Shellfish Immunol. 33, 286–293. doi: 10.1016/j.fsi.2012.05.011
Yilancioglu, K., Cokol, M., Pastirmaci, I., Erman, B., and Cetiner, S. (2014). Oxidative stress is a mediator for increased lipid accumulation in a newly isolated Dunaliella salina strain. PLoS ONE 9:e91957. doi: 10.1371/journal.pone.0091957
Zámocký, M., Gasselhuber, B., Furtmüller, P. G., and Obinger, C. (2014). Turning points in the evolution of peroxidase-catalase superfamily: molecular phylogeny of hybrid heme peroxidases. Cell. Mol. Life Sci. 71, 4681–4696. doi: 10.1007/s00018-014-1643-y
Keywords: hydrogen peroxide, iodide, Tisochrysis lutea, peroxidase, deiodinase, gene expression
Citation: Hernández Javier L, Benzekri H, Gut M, Claros MG, van Bergeijk S, Cañavate JP and Manchado M (2018) Characterization of Iodine-Related Molecular Processes in the Marine Microalga Tisochrysis lutea (Haptophyta). Front. Mar. Sci. 5:134. doi: 10.3389/fmars.2018.00134
Received: 13 January 2018; Accepted: 04 April 2018;
Published: 24 April 2018.
Edited by:
Karla B. Heidelberg, University of Southern California, United StatesReviewed by:
Julian Blasco, Consejo Superior de Investigaciones Científicas (CSIC), SpainGiovanna Romano, Stazione Zoologica Anton Dohrn, Italy
Copyright © 2018 Hernández Javier, Benzekri, Gut, Claros, van Bergeijk, Cañavate and Manchado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Manchado, bWFudWVsLm1hbmNoYWRvQGp1bnRhZGVhbmRhbHVjaWEuZXM=