 Naraporn Somboonna
Naraporn Somboonna Alisa Wilantho
Alisa Wilantho Panan Rerngsamran
Panan Rerngsamran Sissades Tongsima
Sissades Tongsima- 1Department of Microbiology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
- 2Microbiome Research Unit for Probiotics in Food and Cosmetics, Chulalongkorn University, Bangkok, Thailand
- 3Genome Technology Research Unit, National Center for Genetic Engineering and Biotechnology, Pathum Thani, Thailand
Tha Wang coast in Sichang island, Chonburi province, Thailand, serves a major pier, cargo transportation and is populated with anthropogenic activities. Hence, monitoring of environmental status, in part by microbial diversity, is essential. Previously, the coastal marine microbiota of this area in a dry season (February 2011) had been reported, but the database for a wet season is none. As marine microbiota in several sites were reported fluctuate between dry and wet seasons, specifically in the 2011 that one of the largest flood in Thailand history even occurred from July 2011 to January 2012; the present study thereby first revealed the coastal Sichang water microbiota in the 2011 wet season (July 2011) using metagenomic derived 16S rRNA gene sequencing. Wet and dry seasons showed differences in prokaryotic community richness and evenness: community similarity indices by Jaccard, Lennon, and Yue and Clayton were 2.06–58.28%. This resulted in differences in metabolic potentials, for instances the lower metabolisms for energy and nutrient cycling but the greater in pathogenesis in wet season. Our data were also compared against the flooding water microbiota nearby to Chonburi province to observe any possible correlation; and the coastal Sichang was found distinct given diverse sources of flooding water and local contamination might affect the diversity. In conclusion, this wet-season microbiota supported the better knowledge on the seasonal microbiota of coastal Sichang island.
Introduction
Sichang island, Chonburi province, Thailand, situates around an equator, within 23.5 degree north and 23.5 degree south, of the upper Gulf of Thailand, and comprises two coasts. On an east side of Sichang coast, named Tha Wang (TW), is major local and international pier and cargo transportation, whereas on the west side coast named Tham Phang (TP) is quiet with neither pier and cargo transportation nor many human activities. TW also has rather close bay geography surrounding by the mainland of Thailand and two islands named Khaam Yai and Prong islands; and thus minimal water circulation compared with TP. More organic and non-organic wastes (e.g., glass bottles, plastics, biodegradable garbage, metals and hazardous materials) were reported at TW than TP (Piyatiratitivorakul and Rungsupa, personal communications and unpublished data; Chantanawisuti et al., 1996). Associated with these different coastal characteristics, Somboonna et al. (2012) reported dissimilar coastal marine microbial population structures between TW and TP in a dry season of year 2011: TW contain many harsh-living prokaryotic species with possible biotechnology application, meanwhile TP contain natural nutrient cyclers and some with possible pharmaceutical application.
In 2011, Thailand witnessed one largest floods in decades, covering over half of Thailand region (∼60,000 square kilometers), from July 2011 to January 2012. Thailand and other Southeast Asia countries, such as Philippines, Indonesia and Cambodia, generally occur flooding in a wet season due to tropical monsoon (Mhuantong et al., 2015). High water level in a wet season lowers concentration and availability of nutrients for marine microorganisms and organisms. Global ocean microbiome researches reported temperature and seasons as factors for changes of microbial structures, in open oceans and coral reefs (Gilbert et al., 2012; Somboonna et al., 2014; Campbell et al., 2015; Bulan et al., 2018); yet seasonal dynamics of microbial structures in coastal marine sites of the Gulf of Thailand remain limited. This study thereby described the Sichang coastal water microbiota in one period (July 2011) in a wet season of TW and TP (abbreviated TW-wet and TP-wet), and compared with our previous reports of dry season of TW and TP (abbreviated TW and TP) of the same corresponding year and analysis methods, metagenomics derived 16S rRNA gene sequencing (Somboonna et al., 2012).
Bacteria play crucial roles in marine food supplies, as initial producers (e.g., photosynthesis) and final recyclers (e.g., conversion of complex or toxic compounds to simple organic and inorganic substrates) (Moran et al., 2015). The latter is critical for coastal water where anthropogenic pollution is abundant. Our first culture-independent databases benefit marine biologists and environmentalists a better knowledge of microbial ecosystem and metabolic potentials, of wet season.
Materials and Methods
Sample Collection
A minimum of triplicate water samples of TW-wet and TP-wet, at 5–100 cm depth, were collected into sterile glass containers on 17 July 2011, around 12:00–13:00 h. Measurements included GPS, water color, temperature, pH, salinity, and conductivity were recorded (Supplementary Table S1). The geographical location was consistent with Somboonna et al. (2012). The two coastal areas are not vast. All samples were stored at 4°C, and processed immediately upon laboratory arrival within 14 days.
Metagenomic Extraction
Each water sample was poured through four-layered sterile cheesecloths to remove ≥30 μm debris and large-size organisms, followed by a sterile 0.45-micron filter (Merck Millipore, MA, United States) to confine microorganisms of ≥0.45 μm (bacteria and archaea) onto the filter membrane. Then total nucleic acids were isolated using Metagenomic DNA Isolation Kit for Water (Epicentre, WI, United States), following the manufacturer’s instructions, and isolated metagenomes should be randomly sheared and appear around 40 kb in size (Begum and Murray, 2008). The metagenomes were analyzed for metagenomic DNA yield and quality by agarose gel electrophoresis with a fosmid control of around 40 kb (Epicentre) and A260/A280 nanodrop spectrophotometry.
Pyrotagged 16S rRNA Gene Sequencing
Universal prokaryote 338F (forward) and 803R (reverse) primers were used to amplify a 466-nucleotide sequence covering V3–V4 regions of 16S rRNA gene (Baker et al., 2003; Humblot and Guyoet, 2009). The forward and reverse pyrotagged-16S rDNA primers for TW-wet were 5′-TCTCTGTGACTCCTACGGGAGGCAGCAG-3′ and 5′-TCTCT GTGCTACCAGGGTATCTAATC-3′, and TP-wet were 5′-TC TACTCGACTCCTACGGGAGGCAGCAG-3′ and 5′-TCTAC TCGCTACCAGGGTATCTAATC-3′, where italic sequences represent the tagged sequences (Meyer et al., 2008). For each sample, a 50-μl PCR comprised 1× EmeraldAmp®GT PCR Master Mix (TaKaRa, Shiga, Japan), 0.3 μM of each primer, and 100 ng of the metagenomic DNA. PCR conditions were 95°C for 4 min, and 30 cycles of 94°C for 45 s, 50°C for 55 s, and 72°C for 1 min 30 s, followed by 72°C for 10 min. To minimize PCR bias, a minimum of two independent reactions were performed. The PCR products were purified using PureLink®Quick Gel Extraction Kit (Invitrogen, NY, United States), and the products belonging the same sample site were pooled for 175 ng each for pyrotagged TW-wet and TP-wet sequencing. Pyrosequencing was performed on an eight-lane Picotiter plate, using the 454-Life Sciences GS FLX Genome Sequencer system (Roche, Branford, CT, United States), following the manufacturer’s protocols. The datasets were deposited in NCBI Sequence Read Archive (SRA) under an accession number SRP095760.
Bacterial Diversity Analyses
Sequences were categorized TW-wet and TP-wet based on their pyrotag-sequences, then the pyrotagged and primer sequences were removed, and the sequences that do not pass the recommended quality parameters by Mothur (Schloss et al., 2009), including those of less than 100 nucleotides in length and chimera sequences, were removed. The sequences were annotated, visualized and computed for taxonomic compositions using VITCOMIC (Visualization tool for Taxonomic Compositions of Microbial Community) (Mori et al., 2010; Somboonna et al., 2012). BLASTN (Altschul et al., 1997) was performed with E-value ≤1E-08 and other default parameters, against 16S rDNA sequences from NCBI non-redundant (Sayers et al., 2010), RDP (Maidak et al., 2001), and Greengenes (McDonald et al., 2011) databases. Percent relative abundance for each phylum was computed from the frequency of reads in the phylum divided by the total number of identified reads. For community diversity estimates (Jaccard, Lennon, and Yue and Clayton (Thetayc) indices), phylogenetic clustering, and non-metric multidimensional scaling (NMDS) were computed using Mothur with default parameters (Schloss et al., 2009). These 16S rDNA data were compared with those of TW and TP of dry season of the same year (Somboonna et al., 2012), and of Thai flood water areas (Mhuantong et al., 2015). The comparing flood areas locate next and nearby provinces to Chonburi (Bangkok, Nonthaburi, Pathum Thani, and Nakhon Pathom provinces), and the samples were collected 5–150 cm depth in December 2011.
Metabolic Potential Analysis
PICRUSt (Langille et al., 2013) was used to predict metabolic potentials of microbiota into KEGG (Kyoto Encyclopedia of genes and genomes) pathways from the 16S rDNA data. Statistics and visualization were performed at KEGG pathway level 2 using STAMP version 2.1.3 (Parks et al., 2014). Statistic comparison was based on Fisher’s exact test and Benjamini-Hochberg False Discovery Rate (FDR) that yields q-value of <0.05 as significant.
Results
Physical Properties and Metagenomic DNA
Temperature of coastal water of TW-wet and TP-wet were roughly equal as the two sites are only 0.010° latitude and 0.012° longitude apart (Supplementary Table S1). On-site visual observation for water color demonstrated TP-wet water was clearer than TW-wet. The water physical properties exhibited the slightly greater salinity, acidity (lower pH) and conductivity in TW-wet than TP-wet (Supplementary Table S1), partly consistent with previous reports (Somboonna et al., 2012) and the Sichang Marine Science Research and Training Station (SMaRT) monthly records (Rungsupa, personal communication and unpublished data). Comparing between the wet and dry seasons, dissimilarity was denoted, including the higher sea temperature and alkalinity pH while lower salinity and conductivity in the wet season (Supplementary Table S1: TW-wet vs. TW, TP-wet vs. TP). These different water physical properties and monsoon effect in wet season suggested microbial diversity between wet and dry seasons.
The metagenomic DNA appeared in proper size range (>12–40 kb) as described by manufacturer and included a size evaluation via a 40 kb fosmid (Begum and Murray, 2008). The metagenomic DNA yield per ml of seawater of TW-wet was higher than that of TP-wet (Supplementary Figure S1); nonetheless both were averagely half of that reported in a dry season: ∼0.26 ng/ml in wet season vs. ∼0.45 ng/ml in dry season (Somboonna et al., 2012).
Diversity of Microbial Communities and Functional Potentials Between Wet and Dry Seasons
The 16S rDNA profiles demonstrated a wet season had increase in bacterial members belonging Proteobacteria (TW-wet 70.71% vs. TW 56.84%, TP-wet 93.64% vs. TP 50.58%), but Actinobacteria (TW-wet 5.01% vs. TW 23.65%, TP-wet 2.34% vs. TP 41.16%), for both Tha Wang and Tham Phang; and specially for Tha Wang Bacteroidetes also became highly pronounced (TW-wet 24.27% vs. TW 13.68%, TP-wet 4.01% vs. TP 6.01%). Many species of Bacteroidetes are related to municipal wastes, such as feces; hence the finding might reflect Tha Wang’s effect of populated residents and close bay geography.
For diversity of bacteria, the far fewer bacterial diversity was in the wet season (Figures 1A,B). In TW-wet, species such as Bacteroidetes Gramella forsetii (14.51%) remained relatively abundant. This specie has been reported one major marine microbial adaptation, because it can degrade polymeric organic matter and plays a key role in the decomposition of carbon complexes for carbon cycling food chains in several marine ecosystems (Bauer et al., 2006; Langille et al., 2013; Kabisch et al., 2014). Another reserved species in TW-wet are of Proteobacteria in genus Ruegeria, such as R. pomeroyi (1.34%) (Figure 1A). This specie was abundant in coastal elsewhere with versatile utilization ability and roles in cycling of sulfur, carbon, nitrogen and many other compounds (i.e., sulfonate, dimethylsulphoniopropionate, and trimethylamine N-oxide) (Reisch et al., 2013; Rivers et al., 2014; Li et al., 2015). This finding of bacteria with complex organic users and saprophytes were also reported in the 16S rRNA gene sequencing survey in other high-water level areas during the 2011 Thailand flood crisis (Mhuantong et al., 2015). Other abundant species in TW-wet included Pseudoalteromonas haloplanktis (5.54%), Roseobacter denitrificans (5.54%), Marinomonas sp. (5.01%), Aeromonas salmonicida (5.28%), Pectobacterium atrosepticum (5.01%), Pseudomonas aeruginosa (2.11%), and Shigella dysenteriae (0.79%) (Figure 1A). These first prior three species, albeit at the lower percent prevalence than in the dry season, function involve cycling of organic nutrients (Medigue et al., 2005; Swingley et al., 2007; Bai et al., 2014), while the latter four species were reported pathogens of aquatic animals and plants (i.e., starch) (Panda et al., 2016), and thus the KEGG pathways of diseases were denoted in Figure 1C.
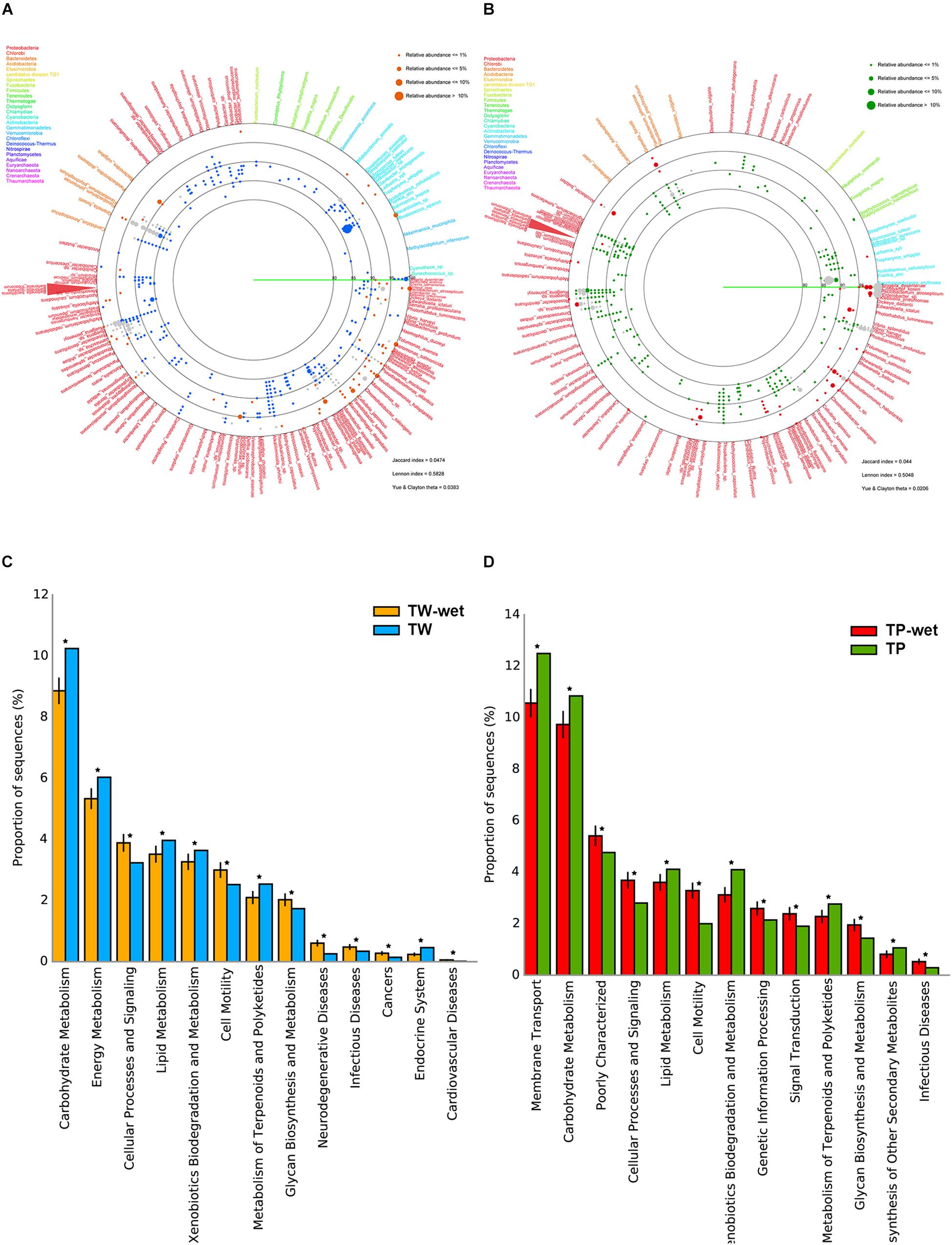
Figure 1. Comparative bacterial diversity (A,B) and potential metabolisms (C,D) between TW-wet against TW, and TP-wet against TP, respectively. In (A,B), each identified read as represented by orange (TW-wet), blue (TW), red (TP-wet), or green (TP) dot, is annotated to genus and species level along the percent sequence similarity between the read and the annotated sequences (80–100%). Dot size refers to percent relative abundance because multiple reads could be annotated the same species, and only ≥0.05% relative abundance was displayed to fit limited diagram space. Gray dot represents conserved species. Sequence is placed on a circular diagram following its relative genetic distances among one another. Different type font color represents species belonging to different phylum. In (C,D), *represents q-value <0.05. See section “Materials and Methods” for details.
In Tham Phang, prevalent species in the wet season were similar to those in TW-wet, including Bacteroidetes Gramella forsetii (TP-wet 2.68%) and Proteobacteria Ruegeria spp. (1.34%). Unlike TP, TP-wet had diminished members of Actinobacteria, whereas microbial pathogens such as Shigella dysenteriae (25.75%), Pectobacterium atrosepticum (16.05%), and Vibrio vulnificus (8.68%) became abundant (Figure 1B). Nonetheless, as the relative amount of microbial metagenomic DNA of TP-wet was much lower than TW-wet (Supplementary Figure S1), a percentage of pathogen in TP-wet (Figure 1D, infectious diseases) might remain minimal in term of the abundance in nature compared with TW-wet. Besides metabolisms of diseases that were high in wet season, other relatively greater metabolisms in TW-wet and TP-wet were cellular processes and signaling, cell motility, and glycan biosynthesis and metabolism (Figures 1C,D).
The diverse differences in the communities between the seasons arouse the low community similarity coefficients by all community similarity formulas, i.e., Jaccard index of 0.0474 for Tha Wang and 0.044 for Tham Phang as reported in the lower right of Figures 1A,B. The closer the community similarity coefficient to 0.000 indicated no community similarity.
Similarity of Microbial Communities With Other Thailand Flood Sites
The TW-wet and TP-wet microbiota were compared with those of reported flood areas in Thailand (Mhuantong et al., 2015). Tha Wang and Tham Phang microbial communities albeit seasons were clustered separated from flood water of different sites albeit next and nearby provinces (Bangkok, Nonthaburi, Pathum Thani, and Nakhon Pathom) (Figure 2). Supportively, in addition to differences by sources of flood water and local contamination, the physical properties between ours and flood water samples were found different by water conductivity and water temperature.
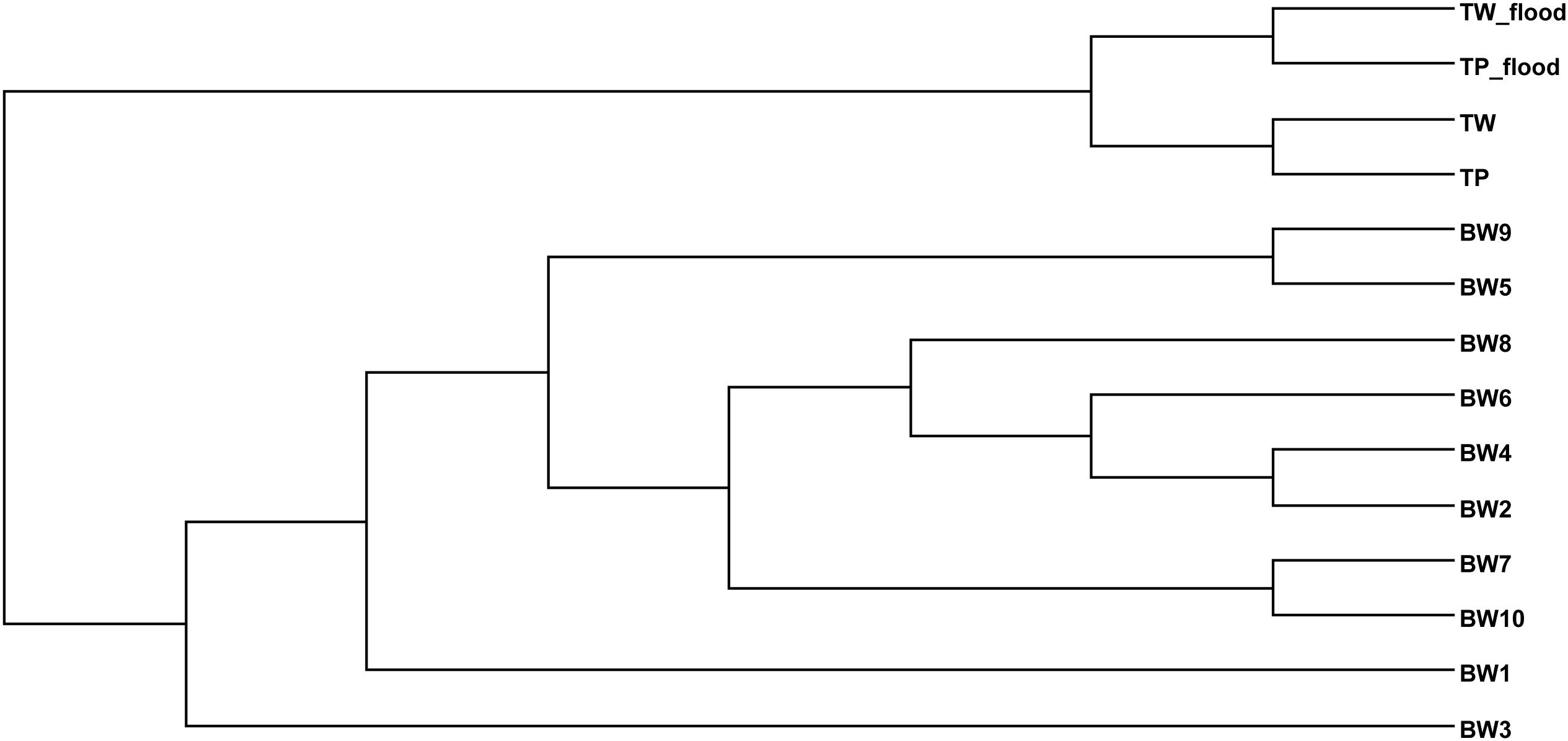
Figure 2. Phylogenetic relationship of bacterial diversity in wet and dry TW and TP, and among diverse flood affected areas of Thailand.
Discussion
This study presented the first study to reveal coastal marine microbiome of the upper Gulf of Thailand, Sichang island, in wet season using metagenomics combined 16S rRNA gene sequencing. This year also signified the 2011 Thailand’s high-water level in decades. As climate change contributes not only sea-level rise from land-to-ocean flux of freshwater and affects global hydrological cycle, but also release of dissolved organic carbon as a result of glacier mass loss (Hood et al., 2015), together these affected diversity of bacteria and their metabolisms (e.g., carbohydrate metabolism and energy metabolism). Comparison between seasons, and with other flood sites from diverse sources of flooding water and local contamination, indicated uniqueness in Tha Wang and Tham Phang microbial ecosystems. The finding of site dependence was consistent with many previous marine microbiome databases. Albeit minimal, a tip of an iceberg of bacteria with infectious diseases metabolism in the wet season suggested an awareness and if flood occurs, a source of nutrients in food webs, and contaminants, in residential and agricultural areas in part support growth of these infectious disease bacteria. Mhuantong et al. (2015) reported that flood crisis disturbed microbial population with overgrowth of pathogens.
Data Availability
The datasets generated for this study can be found in NCBI Sequence Read Archive (SRA), SRP0957.
Author Contributions
NS conceived the study, carried out the molecular biology experiments, participated in and coordinated the data analysis, and drafted the manuscript. AW performed the data analysis. PR collected the samples. ST advised on the data analysis. All authors read and approved the final manuscript.
Funding
This research was supported Research Funds from the Faculty of Science, Chulalongkorn University, under the Project for Interdisciplinary Research, which Targeted to Collaborate with the Leading World Class University (to NS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledged S. Thaniyavarn and K. Kidmoa for experimental support, the National Center for Genetic Engineering and Biotechnology (BIOTEC) and the National Science and Technology Development Agency (NSTDA) for allowing us to compute the data on the server.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00308/full#supplementary-material
FIGURE S1 | Agarose-electrophoretic gels showing (A) TW-wet and (B) TP-wet. Left lane is 80 ng of a 40 kb fosmid control, and right lane is a 1 kb Plus DNA Ladder (Invitrogen) where the top band is at 12 kb.
TABLE S1 | Water sampling sites (GPS) and physical properties.
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Bai, X., Lai, Q., Dong, C., Li, F., and Shao, Z. (2014). Marinomonas profundimaris sp. nov., isolated from deep-sea sediment sample of the Arctic Ocean. Antonie Van Leeuwenhoek 106, 449–455. doi: 10.1007/s10482-014-0213-5
Baker, G. C., Smith, J. J., and Cowan, D. A. (2003). Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 55, 541–555.
Bauer, M., Kube, M., Teeling, H., Richter, M., Lombardot, T., Allers, E., et al. (2006). Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 8, 2201–2213.
Begum, D., and Murray, J. (2008). Direct isolation of metagenomic DNA from environmental water samples. EPICENTRE Forum 15, 7–8.
Bulan, D. E., Wilantho, A., Krainara, P., Viyakarn, V., Chavanich, S., and Somboonna, N. (2018). Spatial and seasonal variability of reef bacterial communities in the upper Gulf of Thailand. Front. Mar. Sci. 5:441. doi: 10.3389/fmars.2018.00441
Campbell, A. M., Fleisher, J., Sinigalliano, C., White, J. R., and Lopez, J. V. (2015). Dynamics of marine bacterial community diversity of the coastal waters of the reefs, inlets, and wastewater outfalls of southeast Florida. Microbiologyopen 4, 390–408. doi: 10.1002/mbo3.245
Chantanawisuti, N., Piyatiratitivorakul, S., and Menasveta, P. (1996). Types and annual distribution of garbages around the shore of Sichang Islands. Thai. J. Aquat. Sci. 2, 108–116.
Gilbert, J. A., Steele, J. A., Caporaso, J. G., Steinbrück, L., Reeder, J., Temperton, B., et al. (2012). Defining seasonal marine microbial community dynamics. ISME J. 6, 298–308. doi: 10.1038/ismej.2011.107
Hood, E., Battin, T. J., Fellman, J., O’Neel, S., and Spencer, R. G. M. (2015). Storage and release of organic carbon from glaciers and ice sheets. Nat. Geosci. 8, 91–96.
Humblot, C., and Guyoet, J.-P. (2009). Pyrosequencing of tagged 16S rRNA gene amplicons for rapid diciphering of the microbiomes of fermented foods such as pearl millet slurries. Appl. Environ. Microbiol. 75, 4354–4361. doi: 10.1128/AEM.00451-09
Kabisch, A., Otto, A., König, S., Becher, D., Albrecht, D., Schüler, M., et al. (2014). Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J. 8, 1492–1502. doi: 10.1038/ismej.2014.4
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, C. Y., Chen, X. L., Shao, X., Wei, T. D., Wang, P., Xie, B. B., et al. (2015). Mechanistic insight into trimethylamine N-oxide recognition by the marine bacterium Ruegeria pomeroyi DSS-3. J. Bacterol. 197, 3378–3387. doi: 10.1128/JB.00542-15
Maidak, B. L., Cole, J. R., Liburn, T. G., Parker, C. T. Jr., Saxman, P. R., Farris, R. J., et al. (2001). The RDP-II (ribosomal database project). Nucleic Acids Res. 29, 173–174.
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2011). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Medigue, C., Krin, E., Pascal, G., Barbe, V., Bernsel, A., Bertin, P. N., et al. (2005). Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 15, 1325–1335.
Meyer, M., Stenzel, U., and Hofreiter, M. (2008). Parallel tagged sequencing on the 454 platform. Nat. Protoc. 3, 267–278. doi: 10.1038/nprot.2007.520
Mhuantong, W., Wongwilaiwalin, S., Laothanacharoen, T., Eurwilaichitr, L., Tangphatsornruang, S., Boonchayaanant, B., et al. (2015). Survey of microbial diversity in flood areas during Thailand 2011 flood crisis using high-throughput tagged amplicon pyrosequencing. PLoS One 10:e0128043. doi: 10.1371/journal.pone.0128043
Moran, M. A., Kujawinski, E. B., Stubbins, A., Fatland, R., Aluwihare, L. I., Buchan, A., et al. (2015). Deciphering ocean carbon in a changing world. Proc. Natl. Acad. Sci. U.S.A. 113, 3143–3151. doi: 10.1073/pnas.1514645113
Mori, H., Maruyama, F., and Kurokawa, K. (2010). VITCOMIC: visualization tool for taxonomic compositions of microbial communities based on 16S rRNA gene sequences. BMC Bioinformatics 11:332. doi: 10.1186/1471-2015-11-332
Panda, P., Vanga, B. R., Lu, A., Fiers, M., Fineran, P. C., Butler, R., et al. (2016). Pectobacterium atrosepticum and Pectobacterium carotovorum harbor distinct, independently acquired integrative and conjugative elements encoding coronafacic acid that enhance virulence on potato stems. Front. Microbiol. 7:397. doi: 10.3389/fmicb.2016.00397
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Reisch, C. R., Crabb, W. M., Gifford, S. M., Teng, Q., Stoudemayer, M. J., Moran, M. A., et al. (2013). Metabolism of dimethylsulphoniopropionate by Ruegeria pomeroyi DSS-3. Mol. Microbiol. 89, 774–791. doi: 10.1111/mmi.12314
Rivers, A. R., Smith, C. B., and Moran, M. A. (2014). An update genome annotation for the model marine bacterium Ruegeria pomeroyi. Stand. Genom. Sci. 9:11. doi: 10.1186/1944-3277-9-11
Sayers, E. W., Barrett, T., Benson, D. A., Bolton, E., Bryant, S. H., Canese, K., et al. (2010). Database resources of the national center for biotechnology information. Nucleic Acids Res. 38, D5–D16. doi: 10.1093/nar/gkp967
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Somboonna, N., Assawamakin, A., Wilantho, A., Tangphatsornruang, S., and Tongsima, S. (2012). Metagenomic profiles of free-living archaea, bacteria and small eukaryotes in coastal areas of Sichang island, Thailand. BMC Genomics 13:S29. doi: 10.1186/1471-2164-13-S7-S29
Somboonna, N., Wilantho, A., Assawamakin, A., Monanunsap, S., Sangsrakru, D., Tangphatsornruant, S., et al. (2014). Structural and functional diversity of free-living microorganisms in reef surface, Kra island, Thailand. BMC Genomics 15:607. doi: 10.1186/1471-2164-15-607
Keywords: microbiome, biodiversity, prokaryote, 16S ribosomal RNA, Sichang, upper Gulf of Thailand
Citation: Somboonna N, Wilantho A, Rerngsamran P and Tongsima S (2019) Marine Bacterial Diversity in Coastal Sichang Island, the Upper Gulf of Thailand, in 2011 Wet Season. Front. Mar. Sci. 6:308. doi: 10.3389/fmars.2019.00308
Received: 26 February 2019; Accepted: 24 May 2019;
Published: 11 June 2019.
Edited by:
Raquel Vaquer-Sunyer, Instituto Mediterráneo de Estudios Avanzados (IMEDEA), SpainReviewed by:
Raghab Ray, The University of Tokyo, JapanRaul Martell-Dubois, National Commission for the Knowledge and Use of Biodiversity (CONABIO), Mexico
Copyright © 2019 Somboonna, Wilantho, Rerngsamran and Tongsima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naraporn Somboonna, TmFyYXBvcm4uU0BjaHVsYS5hYy50aA==