 Mary Beth Johnstone1*†
Mary Beth Johnstone1*† A. P. Wheeler1
A. P. Wheeler1 Elizabeth P. Falwell1†
Elizabeth P. Falwell1† Meg E. Staton2
Meg E. Staton2 Christopher A. Saski3
Christopher A. Saski3 Andrew S. Mount1*
Andrew S. Mount1*- 1Department of Biological Sciences, Clemson University, Clemson, SC, United States
- 2Department of Entomology and Plant Pathology, The University of Tennessee, Knoxville, Knoxville, TN, United States
- 3Department of Plant and Environmental Sciences, Clemson University, Clemson, SC, United States
The proteins derived from the foliated shell layer of the oyster, Crassostrea virginica, are unusually acidic and highly phosphorylated. Here we report the identification of a gene encoding a member of this class of phosphoproteins that we collectively refer to as folian. Using an in silico approach, a virtual probe was constructed from an N-terminal sequence (DEADAGD) determined for a 48 kDa folian phosphoprotein and used to screen an oyster EST databank. A sequence that matched the N-terminus of the 48 kDa protein was found and used to identify the full-length gene from a C. virginica BAC library. The molecular weight of the deduced gene product is 32 kDa and was named folian-cv1. Genomic Southern analysis revealed two variants of the gene. The mature protein is composed of 43.3% Asp, 32.6% Ser, and 9.1% Glu with 37.5% of the amino acids of the protein potentially phosphorylated. The primary sequence of folian-cv1 is organized in blocks, with a short relatively hydrophobic block at the N-terminus and with the remainder containing low complexity regions largely dominated by aspartic acid and serine. Overall, the protein is predicted to be highly disordered. PCR and sequence analyses identified folian-cv1 expression in the mantle and hemocytes. Immuno-histochemical staining of mantle tissue reveals that cells of the shell-facing epithelium and in the periostracal groove secrete a continuous layer of folian-positive material and that folian-positive hemocytes move through the mantle epithelium. The function in shell formation of folian proteins including folian-cv1 is not known. However, based on the complexity of this class of proteins and the two methods of their delivery to the region of shell formation, it is possible they are involved in diverse ways in this process.
Introduction
Molluscan shell is composed of layers of nano- to micro-scaled calcium carbonate crystals embedded in an organic extracellular matrix. This composite structure is responsible for the exceptional toughness of the shell to fracture (Currey, 1999). Broadly speaking the matrix is composed of a variety of macromolecules, including predominately protein, as well as carbohydrates, such as chitin. Among the former are a class of uniquely acidic (anionic at physiological pH) proteins (Weiner and Dove, 2003; Marin et al., 2008). Beyond their role in enhancing the mechanical properties of shell, it has been proposed often that these proteins play a role in controlling the nucleation as well as the microstructure and mineralogy of the mineral phase (Lowenstam, 1981; Addadi and Weiner, 1985, 1989, 2014; Mann, 1988; Wheeler and Sikes, 1989). As evidence of crystal growth control, the microstructures of shell layers are diverse and differ from inorganically grown crystals but generally are conserved within closely related taxonomic groups (Carter, 1980; Carter and Clark, 1985; Lowenstam and Weiner, 1989). For example, the principal shell layer of the Eastern oyster, Crassostrea virginica, is foliated calcite, which is generally prominent among true oysters (Ostreidae) as well as related groups normally included in the order Ostreoida, such as scallops (Checa et al., 2007; Esteban-Delgado et al., 2008).
Most of the studies directed at understanding matrix function have been performed on isolated preparations in vitro or through traditional microscopial techniques. However, more recent studies of matrix gene expression in mineral forming tissues (Zhang et al., 2012; Hüning et al., 2016; Ivanina et al., 2017; Jackson et al., 2017), as well as those using cryo-microscopic techniques (Nudelman et al., 2008) and knockdown studies (Suzuki et al., 2009), among others, show promise in refining our understanding of matrix function. Despite this recent progress, clarity as to the function of individual matrix proteins is still in the formative stages of development.
For some time, several of the authors have analyzed the function, biochemistry and cytology of the extracellular matrix from the inner foliated shell layer of C. virginica (e.g., Wheeler et al., 1987, 1988, 1991; Rusenko et al., 1991; Wheeler, 1992b; Sikes et al., 1998; Mount, 1999; Myers et al., 2007; Johnstone et al., 2008, 2015). These studies showed that the oyster foliated matrix is dominated by highly phosphorylated proteins which are also enriched in aspartic acid, a composition that makes them highly acidic. The oyster phosphoproteins are enigmatic in that extracts from dissolved shell yield soluble forms with a wide range of molecular weight as well as gel-forming insoluble forms, all having similar amino acid compositions. We refer to this family of proteins collectively as folian. Some of these earlier studies also revealed the capacity of the folian proteins to bind to crystals and thereby inhibit and regulate crystal growth in vitro and provided evidence for crystal-matrix interactions in situ. Based on oyster protein structure and activity, synthetic proteins have been produced for use as anti-scalants, dispersants, superabsorbents and many other possible applications (e.g., Wheeler and Koskan, 1993 ; Sikes and Wierzbicki, 1995). Outside of C. virginica, there appears to be a correlation between the foliated microstructure and the presence of highly phosphorylated matrix proteins (Borbas et al., 1991; Sarashina and Endo, 2001; Samata et al., 2008). However, no causal relationship between this highly phosphorylated matrix and the formation of the foliated microstructure has been established. One of the limitations to understanding oyster matrix function is that previous studies were performed using preparations of varying degrees of heterogeneity. In fact, to date no individual folian protein from C. virginica has been fully characterized for functional assessment. Accordingly, in this study, using an in silico approach, we report the complete sequence of one gene from this class which we have named folian-cv1.
Traditionally, it has been assumed that most or all components necessary for mineralization are secreted by the mantle into the extrapallial space (Lowenstam and Weiner, 1989; Simkiss and Wilbur, 1989; Wheeler, 1992a). In fact, immuno-cytochemical studies have demonstrated secretion of matrix proteins by the mantle of C. virginica (Kawaguchi and Watabe, 1993; Myers et al., 2007; Johnstone et al., 2008) and their inclusion in mineralized layers during shell formation (Kawaguchi and Watabe, 1993). A special class of cells in the outer mantle epithelium has been identified as the likely candidate for secretion of folian proteins (Myers et al., 2007). More recently, hemocytes have also been implicated in shell formation or regeneration (Mount et al., 2004; Kádár, 2008; Li et al., 2016; Ivanina et al., 2017; Huang et al., 2018). In addition, they have been shown to form mineral ex vivo on surfaces, a process which has potential anti-fouling, protective coating and cellular adhesive applications (Mount et al., 2016). In the oyster, hemocytes are immuno-positive for folian proteins (Johnstone et al., 2008) and a class of these cells which contain calcium carbonate crystals has been demonstrated in association with regenerating shell (Mount et al., 2004) or shell formed on implants (Johnstone et al., 2015). In this study, immuno-histochemical staining of mantle tissue confirms the secretion of folian from mantle cells and the movement of folian-positive hemocytes through the mantle epithelium toward the sites of mineralization. The expression of folian-cv1 by mantle and hemocytes was confirmed by PCR. It therefore can be deduced that folian-cv1 is directly active in shell formation. However, because folian proteins are diverse and produced not only by both mantle and hemocytes but other tissues in the oyster as well (Johnstone et al., 2008), it is possible that folian, and by extension folian-cv1, may have multiple and, in some cases, indirect roles in mineralization, such serving as a part of the scaffolding for cell attachment (Chan et al., 2018), and possibly function in other processes as well.
Materials and Methods
Shell Protein Extraction and Purification
Shells from live specimens of the Eastern oyster, Crassostrea virginica, were collected from Sixty Bass Creek of North Inlet estuary, Georgetown, SC, United States. The shell proteins were extracted following protocols described in Wheeler et al. (1988) and Johnstone et al. (2008). Specifically, the shells were scrubbed under tap water and the outer shell layers, including the periostracum and the underlying prismatic layer, were removed with a sanding tool. Shell pieces composed of foliated mineral, free of myostracum and conchiolin patches, were selected and ground into powder. Protein was extracted from the CaCO3 phase by dissolving 25 g of the ground foliated shell in 750 ml of 17% ethylenediamine tetraacetic acid (EDTA), pH 8 at 4°C until the mineral was fully dissolved (about 48 h). The resulting shell extracellular matrix suspension was centrifuged at 27,000 × g for 30 min to pellet the insoluble matrix. The supernatant containing the soluble matrix was decanted, concentrated to approximately 50 ml, then dialyzed against 1 L of 10 mM NaCl using a Millipore Minitan tangential flow filtration apparatus with a molecular weight exclusion limit of 10 kDa. The resultant dialysate was further dialyzed against distilled water and lyophilized. Soluble matrix stock solutions were made by reconstituting the protein in distilled water to a concentration of 5 mg ml−1 based on total weight of the dried material. The pellet containing the insoluble matrix portion of the shell extract was reconstituted with 2 ml of 8 M urea and agitated end-over-end overnight at 4°C. The urea solubilized matrix suspension was centrifuged at 27,000 × g for 30 min to pellet the residual insoluble matrix. The supernatant containing the urea soluble matrix extract was dialyzed against distilled water and the dialysate concentrated using Microcon concentrators (Millipore).
Soluble matrix proteins were purified electrophoretically using protocols outlined in Johnstone (2008) by applying 2 mg of dry weight extract to 12% Tris-glycine sodium dodecyl sulfate polyacrylamide (SDS-PAGE) preparative gels. Protein bands were visualized by staining with 0.3 M CuCl2, and bands of interest were excised from the gel, de-stained for approximately 30 min in three washes of 50 mM EDTA, pH 8 and eluted using an electro-elution module (Bio-Rad) according to manufacturer’s protocols. Following electro-elution, protein was dialyzed against distilled water using Microcon concentrators or dialysis cassettes (Pierce), both having a MW cut-off of 10–12 kDa. Purified protein was lyophilized and stored at −20°C. Wherever protein quantities are reported they were determined using the BCA (Pierce) assay according to the manufacturer’s protocol.
Antibody Production
Antibody probes were prepared using two strategies. For Western analyses comparing the reactivity of soluble matrix proteins to antibodies made to 48 and 55 kDa proteins, 100 μg each of preparative SDS-PAGE-purified 48 and 55 kDa protein was eluted from gels, reconstituted in 200 μl of Tris buffered saline, pH 7.5 (TBS) and emulsified with an equal volume of complete Freud’s adjuvant. The emulsion for each eluted band was injected subcutaneously into two New Zealand White rabbits. After 3 weeks, the rabbits were boosted with an additional 100 μg of protein in the same manner. Sera containing anti-48 kDa or anti-55 kDa antibodies were harvested 6 weeks after the boosts and were stored in 1 ml aliquots at −20°C.
For immuno-histochemical analyses, an affinity purified antibody preparation made against the 48 kDa protein was produced by applying anti-sera generated against whole soluble matrix to a column packed with resin linked to gel purified 48 kDa protein (Johnstone et al., 2008). This method produced similar banding patterns on Western blots to that for the antibody produced as described above, but it was a more efficient way to produce sufficient antibody for these analyses.
SDS-PAGE and Western Analyses
Thirty-five μg of water soluble and urea-solubilized matrix were resolved in duplicate on gradient (4-20%) Tris-Glycine ready gels (Novex) in the presence of sodium dodecyl sulfate (SDS). One gel was stained with Stains-all (Sigma) to reveal the positions of acidic and phosphorylated proteins according to methods described by Campbell et al. (1983) as modified by Myers et al. (1996). The duplicate gel was transferred onto nitrocellulose for 1 h at 100 v in buffer containing 25 mM Tris, 192 mM glycine, 20% v/v methanol at pH 8.3 using a mini-transfer unit (Invitrogen). Following transfer, the membrane was rinsed once with TBS, then placed into 50 ml of TBS containing 3% bovine serum albumin (BSA) and 1% goat serum (blocking buffer) and allowed to incubate overnight at 4°C. After blocking, membranes were incubated in 25 ml of solution containing either the anti-55 kDa or anti-48 kDa sera diluted 1:800 in BS containing 3% BSA (Western buffer) for 2 h at room temperature. Membranes were washed 3X in 50 ml of TBS containing 1% BSA and 0.05% Tween-20 (wash buffer) for 5 min each. The membrane was incubated in 50 ml of alkaline phosphatase conjugated goat anti-rabbit IgG (Sigma) diluted to 1:30,000 in Western buffer for 1.5 h, and then washed 3X for 5 min each in wash buffer. A final rinse was carried out in TBS and color was developed with BCIP/NBT (Sigma). Development was stopped by repeated washing in distilled water over 10 min. All incubations and washes were carried out with continuous gentle shaking at room temperature. Molecular weight estimates of bands were determined from plots of the log MW of the SeeBlue standards (Novex) as a function of their relative migration distances.
Amino Acid Analyses
Amino acid analyses were conducted following the method of Waite and Benedict (1984) to optimize recovery of dihydroxyphenylalanine (dopa). Briefly, 10 μg of dry weight protein was re-suspended in 6 N HCL containing 5% v/v phenol. Hydrolysis was carried out in vacuo at 110°C for 24 h. Following hydrolysis, samples were dried in a Speed Vac apparatus, re-suspended in 150 μl of sample buffer and analyzed on a Beckman 6300 Autoanalyzer with post-column ninhydrin detection. All reagents used for the analyses were obtained from the manufacturer.
Phosphate and Carbohydrate Analyses
Protein-bound phosphate was determined by the spectrophotometric method of Eisenreich et al. (1975). Absorbance was determined at 880 nm against a standard range of 0.07–5.0 μg ml−1 total phosphate. Carbohydrate content was analyzed by two methods. A weight estimation was obtained using the spectrophotometric Glycoprotein Carbohydrate Estimation Kit (PIERCE) according to manufacturer’s protocol. Absorbance was read at 550 nm and is proportional to the percentage of carbohydrate component in the protein. A standard curve was generated using proteins of known total carbohydrate content. Additionally, the DIG Glycan/Protein Double labeling kit (Boehringer-Mannheim) was used to qualitatively label both glycosylated and non-glycosylated protein from blots following separation on SDS-PAGE gels following manufacture’s protocols. Once blots identifying glycoproteins were photographed, the blots were re-stained to identify all proteins.
Edman Sequencing
Prior to sequencing, approximately 200 μg of whole soluble matrix protein and protein solubilized with urea from insoluble matrix protein were resolved on a 4–20% Tris-glycine SDS gel, blotted onto Immobilon CD membrane (Milipore) and stained with a stain developed specifically for Immobilon CD according to the manufacturer’s directions. Protein bands of interest stained white against a purple background. Two bands with estimated molecular weights of 48 and 55 kDa were excised from the blot, rinsed three times in distilled H20 for 10 min each, air-dried and submitted to the Proteomics Center at the Medical University of South Carolina for sequencing.
Probing of BAC Library
Using amino acid sequence data deduced from the N-terminal sequence of the 48 kDa protein corresponding to DEADAGD, a perl script was developed to convert the amino acid sequence into a regular expression encompassing all possible codon combinations, which were used to scan all molluscan nucleotide sequences in the National Center for Biotechnology Information database (NCBI)1. All candidate genes with the potential to create the target amino acid sequence were examined further by performing a 6-frame translation and inspecting start codons, stop codons, and acidic characters. A single candidate transcript sequence was identified and Primer3 (Rozen and Skaletsky, 2000) was used to develop primers for this transcript.
The folian-cv1 probe was developed by PCR amplification with the candidate primers and surplus genomic DNA (template) from a BAC library previously constructed using a South Atlantic population of C. virginica collected from Wadmalaw Sound, South Carolina (Cunningham et al., 2006). The Advantage2 polymerase from Clonetech was used and cycled 25 times as follows: 95 C for 5 min; 60°C for 30 s; 68°C for 1 min. A final extension was carried out at 72°C for 10 min and the products were held at 4°C. The raw PCR products were removed of primer-dimers and unincorporated dNTPs by ethanol precipitation and their size validated by agarose gel electrophoresis on a 1.2% Tris-acetate-EDTA (TAE) mini-gel. Verified amplification products were eluted from the gel and ligated into the EcoRV site of the pGEM-Teasy vector (Promega) for Sanger sequence verification. Confirmed PCR products were purified and 100 ng was used to construct probes that were radiolabeled with dATP∗, using the DECAprimeTMII random-primer labeling kit (Ambion) according to the manufacturer’s protocols. Three BAC macroarrays from the BAC library (CVBa) were pre-hybridized overnight in 50 ml hybridization buffer (0.25 M Na2HPO4, 7% SDS, 1 mM EDTA and 1% w/v BSA, pH 7.2) at 65°C in a hybridization oven. Radio-labeled probe was then added and hybridization was performed overnight at 65°C. Filters were exposed to phosphor storage screens (GE Healthcare) overnight. Positively identified BAC clones were PCR verified and the complete gene sequence recovered by custom oligo design and direct BAC sequencing with standard Sanger sequencing procedures. Individual sequences across each BAC were assembled by Phrap (Gordon et al., 1998).
Sequence Annotation and Proteomic Analysis
The genomic sequences encompassing the folian-cv1 gene and upstream regulatory elements were annotated by a series of software applications and final manual examination. BLASTn (Altschul et al., 1990) was used to align the candidate transcript to the genomic sequence. GeneMark (Lomsadze et al., 2005) and FGENEsH (Burge and Karlin, 1997) were used to determine start/stop codons and open reading frames, and to discover nearby flanking genes. Splice sites (exon/intron boundaries) were determined by the Drosophila Neural Network Splice Site Prediction tool, running NNSPLICE 0.9 (Reese et al., 1997). A final amino acid sequence for folian-cv1 was assembled from all of this information and was input to InterProScan to identify any previously reported protein signatures (Zdobnov and Apweiler, 2001). Additional proteomic analyses were conducted using the tools and servers available on the ExPASy Proteomics Server2. Folian-cv1 BAC isolates were confirmed by searching the NCBI database using the Genome Browser interface and tBLASTn query tool3. Multiple protein sequence alignment was performed with Clustal Omega sequence alignment tool; pairwise protein alignment was performed with the Needleman–Wunsch alignment tool. Both tools are provided online by the European Bioinformatics Institute (EMBL-EBI)4.
Southern Hybridization
Gene copy number for folian-cv1 in the C. virginica genome was determined by fully digesting approximately 10 μg of genomic DNA with 2 complimentary 6-base cutters (BamHI and HindIII) and resolving the fragments by pulsed-field gel electrophoresis. Digested genomic DNA was denatured and transferred to positively charged nylon membrane (GE Healthcare Life Sciences) using downward capillary methods following published procedures (Brown, 1999). Hybridization was conducted as previously described (see BAC probing methodology) using amplification products derived from the folian-cv1 gene.
PCR Analyses
PCR primers for folian-cv1 were designed using the NCBI primer blast tool5. A forward primer (5′-GACACCGATGGATATGGCCC-3′) corresponding to the 127–146 bp region of folian-cv1 and a reverse primer (5′-TCATCGGCACTTGAGTCGTC-3′) corresponding to the 263-244 bp region were purchased from Integrated DNA Technologies (IDT). These primers had a predicted melting temperature (Tm) of 58 and 57°C respectively. The expected length of the target folian-cv1 product was 135 bp. Two ml of hemolymph was obtained from the adductor muscle of 5 oysters using a 21-gauge needle and syringe and spun at 6000 × g for 5 min to pellet hemocytes. The resulting pellets were combined into one tube in RNAlater (Life Technologies). RNA was extracted from hemocyte pellets and pieces of mantle excised from the edge, using the RNeasy mini kit (Qiagen) according to the manufacturer’s protocol. Extracted RNA was quantified using an Eppendorf Biophotometer. Following normalization, RNA was incubated at 65°C for 5 min and reverse transcribed for 1 h at 37°C using an RT mastermix comprised of RNA free water, 5x MMLV Buffer (Promega), 10 mM dNTP mix (Qiagen), 50 ng/μL random primers (Promega) and Moloney murine leukemia virus (MMLV) RT enzyme (Promega). Finally, tubes were incubated at 95°C for 10 min. For PCR amplification 100 ng of cDNA was combined with 47.5 μL PCR mastermix (Qiagen) comprised of sterile filtered water, 10x PCR buffer, 10mM dNTP mix (Qiagen), 10 μM forward primers, 10 μM reverse primers and Taq Polymerase. A cDNA-free control was also run. PCR was performed on an Applied Biosystems Veriti Thermal Cycler with an initial denaturation at 94°C for 3 min followed by 25–35 cycles at 94°C for 30 s, 60°C for 30 s, and 72 C for 1 min. A final extension was carried out at 72°C for 10 min and the products were held at 4°C. Products were run on a 2% agarose gel with Tris-acetate-EDTA buffer (TAE) at pH 8 in sample buffer (Promega) including 10 μl of 10 mg ml−1 ethidium bromide. The target sequence was validated using ExoSAP-IT® reagent (Affymetrix) followed by Sanger sequencing contracted through the Clemson University Genomics Institute.
For qPCR, the collection of hemocytes and extraction of RNA were as described above. Three separate collections were analyzed. Comparison of the expression of folian-cv1 was made to cv-actin and cv1-MMP (matrix metalloproteinase) using cv-18S rRNA as a reference gene. Cv-actin served as an example of a housekeeping gene and cv1-MMP is a gene belonging to a class that has been linked to both immunity (Bonnans et al., 2014) and biomineralization (Luke and William, 2001; Ortega et al., 2003) and has been identified in oyster hemocytes (Falwell, 2015). The folian-cv1 primer set was described above. Primers for the other genes were as follows with Tm in parentheses: cv-actin forward, 5′-TTGGACTTCGAGCAGGAGATGGC-3′ (61 C) and cv-actin reverse, 5′-ACATGGCCTCTGGGCACCTGA-3′ (63 C); cv1-MMP forward, 5′-ATGGTTCATTGTATGCGACCCGGA-3′ (61 C) and cv1-MMP reverse, 5′-TTGGGCGCTCTCTTGTACACCTTT-3′ (61C); cv-18S rRNA forward, 5′-CTGGTTAATTCCGATAACGAACGAGACTCTA-3′ (59 C) and cv-18S rRNA reverse, 5′-TGCTCAATCTCGTGTGGCTAAACGCCACTTG-3′ (65 C). The sequences for cv-actin and cv1-MMP were obtained from the Gomez-Chiarri lab and those for cv-18S rRNA from those provided by Jing et al. (2011). Primers were validated as described above for folian-cv1 and their concentrations optimized for 1-step qPCR.
For all primer sets, a mastermix containing SYBR®Green 1 dye and buffers, a RT enzyme mix, and optimized dilutions of primers were prepared according to the instrument manufacturer’s recommendations. The reactions were carried out in a total volume of 25 μL with 1 μL of target RNA on a BioRad iCycler iQ5 with the following program: holding at 48°C for 30 min, holding at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15 s and extension at 60°C for 1 min. Seventy cycles of the melt curve were run as follows: denaturation at 95°C for 15 s, annealing at 60 C for 15 s and extension at 95 C for 15 s. Melt curve predictions were performed with uMelt software6.
Expression for genes of interest was normalized to that of the reference gene cv-18S rRNA for each hemocyte sample collection. The formula used for calculating normalized gene expression (NE), based on Muller et al. (2002) as modified by Roling et al. (2004) is:
E is the efficiency of amplification for a particular gene, Ct is the threshold cycle, “ref” is cv-18s rRNA and “goi” is one of the genes of interest, folian-cv1, cv1-MMP or cv-actin. The null hypothesis that the qPCR expression means of the three genes of interest were equal was evaluated with an Analysis of Variance Test (ANOVA). The three follow-up null hypotheses that the qPCR expression means of pairs of the genes of interest were equal was evaluated using Fisher’s Protected Least Significant Difference Test.
Histochemistry and Immuno-Histochemistry
Prior to histochemical analysis, oysters were perfused and fixed intact with 4% paraformaldehyde in phosphate buffered saline (PBS), pH 7.4. PBS was adjusted using 1M NaCl, to match osmolality of the holding tank at the time of tissue harvest. Tissue was embedded in ImmunobedTM (Poly Sciences) and sectioned at 1–1.5 μm. To assess general tissue morphology and integrity, sections were stained using a solution that contained equal parts 1% Azure II in double distilled water and 1% Methylene Blue in 1% sodium borate. The working solution was filtered through a 0.22 μm syringe filter and applied directly to dried mounted sections for 10–30 s. The slides were rinsed with doubly- distilled water and dried with a jet of air.
Immuno-histochemical staining was carried out using the affinity purified anti-48 kDa antibody as previously described (Johnstone et al., 2008). Antibodies were diluted in PBS containing 1% BSA and 0.1% Triton X100. This buffer was also used as the wash buffer. The anti-48kDa antibody was diluted to 1:700. Prior to staining, slides were rehydrated through a graded series of alcohol solutions in distilled water, starting with 100% EtOH and ending with PBS for 2 min each. Slides were blocked in PBS buffer containing 5% BSA, 10% normal goat serum (Sigma) and 0.1% Triton X 100, for 6 h at room temperature. After a brief rinse in PBS, slides were incubated in primary antibody solutions overnight at 4°C. Washes following the primary and secondary incubations were carried out 3X for 10 min each. Immuno-reactivity was detected using Alexafluor 488 goat anti-rabbit fluorescent antibody at a concentration of 6 μg ml-1. All incubations were carried out in a covered humid chamber with gentle agitation. Sections were mounted in a 1:1 solution of glycerol and PBS and then examined with a Zeiss Axiovert 135 inverted fluorescent microscope equipped with a Diagnostic Instruments SPOT-RT Cooled Color CCD camera.
Confocal Laser Scanning Microscopy (CLSM) of Live Mantle and Hemocytes
To observe potential channels in the mantle, an approximately 1 cm2 piece of live tissue was excised from the mantle edge, mounted in a depression slide in filtered seawater and covered by a 1.5 cover glass. Autofluorescence was observed directly with a Zeiss 510 CLSM at 488 nm excitation using a ZeissPlan Neofluar 40X/1.3 oil DIC objective.
In an alternative experiment, 600 μl of hemolymph was collected from the adductor muscle using a sterile 21-gauge needle affixed to a sterile 3 ml plastic syringe. The adductor muscle was accessed by notching the oyster shell with a diamond blade saw and the oyster was immediately placed into a holding vessel containing 2 L of fresh aerated seawater. The hemolymph was transferred into a sterile 1.5 ml plastic microfuge tube and the hemocytes were labeled in suspension by adding 50 μl of 2 μM Calcein AM ester. The preparation was mixed gently and incubated at room temperature for 30 min. Following incubation, the preparation containing the labeled hemocytes was re-injected into the adductor muscle using a sterile syringe. After 2 min, the oyster was returned to the holding vessel for 60 min to allow time for the hemocytes to circulate throughout the hemocoel. Following incubation, a piece of mantle was collected and observed at excitation wavelengths of 488 and 594 nm. A 60-slice image stack was then post-processed using the Quorum Technologies Volocity visualization software to show rotation of the outer shell facing mantle epithelium about two axes.
Results
Identification and Characterization of the Folian Protein Family
Proteins extracted from the foliated shell layer were visualized by Stains-all following SDS-PAGE. This cationic stain has been successful in identifying highly anionic proteins and phosphoproteins derived from shell, tooth and bone (Campbell et al., 1983; Myers et al., 1996). Proteins enriched in negatively charged groups stain a deep purple color while proteins with less of, or lacking, these groups stain pink, and the color fades quickly upon exposure to light. Figures 1A,B, show the electrophoretic patterns of the EDTA soluble matrix proteins (SM) and proteins rendered soluble after extraction of the EDTA insoluble matrix protein (IM) by urea (uSM). The SM extract pictured in Figure 1A resulted in two prominent bands at 48 and 55 kDa and two adjacent minor bands at 43 and 63 kDa. In addition, staining spanned nearly the entire length of the gel, with a relatively discrete set of bands from 5 to 50 kDa. In a separate shell extract (Figure 1B), only the 48 kDa band was prominent. However, a 55 kDa band is clearly visible in the uSM fraction of this extract, suggesting that the protein(s) in this band may be more tightly bound to the insoluble structures than those of the 48 kDa protein.
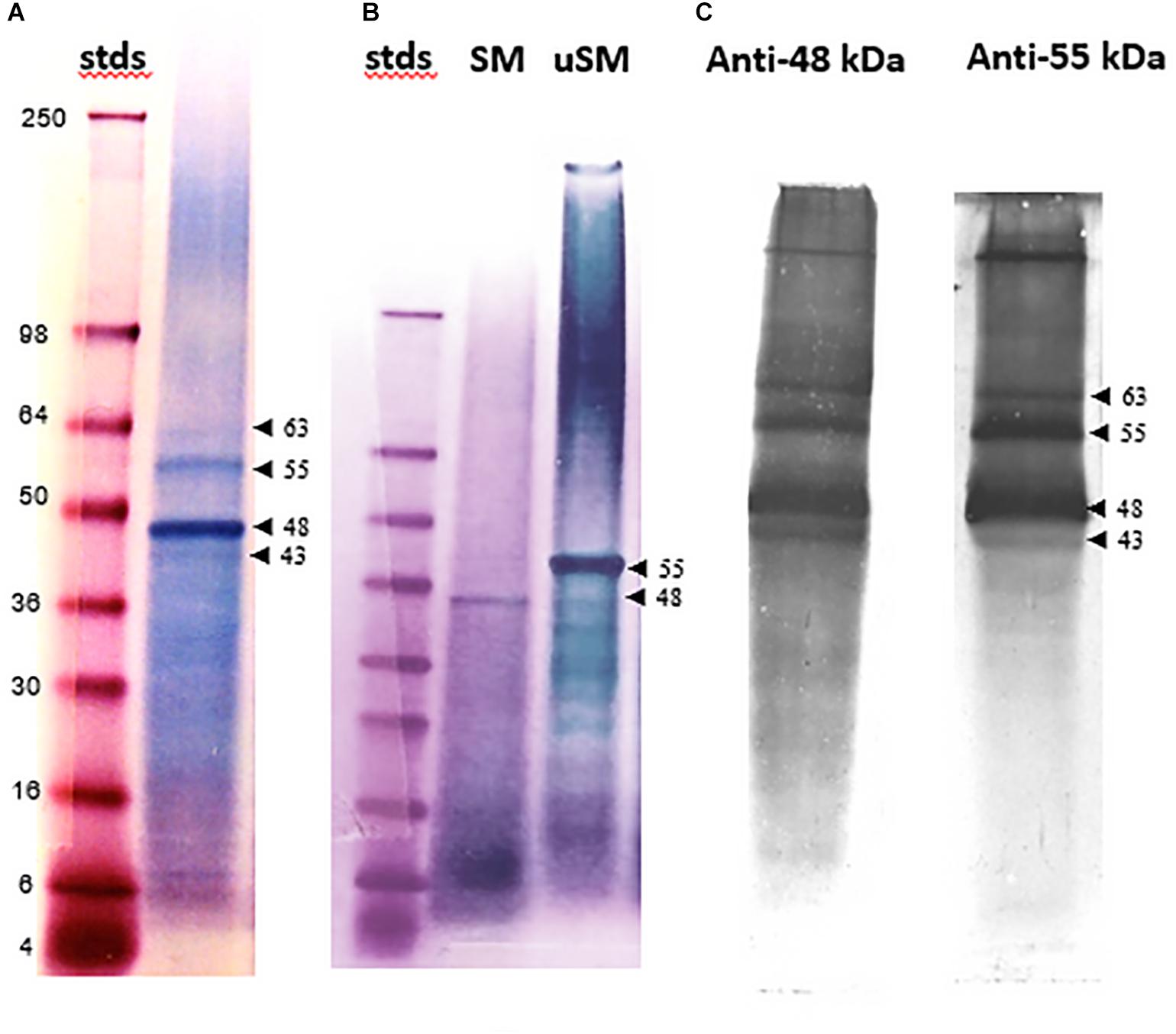
Figure 1. SDS-PAGE and Western analyses of soluble shell matrix proteins. (A) Thirty-five μg of EDTA soluble matrix (SM) was resolved on a 4–20% gradient Tris-glycine gel and stained with Stains-all. Standards (stds) were SeeBlue protein molecular weight markers labeled in kDa. Two prominent bands appear at 55 and 48 kDa while two adjacent minor bands occur at 63 and 43 kDa (arrow heads). (B) Thirty-five μg SM and urea-solubilized matrix (uSM) were electrophoresed and stained as in (A). The position of the 55 and 48 kDa bands are indicated with arrow heads. While the 55 kDa band is not prominent in this preparation of SM, it is clearly the dominant band upon urea extraction of the insoluble matrix from this preparation. (C) Western analyses of SM extract immuno-stained separately with anti-48 kDa and anti-55 kDa antibodies. Both lanes contained 50 μg of SM and were resolved on 12% Tris-glycine SDS gels. All four of the Stains-all positive bands identified in (A) are immuno-reactive with the anti-48 and anti-55 kDa antibodies (arrow heads). An additional high molecular protein that exceeded 98 kDa was also immune-reactive. All matrix loads are in quantities of protein as determined by BCA analysis. For these analyses, only lanes showing data pertinent to this study are presented.
Western analyses indicated that antibodies made against either the 48 or 55 kDa bands reacted with a wide range of molecular weight classes from the SM (Figure 1C). Each antibody recognized four discrete bands corresponding to the Stains-all positive bands following gradient SDS-PAGE, including highly reactive proteins at 48 and 55 kDa and more moderately reactive bands at 43 and 63 kDa. The anti-48 kDa antibody identified a broad low molecular weight continuum of proteins, much like Stains-all, while the anti-55 kDa antibody did not react broadly with proteins in this region. In addition to the Stains-all bands, a high molecular weight band was also immuno-reactive. The significant cross-reactivity observed among this class of proteins demonstrates that they share common epitopes and suggests that they are related.
The electrophoretically separated and blotted proteins of SM reveal a range of molecular weight bands that are glycosylated, including the 48 kDa band (Figure 2A). Some of these bands, such as one that is about 34 kDa, are not evident with Stains-all staining. The highly variable level of glycosylation becomes evident when the blots are re-stained for protein (Figure 2B). The prominent 48 kDa band would be classified as moderately glycosylated given the color shift from blue to more of a brown color in the band after re-staining. This color shift is not evident for the 34 kDa band or some of the higher molecular weight bands, suggesting they are more highly glycosylated than the 48 kDa band. However, the color shift is more pronounced in some of the lower molecular weight bands, suggesting they have lower levels of glycosylation than the 48 kDa band.
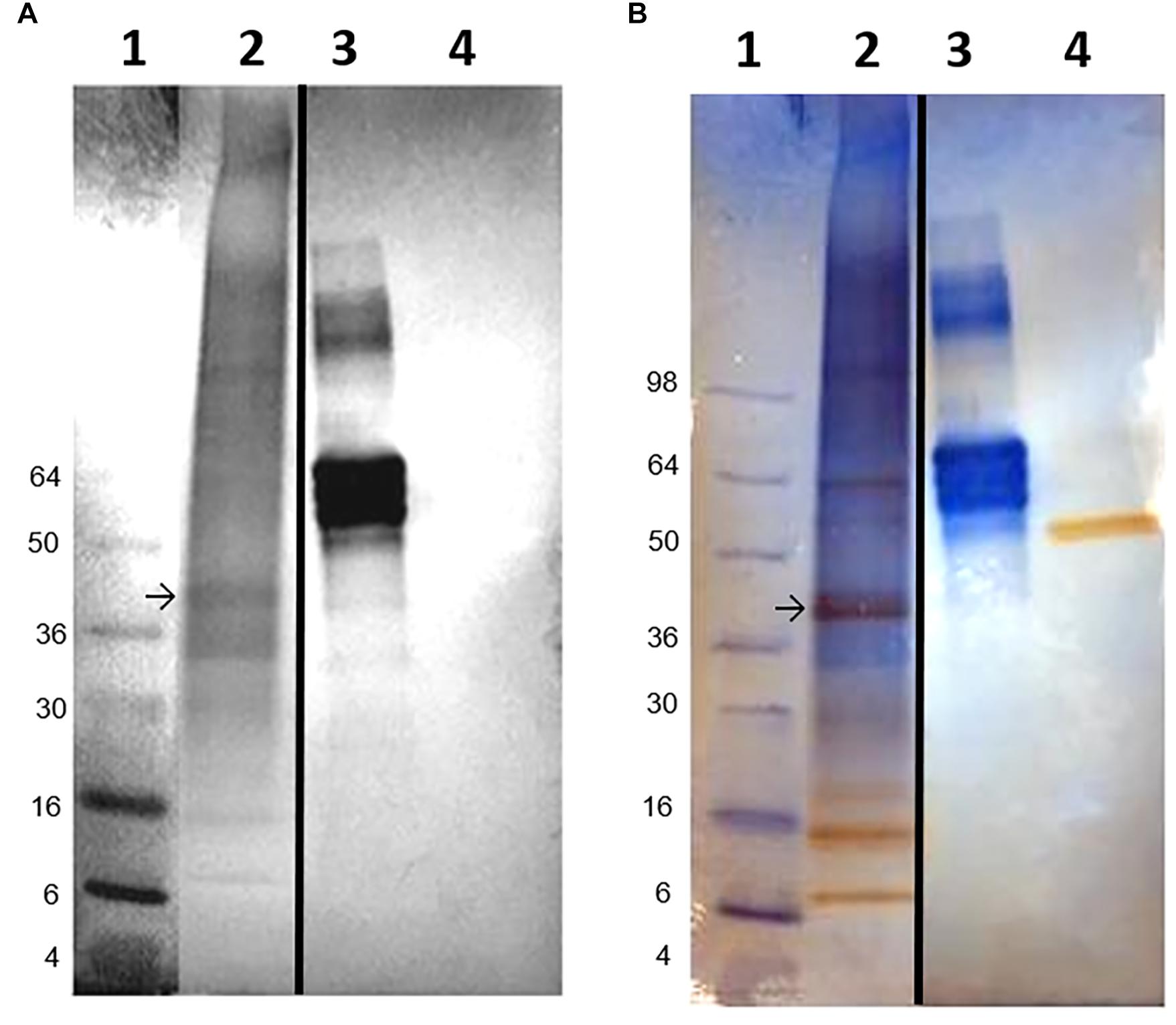
Figure 2. Glycoprotein detection in soluble shell matrix proteins. (A) Twenty-five μg (dry weight) of each protein was resolved using a 10% Tris-glycine gel and transferred onto nitrocellulose. The blot was stained to identify glycosylated protein. Lane 1- SeeBlue protein molecular weight standards labeled in kDa. Lane 2- SM. Lane 3- Fetuin (glycosylated protein control). Lane 4- Creatinase (non-glycosylated protein control). The intensity of staining is proportional to the amount of carbohydrate present on each protein. SM appears to contain a broad range of lightly glycosylated proteins. The 48 kDa band is indicated with an arrow and shifted to a lower apparent molecular weight following blotting when compared to Figure 1A. This shift was observed commonly following blotting. A band identified at approximately 34 kDa was not identifiable in Figure 1A following Stains-all staining. Stained gels were photographed in black and white to enhance detectability of faint blue bands. (B) The same blot as in (A) was re-stained for protein to distinguish non-glycosylated proteins from glycosylated proteins. In lane 3, fetuin remains blue and is indicative of its heavy glycosylation. Creatinase is now apparent in lane 4. In lane 1, the 48 kDa band shifts toward brown staining indicating less glycosylation than the 34 kDa band which remains blue. For this analysis, only lanes showing data pertinent to this study are presented. Lanes 1 and 2 were cropped from the original blot and digitally placed next to lanes 3 and 4 (controls) which were also cropped from the original image; the splice sites are demarcated by a black line. In (A), the contrast in Lane 1 was digitally adjusted to visually discern the protein standards.
Amino acid analysis confirmed the close relation among the matrix protein fractions (Table 1). A comparison of the 48 and 55 kDa proteins shows that they have a markedly similar overall amino acid composition, with the common distinguishing characteristic of being approximately 80% asp, ser and gly, and having a nearly identical overall hydrophobicity. The overall similarity of their amino acid composition was supported using the Cornish-Bowden index (Cornish-Bowden, 1983) from which it could be concluded that these fractions have a 95% certainty of being related (Supplementary Table 1). Using the same comparisons, there is also a relatively close correspondence of the composition of both the 48 and 55 kDa proteins with that of the whole SM extract. IM is also similar enough in its composition to the soluble fractions as to be weakly related to them. Further, like SM, IM is highly phosphorylated and moderately glycosylated. Despite their otherwise high degree of similarity, the folian proteins from the 48 and 55 kDa bands are clearly distinct in their amino terminal regions (Table 2). Amino terminal regions obtained from the 55 kDa band are nearly identical regardless of their origin from SM or urea solubilized from IM (uSM) (Table 2), suggesting they may contain identical proteins.
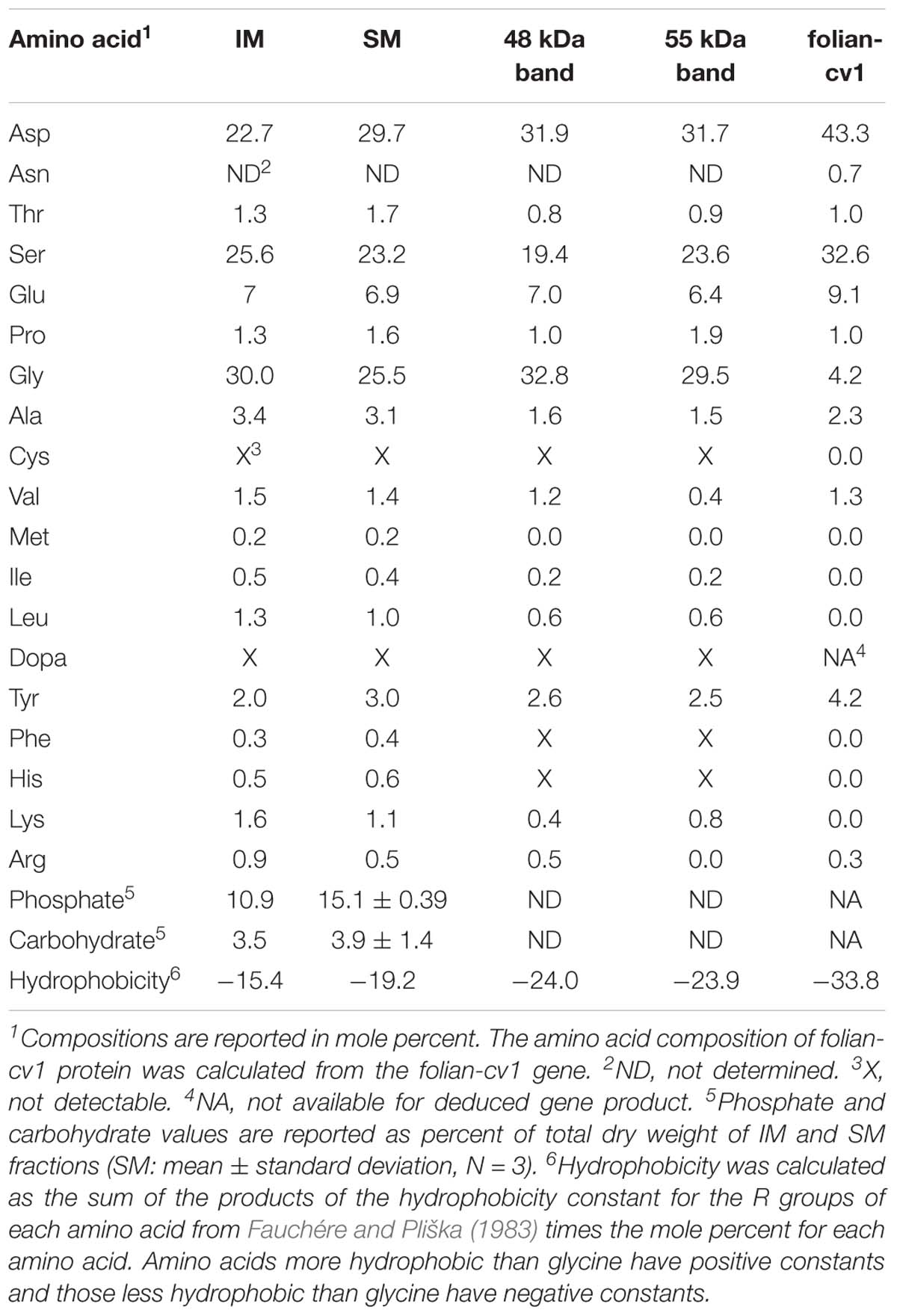
Table 1. Amino acid compositions of shell matrix fractions compared with the deduced amino acid composition for folian-cv1.
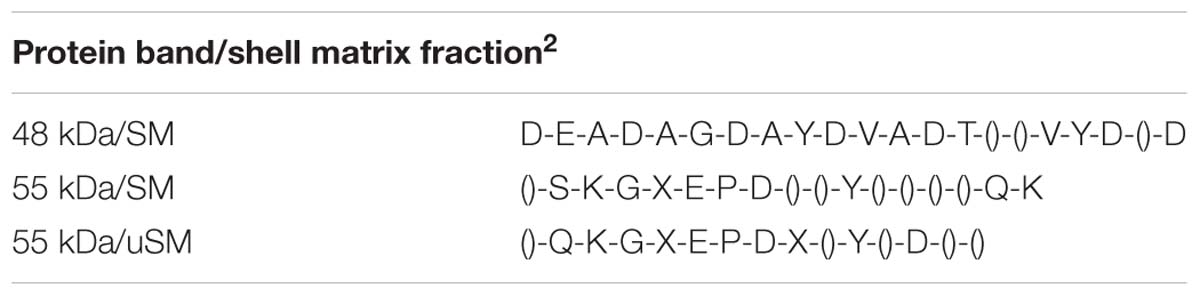
Table 2. Amino-terminal sequences for the 48 and 55 kDa bands1.
Identification of the Folian-cv1 Gene
Probing the C. virginica BAC library with a nucleotide sequence based on an EST that encoded the N-terminal sequence of the 48 kDa band resulted in 36 positive BAC clones. Genomic Southern analysis revealed that the folian-cv1 gene occurred at two independent loci (Supplementary Figure 1). PCR analysis of the 36 BAC clones identified representative BACs harboring the two variants, which were used for direct BAC sequencing to determine the full length of the coding and regulatory sequences. The first region contained 1716 bp and the second, 2604 bp. The 1716 bp region aligned within the 2604 bp region with greater than 97% identity. Each region encompassed an entire gene, with a transcriptional start site, a start codon, two exons defined by splice boundaries, a stop codon, and a putative poly-A site (Supplementary Figure 2). The deduced transcribed products resulted in two nearly identical gene variants differing by 4 point mutations (Figure 3); three of these mutations result in amino acid substitutions which are discussed further below. The gene and deduced gene product were named folian-cv1 for the shell layer and species from which they were derived.
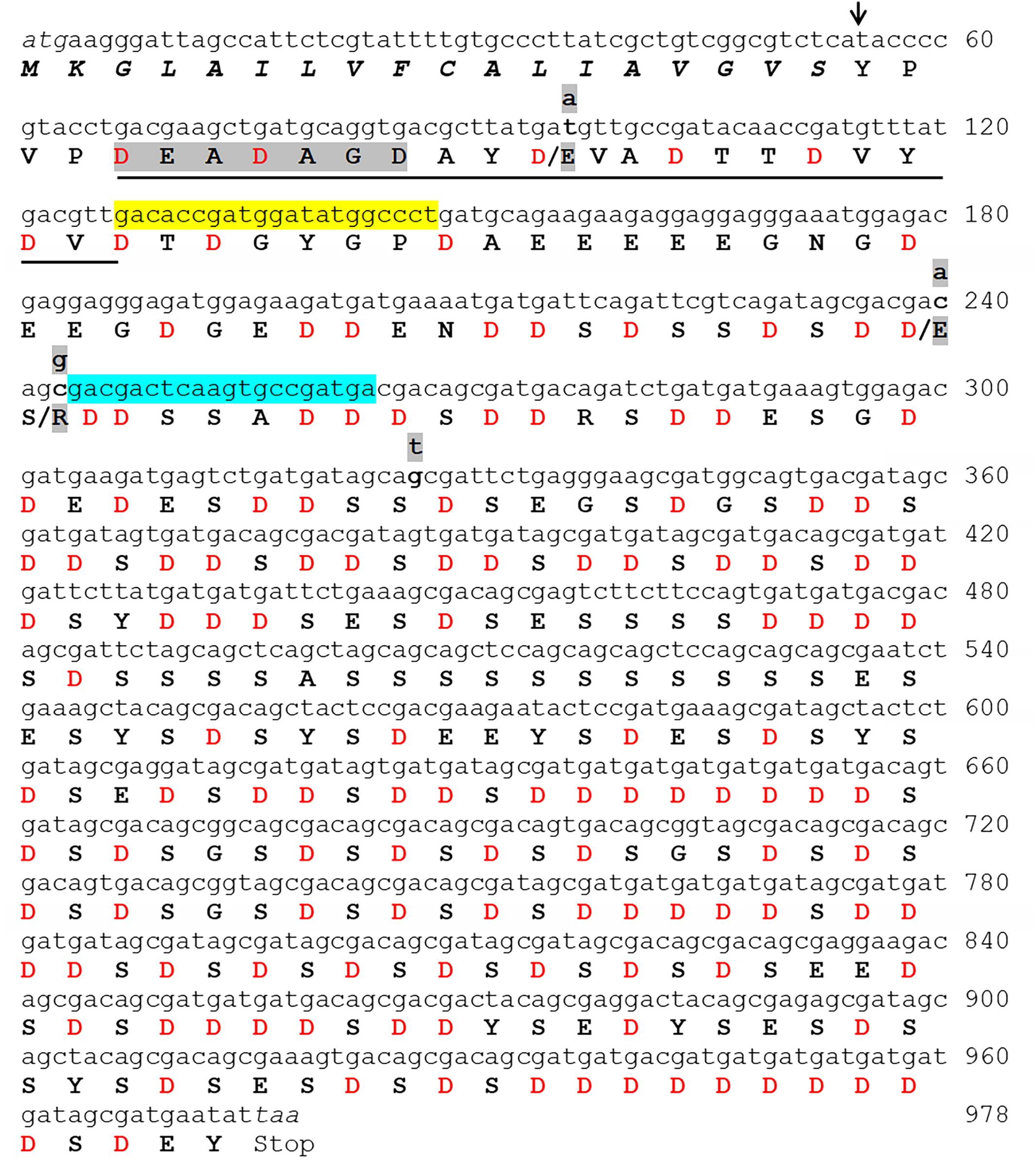
Figure 3. The complete folian-cv1 gene and deduced amino acid sequence. Two copies of folian-cv1, identical with the exception of four point mutations, were identified. The full sequence is shown for copy 1. The point mutations shown in copy 2 occur in the N-terminal region of the protein and are indicated in lower case highlighted letters above the DNA sequence. Three of the four mutations resulted in an amino acid change indicated by the highlighted amino acids following the “/”. Mutations at positions 96 and 240 result in a substitution for Asp to Glu. The mutation located at position 243 resulted in a change from serine to arginine. The putative signal sequence starting with the ATG start codon appears in italics. The putative cleavage site of the signal sequence is indicated with an arrow (↓) and cleaves between the “S” and “Y” residues. The deduced amino acid sequence minus the signal sequence encodes a 307 amino acid product enriched in aspartic acid and serine. The underlined sequence matches well with the N-terminal sequence identified by Edman sequencing (Table 2) and the gray highlighted sequence “DEADAGD” was used to design the in silico probe which identified the transcript used to screen the BAC library. The yellow highlighted region from 127 to 146 bp and the blue highlighted region from 263 to 244 bp indicate the regions that were used to construct forward and reverse primers respectively for subsequent PCR analyses.
The two variants of the folian-cv1 gene identified from BAC cloning were verified by searching the recently released C. virginica genome constructed from a Delaware population of oyster (Gomez-Chiarri and McDonnell, 2017). Using BlastX tools and the folian-cv1 deduced protein sequence as a virtual probe, two variations of folian-cv1 were identified on chromosome 1 at two distinct loci (NCBI accession number NC_035780; gene ID: 111101504 and NCBI accession number NC_035780; gene ID: 111119246). The two Delaware genome isolates (1504 and 9246) demonstrate a slightly higher level of heterogeneity compared with the two South Carolina BAC isolates reported in Figure 3, differing not only by substitutions but by gap regions or insertions as well (Supplementary Figure 3). Alignment of copy 1 of the folian-cv1 BAC isolate derived from the South Carolina population with the two variants identified from the Delaware genome revealed strong sequence agreement between the two populations, differing slightly in length and composition due to gap regions or insertions, and substitutions (Supplementary Figure 3). Some of the sequence variations between the populations likely reflects their geographic isolation. Because only two variants of folian-cv1 were found in either population and were localized on chromosome 1 in the Delaware population, it is likely these copies result from gene duplication.
Structure of the Folian-cv1 Deduced Protein
The complete gene and deduced folian-cv1 protein of 325 amino acids is shown in Figure 3. The only domain recognized by InterProScan software was a putative signal peptide (italicized) and signal cleavage site (black arrow), which indicates the protein is secreted (Zdobnov and Apweiler, 2001). After the first three amino acids following the signal sequence, the subsequent sequence (underlined) matches that obtained by Edman chemistry for the 48 kDa protein as reported in Table 2. The four point mutations that distinguish the two copies of folian-cv1 occur toward the N-terminus. Two mutations at nucleotide positions 96 and 240 result in substitutions for aspartic acid to glutamic acid, thus resulting in no change in charge. A mutation at position 243 results in a substitution for serine to arginine, and potentially a change to a positively charged group from a negatively charged group if the serine were phosphorylated. The fourth point mutation at 326 does not produce an amino acid substitution.
Removal of the 18 amino acid signal sequence results in a 307 amino acid protein with a calculated molecular weight of 32 kDa. The secreted form of folian-cv1 is highly acidic, as approximately 52% of its amino acid composition is comprised of aspartic acid (132 residues) and glutamic acid (28 residues) (Table 1). While its high proportion of aspartic acid and serine and its low hydrophobicity are comparable to other folian fractions, it is distinctive in having a considerably lower glycine content than these fractions. This difference, in large measure, explains its low relatedness to the other fractions when compared by the Cornish-Bowden index (Supplementary Table 1). The theoretical pI for folian-cv1 is 2.19; however, the possibility for additional anionic charge is present in the 115 potential phosphorylation sites (100 for Ser, 12 for Tyr, and 3 for Thr) predicted by NetPhos 3.17. Bearing in mind that the protein-bound phosphate would have a second ionizable group with a reported pKa near neutrality (Lee et al., 1977), then more than half of these groups would be doubly ionized at physiological or seawater pH. Accordingly, assuming full theoretical phosphorylation and 1.5 charges per phosphate group, the net charge of folian-cv1 under these conditions would be −332.5 per molecule, or just over a −1 charge per amino acid residue. If all these sites were phosphorylated, the protein would have an isoelectric point of 0.368. A high level of phosphorylation would be consistent with the phosphate content of the whole matrix fractions. If the actual level of phosphorylation of folian-cv1 were, at a minimum, the same by weight as for the whole soluble extract, then approximately 50% of the theoretical sites would be phosphorylated. Under these circumstances, the pI would still be less than 0.7. Twenty-eight potential mucin type, O-glycosylation sites were predicted, with 25 of these sites in the region including residues 153–188 and three additional sites in the region including residues 234-238 (NetOGlyc 3.1 Server9). Some level of glycosylation would be consistent with the presence of carbohydrate in the whole matrix fractions (Table 1) and the apparent glycosylation of the 48 kDa band (Figure 2). Four of the 13 tyrosine residues are potentially sulfated including residue positions 40, 291, 295, and 32510.
Notable features of the folian-cv1 primary structure are shown in Figure 4, which illustrates its repetitive and modular structure. As expected from its composition, much of the primary structure is dominated by aspartic acid and serine residues, which are concentrated from position 71 on. These two amino acids appear together in various configurations. Conspicuous arrangements are 7 tandem repeats of DDS and 8 tandem repeats of DS. An additional DS-rich region is made up of 3 tandem repeats of DSDSGSDSDS. There are multiple poly-D blocks, six of which have runs of 4–10 residues. These are bracketed by S residues and are located from the middle of the protein to near the C-terminus. A ser-rich region has poly-S blocks totaling 19 residues near the middle of the protein. There are two tyrosine-rich blocks containing YS or SYSD repeats, one following the ser-rich region and another located near the C-terminus. With only a few exceptions, the hydrophobic residues (other than those associated with the signal sequence) are near the N-terminus giving the region from position 19–51 a hydrophobicity index of +14.7 compared to −33.1 for that of the whole post-signal sequence protein (see Table 1). This region is punctuated with regularly spaced asp residues in D-X-X-D and D-X-D arrangements. A glu-rich region, containing a 5-residue poly-E block, occurs between the hydrophobic N-terminus and the asp-ser-rich region.
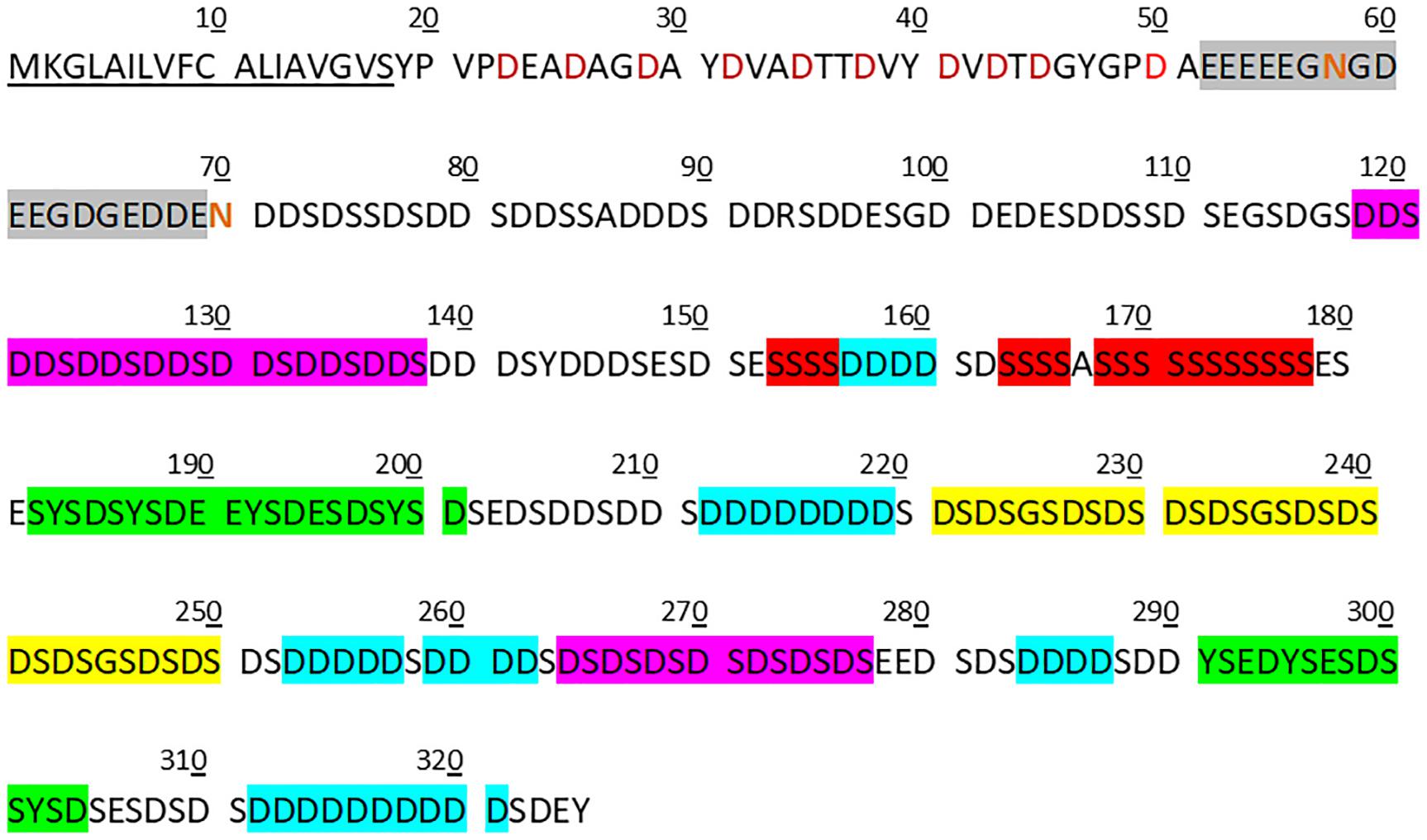
Figure 4. Features of copy 1 of the folian-cv1 primary structure. The secreted protein can be divided approximately into three distinct regions following the signal peptide (underlined). The first is a relatively hydrophobic N-terminal region (33 residues in length) with regularly spaced asp residues (red symbols). This is in turn followed by a short glu-rich region (18 residues in length; gray box) which contains a 5-residue glu run and has the two asparagine residues in the protein (red symbols) in or immediately following the run. The remainder of the protein (255 residues) is generally asp-ser-rich. Notable sub-regions in the asp-ser-rich region include one that is exclusively DDS (7 tandem repeats in residues 118–138) and one that is exclusively DS (8 tandem repeats in residues 262–277) (pink boxes). In addition, there are three 10-residue tandem repeats for which each repeat has a single GS bracketed by two DS pairs (yellow box). Other repeat regions include poly-D runs (those runs greater than three shown in blue boxes), which are bracketed by S, poly-S runs (red boxes) and those that are Y-rich containing YS or SYSD sequences (green boxes). Nearly all of the ser, tyr, and thr were predicted to be possible phosphorylation sites using NetPhos 3.1 (www.cbs.dtu.dk/services/NetPhos).
Folian-cv1 Gene Expression in Hemocytes and Mantle
PCR analyses of mantle and hemocyte tissue demonstrated that these tissues expressed folian-cv1 (Figure 5A), which was validated by sequence analysis (Figure 5B). The results of qPCR analysis of gene expression in hemocytes for the housekeeping gene cv-actin and cv1-MMP, a gene implicated in immunity as well as biomineralization, are compared to that of folian-cv1 in Figure 5C. ANOVA suggested that their expression relative to that of cv-18s rRNA was different (p = 0.0217). Subsequent pairwise comparisons suggested that the relative expression of both folian-cv1 and cv1-MMP was different from that of actin (p = 0.0125 for both comparisons), but that the expression of folian-cv1 and cv1-MMP were not statistically different (p = 0.992). Based on the averages, the expression of cv-actin is 2–3 orders of magnitude higher than either cv1-MMP and folian-cv1, and the expression of the latter pair are nearly identical.
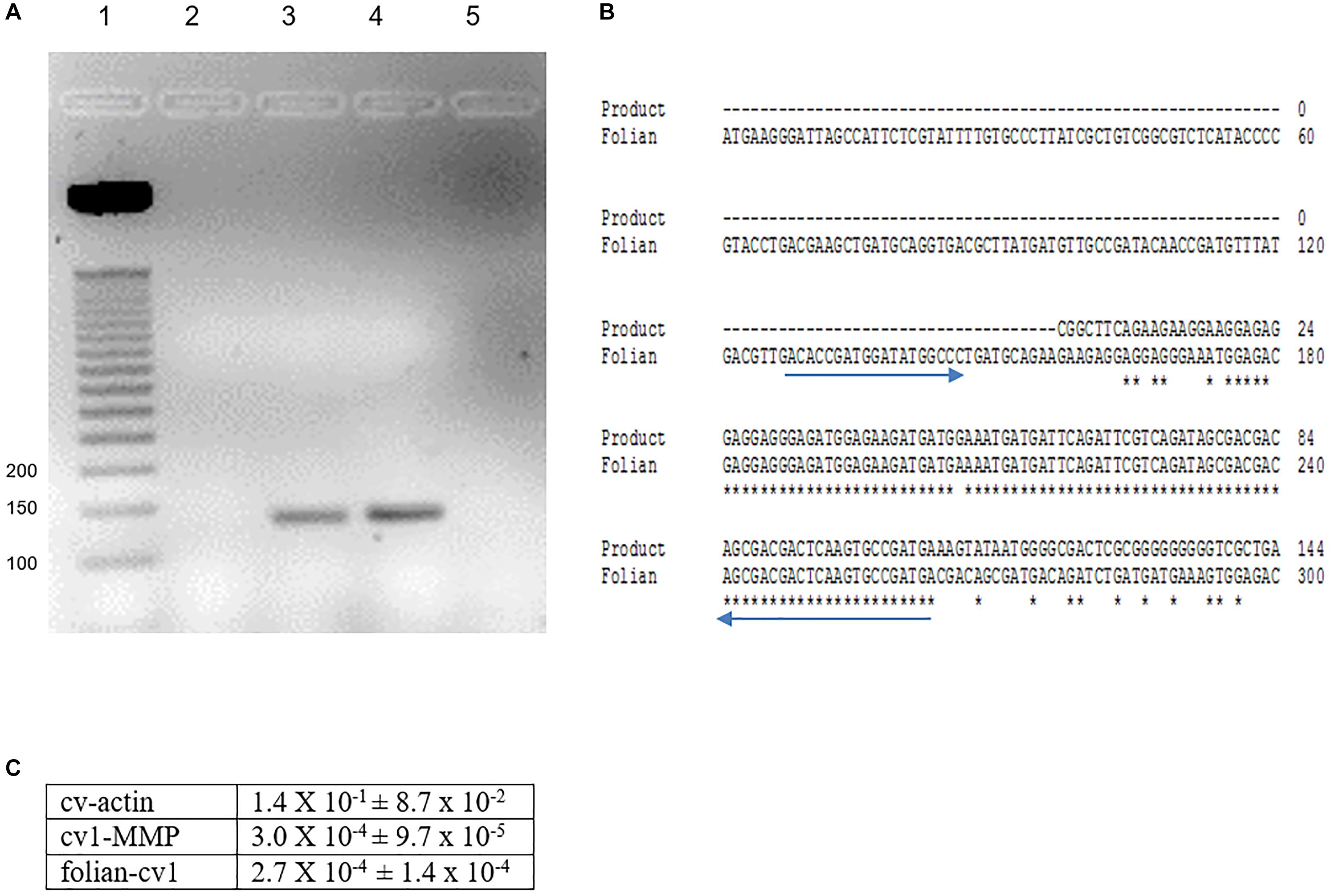
Figure 5. Identification of folian-cv1 gene expression in mantle tissue and hemocytes. (A) PCR products amplified from mantle and hemocytes using folian-cv1 forward and reverse primers resolved on a 2% agarose gel and stained with EtBr. Lane 1- standards labeled in base pairs; Lane 2- negative control, no template; Lane 3- hemocytes; Lane 4- mantle tissue; Lane 5-blank. The products are similar in size to the length of the region delimited by the primers (135 bp). (B) Sequence validation of PCR target. The PCR product from hemocytes was sequenced and aligned with the folian-cv1 gene sequence. Primers indicated with blue arrows. High fidelity matching occurred between nucleotides 159 and 266 on the folian-cv1 sequence. (C) Comparison of qPCR expression for cv-actin, cv1-MMP and folian-cv1 in hemocytes. Values are relative to cv-18S rRNA expression (Mean ± standard deviation; N = 3 hemocyte extracts).
Immuno-Histochemical Localization of Folian Proteins in the Mantle and Evidence for Hemocyte Movement Through the Mantle Using Confocal Imagining
The mantle organ lies in close approximation to the entire inner shell surface, separated by a nominal extrapallial space (Simkiss and Wilbur, 1989). In C. virginica, as is the typical case for bivalves, the organ elaborates into three lobes at its margin (Ebel and Scro, 1996; Figure 6A from Johnstone et al., 2008). The outer lobe and the general shell-facing outer epithelium of the mantle are generally considered responsible for the formation of shell layers in the extrapallial space. The outer and middle lobes form the periostracal groove (PG) which at its base secretes periostracum. During shell formation this material nominally completes the isolation of the extrapallial space at the shell edge and has been reported as the primary surface on which the mineral layers form in this oyster and other molluscs (Galstoff, 1964; Saleuddin and Petit, 1983; Checa, 2000).
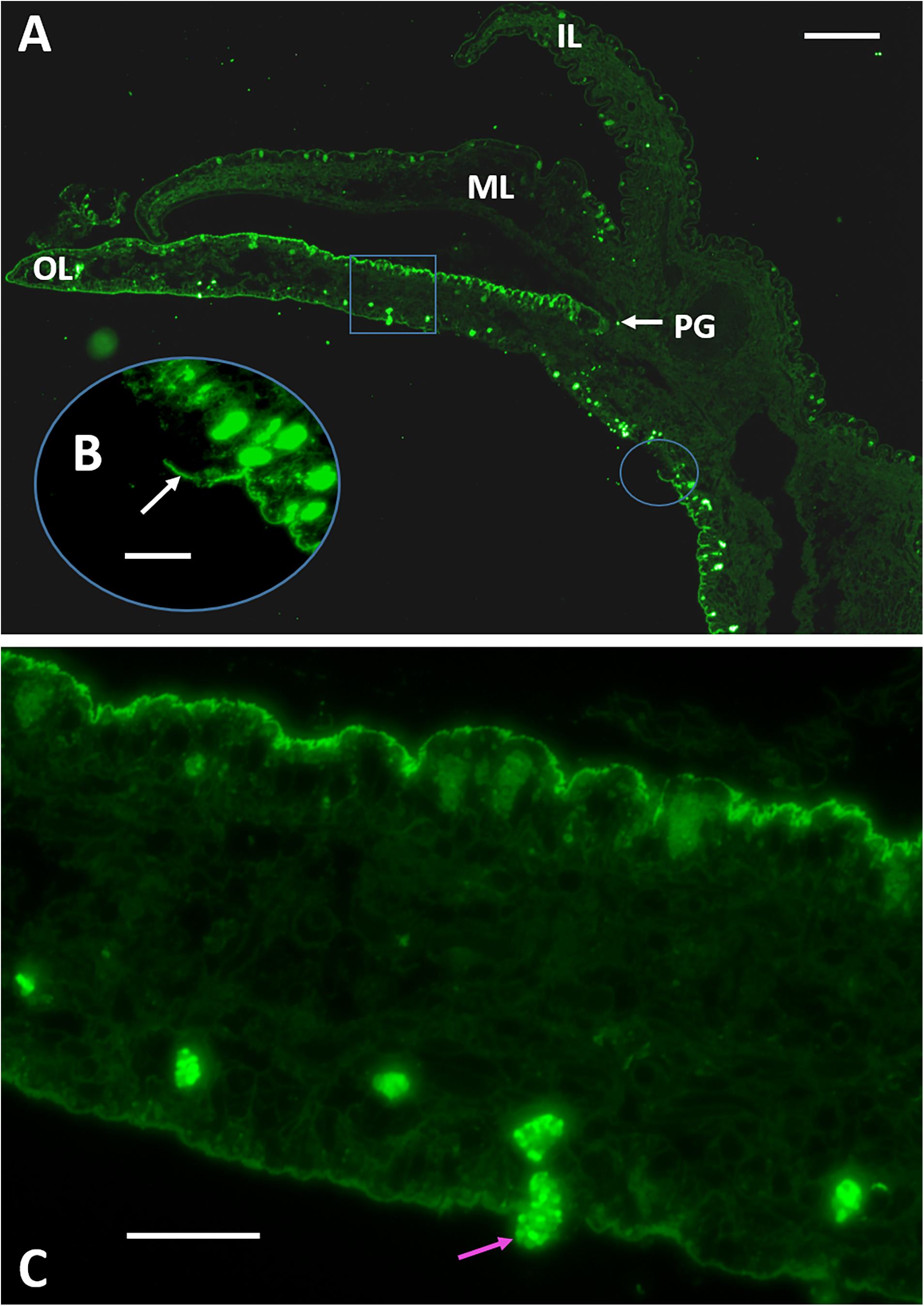
Figure 6. Immuno-fluorescence microscopy of transverse sections of the mantle organ using an anti-48 kDa affinity purified antibody. (A) The mantle edge elaborates into three lobes; the inner lobe (IL) faces the body of the animal and the outer lobe (OL) faces the shell and joins to the middle lobe (ML) at its base. The space between the OL and ML is termed the periostracal groove. Folian-positive material localizes along the outer lobe epithelium. Scale bar 125 μm. (B) Oval inset indicated in (A) showing the sheet-like character of the folian material where it is broken and lifted from the epithelium surface (white arrow). Scale bar 40 μm. (C) Square inset indicated in (A) showing folian positive hemocytes regularly spaced along the length and near the surface of the outer lobe on the shell-facing side. A folian-positive hemocyte is shown moving through the epithelium into the extrapallial cavity (pink arrow). Scale bar 40 μm. Micrograph in (A) without insets from Johnstone et al. (2008).
Previously, immuno-histochemical staining showed that folian protein-positive cells were by far most concentrated along the outer lobe of the mantle epithelium (Figure 6A). In this study, we expand the analysis of this section by showing that the folian proteins are an integral part of what appears to be a structurally cohesive sheet, which is evidenced by regions where the structure is broken and lifted off the surface (Figure 6B). This sheet extends across the outer lobe surface in the direction of the adductor muscle further than previously reported (Supplementary Figure 4). Further analysis of Figure 6A also reveals folian-positive hemocytes in numerous locations, some of which appear to be moving through the outer lobe into the extrapallial space (Figure 6C). A confocal stack of a living outer lobe outer epithelium shows channels approximately 10 μm in diameter that span the entire thickness of the epithelial layer (Supplementary Figures 5A–E). These channels could act as conduits for cell movement into the extrapallial space evident in Figure 6A. A 3-D rendering of a confocal stack of a living shell-facing mantle epithelium shows a Calcein-stained hemocyte moving through the mantle, presumably within a channel (Supplementary Figures 6A–L). The confocal stack of Supplementary Figure 6 can be dynamically viewed in 3D in the Supplementary Video 1.
At higher magnification, immuno-staining shows folian localizes heavily inside cells lining the outer lobe epithelium and at the outer lobe epithelial surface in the PG (Figure 7A). A different material, presumed to be nascent periostracum), is not appreciably immuno-reactive and in general is spatially distinct from the folian-positive material. Folian is secreted nearly but not completely to the base of the PG. Some faint staining at the base of the PG may be due to displacement of material by the mantle, which is highly mobile (Galstoff, 1964). A section analyzed at less saturation shows distinct folian-positive granules inside secretory cells (Figure 7B). Non-reactive cells appear black and are concentrated near the base of the PG.
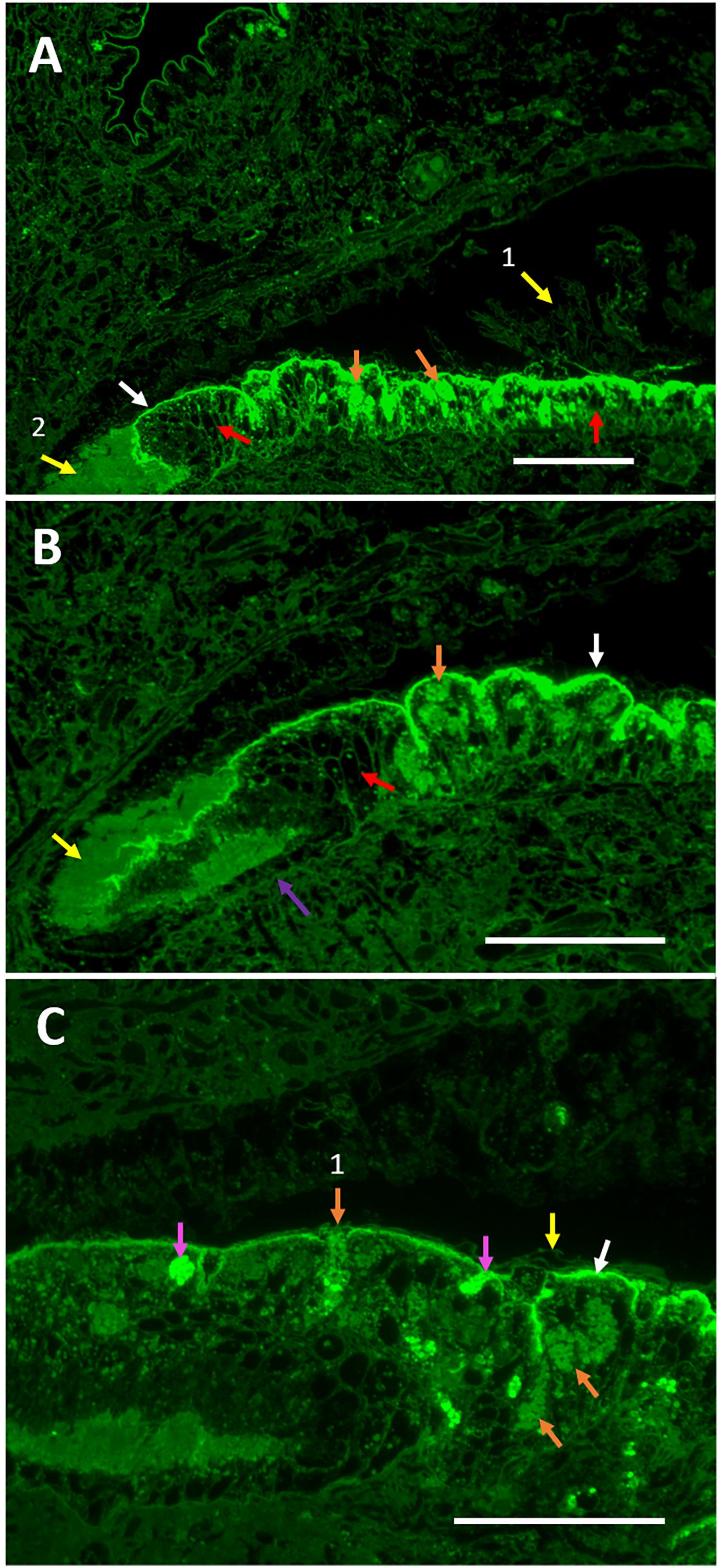
Figure 7. Folian immuno-reactivity in the periostracal groove region between the outer and middle mantle lobes. (A) Intensely staining folian-positive material localizes along the surface of the outer lobe epithelium (white arrow) along with but separate from a different material presumed to be periostracal material (yellow arrow 1). It is presumed that the material at the very base of the groove is periostracal material that collected there and is moved distally to the shell edge during shell formation (yellow arrow 2). Numerous folian-positive secretory cells (orange arrows) are visible and interspersed with non-staining cells which appear black (red arrows). Scale bar 140 μm. (B) Increased magnification shows the groove appears to be lined with simple cuboidal cells (purple arrow) adjacent to the non-staining secretory cells evident on the distal end of the groove (red arrow). At this lower saturation, folian-positive granules are visible (orange arrow) within cells. Periostracal material (yellow arrow) and folian-positive material (white arrow) are evident. Scale bar 120 μm. (C) Additional magnification shows intensely folian-positive cells (pink arrows), possibly hemocytes, which appear to be moving through the epithelium. Secretory cells containing folian-positive granules are visible (orange arrows), one of which appears to be actively secreting product (orange arrow 1). Periostracal material (yellow arrow) is distinct from folian-positive material and appears to float above the folian sheet (white arrow). Scale bar 70 μm).
The diversity of secretory cells and their products are evident in an Azure II-stained companion section from the same region (Supplementary Figure 7A), which also demonstrates that the integrity of the tissue was retained during fixation and sectioning. While it is impossible to specifically correlate the cells in the Azure II-stained sections with the folian-positive cells, some of the lighter staining cells in the Azure-II sections are distributed more or less like the folian-positive cells and appear to have a morphology and vesicle size described by Myers et al. (2007) for phosphoprotein secreting cells, Other, darker staining secretory and cuboidal cells are concentrated near the base of the periostracal groove and appear to be the non-folian staining cells from Figure 7B. It could be hypothesized that these secretory cells are involved in the production of periostracum.
Figure 7C shows two folian-positive cells of interest: one that appears to be a secretory cell releasing folian material (perhaps still in vesicles) into the PG, and a second appears to be a hemocyte en route to the PG. Hemocytes are abundant in the tissue of this region (Supplementary Figure 7B) and can be seen associated with periostracal material (Supplementary Figure 7C).
Discussion
In this study, we report the identification of a gene encoding a highly acidic protein that is part of a diverse family of similar extracellular matrix proteins from the foliated shell layer of the Eastern oyster, Crassostrea virginica. Because of its microstructural origin, we refer to the family as folian and have named this gene folian-cv1. The gene was elucidated starting with the N-terminus sequenced from an electrophoretic band of approximately 48 kDa. The deduced protein has an amino acid composition that is 76% asp and ser and has a molecular weight of 32 kDa, excluding the signal peptide and any post-translational modifications. If fully predicted phosphorylation were included, the molecular weight would be approximately 41 kDa, more closely matching the apparent molecular weight of the electrophoretic band. If the predicted O-glycosylation sites indeed contain carbohydrate, the match between theoretical and observed molecular weight would be even closer. The 48 kDa band and a second prominent 55 kDa band were initially identified by Myers et al. (1996) as containing prominent acidic phosphoproteins based on their Stains-all staining characteristics. Here, the 48 and 55 kDa proteins are shown to be very similar in amino acid composition, with asp, ser, and gly constituting in excess of 80% of the molar content. Moreover, their compositions are very similar to the highly phosphorylated whole EDTA-soluble shell extracellular matrix extract.
Although there are clear defining similarities among the various folian proteins, it is equally clear that there is an underlying diversity and complexity as well. For example, the 48 and 55 kDa bands have proteins that are distinct in their N-terminal regions and their solubility following shell dissolution in EDTA. Further, the composition of the folian-cv1, with its comparably lower gly and concomitantly higher ser and asp content, is distinct from the composition of the 48-kDa band. This observation suggests that there are other proteins in this band, especially those that are high in glycine. Further evidence for other proteins co-migrating with folian-cv1 come from mass spectroscopy results in Johnstone et al. (2008), who revealed specific sequences from the 48 kDa band that are not present in folian-cv1. Additional diversity of folian proteins was evident in this study, as other electrophoretic bands were identified by Western analysis or staining for phosphoprotein and carbohydrate. In the latter case, it was evident that the level of glycosylation varied from band to band.
The complexity of the phosphoprotein components of C. virginica foliated shell has been documented extensively in earlier studies. For example, the principal component of the EDTA-insoluble extracellular foliated shell matrix is a hydrogel-forming aspartate-, glycine-, and phosphoserine-rich complex (Rusenko, 1988; Wheeler, 1992b; Wheeler and Koskan, 1993; see also Table 1) whose water absorption capacity is similar to that of commercial superabsorbents (Mount, 1999). Based on gel permeation studies, the whole EDTA-soluble extract has components that range from approximately 15–20,000 kDa (Rusenko et al., 1991). The various broad molecular weight classes have amino acid compositions nearly identical to each other (Wheeler et al., 1988) and to that reported herein for the whole soluble matrix fraction. Sub-fractionation of the lower molecular class (∼15–600 kDa) by reverse phase HPLC yields two classes of protein, with the principal one being very hydrophilic (RP-1) and having a composition and level of phosphorylation nearly identical to the extracted matrix fractions described in this study (Rusenko et al., 1991). Using chemical degradation analyses, Rusenko et al. (1991) and Donachy et al. (1992) demonstrated that RP-1 and other matrix fractions have runs of aspartic acid much like folian-cv1.
While the origin of the complexity of the folian family of phosphoproteins is not known, prior to acquiring their final form in the shell, matrix proteins may sustain significant post-translational processing. For example, crosslinking of lower molecular weight proteins into the higher molecular weight and insoluble fractions seems likely. The chemical origin of any cross-links of this matrix remains subject to speculation (Wheeler et al., 1988). Comparable to C. virginica, Samata et al. (2008) also reported a gel-like insoluble matrix from the shell of the Iwagaki oyster, Crassostrea nippona. This complex was rendered largely soluble by reducing disulfide linkages, yielding what appears to be a single electrophoretic band of 52 kDa. These observations led the authors to deduce that the matrix could be largely a polymer of this solubilized protein. In contrast, Mount (1999) was able to render soluble only a small percentage of C. viginica insoluble matrix with reduction of these linkages. This is consistent with analyses which show that matrix protein fractions of C. virginica either have considerably lower or, in some studies including the current one, undetectable levels of cys (Wheeler et al., 1988; Rusenko et al., 1991; Kawaguchi and Watabe, 1993). Further, the deduced sequence of folian-cv1 contains no cys.
Non-covalent bonds are another potential source of stabilization of matrix complexes. From the results of the urea extraction of insoluble matrix in this study, it appears that some of the soluble matrix proteins, and in particular those of the 55 kDa band, can be associated with the insoluble matrix in this way. However, these types of interactions do not account for the majority of the insoluble complex, as Mount (1999) showed that only a few percent of the C. virginica insoluble matrix was rendered soluble when treated with chaotropic agents such as urea and SDS. Interestingly, Kawaguchi and Watabe (1993) were able to solubilize much of this matrix with acetone, suggesting the involvement of lipids or perhaps highly hydrophobic regions of the protein in stabilizing the gel.
In addition to cross-linking, post-translation heterogeneity of folian proteins could result from proteolytic cleavage or processing steps at the gene level, such as exon shuffling and gene duplication, recruitment and replication, and slippage (Kocot et al., 2016; Jackson et al., 2017). Some of these genetic processes may explain what appears to be the insertion of significant gly-rich regions into many of the folian proteins and the variants of folian-cv1 from the two populations of oysters as identified in this study.
An overarching question about shell proteins is to what extent their properties are consistent for species that from the same microstructure. In this regard, a comparative analysis of the composition and structure of proteins from foliated structures can be found in the Supplementary Discussion and Supplementary Tables 2, 3. What is perhaps among the most noteworthy of the similarities among these foliated proteins is their high theoretical or actual levels of phosphorylation (Borbas et al., 1991; Sarashina and Endo, 1998, 2001; Johnstone, 2008; Samata et al., 2008; Table 1 and Supplementary Table 3). As the highly phosphorylated matrix proteins described in these various studies come from species that fall within the same order (Ostreoida), it is tempting to speculate that phosphorylation could represent the preservation of an ancestral property necessary for production of the foliated microstructure in this taxon.
It is not known with any certainty what specific role these phosphoproteins have in directing the formation of folia, but there efficacy in controlling mineral growth in vitro has been established. Most models of crystal regulation have some direct interaction between matrix proteins and forming mineral. In this context, it has been demonstrated repeatedly that soluble matrix proteins, like most polyanions, can bind to crystals and their nuclei thereby inhibiting their growth (e.g., Wheeler et al., 1991). Using in vitro crystal growth assays, Borbas et al. (1991) have shown that highly phosphorylated foliated matrices are better inhibitors of calcite growth than less phosphorylated matrices from other microstructures or dephosphorylated C. virginica matrix. Further, the addition of even a single phosphoserine to a polyaspartate molecule greatly increases its capacity to inhibit calcite nucleation (Sikes et al., 1991). Recently, Du et al. (2018) demonstrated that an anti-phosphoserine/threonine/tyrosine antibody injected into the extrapallial cavity of Pinctada fucata effected abnormal prismatic and nacreous layer formation. Further, the authors showed that enzymatic dephoshorylation of matrix proteins reduced their in vitro efficacy for inhibiting calcite growth, modifying calcite crystal morphology and occlusion in growing calcite crystals. It should be noted that the proteins from the shell layers of P. fucata are moderately phosphorylated compared to those from foliated shell, suggesting that phosphorylation of C. virginica shell proteins would be equally, if not more critical for regulating shell formation in this latter species.
In addition to phosphorylation, the primary sequences of oyster folian proteins, in particular their polyaspartate regions, would clearly have an impact on crystal growth regulation. This was demonstrated in a study of direct crystal binding as correlated with inhibition of calcite crystal growth by the oyster folian reverse phase fraction RP-1 and synthetic peptides (Wheeler et al., 1991). Of all the synthetic peptides tested, polyaspartate could bind to the most sites on crystals and resulted in complete inhibition of crystal growth, whereas peptides with combinations of ser and gly inserted between aspartic acids reduced their binding capacity on the crystals with the same fractional loss of inhibitory activity. Like polyaspartate, RP-1 could render complete inhibition of calcite growth when bound at the capacity of the crystals for the protein. However, it had a higher affinity and lower capacity than the polyaspartate, indicating multiple binding sites and, because of its molecular size, coverage of multiple growth sites. It is not immediately obvious what, if any, role the relatively hydrophobic N-terminus of folian-cv1 might be in the control of crystal growth. However, Sikes et al. (1991) demonstrated that the addition of hydrophobic tails to polyaspartate enhanced its ability to inhibit nucleation, presumably by creating a diffusion barrier for the lattice ions to the surface of the forming crystal.
An often-cited role for matrix-biomineral interaction is the control of crystal morphology (Addadi and Weiner, 1989). One reason this function has been promoted results from the presence of crystal faces in shell mineral atypical of inorganically grown calcite, including those of foliar laths (e.g., Checa et al., 2007). In fact, matrix soluble proteins from other microstructures operating from solution can specifically affect crystal morphology and mineralogy (Berman et al., 1988; Belcher et al., 1996). However, Berman et al. (1988) and Addadi and Weiner (1989) pointed out that highly anionic molluscan soluble proteins similar to folian-cv1 are typically adsorbed non-specifically to crystal surfaces. Such non-specific binding could lead to the steps on calcite crystals observed by Wheeler and Sikes (1984) when they were grown in the presence of soluble matrix from C. virginica. Nevertheless, Sikes and Wierzbicki (1995) provided evidence that oyster folian RP-1 proteins selectively adsorbed to these steps, forming crystallographic faces arguably like those found on the surface of folia. Samata et al. (2008) has also shown changes in morphology of crystals grown in vitro with the C. nippona protein solubilized from insoluble matrix. Under select media conditions, structures that resemble foliar laths were observed in their assays.
In addition to operating from solution, earlier models suggest that insoluble proteins form a framework to which crystal growth-controlling anionic proteins attach (e.g., Weiner et al., 1983). In fact, soluble proteins when immobilized in vitro are capable of producing crystal morphologies representative of those that grow in vivo (Belcher et al., 1996; Falini et al., 1996). The association of soluble proteins with insoluble matrix in oyster is suggested by the release of the 55 kDa protein from the insoluble phase with urea and the 48 kDa antibody-reactive sheet discussed below.
Calcium binding by matrix proteins has often been invoked as playing a role in the formation of biomineral. The sequence of folian-cv1 contains no high affinity binding sites for calcium such as might be found in some intracellular calcium-binding proteins. However, its polyanionic structure, especially due to runs of aspartic acid, is likely conducive to low affinity high capacity calcium binding typical of weak carboxylic acids (Williams, 1980), including some asp-rich proteins, such as the sarcoplasmic reticulum calcium-reservoir protein calsequestrin (Milner et al., 1992; Shin et al., 2000; Beard et al., 2004). Citing domains similar to calsequestrin, Gotliv et al. (2005) postulate calcium binding for asprich, a matrix protein from the calcitic prismatic layer of Atrina rigida. Such binding by matrix proteins has been invoked specifically in the process of inducing nucleation and oriented crystal growth in molluscs (e.g., Crenshaw, 1972b; Greenfield et al., 1984; Addadi and Weiner, 1985, 1989). The role of calcium binding in these processes was called into question by Wheeler et al. (1987), who demonstrated that much of the calcium binding in EDTA-extracted matrices was due to residual chelating agent. This finding is supported by chemical and histological observations of matrix preparations (Wada, 1980; Samata and Matsuda, 1986; Pereira-Mouriès et al., 2002; Nudelman et al., 2006). In addition, even the high capacity, low affinity binding of EDTA-free oyster soluble matrix protein was nearly eliminated at the ionic strength equivalent of that of seawater or the extrapallial fluid (Wheeler et al., 1987). EDTA-free oyster insoluble matrix also showed little calcium binding in high ionic strength solutions (Mount, 1999). It should be noted that binding studies conducted in vitro may not represent conditions in mineralizing compartments, including those created by matrix, the mantle epithelium or in vesicles. In this context, Crenshaw (1972a) identified some non-dialyzable calcium when extrapallial fluid was dialyzed against seawater, suggesting that proteins in this mineralization compartment retained some calcium binding activity.
Higher order structures of matrix proteins may have functional significance as well. For example, Addadi and Weiner (1989) provide models, based on crystal growth assays, that support the idea that polyanionic β-pleated sheets would present the optimal stereochemical configuration of protein carboxylate groups to stabilize growth axes and crystal surfaces that are typical of molluscan biominerals. The β configuration for both insoluble matrix (immobilized with and without fixation of the soluble components) and soluble matrix proteins upon binding calcium is supported by X-ray and FTIR analyses, including in some cases matrix from the genus Crassostrea (Weiner and Traub, 1980; Wourms and Weiner, 1986). Lee and Choi (2007) further demonstrated significant β structure in C. gigas soluble matrix using circular dichroism analyses.
Predictions of secondary structure for folian-cv1 using the algorithms PSIPRED, http://bioinf.cs.ucl.ac.uk/psipred (Buchan et al., 2013; Jones, 1999), MESSA, http://prodata.swmed.edu/MESSA/ (Cong and Grishin, 2012), APSSP2, http://crdd.osdd.net (Raghava, 2000), CFSSP, http://www.biogem.org/tool (Chou and Fasman, 1974, 1978; Ashok Kumar, 2013), revealed that there was a high probability that either all or the majority of this protein was a random coil, especially in the asp-ser-rich region. Any exceptions to the random coil structure revealed by these programs occurred in the hydrophobic or glu-rich regions near the N-terminus. Not unexpected by the low complexity, random coil characteristics of folian-cv1, PSIPRED predicts that the vast majority of the structure has a very high probability of intrinsic disorder. Some of the work cited above suggests that the flexibility that comes with disorder might result in induced structures depending on such factors as whether or not the protein is immobilized to insoluble components, is calcium loaded or is interacting with crystals. As an example of an induced secondary structure, the longer runs of aspartic acid may take on β-sheet-like configurations when binding to crystals, as suggested in models and AFM observations for polyaspartate by Sikes and Wierzbicki (1995).
Specialized sequences have been identified in matrix proteins, which may be significant for their higher order structure and function. For example, using various modeling programs, Evans (2012) has revealed cross-β aggregation regions for numerous nacre matrix proteins that could function in the aggregation of matrix proteins. However, using TANGO algorithms, http://tango.crg.es/about.jsp (Linding et al., 2004), no such aggregation regions were identified for folian-cv1. In addition, loop structures have been predicted for much of MPP1 (Samata et al., 2008). These correspond to the phosphorylated SG domains with DE domain inserts. These sequences together would give a regular pattern of the phosphate and carboxylate anions, which the authors suggest could participate in crystal nucleation. This arrangement would appear not applicable to folian-cv1, as the serine in folian-cv1 is not in domains of SG. Glycine loops have also been predicted for Lustrin (Jackson et al., 2017) in which GS rich domains are interspersed with aromatic domains. The authors suggest such loops contribute to the elastic properties of matrix. Again, neither of these regions exist for folian-cv1. Despite lacking the loop forming characteristics described for MPP1 and Lustrin, CFSSP evaluation predicts that the sequence of folian-cv1 is in fact punctuated at irregular intervals with turns. However, any significance for such structures would be hard to predict.
Although from the earlier sections in this discussion it can be concluded that the biochemical relationship and higher structural order of the folian components of C. virginica remain unresolved, the immuno-histological studies presented herein suggest that at least some of the soluble matrix components are included in a cohesive sheet-like structure upon secretion. This extracellular structure is present throughout the region of the mantle that would interface with forming mineral. An association of soluble components with more insoluble ones is consistent with the immuno-histochemical studies of Kawaguchi and Watabe (1993) who showed binding of soluble components to an insoluble matrix in C. virginica shell.
In TEM studies of nacre, the formation of interlamellar sheets is quite evident (Bevelander and Nakahara, 1969; Nakahara et al., 1980) and these authors consider them as significant in defining the limits or compartments of crystal growth. Hydrophobic (silk, fibroin-like) insoluble matrix of nacre appears to be gel-like and is hypothesized to be displaced from the compartments into the intercrystalline matrix by growing crystals (Nudelman et al., 2008). These authors envision the gel as containing acidic proteins which inhibit nucleation events in the general matrix compartment, confining crystal nucleation to specific sites that have been identified on the interlamellar matrix (Crenshaw and Ristedt, 1976; Nudelman et al., 2006).
In earlier TEM studies, Watabe (1965) also described interlamellar sheets for nacre, but he did not identify such sheets in the lamellae of the foliated layer of C. virginica. It is possible that any sheets in the foliated lamellae of the oyster do not remain identifiable as such once they are included into mineral layers. Accordingly, what may represent an alternate final arrangement of the secreted matrix was suggested by Sikes et al. (1998) from AFM studies on what would be interlamellar surfaces of isolated pieces of folia. The authors revealed extensive globular structures on this surface that contained both mineral, and, based on their sensitivity to proteolysis and reactivity to an anti-folian (48 kDa) antibody, they contained matrix protein as well (Sikes et al., 1998).
Watabe (1965) did identify a thinner intercrystalline matrix that surrounds the individual folia and Kawaguchi and Watabe (1993) argued that soluble proteins are attached to this matrix in a way that brings them in contact with crystals. In contrast to nacre, there is no evidence of silk-like proteins in the C. gigas transcriptome (Zhang et al., 2012). However, as mentioned above, the insoluble folian matrix of C. virginica is in fact a gel-forming material. The gel-forming properties of oyster insoluble matrix combined with the proposed association of soluble proteins with the insoluble matrix may be analogous to and have a similar inhibitory function as described above for nacre. Consistent with this inhibitory hypothesis, isolated insoluble matrix gel from C. virginica does not lower the supersaturation required for nucleation in vitro (Mount, 1999). Also, crystal growth does not occur readily on the surface of foliated shell pieces with matrix intact compared to the facile nucleation that occurs on these surfaces from which the matrix was removed (Sikes et al., 2000).
Using the results of cryo-transmission electron microsocopy, Levi-Kalisman et al. (2001) postulate that the interlamellar sheets of nacre are made up largely of chitin, which is perhaps associated with acidic proteins. In contrast to this hypothesis, using multiple analytical techniques, Agbaje et al. (2018) were unable to demonstrate significant chitin in nacre or other microstructures, but they did not include shells with foliated microstructures in their study. Indirect evidence exists that chitin is present in this microstructure. For example, Chan et al. (2018), using chitin-specific probes, has demonstrated chitin associated with shell regeneration in C. virginica, and in particular with hemocytes involved in this process. Further, among all C. gigas tissues, chitin synthase is almost exclusively expressed in mantle, and chitin-binding domains are enriched in shell proteins (Zhang et al., 2012). As yet, no chitin-binding domains have been identified in the folian proteins and are not present in folian-cv1.
From this study, it is evident that the cells involved in the production of the antibody-positive sheet material by the outer mantle epithelium are a fraction of the pseudostratified secretory cells of this mantle region. In TEM studies, Myers et al. (2007) have shown that a class of folian-producing cells are in fact morphologically distinct from the many other secretory cells in this region and have confirmed that they produce folian using the traditional membrane-associated pathway for a secretory protein. As discussed earlier, it is evident that the mineralizing matrix is a complex of proteins, making it possible that they are likely assembled in a variety of cellular and extracellular sites. To this point, Kawaguchi and Watabe (1993) have shown that various oyster matrix proteins are synthesized separately and then assembled either in secretory vesicles or at the mineralization front.
Consistent with earlier studies (Johnstone et al., 2008), the immuno-cytochemistry along with the quantitative PCR results of this study confirm that folian proteins, and specifically folian-cv1, are synthesized within hemocytes, a class of cells that function in non-specific immunity. The granulocytic sub-set of these cells have been shown to function in C. virginica shell formation during experimental induced regeneration of shell by delivering crystals to the mineralization front (Mount et al., 2004). Further, hemocytes participate in mineral formation on implants placed between the outer mantle epithelium and the shell of this oyster (Johnstone et al., 2015). Hemocytes have been shown to be active in shell regeneration of other molluscs as well (e.g., Kádár, 2008; Cho et al., 2011; Li et al., 2016). Further, Huang et al. (2018) have demonstrated the presence of hemocytes with calcium-rich vesicles and crystals in the extrapallial cavity during normal shell formation of Pinctada fucata. Although, in this latter study most shell matrix proteins were not shown to be expressed in hemocytes, Ivanina et al. (2017) have isolated classes of hemocytes from C. gigas that are distinct from the immune response cells and which express genes for both lattice ion transport and shell extracellular matrix production. Of interest in this regard, Patel (2004) has demonstrated that hemocytes from C. virginica release collagen along with folian in vitro. In fact, collagen fibers have been identified on newly regenerated oyster shell (unpublished observations) and a proteomic analysis of C. gigas shell matrix, Zhang et al. (2012) showed that collagen was identified in shell along with other extracellular matrix proteins. As mentioned above, Chan et al. (2018) has demonstrated that hemocytes from C. virginica produce chitin that appears to be involved in shell mineralization.
From the current study, immune-reactive hemocytes appear to move directly from the blood across mantle epithelium into the area of shell layer formation. This movement across the epithelium was described for C. virginica by Galstoff (1964), who used the term diapedesis, implying an analogy to the process of extravasation of leukocytes across blood vessels in vertebrates as part of their immune response. Whether hemocyte movement involves the complex steps found in this immune response is not clear at this time. The results of this study demonstrate channels through which the hemocytes appear to move, suggesting that the process of their passage might be relatively passive. Johnstone et al. (2015) present SEM images of the surface of the outer mantle facing the edge of the shell that appears to be punctuated regularly with pores. The number of these putative pores is greatest at the edge, where the rate of shell formation is the highest, and decreases markedly away from the edge in areas where the rate of shell formation is less. Alternatively, Li et al. (2016) described the hemocyte movement during shell regeneration in the pearl oyster, P. fucata, as a release from secretory cavities. Earlier, Neff (1972a) showed hemocytes with calcium-rich granules positioned under the outer mantle epithelium of the quahog, Mercenaria mercenaria, and speculated that the cells delivered these granules to the mantle via the intercellular space. He further speculated that the granules were matrix protein-calcium complexes. Hemocyte mobilization toward the mineralization front may account for the decrease in folian levels observed in these cells collected from the adductor muscle during experimentally induced shell regeneration (Johnstone et al., 2008). That is, the folian-loaded cells may be selectively recruited to the mineralization front by factors yet unknown, thereby depleting this class of cells in other tissues. Once there, they can aggregate on the substrate in a process that resembles wound healing (Johnstone et al., 2008). In fact, hemocytes were shown to be aligned with matrix fibers during shell regeneration (Mount et al., 2004) suggesting these structures provide recognition or binding sites for the cells.
Beyond the outer mantle epithelium and hemocytes, folian proteins are expressed on the inner mantle epithelium of oyster (Myers et al., 2007) and in a number of other tissues as well (Johnstone et al., 2015), suggesting the possibility that other tissues are involved in the production of shell proteins. Wang et al. (2013) suggests that such proteins could be delivered from tissues to the mineralization front by hemocytes, which, as mentioned above, are present at the sites of shell mineralization. Alternatively, these proteins might have functions that are not directly related to interactions with shell mineral. As an example, bone morphogenic proteins, regulators of orthotopic bone formation, are also present at the sites of vascular calcification (Hruska et al., 2005). Alternatively, rather than promoting mineralization, perhaps folian proteins in these non-shell forming tissues simply prevent ectopic calcification.
Conclusion and Prospectus
Folian-cv1 is a deduced highly acidic phosphoprotein protein from the foliated shell microstructure of the oyster Crassostrea virginica. Its relationship to the apparent myriad of other foliated shell phosphoproteins found in this species (folian family) is unknown and will require detailed biochemical and molecular biological analysis. Of especial interest in this regard is the relatively high glycine content of the general folian class compared to folian-cv1, a situation that poises a genetic dilemma at this time. Nonetheless, the cross reactivity of folian antibodies suggest that the members of the folian family have epitopes in common and thus are related on some level. Folian-cv1 has minimal, if any, identifiable higher order structure, has no known functional domains and is intrinsically disordered. Disorder may be of value if the protein needs to conform to crystal or macromolecular surfaces. Although its specific functions are unknown, because it was deduced from a shell protein and is expressed in mantle, it seems likely that it plays a direct role in shell formation. Given its primary structure and composition, it is likely to have the capacity for crystal growth regulation. Further implicating this class in shell formation, folian proteins are clearly secreted by the shell-forming epithelia, forming a sheet-like structure on their surface. Folian-cv1 is also expressed in hemocytes, expanding the idea that these cells are directly involved in shell formation. It appears that they carry folian proteins through mantle epithelial pores from the hemolymph to the site of mineral formation. More indirect involvement of folian proteins in shell formation or their involvement in non-shell forming process cannot be ruled out at this time. An understanding of folian-cv1 function may be advanced through the use of antibodies made to a recombinant protein in order to localize this protein in shell, cells and secretory products. Also, the change of expression during larval development, especially during the transition from aragonite to calcite shell, and following induced shell repair may provide functional insights, especially if studied in combination with knock-down studies. While folian-cv1 is unique, other bivalves that secrete the foliated microstructure have genes for phosphoproteins which appear to be homologous to varying degrees. A more thorough study of folian-type proteins from a range of species in the order Ostreoida may provide insight into their functions, the origins and expansion of the foliated microstructure and the reasons why this class of proteins is not more rigidly conserved.
Data Availability
The datasets generated for this study can be found in GenBank, with the following accession numbers: Folian cv-1, variant 1: MN108493, Folian cv-1 variant 2: MN108494.
Author Contributions
MJ contributed to the data acquisition and analysis, drafting the manuscript, major editing, and critical review. AW contributed to the data analysis, drafting the manuscript, major editing, and critical review. EF, CS, and AM contributed to the data acquisition and analysis, narrative contribution, and critical review. MS contributed to the data acquisition and analysis, narrative contribution, and review. All authors read and approved the final manuscript.
Funding
This study was supported by the South Carolina Sea Grant Consortium and the United States Airforce Office of Scientific Research project # AFOSR-AF29550-06-1-0133.
Conflict of Interest Statement
EF is currently employed by a company, Academic Analytics. However, her contribution to the manuscript was carried out while employed at Clemson University.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We extend our gratitude to Marta Gomez-Chirarri for her contribution of MMP data and her expertise in the Crassostrea virginica genome and to Jan Lay from the Clemson Computing and Information Technologies and Vera Chan for their help with image layout and quality.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00366/full#supplementary-material
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/nucest
- ^ http://www.expasy.ch/tools/
- ^ https://www.ncbi.nlm.nih.gov/genome/gdv/
- ^ www.ebi.ac.uk/services
- ^ http://www.ncbi.nlm.nih.gov/tools/primer-blast/
- ^ https://www.dna.utah.edu/umelt/um.php
- ^ www.cbs.dtu.dk/services/NetPhos
- ^ https://scansite4.mit.edu/webservice/
- ^ http://www.cbs.dtu.dk/services/NetOGlyc-3.1/
- ^ https://web.expasy.org/sulfinator/
References
Addadi, L., and Weiner, S. (1985). Interactions between acidic proteins and crystals: Stereochemical requirements in biomineralization. Proc. Nat. Acad. Sci. U.S.A. 82, 4110–4114. doi: 10.1073/pnas.82.12.4110
Addadi, L., and Weiner, S. (1989). “Stereochemical and structural relations between macromolecules and crystals in biomineralization,” in Biomineralization: Chemical and Biochemical Perspectives, eds S. Mann, J. Webb, and R. J. P. Williams (Weinheim, FRG: VCH Publishers), 133–156.
Addadi, L., and Weiner, S. (2014). Biomineralization: mineral formation by organisms. Phys. Scr. 89:098003. doi: 10.1088/0031-8949/89/9/098003
Agbaje, O. B. A., Ben Shir, I., Zax, D. B., Schmidt, A., and Jacob, D. E. (2018). Biomacromolecules within bivalve shells: is chitin abundant? Acta Biomater. 80, 176–187. doi: 10.1016/j.actbio.2018.09.009
Aguilera, F., McDougall, C., and Degnan, B. (2017). Co-option and de novo gene evolution underlie molluscan shell diversity. Mol. Biol. Evol. 34, 799–792. doi: 10.1093/molbev/msw294
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ashok Kumar, T. (2013). CFSSP: chou and fasman secondary structure prediction server. Wide Spectr. 1, 15–19. doi: 10.5281/zenodo.50733
Beard, N. A., Laver, D. R., and Dulhunty, A. F. (2004). Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 85, 33–69. doi: 10.1016/j.piomolbio.2003.07.001
Belcher, A. M., Wu, X. H., Christensen, R. J., Hansma, P. K., Stucky, G. D., and Morse, D. E. (1996). Control of crystal phase switching and orientation by soluble mollusk-shell proteins. Nature 381, 56–58. doi: 10.1038/38105a0
Berman, A., Addadi, L., and Weiner, S. (1988). Interactions of sea-urchin skeleton macromolecules with growing calcite crystals-a study of intracrystalline proteins. Nature 331, 546–548. doi: 10.1038/331546a0
Bevelander, G., and Nakahara, H. (1969). An electron microscope study of the formation of the nacreous layer in the shell of certain bivalve molluscs. Calc. Tiss. Res. Int. 3, 84–92. doi: 10.1007/BF02058648
Bonnans, C., Chou, J., and Werb, Z. (2014). Remodeling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell. Biol. 15, 786–801. doi: 10.1038/nrm3904
Borbas, J. E., Wheeler, A. P., and Sikes, C. S. (1991). Molluscan shell matrix phosphoproteins: correlation of degree of phosphorylation to shell mineral microstructure and to in vitro regulation of mineralization. J. Exp. Zool. 258, 1–13. doi: 10.1002/jez.1402580102
Brown, T. (1999). Analysis of DNA sequences by blotting and hybridization. Curr. Protoc. Mol. Biol. 9, 1–20.
Buchan, D. W. A., Minneci, F., Nugent, T. C. O., Bryson, K., and Jones, D. T. (2013). Scalable web services for the PSIPRED protein analysis workbench. Nucl. Acids Res. 41, 340–348. doi: 10.1093/nar/gkt381
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Campbell, K. P., MacLennan, D. H., and Jorgensen, A. O. (1983). Staining for the Ca2+-binding proteins, calsequestrin, calmodulin, troponin C, and S-100, with the carbocyanine dye “Stains-all”. J. Biol. Chem. 258, 11267–11273.
Carter, J. G. (1980). “Selected mineralogical data for the bivalvia,” in Skeletal Growth of Aquatic Organisms: Biological Records of Environmental Change, eds D. C. Rhoads and R. A. Lutz (New York, NY: Plenum Press), 627–643.
Carter, J. G., and Clark, G. R. (1985). “Classification and phylogenetic significance of molluscan shell microstructure,” in Mollusks: Notes for a Short Course, ed. T. W. Broadhead (Knoxville TN: Univ. of Tennessee, Dept. of Geological Studies, Studies in Geology).
Chan, V. B. S., Johnstone, M. B., Wheeler, A. P., and Mount, A. S. (2018). Chitin facilitated mineralization in the Eastern oyster. Front. Mar. Sci. 5:347. doi: 10.33891/fmars.2018.00347
Checa, A. (2000). A new model for periostracum and shell formation in Unionidae (Bivalvia, Mollusca). Tissue Cell 32, 405–416. doi: 10.1054/tice.2000.0129
Checa, A. G., Esteban-Delgado, F. J., and Rodríquez-Navarro, A. B. (2007). Crystallographic structure of the foliated calcite of bivalves. J. Struct. Biol. 157, 393–402. doi: 10.1016/j.jsb.2006.09.005
Cho, S. M., Lee, Y. M., and Jeong, W. G. (2011). Effect of polycyclic aromatic hydrocarbon (PAH) on shell repair in the Pacific Oyster, Crassostrea gigas. Korean J. Malacol. 27, 35–42. doi: 10.9710/kjm.2011.27.1.035
Chou, P. Y., and Fasman, G. D. (1974). Prediction of protein conformation. Biochemistry 13, 222–245. doi: 10.1021/bi00699a002
Chou, P. Y., and Fasman, G. D. (1978). Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. 47, 145–148.
Cong, Q., and Grishin, N. V. (2012). MESSA: Meta-server for protein sequence analysis. BMC Biol. 10:82. doi: 10.1186/1741-7007-10-82
Cornish-Bowden, A. (1983). Relating proteins by amino acid composition. Meth. Enzymol. 91, 60–75. doi: 10.1016/S0076-6879(83)91011-x
Crenshaw, M. A. (1972a). The inorganic composition of molluscan extrapallial fluid. Biol. Bull. 143, 506–512. doi: 10.2307/1540180
Crenshaw, M. A. (1972b). The soluble matrix from Mercenaria mercenaria shell. Biomineralization 6, 6–11.
Crenshaw, M. A., and Ristedt, H. (1976). “The histochemical localization of reactive groups in septal nacre from Nautilus pompilius L,” in The Mechanisms of Mineralization in the Invertebrates and Plants, eds N. Watabe and K. M. Wilbur (Columbia, SC: University of South Carolina Press), 355–367.
Cunningham, C., Hikima, J., Jenny, M. J., Chapman, R. W., Fang, G. C., Saski, C., et al. (2006). New resources for marine genomics: bacterial artificial chromosome libraries for the Eastern and Pacific oysters (Crassostrea virginica and C. gigas). Mar. Biotechnol. 8, 521–533. doi: 10.1007/S10126-006-6013-9
Currey, J. D. (1999). The design of mineralized hard tissues for their mechanical functions. J. Exp. Biol. 202, 3285–3294.
Donachy, J. E., Drake, B., and Sikes, C. S. (1992). Sequence and atomic-force microscopy analysis of a matrix protein from the shell of the oyster Crassostrea virginica. Mar. Biol. 114, 423–428. doi: 10.1007/BF00350033
Du, J., Xu, G., Liu, C., and Zhang, R. (2018). The role of phosphorylation and dephosphorylation of shell matrix proteins in shell formation: an in vivo and in vitro study. Crystal Eng. Comm. 20, 3905–3916. doi: 10.1039/c8ce00755a
Ebel, A. E., and Scro, R. (1996). “General anatomy,” in The Eastern Oyster: Crassostrea virginica, eds V. S. Kennedy, R. I. E. Newell, and A. F. Eble (College Park, MD: Maryland Sea Grant), 19–73.
Eisenreich, S. J., Bannerman, R. T., and Armstrong, D. E. (1975). A simplified phosphorus analysis technique. Environ. Lett. 9, 43–53. doi: 10.1080/00139307509437455
Esteban-Delgado, F. J., Harper, E. M., Checa, A. G., and Rodríguez-Navarro, A. B. (2008). Origin and expansion of foliated microstructure in pteriomorph bivalves. Biol. Bull. 214, 153–165. doi: 10.2307/25066672
Evans, J. S. (2012). Aragonite-associated biomineralization proteins are disordered and contain interactive motifs. Bioinformatics 28, 3182–3185. doi: 10.1093/bioinformatics/bts604
Falini, G., Albeck, S., Weiner, S., and Addadi, L. (1996). Control of aragonite or calcite polymorphism by mollusk shell macromolecules. Science 271, 67–69. doi: 10.1126/science.271.5245.67
Falwell, E. P. (2015). The Role of Folian and Collagenases on Shell Repair in the Eastern oyster, Crassostrea virginica. Ph.D. dissertation. Clemson: Virginia Polytechnic and State University.
Fauchére, J.-L., and Pliška, V. (1983). Hydrophobic parameters of amino-acid side chains from the partitioning of N-acetyl-amino-acid amines. Eur. J. Med. Chem. Chim. Ther. 18, 369–375.
Galstoff, P. S. (1964). The American Oyster, Crassostrea virginica (Gmelin). Fishery Bulletin 64. Washington, D.C: U. S. Fish and Wildlife Services.
Gomez-Chiarri, M., and McDonnell, W. W. (2017). Crassostrea virginica sequence assembly Version 3.0.
Gordon, D., Abajian, C., and Green, P. (1998). Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202. doi: 10.1101/gr.8.3.195
Gotliv, B. A., Kessler, N., Sumerel, J. L., Morse, D. E., Tuross, N., Addadi, L., et al. (2005). Asprich: a novel aspartic acid-rich protein family from the prismatic shell matrix of the bivalve Atrina rigida. Chembiochem 6, 304–318. doi: 10.1002/cbic.200400221
Greenfield, E. M., Wilson, D. C., and Crenshaw, M. (1984). Ionotropic nucleation of calcium carbonate by molluscan matrix. Am. Zool. 24, 925–932. doi: 10.1093/icb/24.4.925
Hruska, K. A., Suresh, M., and Saab, G. (2005). Bone morphogenetic proteins in vascular calcification. Circ. Res. 97, 105–114. doi: 10.1161/01.RES.0000017551.53833.6c
Huang, J., Li, S., Liu, Y., Liu, C., Xie, L., and Zhang, R. (2018). Hemocytes in the extrapallial space of Pinctada fucata are involved in immunity and biomineralization. Sci. Rep. 8:4657. doi: 10.1038/s41598-018-22961-y
Hüning, A. K., Lange, S. M., Ramesh, K., Jacob, D. E., Jackson, D. J., Panknin, U., et al. (2016). A shell regeneration assay to identify biomineralization candidate genes in mytilid mussels. Mar. Genomics 27, 57–67. doi: 10.1016/j.margen.2016.03.011
Ivanina, A. V., Falfushynska, H. I., Beniash, E., Piontkivska, H., and Sokolova, I. M. (2017). Biomineralization-related specialization of hemocytes and mantle tissues of the Pacific oysters Crassostrea gigas. J. Exp. Biol. 220, 3209–3221. doi: 10.1242/jeb.160861
Jackson, D. J., Reim, L., Randow, C., Cerveau, N., Degnan, B., and Fleck, C. (2017). Variation in orthologous shell-forming proteins contribute to molluscan shell diversity. Mol. Biol. Evol. 34, 2959–2969. doi: 10.1093/molbev/msx232
Jing, X., Espinosa, E. P., Perrigault, M., and Adam, B. (2011). Identification, molecular characterization and expression analysis of a mucosal C-type lectin in the eastern oyster, Crassostrea virginica. Fish. Shellfish Immunol. 30, 851–858. doi: 10.1016/j.fsi.2011.01.007
Johnstone, M. B. (2008). A Comparative Study of Molluscan Shell Proteins With an Emphasis on the Structure of Two Phosphoproteins Derived From the Eastern Oyster, Crassostrea virginica. Ph.D. dissertation, Clemson University, Clemson, SC.
Johnstone, M. B., Ellis, S., and Mount, A. S. (2008). Visualization of shell matrix proteins in hemocytes and tissues of the Eastern oyster. Crassostrea virginica. J. Exper. Zool. 310B, 227–239. doi: 10.1002/jez.b.21206
Johnstone, M. B., Gohad, N. V., Falwell, E. P., Hansen, D. C., Hansen, K. M., and Mount, A. S. (2015). Cellular orchestrated biomineralization of crystalline composites on implant surfaces by the eastern oyster, Crassostrea virginica (Gmelin, 1791). J. Exp. Biol. Ecol. 463, 8–16. doi: 10.1016/j.jembe.2014.10.014
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202. doi: 10.1006/jmbi.1999.3091
Kádár, E. (2008). Hemocyte response associated with induction of shell regeneration in the deep-sea vent mussel Bathymodiolus azoricus (Bivalvia: Mytilidae). J. Exp. Mar. Biol. Ecol. 362, 71–78. doi: 10.1016/jembe.2008.05.014
Kawaguchi, T., and Watabe, N. (1993). The organic matrices of the shell of the American oyster Crassostrea virginica Gmelin. J. Mar. Biol. Ecol. 170, 11–28. doi: 10.1016/0022-0981(93)90126-9
Kocot, K. M., Aguilera, F., McDougall, C., Jackson, D. J., and Degnan, B. M. (2016). Seashell diversity and rapidly evolving secretomes: insights into the evolution of biomineralization. Front. Zool. 13:23. doi: 10.1186/s12983-016-0155-z
Lee, S. L., Veis, A., and Glonek, T. (1977). Dentin phosphoprotein: an extracellular calcium-binding protein. Biochemistry 16, 2971–2979. doi: 10.1021/bi00632a026
Lee, S. W., and Choi, S. C. (2007). The correlation between organic matrices and biominerals (myostracal prism and folia) of the adult oyster shell. Crassostrea gigas. Micron 38, 58–64. doi: 10.1016/j.micron.2006.03.018
Levi-Kalisman, Y., Falini, G., Addadi, L., and Weiner, S. (2001). Structure of the nacreous organic matrix of a bivalve mollusk shell examined in the hydrated state using cryo-TEM. J. Struct. Biol. 135, 8–17. doi: 10.1006/jsbi.2001.4372
Li, S., Liu, Y., Liu, C., Huang, J., Zheng, G., and Xie, L. (2016). Hemocytes participate in calcium carbonate crystal formation, transportation and shell regeneration in the pearl oyster Pinctada fucata. Fish Shellfish Immunol. 51, 263–270. doi: 10.1016/j.fsi.2016.02.027
Linding, R., Schymkowitz, J., Rousseau, F., Diella, F., and Serrano, L. (2004). A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 342, 345–353. doi: 10.1016/j.jmb.2004.06.088
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506. doi: 10.1093/nar/gki937
Lowenstam, H. A. (1981). Minerals formed by organisms. Science 211, 1125–1131. doi: 10.1126/science.7008198
Lowenstam, H. A., and Weiner, S. (1989). On Biomineralization. New York, NY: Oxford University Press.
Luke, G. H., and William, J. L. (2001). Inhibitors of procollagen C-terminal proteinase block gastrulation and spicule elongation in the sea urchin embryo. Dev. Growth Diff. 43, 415–424. doi: 10.1046/j.1440-169x2001.00589.x
Mann, S. (1988). Molecular recognition in biomineralization. Nature 332, 119–124. doi: 10.1038/332119a0
Marin, F., Luquet, G., Marie, B., and Medakovic, D. (2008). Molluscan shell proteins: primary structure, origin, and evolution. Curr. Top. Dev. Biol. 80, 209–276. doi: 10.1016/S0070-2153(07)80006-8
Milner, R. E., Famulski, K. S., and Michalak, M. (1992). Calcium binding proteins in the sarcoplasmic/endoplasmic reticulum of muscle and nonmuscle cells. Mol. Cell. Biochem. 112, 1–13. doi: 10.1007/BF00229637
Mount, A. S. (1999). Nucleation of Calcite in the Eastern oyster, Crassostrea virginica: Biochemical and Functional Studies of the Organic Matrix From Foliated Shell. Ph.D. dissertation, Clemson University, Clemson, SC.
Mount, A. S., Gohad, N. V., Hansen, D. C., Hansen, K. M., and Johnstone, M. B. (2016). Deposition of Nanocrystalline Calcite on Surfaces by a Tissue and Cellular Biomineralization. Available at: https://www.google.com/patents/US9371451 (accessed June 21, 2016).
Mount, A. S., Wheeler, A. P., Paradkar, R. P., and Snider, D. (2004). Hemocyte-mediated shell mineralization in the Eastern oyster. Science 304, 297–300. doi: 10.1126/science.1090506
Muller, P. Y., Janovjak, H., Miserez, A. R., and Dobbie, Z. (2002). Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32, 1372–1374.
Myers, J. M. (1999). Studies on the Secretory Epithelium of the Mantle From the Eastern oyster, Crassostrea virginica, in Relation to Shell Growth. Ph.D. dissertation, Clemson University, Clemson, SC.
Myers, J. M., Johnstone, M. B., Mount, A. S., Silverman, H., and Wheeler, A. P. (2007). TEM immunocytochemistry of a 48 kDa MW organic matrix phosphoprotein produced in the mantle epithelial cells of the Eastern oyster (Crassostrea virginica). Tissue Cell 39, 247–256. doi: 10.1016/j/tice.2007.03.006
Myers, J. M., Veis, A., Sabsay, B., and Wheeler, A. P. (1996). A method for enhancing the sensitivity and stability of Stains-All for phosphoproteins separated in sodium dodecyl sulfate-polyacrylamide gels. Anal. Biochem. 240, 200–302. doi: 10.1006/abio.1996.0361
Nakahara, H., Kakei, M., and Bevelander, G. (1980). Fine structure and amino acid composition of the organic “envelope” in the prismatic layer of some bivalve shells. Venus 39, 167–177.
Neff, J. M. (1972a). Ultrastructure of the outer epithelium of the mantle in the clam Mercenaria mercenariain relation to calcification of the shell. Tissue Cell 4, 591–600. doi: 10.1016/S0040-8166(72)80032-6
Neff, J. M. (1972b). Ultrastructure studies of periostracum formation in the hard shelled clam Mercenaria mercenaria (L). Tissue Cell 4, 311–326. doi: 10.1016/S0040-8166(72)80050-8
Nudelman, F., Gotliv, B. A., Addadi, L., and Weiner, S. (2006). Mollusk shell formation: Mapping the distribution of organic matrix components underlying a single aragonitic table in nacre. J. Struct. Biol. 153, 176–187. doi: 10.1016/j.jsb.2005.09.009
Nudelman, F., Shimoni, E., Klein, E., Rousseau, M., Bourrat, X., Lopez, E., et al. (2008). Forming nacreous layer of the shells of the bivalves Atrina rigida and Pinctada margaritifera: an environmental- and cryo-scanning electron microscopy study. J. Struct. Biol. 162, 290–300. doi: 10.1016/j.jsb.2008.01.008
Ortega, N., Behonick, D., Stickens, D., and Werb, Z. (2003). How proteases regulate bone morphogenesis. Ann. N.Y. Acad. Sci. 995, 109–116. doi: 10.1111/j.1749-6632.2003.tb03214.x
Patel, S. V. (2004). A Novel Function of Invertebrate Collagen in the Biomineralization Process During the Shell Repair of Eastern oyster, Crassostrea virginica. M.S. thesis, Clemson University, Clemson, SC.
Pereira-Mouriès, L., Almeida, M.-J., Ribeiro, C., Peduzzi, J., Barthélemy, M., Milet, C., et al. (2002). Soluble silk-like organic matrix in the nacreous layer of the bivalve Pinctada maxima. Eur. J. Biochem. 269, 4994–5003. doi: 10.1046/j.1432-1033.2002.03203.x
Raghava, G. P. S. (2000). Protein secondary structure prediction using nearest neighbor and neural network approach. CASP 4, 75–76.
Reese, M. G., Eeckman, F. H., Kulp, D., and Haussler, D. (1997). Improved splice site detection in Genie. J. Comput. Biol. 4, 311–323. doi: 10.1089/cmb.1997.4.311
Roling, J. A., Bain, L. J., and Baldwin, W. S. (2004). Differential gene expression in mummichogs (Funulus heteroclitus) following treatment with pyrene: comparison to a creosote contaminated site. Mar. Environ. Res. 57, 377–395. doi: 10.1016/j.marenvires.2003.11.001
Rozen, S., and Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Meth. Mol. Biol. 132, 365–386. doi: 10.1385/1-59259-192-2:365
Rusenko, K. (1988). Studies on the Structure and Function of the Shell Matrix Proteins from the American oyster, Crassostrea virginica. Ph.D. dissertation, Clemson University, Clemson, SC.
Rusenko, K. W., Donachy, J. E., and Wheeler, A. P. (1991). “Purification and characterization of a shell matrix phosphoprotein from the American oyster,” in Surface Reactive Peptides and Polymers ACS Symposium Series 444, eds C. S. Sikes and A. P. Wheeler (Washington, DC: ACS), 107–124. doi: 10.1021/bk-1991-0444.ch008
Saleuddin, A. S. M., and Petit, H. P. (1983). “The mode of formation and the structure of periostracum,” in The Mollusca, Vol. 4, eds K. M. Wilbur and A. S. M. Saleuddin (New York: Academic Press), 199–234. doi: 10.1016/b978-0-12-751404-8.50013-x
Samata, T., Ikeda, D., Kajikawa, A., Sato, H., Nogawa, C., Yamada, D., et al. (2008). A novel phosphorylated glycoprotein in the shell matrix of the oyster Crassostrea nippona. FEBS J. 275, 2977–2989. doi: 10.1111/j.1742-4658.2008.06453.x
Samata, T., and Matsuda, M. (1986). Contaminating peptides widely present in ion-exchanged water, reagents, experimental implements and natural sample. Comp. Biochem. Physiol. 84B, 531–535. doi: 10.1016/0305-0491(86)90118-5
Sarashina, I., and Endo, K. (1998). Primary structure of a soluble matrix protein of the scallop shell: implications for calcium carbonate biomineralization. Am. Mineral. 83, 1510–1515. doi: 10.2138/am-1997-11-1239
Sarashina, I., and Endo, K. (2001). The complete primary structure of molluscan shell protein 1 (MSP-1), an acidic glycoprotein in the shell matrix of the scallop Patinopecten yessoensis. Mar. Biotechnol. 3, 362–369. doi: 10.1007/s10126-001-0013-6
Shin, D. W., Ma, J., and Kim, D. H. (2000). The asp-rich region of the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 486, 178–182. doi: 10.1016/S0014-57193(00)02246-8
Sikes, C. S., Wheeler, A. P., Wierzbicki, A., Dillaman, R. M., and DeLuca, L. (1998). Oyster shell protein and atomic force microscopy of oyster shell folia. Biol. Bull. 194, 304–316. doi: 10.2307/1543100
Sikes, C. S., Wheeler, A. P., Wierzbicki, A., Mount, A. S., and Dillaman, R. M. (2000). Nucleation and growth of calcite on native versus pyrolyzed oyster shell folia. Biol. Bull. 198, 50–66. doi: 10.2307/1542803
Sikes, C. S., and Wierzbicki, A. (1995). “Mechanisms of regulation of crystal growth in selected biological systems,” in Mineral Scale Formation and Inhibition, ed. Z. Amjad (New York, NY: Plenum Press), 183–206. doi: 10.1007/978-1-4899-1400-2_16
Sikes, C. S., Yeung, M. L., and Wheeler, A. P. (1991). “Inhibition of calcium carbonate and phosphate crystallization by peptides enriched in aspartic acid and phosphoserine,” in Surface Reactive Peptides and Polymers, eds C. S. Sikes and A. P. Wheeler (Washington, DC: American Chemical Society), 72–84.
Simkiss, K., and Wilbur, K. M. (1989). Biomineralization: Cell Biology and Mineral Deposition. San Diego, CA: Academic Press.
Suzuki, M., Saruwatari, K., Kogure, T., Yamamoto, Y., Nishimura, T., Kato, T., et al. (2009). An acidic matrix protein, Pif, is a key macromolecule for nacre formation. Science 325, 1388–1390. doi: 10.1126/science.1173793
Wada, K. (1980). “Initiation of mineralization in bivalve molluscs,” in The Mechanisms of Biomineralization in Animals and Plants, eds M. Omori and N. Watabe (Tokyo: Tokai University Press), 79–92.
Waite, J. H., and Benedict, C. V. (1984). Assay of dihydroxyphenylalanine (dopa) in invertebrate structural proteins. Meth. Enzymol. 107, 297–413. doi: 10.1016/0076-6879(84)07028-2
Wang, X., Li, L., Zhu, Y., Du, Y., Song, X., and Chen, Y. (2013). Oyster shell proteins originate from multiple organs and their probable transport pathway to the shell formation front. PLoS One 8:e66522. doi: 10.1371/journal.pone.0066522
Watabe, N. (1965). Studies on shell formation XI. Crystal-matrix interactions in the inner layer of mollusk shells. J. Ultrastruct. Res. 12, 351–370. doi: 10.1016/S0022-5320(65)80104-6
Weiner, S., and Dove, P. M. (2003). An overview of biomineralization processes and the problem of the vital effect. Rev. Mineral. Geochem. 54, 1–29. doi: 10.2113/0540001
Weiner, S., and Traub, W. (1980). X-ray diffraction study of the insoluble organic matrix of mollusk shells. FEBS Lett. 111, 311–316. doi: 10.1016/0014-5793(80)80817-9
Weiner, S., Traub, W., and Lowenstam, H. A. (1983). “Organic matrix in calcified exoskeleton,” in Biomineralization and Biological Metal Accumulation, eds P. Westbroek and E. W. de Jong (Dordrecht Holland: D. Reidel Publishing Co.), 205–224. doi: 10.1007/978-94-009-7944-4_18
Wheeler, A. P. (1992a). “Mechanisms of molluscan shell formation,” in Calcification in Biological Systems, ed. E. Bonucci (Boca Raton, FL: CRC Press), 179–216.
Wheeler, A. P. (1992b). “Phosphoproteins of oyster (Crassostrea virginica) shell organic matrix,” in Hard Tissue Mineralization and Demineralization, eds S. Suga and N. Watabe (Tokyo: Springer-Verlag), 171–187. doi: 10.1007/978-4-431-68183-0_11
Wheeler, A. P., and Koskan, L. (1993). “Large scale thermally synthesized polyaspartate as a biodegradable substitute in polymer applications,” in Proceedings of the Biomolecular Materials MRS Symposium, Vol. 292, eds C. Viney, S. K. Case, and J. H. Waite (Pittsburg), 277–283.
Wheeler, A. P., Low, K. C., and Sikes, C. S. (1991). “CaCO3 crystal-binding properties of peptides and their influence on crystal growth,” in Surface Reactive Peptides and Polymers, eds C. S. Sikes and A. P. Wheeler (Washington, DC: American Chemical Society), 72–84. doi: 10.1021/bk-1991-0444.ch006
Wheeler, A. P., Rusenko, K. W., George, J. W., and Sikes, C. S. (1987). Evaluation of calcium binding by molluscan shell organic matrix and its relevance to biomineralization. Comp. Biochem. Physiol. 87B, 953–960. doi: 10.1016/0305-0491(87)90418-4
Wheeler, A. P., Rusenko, K. W., Swift, D. M., and Sikes, C. S. (1988). Regulation of in vitro and in vivo CaCO3 crystallization by fractions of oyster shell organic matrix. Mar. Biol. 98, 71–80. doi: 10.1007/BF00392660
Wheeler, A. P., and Sikes, C. S. (1984). Regulation of carbonate calcification by organic matrix. Am. Zool. 24, 933–944. doi: 10.1093/icb/24.4.933
Wheeler, A. P., and Sikes, C. S. (1989). “Matrix-crystal interactions in CaCO3 biomineralization,” in Biomineralization: Chemical and Biochemical Perspectives, eds S. Mann, J. Webb, and R. J. P. Williams (Weinheim, FRG: VCH Publishers), 95–131.
Williams, R. J. P. (1980). “A general introduction to the special properties of the calcium ion and their deployment in biology,” in Calcium-Binding Proteins: Structure and Function, eds F. L. Siegel, E. Carafoli, R. H. Kretsinger, D. H. MacLennan, and D. H. Wasserman (New York: Elsevier), 3–10.
Wourms, D., and Weiner, S. (1986). Mollusk shell organic matrix: Fourier transform infrared study of the acidic macromolecules. J. Exp. Zool. 237, 11–20. doi: 10.1002/jez.1402370104
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Keywords: oyster, Crassostrea, shell formation, matrix phosphoproteins, hemocytes, mantle, shell protein evolution
Citation: Johnstone MB, Wheeler AP, Falwell EP, Staton ME, Saski CA and Mount AS (2019) Folian-cv1 Is a Member of a Highly Acidic Phosphoprotein Class Derived From the Foliated Layer of the Eastern Oyster (Crassostrea virginica) Shell and Identified in Hemocytes and Mantle. Front. Mar. Sci. 6:366. doi: 10.3389/fmars.2019.00366
Received: 11 February 2019; Accepted: 13 June 2019;
Published: 02 July 2019.
Edited by:
Pei-Yuan Qian, The Hong Kong University of Science and Technology, Hong KongReviewed by:
Yang Zhang, South China Sea Institute of Oceanology (CAS), ChinaVengatesen Thiyagarajan (Rajan), The University of Hong Kong, Hong Kong
Copyright © 2019 Johnstone, Wheeler, Falwell, Staton, Saski and Mount. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Beth Johnstone, bWJqQGNsZW1zb24uZWR1; Andrew S. Mount, bW91bnRAY2xlbXNvbi5lZHU=
†Present address: Mary Beth Johnstone, Department of Genetics and Biochemistry, Clemson University, Clemson, SC, United States; Elizabeth P. Falwell, Academic Analytics, LLC, Charlotte, NC, United States