 Luis Felipe Muriel-Millán1
Luis Felipe Muriel-Millán1 José Luis Rodríguez-Mejía1
José Luis Rodríguez-Mejía1 Elizabeth Ernestina Godoy-Lozano1†
Elizabeth Ernestina Godoy-Lozano1† Nancy Rivera-Gómez1†
Nancy Rivera-Gómez1† Rosa-María Gutierrez-Rios1
Rosa-María Gutierrez-Rios1 Daniel Morales-Guzmán2
Daniel Morales-Guzmán2 María R. Trejo-Hernández2
María R. Trejo-Hernández2 Alejandro Estradas-Romero1†
Alejandro Estradas-Romero1† Liliana Pardo-López1*
Liliana Pardo-López1*- 1Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
- 2Centro de Investigación en Biotecnología, Universidad Autónoma del Estado de Morelos, Cuernavaca, Mexico
Indigenous bacterial populations play an important role in the restoration of crude oil-polluted marine environments. The identification and characterization of these bacteria are key in defining bioremediation strategies for the mitigation of possible future oil spills. In this work, we characterized Pseudomonas aeruginosa strain GOM1, which was isolated from the water column in the southwestern Gulf of Mexico. Phylogenetic analysis revealed that GOM1 strain was most closely related to P. aeruginosa WC55, a strain isolated from the northern Gulf of Mexico after the Deepwater Horizon oil spill. The hydrocarbon-degrading capacity of P. aeruginosa GOM1 was investigated using various approaches. This strain degraded 96% of the aliphatic fraction (C12–C38) of crude oil during a 30-day incubation period, exhibiting a high activity on long-chain alkanes, and expressing alkane hydroxylases AlkB1, AlkB2 and AlmA. Addition of nitrogen and phosphate to seawater culture medium enhanced hexadecane degradation by GOM1. Additionally, the strain exhibited high surfactant/rhamnolipid production and emulsifying activity when grown in a complex medium in the presence of hexadecane. Comparisons of growth kinetics, hydrocarbon degradation and gene expression between GOM1 and the closely related P. aeruginosa laboratory strain PAO1 revealed that the marine isolate is better adapted to degrade alkanes. Taken together, our results place P. aeruginosa GOM1 as a potentially effective candidate to be included in a consortium for use in the bioremediation of oil-polluted sites.
Introduction
Oil spills severely affect many different ecosystems due to the high toxicity and recalcitrance of crude oil in the environment. Various strategies have been used for the restoration of crude oil-contaminated sites, among which bioremediation is considered a more efficient and eco-friendly alternative, compared to physical and chemical treatments (Mapelli et al., 2017). Natural bioremediation is usually done by endogenous microbial populations (fungi, microalgae, and bacteria) present in crude oil-polluted sites, playing an important role in the degradation of contaminants and thus contributing to the restoration of the environment. Traditional culturing methods and metagenomic analyses of bacterial communities from contaminated sites have been used to understand how crude oil biodegradation is accomplished (Lamendella et al., 2014; Xue et al., 2015). Several species from the genera Pseudomonas, Alcanivorax, Acinetobacter, Marinobacter, Bacillus, Dietzia, Rhodococcus, among others, have been reported as able to degrade aromatic and/or aliphatic hydrocarbons (Wentzel et al., 2007; Ghosal et al., 2016). Usually, marine environments hold hydrocarbon-degrading bacteria in low abundance (Godoy-Lozano et al., 2018), but oil spills trigger blooms of such populations, exhibiting a bacterial succession according to the type of hydrocarbons present. For instance, in the Deepwater Horizon (DWH) spill that occurred in the northern Gulf of Mexico in 2010, Oceanospirillales and Pseudomonas taxa dominated the microbial community when the concentration of n-alkanes and cycloalkanes was highest at depth (900–1300 m) during the initial phase of the spill. On the other hand, Colwellia and Cycloclasticus taxa were enriched at high concentrations of aromatic hydrocarbons, which occurred 43 days after the spill once the partial capture of oil and gas was performed (Dubinsky et al., 2013).
Aromatic hydrocarbons (AHs) are molecules composed of one or more benzene rings and can constitute between 0 and 60% of crude oil (Hollebone, 2015). The study of AH biodegradation in marine environments is relevant due to these molecules being highly toxic, mutagenic and carcinogenic, and may affect wildlife and human’s health via food chain (Tiwari et al., 2017; Poirier et al., 2019). In general, aerobic degradation of AH involves two major steps, known as the upper and the lower pathways. In the upper pathway, mono- or dioxygenases carry out hydroxylation and dehydrogenation reactions of the aromatic ring(s), transforming it (them) into a catechol molecule (or a closely related compound). In the lower pathway, the catechol is cleaved by a second dioxygenase and then transformed by subsequent reactions, resulting in products which are incorporated into the Tricarboxylic acid cycle (George and Hay, 2011).
Alkanes also represent an important fraction of crude oil, which can reach up to 90%, depending on the petroleum source (Hollebone, 2015). Given their low solubility and reactivity, alkane degradation represents a challenge to bacteria. Nonetheless, some bacteria have the metabolic capacity to use alkanes as carbon and energy sources (Rojo, 2009). The bacterial aerobic degradation of alkanes involves an initial hydroxylation step, catalyzed by alkane hydroxylases (AHs), of which the membrane-embedded AlkB enzymes are the most widely distributed among bacteria from different environments (Nie et al., 2014).
Pseudomonas putida GPo1 is one of the best-characterized alkane-degrading strains. This Gram-negative bacterium can oxidize n-alkanes ranging from C5 to C13, but is unable to metabolize longer-chain alkanes (van Beilen et al., 2005). Nevertheless, other Pseudomonas species have been reported as being capable of degrading a wide range of aliphatic hydrocarbons. One of these is P. aeruginosa, which is widely distributed in different ecosystems. While this species has been identified as an opportunistic pathogen for humans, several strains of P. aeruginosa that were isolated from crude oil-polluted environments, have been found to have a high alkane-degrading capacity (Chaerun et al., 2004; Liu et al., 2014; Thomas et al., 2014). Unlike P. putida, which contains one AlkB, P. aeruginosa has two homologous AlkB hydroxylases; these are differentially induced depending on the length of the hydrocarbon chain present, thus allowing this species to degrade a wide range of alkanes (Marín et al., 2003; Liu et al., 2014). In addition, P. aeruginosa synthesizes rhamnolipids, which are amphipathic molecules composed of one or two (L)-rhamnose- moieties linked to β-hydroxylated fatty acid chains (Chong and Li, 2017). Rhamnolipids are biosurfactants that promote the solubilization of hydrocarbons (including long-chain alkanes), enhancing their biodegradation (Zhang and Miller, 1995). Alkane uptake in P. aeruginosa also induces the expression of proteins involved in various other cellular processes, such as chemotaxis, membrane transport, biofilm formation, stress response and secretion, suggesting that alkane consumption is a complex process in this species (Liu et al., 2015).
While P. aeruginosa strains isolated from different habitats tend to have a high degree of genomic conservation, they can exhibit differences in gene expression and in a variety of phenotypes, including surfactant production, motility, and antibiotic susceptibility, among others (Grosso-Becerra et al., 2014). Indeed, some environmental isolates of P. aeruginosa have greater alkane-degradation capacities than clinical strains (Smits et al., 2003; Gunasekera et al., 2017), suggesting an evolutionary adaptation of the former to deal with unique adverse conditions, such as the presence of crude oil contaminants. However, it is not entirely clear which genes enable the fast growth of environmental strains under such conditions, compared to that of clinical strains (Grady et al., 2017).
In this study, we report the isolation and characterization of the P. aeruginosa marine strain GOM1, obtained from the water column in the southwestern Gulf of Mexico (swGoM). Phylogenetic analysis revealed that GOM1 is a close relative of P. aeruginosa WC55, a strain isolated from the water column in the northern Gulf of Mexico after the DWH oil spill (Thomas et al., 2014). The GOM1 strain was able to use heavy, medium and light-crude oils as its sole carbon source, showing a high activity on long-chain alkanes. The alkane degradation activity of this strain in seawater was enhanced by the addition of ammonium and phosphate. A comparison of GOM1 against the P. aeruginosa PAO1 laboratory strain, revealed an enhanced adaptation of the former to degrade hexadecane. These results place P. aeruginosa GOM1 as a potentially strong candidate for the design of a bacterial consortium to be used in the bioremediation of crude oil contaminants.
Materials and Methods
Culture Media and Hydrocarbons
Bushnell Hass (BH) medium from Sigma-Aldrich (St. Louis, MO, United States) was used for the inoculation of seawater samples and the isolation of bacterial consortia. The isolation of individual bacterial species was done in Luria-Bertani (LB) plates. The growth and hydrocarbon-degrading capacity of the GOM1 strain was evaluated in basal mineral medium (BM) with the following composition in g L–1: 0.8 K2HPO4, 0.2 KH2PO4, 0.3 NH4Cl, 0.19 Na2SO4, 0.07 CaCl2, 0.005 FeSO4.7H2O, 0.16 MgCl2, and 0.0002 Na2MoO4. Mexican crude oils (light, API 40°; medium, API 16–20°; and heavy, API 7–10°) were sterilized by autoclaving at 121°C and 15 psi for 15 min. A crude oil-kerosene mixture (1:10) was sterilized by 0.22 μm-membrane filtration. All n-alkanes were purchased from Sigma-Aldrich and were 99% pure (except octane, which was 98%). For detection of rhamnolipid production and surfactant activity, the GOM1 strain was grown in Peptone and Yeast extract (5 g L–1 each) (PY) medium supplemented with different carbon sources (2%). The concentration of liquid (light crude oil, crude oil-kerosene mixture, and C8-C16 alkanes) and solid/viscous (glucose, sodium succinate, medium and heavy crude oils, and C18-C28 alkanes) carbon sources used in this study is expressed as %v/v and %w/v, respectively.
Isolation of GOM1 Strain
Between February and March of 2016, different seawater samples were collected from the southwestern Gulf of Mexico using a Rosette device fitted with 12 Niskin-bottles of 20-L capacity. One hundred liters were pumped with a Cole-Palmer Masterflex® peristaltic pump l/S 77200-62 and concentrated to 2 l with a KrosFlo® tangential flow filtration system (filtration surface area: 2.55 m2). A 500-μL sample of concentrated seawater was inoculated in 25 mL of BH medium supplemented with sterile hydrocarbon mixture (final concentration: 0.01% medium crude oil, 0.1% kerosene). The cultures were incubated for 4 weeks in a cold room, followed by incubation at room temperature for three more weeks. Then, 10 mL of each culture were centrifuged at 6000 rpm for 20 min at 22°C. The cellular pellets were washed three times with sterile 10 mM MgSO4 and resuspended in 1 mL of the same solution. A 250-μL aliquot of each cellular suspension was inoculated into 25 mL of BM medium supplemented with the mixture of medium crude oil and kerosene (final concentration: 0.01%; 0.1%, respectively). The cultures were incubated for 7 days at 30°C, 180 rpm.
The selected B9 consortium was collected at a depth of 55 m, off the coast of Tamaulipas in the Perdido Escarpment area (25° 38.199′ N; 95° 1.283′ W). For the identification of bacterial isolates of the B9 consortium, a 100-μL aliquot of the first culture was used for serial dilutions, and aliquots (100 μL) of 10–4 to 10–6 dilutions were plated on solid LB medium. The plates were incubated for 2 days at 30°C. Eight morphologically-distinct colonies were observed, all of which were newly streaked in LB plates for purification.
Nucleic Acid Extraction and Sequencing
Total DNA was extracted from the GOM1 strain, which was cultured in BM medium with 0.5% glucose, using the Quick-DNATM Miniprep Kit from Zymo Research (Irvine, CA, United States). A sequencing library was prepared using the Illumina NextSeq500 platform, following the vendor’s protocol (San Diego, CA, United States). Raw sequences were made available in the SRA database under accession number PRJNA517612.
Genome Assembly, Functional and Taxonomic Annotations
Raw sequences were filtered with a quality ≥Q20 and were used for de novo assembly with ABySS v1.5.2 (Simpson et al., 2009) with a k-mer size of 45. A high-quality draft genome was obtained using REAPR v1.0.18 (Hunt et al., 2013), BESST v2.2.5 (Sahlin et al., 2014), GapFiller v1-10 (Nadalin et al., 2012), and iCORN 2 v0.95 (Otto et al., 2010) for misassembly error correction, scaffolding, scaffold gap filler and corrected scaffolds, respectively. Genes were predicted with GeneMarkS v4.32 (Borodovsky and Lomsadze, 2014) and annotated with an adaptation of Trinotate pipeline1. Taxonomic annotation was performed with Kraken v0.10.5-beta (Valenzuela-González et al., 2016), MetaPhlAn2 v2.2.0 (Truong et al., 2015), and Phyla AMPHORA v2 (Wang and Wu, 2013). The 16S rRNA gene was also predicted with RNAmmer v1.2 (Lagesen et al., 2007) and annotated with the NCBI Bacterial 16S Ribosomal RNA RefSeq database (Bioproject PRJNA33175). NCBI BLAST+ (Camacho et al., 2009) and BRIG (Alikhan et al., 2011) were used for visualization and comparison of the whole genome of P. aeruginosa GOM1 with selected P. aeruginosa genomes (Supplementary Table S1). The whole-genome comparison was performed using the Microbial Genome Atlas (MiGA) (Rodriguez-R et al., 2018) against the NCBI prokaryotic databases. MiGA analysis established species identity by calculating the average nucleotide identity (ANI) and average amino acid identity (AAI) with the genomes in the database. The genome has been deposited into DDBJ/ENA/GenBank under accession number SDVY00000000.
Pan-Genome Analysis
The pan-genome analysis was performed with GET_HOMOLOGUES version 02012019 (Contreras-Moreira and Vinuesa, 2013) using the 26 genomes described in Supplementary Table S1. The COGS and OMCL algorithms were employed, and the intersection was used to obtain the core, softcore, shell and cloud sizes. In addition, P. aeruginosa PAO1 and P. aeruginosa GOM1 were compared using the OMCL algorithm.
Phylogenetic Analysis
Phylogenetic analysis was performed with concatenated amino acid sequences of 14 proteins (namely RpoS, RpoD, RpoH, MutS, MulT, GyrA, GyrB, RecA, AcsA, AroE, GuaA, NouD, PpsA, and TrpE) (Khan et al., 2008) from 26 P. aeruginosa strains. This analysis included clinical and environmental strains with or without hydrocarbon-degrading ability experimentally demonstrated (Supplementary Table S1), as well as three P. putida strains and three Escherichia coli strains as the outgroup. The dendrograms were generated by maximum parsimony (MP), neighbor-joining (NJ), and minimum evolution (ME). Evolutionary distances were computed using the JTT matrix-based method with bootstrap analysis (1000 replications). Evolutionary analyses were conducted in MEGA7 (Kumar et al., 2016). The evolutionary history was inferred using the Minimum Evolution method (Rzhetsky and Nei, 1992).
Bioinformatic Identification of alkB1, alkB2, almA and Other Genes Implicated in the Degradation of Aromatic Compounds
We performed a bioinformatic search in the GOM1 genome for homologous genes encoding putative enzymes capable of degrading aromatic or aliphatic hydrocarbons present in crude oil. We compared our annotated genes with the list of xenobiotic and lipid pathways described in the KEGG database 2017 (Kanehisa et al., 2017). From this comparison, we selected the genes involved in oil hydrocarbon degradation pathways, identifying those involved in alkane and benzoate degradation. Since we wanted to investigate whether the corresponding gene products are potentially functional, we also conducted an in-depth inspection to determine whether the genomic organization and genes are preserved, compared to those found in other members of the Pseudomonas genus.
Using the Operon mapper tool (Taboada et al., 2018), we searched for homologs of the proteins AlkB1, AlkB2 and AlmA, and determined their genomic organization. The algorithm also assigned Clusters of Orthologous Genes (COGs), which are useful for comparisons with homologous proteins from other Pseudomonas genomes. In order to verify the conservation of alkB1, alkB2, and almA in other strains, we extracted 103 available proteomes of fully sequenced Pseudomonas from the KEGG database version 2017 (Kanehisa et al., 2017) and extracted the genomic contexts of coding proteins using Perl script2. We compared the COGs and protein annotations of the genomic context of our strain with each of the 103 extracted Pseudomonas genomes, using a homemade JavaScript algorithm that allows to graphically inspect the genomic context. The COG annotation for the extracted Pseudomonas genomes was also taken from the KEGG database and organized using Perl scripts3.
A bank of amino acid sequences of enzymes involved in the different degradation pathways of aromatic compounds deposited in the KEGG database was constructed. Using these sequences, a search was made on the GOM1 genome using blastp with parameters by default with an E-value less than 1 × 10E-35. All hits were taken above 40% identity and these were mapped on the different degradation pathways using KEGG mapper.
Growth in Mineral Medium and Seawater With Hydrocarbons
Pseudomonas aeruginosa GOM1 strain was grown in 250-mL flasks containing 50 mL of BM medium with glucose (0.5%) for 24 h at 30°C. The cells were harvested by centrifugation and washed three times with 50 mL sterile 10 mM MgSO4, and resuspended in 5 mL of the same solution. Flasks containing 50 mL of BM medium with 0.5% hydrocarbons (crude oils or pure alkanes) were inoculated with an aliquot of pre-inoculum to obtain a final optical density (OD) at 600 nm of approximately 0.05 in the medium. Negative controls consisted of inoculated BM media without any carbon source. Bacterial growth was monitored measuring the OD600nm of the cultures and/or their total protein content, measured by the Lowry method (Lowry et al., 1951).
To culture P. aeruginosa GOM1 under conditions mimicking the marine environment, surface seawater from the Gulf of Mexico was sterilized by 0.22 μm-membrane filtration and supplemented with hexadecane (0.5%), NH4Cl and phosphates (K2HPO4 and KH2PO4) at a C:N:P molar ratio of 100:10:1, as previously reported (Elango et al., 2014).
Extraction of Hydrocarbons and Analysis by Gas Chromatography
The total cultivated growth medium volume was collected for both the abiotic control (growth medium supplemented with hydrocarbons, and without bacterial inoculum) and the biotic samples. All glassware used for hydrocarbon extraction was previously washed twice with dichloromethane. Hydrocarbon extraction was carried out using HPLC-grade dichloromethane from BDH (Radnor, PA, United States), which was added at a ratio of 1:1 (v/v), mixed by manually swirling the flask in order to homogenize the mixture, and allowed to stand for 12–14 h for phase separation. The lower organic solvent phase was recovered by pipetting, and transferred to another flask. For optimal hydrocarbon recovery, the aqueous phase was then centrifuged at 4°C in 50-mL glass tubes at 2,000 rpm for 10 min; the residual organic solvent was also recovered by pipetting, and pooled with the first volume obtained. The total recovered volume of the organic solvent/hydrocarbon phase was left uncovered in a fume hood for 48 h, allowing complete evaporation of the dichloromethane solvent. The remaining hydrocarbon fraction was recovered by dissolving it in 1 mL of dichloromethane, and then passed through a sodium sulfate column built in a 9-inch Pasteur pipette, to remove any possible water residue. The volume was harvested in a 1-mL Wheaton glass vial and allowed to stand in a fume hood until the dichloromethane was completely evaporated.
The extracted samples were analyzed in a Hewlett Packard HP5890 Gas Chromatography Series II equipped with a flame ionization detector (FID) and a rubber capillary column of Phenomenex ZB-5 (30 m long × 0.32 mm inner diameter; 0.25 μm film). Sample injection was carried out with split 1:10 using the following conditions: Helium as carrier gas, injector temperature 270°C, detector temperature 330°C, in the furnace, initial temperature 60°C, which was held for 2 min; ramp1 from 6°C/min up to 250°C, ramp2 from 12°C/min up to 320°C, isotherm 10.5 min; execution time 50 min.
Cellular Adherence to Alkanes
Cellular hydrophobicity was measured following a previously reported method (Rosenberg et al., 1980). Briefly, GOM1 strain was cultured in BM medium with either light crude oil or glucose (0.5%) at initial OD600nm of approximately 0.05. After 7 days of incubation, the cells were harvested by centrifugation for 15 min at 6,000 rpm at room temperature, and washed two times with PUM buffer (pH:7.1, in g L–1: 22.2 K2HPO4, 7.26 KH2PO4, 1.8 urea and 0.2 MgSO4). Harvested cells were then resuspended in PUM buffer adjusting to an initial OD550nm between 0.5 and 0.6 (OD550nm = 0.539 and 0.514 for cells grown with glucose and light crude oil, respectively). Cellular suspensions with similar ODs have been previously used for the measurement of cellular hydrophobicity in other strains (Mnif et al., 2009; Sotirova et al., 2009).
A 1.2-mL aliquot of cellular suspension and 200 μL of n-alkane were placed in a round-bottom test tube and vigorously mixed by vortexing for 2 min, then incubated at room temperature for 1 h. The lower aqueous phase of the mixture was carefully pipetted to measure its OD. Cellular hydrophobicity was expressed as percentage of cells adhered to hydrocarbons, calculated as follows:
Detection of Rhamnolipid Production by the GOM1 Strain
The GOM1 strain was grown in PY liquid medium for 24 h at 30°C, 180 rpm. A 500-μL aliquot of this culture was used to inoculate flasks containing 50 mL of either PY liquid medium without any additional carbon source, or PY medium supplemented with 2% of a carbon source (succinate, glucose, light crude oil, or hexadecane). The cultures were incubated for 5 days at 30°C, 180 rpm. The cultures were then centrifuged at 6000 rpm and the supernatants were separated for further analysis.
Production of rhamnolipids was detected using the method reported by Siegmund and Wagner (1991), which consists of the formation of a dark blue precipitate due to the interaction between the cationic detergent Cetyl Trimethyl Ammonium Bromide (CTAB) and the anionic surfactant (rhamnolipids) in the presence of methylene blue dye. CTAB plates consisted of BM medium (see above), 1.2% (w/v) bacteriological agar, 0.02% (w/v) CTAB and 0.0005% (w/v) methylene blue. Wells were made on the agar plates using a flamed cork borer (5 mm diameter). An aliquot of cell-free supernatant (80 μL) was dispensed in each well and the plates were incubated for 24 h at room temperature (25°C) followed by an additional incubation for 12 h at 8°C.
Determination of the Surface Tension and Emulsification Index of GOM1’s Culture Supernatants
Surface tension (ST) was measured using 3 mL of cell-free supernatants (the same supernatants used for detection of rhamnolipid production). The samples were placed in a fireproof sample vessel (30 × 30 mm) and measured with a digital tensiometer K20S (Krüss, Hamburg, Germany) by using the Wilhelmy plate method (Hassanshahian, 2014). The validity of the ST readings was checked with pure water (72.8 ± 0.2 mN/m) before each reading. Culture media and distilled water were used as controls.
Determination of the emulsification index was carried out according to the Cooper and Goldenberg (1987) protocol. It consisted of mixing 6 mL of kerosene (Pochteca, Mexico) and 4 mL of cell-free supernatant in a round-bottom test tube. The solution was vigorously mixed by vortexing for 2 min, then the emulsion was left to settle during 24 h and the emulsification index was calculated as follows:
Quantitative Real Time PCR (qRT-PCR)
Total RNA was isolated from cells grown in BM medium containing either n-alkanes (C8–C28) or glucose (at final concentration of 0.5%) at mid-exponential phase of growth (OD600nm: 0.2–0.3), using the RNA MicroPrepTM Kit from Zymo Research and following the manufacturer’s instructions. Contaminant DNA was removed by treating the RNA sample with DNAse I from Thermo Scientific (Waltham, MA, United States). For cDNA synthesis, 200 ng of DNAse-treated RNA were used for reverse transcription reactions using the RevertAid First Strand cDNA synthesis Kit (Thermo Scientific) and the specific reverse oligonucleotides (Supplementary Table S2). The qRT-PCRs assays were carried out with SYBR green mix (Thermo Scientific) in a 96 Light Cycler (Roche) using the following program: 1 cycle of denaturing at 95°C for 10 min, 40 cycles of denaturing at 95°C for 30 s, and annealing and elongation at 60°C for 60 s, followed by a melting curve from 60 to 95°C. The 16S gene was used as internal control to normalize the expression levels of alkB1, alkB2, and almA. Analyses were made from three independent experiments (biological replicates), each one performed in triplicate (technical replicates). The relative levels of tested genes were calculated by the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Statistical Analysis
Data were analyzed by unpaired two-tailed Student’s t-test using GraphPad Prism version 6.01 (GraphPad Software Inc). Significant differences were considered at p < 0.05.
Results
Isolation and Identification of the GOM1 Strain
To isolate hydrocarbon-degrading bacteria indigenous to the swGoM, we inoculated water samples collected from different sites into BH medium containing hydrocarbons as the sole carbon source (see section Material and Methods). Fourteen bacterial consortia were isolated and newly grown in BM medium supplemented with a crude oil-kerosene mixture. Among these, the B9 consortium exhibited the greatest growth (measured by protein concentration) (Supplementary Figure S1A). From the B9 consortium, eight different isolates were identified based on their distinct colony morphology after culturing in LB plates for 48 h at 30°C. The capacity of each isolate to use crude oil and kerosene as the sole carbon source was tested in BM medium. Isolates 1 and 8 exhibited the greatest growth (Supplementary Figure S1B), but of these two, only isolate 1 was able to grow in BM medium containing light crude oil as the sole carbon source (data not shown). Moreover, comparison of growth kinetics revealed that isolate 1 exhibited a growth pattern comparable to that of the complete bacterial consortium (Supplementary Figure S1C). Therefore, isolate 1 was selected for further characterization and was named GOM1.
Sequencing of the GOM1 Genome
Genome analysis was performed with 17,031,167 paired reads with an estimated coverage of ∼193X. The assembly of genome sequences was performed with a k-mer size of 45. The final assembly was assembled with 81 contigs with an estimated length of 7,146,312 bp (Figure 1A). The length average of the scaffolds was 88,226 bp and the largest contig size was 714,401 bp with N50 of 217,999 bp content in 10 scaffolds and N90 of 57,864 bp content in 33 scaffolds. The GC content (65.98%) was very similar to that of several P. aeruginosa strains. A total of 6,592 protein-coding genes were predicted. Seven copies of 16S rRNAs were found, each with a length of 1,524 bp. All 16S rRNAs copies had 99.93% identity with a partial sequence from P. aeruginosa strain DSM 50071 (NCBI: NR_117678.1), 100% identity with P. aeruginosa strain PACS2 (Greengenes: 4485291; NCBI: NZ_AAQW01000001.1), P. aeruginosa strain PA99 (Silva 132: JARJ01000075.3682.5217; NCBI: JARJ01000075.1) and with P. aeruginosa strain PAO1_Orsay (RDP: S004508083; NCBI: NZ_LN871187.1). Taxonomic annotations were performed using different softwares based in k-mers and single-copy markers. All results indicated that the genome corresponds to Pseudomonas aeruginosa (Supplementary Table S3). According to the MiGA genome comparison results, the closest genome in the NCBI Prok database was that of P. aeruginosa USDA-ARS-USMARC-41639 (NCBI: NZ_CP013989) with a 99.46% of ANI and 99.37% of AAI. The percentage of proteins predicted shared with this genome was 91.63%. We also evaluated the quality of the genome and found a completeness of 91.9% (102/111 essential genes). The missing genes were phosphoglycerate kinase (PGK), glycine-tRNA ligase (TIGR00389), proline-tRNA ligase (TIGR00408), phenylalanine-tRNA ligase beta subunit (TIGR00471), Na+/H+ antiporter NhaD family (TIGR00775), preprotein translocase SecG subunit (TIGR00810), preprotein translocase SecE subunit (TIGR00964), DNA primase (TIGR01391), and DNA-directed RNA polymerase gamma subunit (TIGR02387).
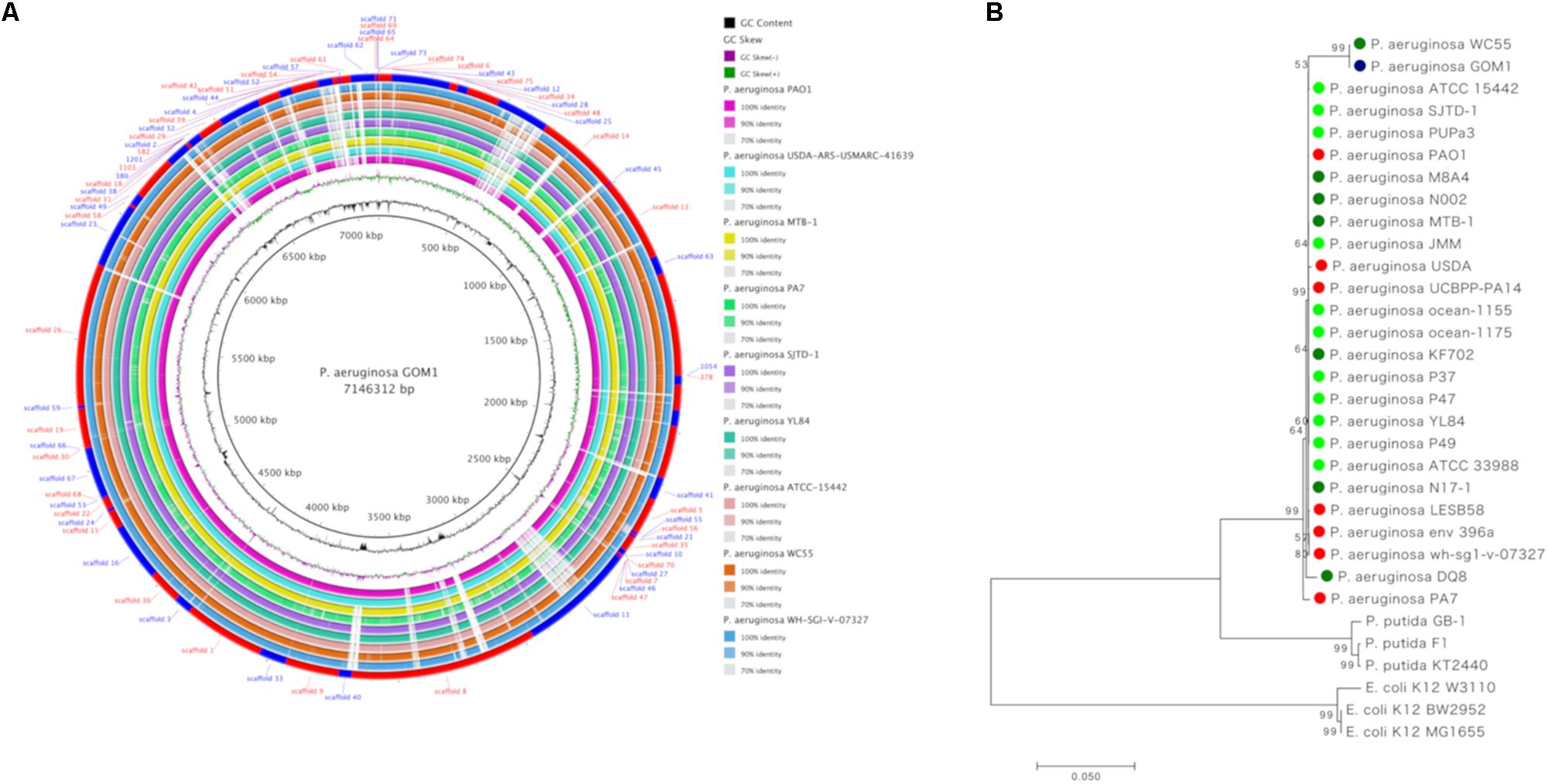
Figure 1. Whole genome sequence analysis and comparison of P. aeruginosa GOM1 with other P. aeruginosa strains. (A) Circular diagram of P. aeruginosa GOM1 showing (from inner to outer),% G + C, GC skew and the homology based on BLAST analysis of P. aeruginosa GOM1 against 9 P. aeruginosa genomes (refer color-coded legend). The outer circle shows the location of scaffolds. (B) Tree of fourteen concatenated markers from P. aeruginosa GOM1 and other P. aeruginosa strains, the rooted tree with P. putida and E. coli. The circle’s colors indicate the lifestyle: red: clinical isolate; dark green: involved in degradation; light green: environmental isolate, and blue: P. aeruginosa GOM1 isolated from the swGoM. The evolutionary history was inferred using the Minimum Evolution method.
For whole genome comparisons we used reference genomes of P. aeruginosa strains isolated from diverse habitats including clinical strains (PAO1, USDA-ARS-USMARC-41639, WH-SGI-V-07327, and PA7), environmental strains (ATCC 15442 and YL84), a strain isolated from chlorinated organic insecticide-contaminated soil (MTB-1), and strains capable of degrading hydrocarbons (SJTD-1 and WC55) (Figure 1A).
We used 27 P. aeruginosa strains for the pan-genome analysis that included P. aeruginosa GOM1. The cloud size was 3386 cluster genes, the shell size was 1607 cluster genes, the softcore size was 5069 cluster genes, and the core size was 3580 cluster genes (Supplementary Figure S2A). The core was contained in all 27 genomes analyzed, the soft core was in 26 genomes, and the shell was in 2 genomes (Supplementary Figure S2B).
Phylogenetic Analysis
According to the various initial analyses (16S rRNAs and MiGA), strain GOM1 is a close relative of several other P. aeruginosa strains (Figure 1A). To further explore its degree of evolutionary closeness to them, we performed a phylogenetic analysis (Figure 1B) that compared fourteen concatenated markers (Khan et al., 2008) from strains with hydrocarbon-degrading potential, as well as environmental and clinical strains (Supplementary Table S1). As described in the section “Materials and Methods,” different methodologies were used for the construction of the tree, and we selected the topology generated by the Minimum Evolution method of the package MEGA version 7. We observed that P. aeruginosa GOM1 is closest to P. aeruginosa WC55, a strain identified in the water column of the northern Gulf of Mexico after the DWH spill (Thomas et al., 2014). Close to both the GOM1 and WC55 strains is the non-pathogenic P. aeruginosa ATCC 15442 strain, which was isolated from an animal room water bottle (Figure 1B). Other P. aeruginosa strains that have been isolated from oil-contaminated sites fall in the same node (Figure 1B), including SJTD-1 (Liu et al., 2012, 2014), N002 (Roy et al., 2013), and M8A4 (Vives-Flórez and Garnica, 2006). The P. aeruginosa PUPa3 strain is a plant growth-promoting strain isolated from the rhizosphere (Uzelac et al., 2014). Finally, in this same node, we found the P. aeruginosa laboratory strain PAO1 (Holloway, 1955).
Genomic Organization of Genes Related to Alkane and Aromatic Compound Degradation in P. aeruginosa GOM1
To investigate the potential of the GOM1 strain to degrade both alkanes and AHs, we explored its genome in search for genes related to the corresponding degradation pathways. For alkane metabolism, we identified that the alkane hydroxylase genes alkB1, alkB2, and almA are present in P. aeruginosa GOM1 (see section Materials and Methods). In order to compare the genomic organization of alkB1, alkB2 and almA, we made an operon prediction and assigned the genomic context to these genes as described in Section “Materials and Methods,” then compared them with the genomic contexts and operon organization of the 103 fully sequenced Pseudomonas strains stored in the 2017 version of the KEGG database (Kanehisa et al., 2017). We found that the alkB2 gene, encoding alkane 1-monooxygenase 2 and located in scaffold 17, is not within an operon, but it is rather divergently transcribed from a putative transcriptional regulator of the GntR family. This gene has homologs in twelve of the analyzed Pseudomonas strains (Supplementary Table S4), all of them sharing an almost identical genomic context. The homolog of alkB2 (pae-PA1525) is found in the reference genome of P. aeruginosa PAO1. Similarly, we found homologs of this protein in Pseudomonas citronellolis P3B5 (pcq-PcP3B5_23990) and Pseudomonas knackmussii B13 (pkc-PKB_2028). The genomic contexts surrounding the homologs of alkB2 have, immediately downstream, genes encoding the N- and C-terminal subunits of a xanthine dehydrogenase (xdhA and xdhB, respectively) in eight of the available Pseudomonas strains. This context more closely resembles that of P. aeruginosa PAO1. In order to determine if AlkB2 protein from the strains sharing a similar genomic context, is closely related, we constructed a phylogenetic tree (Supplementary Figure S3), which shows that the AlkB2 from our strain is closely related to P. aeruginosa YL84, P. aeruginosa RP73 and P. aeruginosa c7447m, but it is also close to P. citronellolis P3B5 and P. knackmussii B13.
GOM1 contains a second copy of the alkane-1-monooxygenase gene (alkB1), which, just as alkB2, is not within an operon. The genomic context surrounding alkB1 is different from that of alkB2, containing (upstream and on the opposite strand) a predicted oxidoreductase related to nitroreductase. Also, upstream we found other apparently unrelated genes, encoding a drug-permease protein and a transcriptional regulator of the Lrp family. Downstream, alkB1 is followed by genes encoding two chemotaxis proteins and a histidine kinase, none of them organized within an operon. This context is conserved in nine P. aeruginosa strains (Supplementary Table S4). The phylogenetic tree (Supplementary Figure S3) clearly shows that these genes are duplications exhibiting very little divergence.
Flavin-binding monooxygenase AlmA is an alkane hydroxylase involved in the degradation of long-chain alkanes (Liu et al., 2014). We identified that scaffold 13, encompassing 166,144 bp is duplicated in scaffold 32 (Supplementary Figure S4) and these duplicated regions hold two almA copies, exhibiting exactly the same genomic context, both being organized in an operon with a Kef-type K+ transport system, membrane component, belonging to COG0475 (Supplementary Table S4). It is also remarkable that 91 genes up and 64 genes downstream from the two copies of almA conserve the same genomic context.
We did not find genes associated to AH degradation in GOM1’s genome. However, we identified enzymes involved in the degradation of benzoate (Supplementary Table S5), an aromatic compound produced during the degradation of toluene (Ramos et al., 1997). In addition, enzymes necessary for the degradation of aminobenzoate and fluorobenzoate were also detected (Supplementary Table S5). Thus, the GOM1 strain appears to have the capability to degrade some aromatic compounds, but not polyaromatic hydrocarbons (PAHs). Therefore, we carried out a functional characterization focusing on the alkane-degrading capacity of P. aeruginosa GOM1.
P. aeruginosa GOM1 Growth With Crude Oils as the Sole Carbon and Energy Source
To investigate the metabolic capacity of P. aeruginosa GOM1 to use petroleum as the sole carbon and energy source, the strain was cultured in the presence of three different crude oils, differing in API gravity. As seen in Figure 2A, GOM1 was able to grow using all three crude oils tested as its sole carbon source, as measured by an increase in OD600nm, with greater growth observed by the fifth day of incubation in the presence of both light and medium, compared to heavy crude oil. Similar results were obtained through protein quantification assays (Figure 2B). While protein concentration during the first 2 weeks of incubation was generally greater in the cultures grown with light and medium crude oils, by day 18 the protein concentration was similar in all three crude oil cultures. These results indicate that P. aeruginosa GOM1 is able to use all three tested crude oils as its sole carbon and energy source.
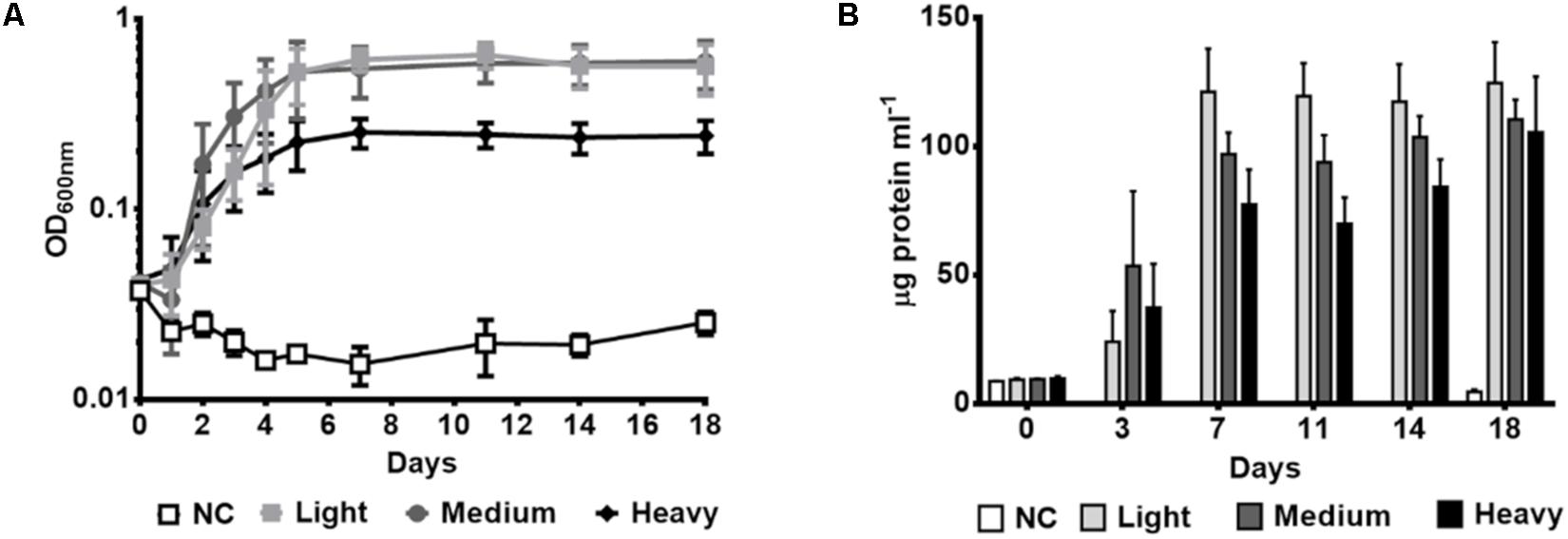
Figure 2. Growth of P. aeruginosa GOM1 using light, medium or heavy crude oils as sole carbon source. Growth kinetics were monitored by measuring (A) optical density at 600 nm and (B) protein concentration (μg protein mL–1). NC: Negative control (inoculated BM medium without any carbon source). The strain was cultured in BM medium with 0.5% of light, medium or heavy crude oils. The data represent the average of three independent determinations. Error bars indicate the standard deviation.
Hydrocarbon Degradation by P. aeruginosa GOM1
In order to determine the hydrocarbon-degradation capacity of P. aeruginosa GOM1, cultures of this strain in BM medium containing 0.5% of light crude oil were incubated for 30 days, and then subjected to extraction of total hydrocarbons for GC analysis. Figure 3A shows the light crude oil hydrocarbon profile extracted from the abiotic control, which was used as a reference. A comparison with the profile obtained from the P. aeruginosa GOM1 cultures (Figure 3B) indicates that this strain was metabolically active on the aliphatic fraction of crude oil, degrading a total of 96% of alkanes (C12–C38) (Table 1).
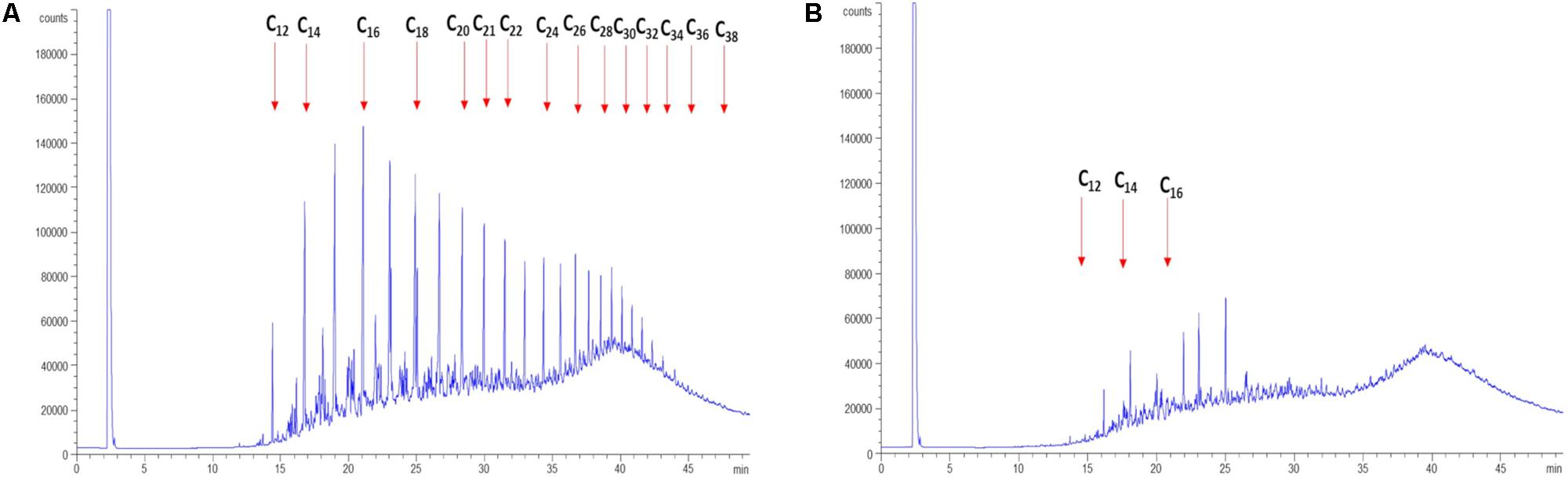
Figure 3. Degradation of light crude oil by P. aeruginosa GOM1. GC profiles of light crude oil in (A) abiotic control and (B) GOM1 strain culture after 30 days of incubation at 30°C. BM medium with 0.5% light crude oil was used. Data are representative of three independent experiments.
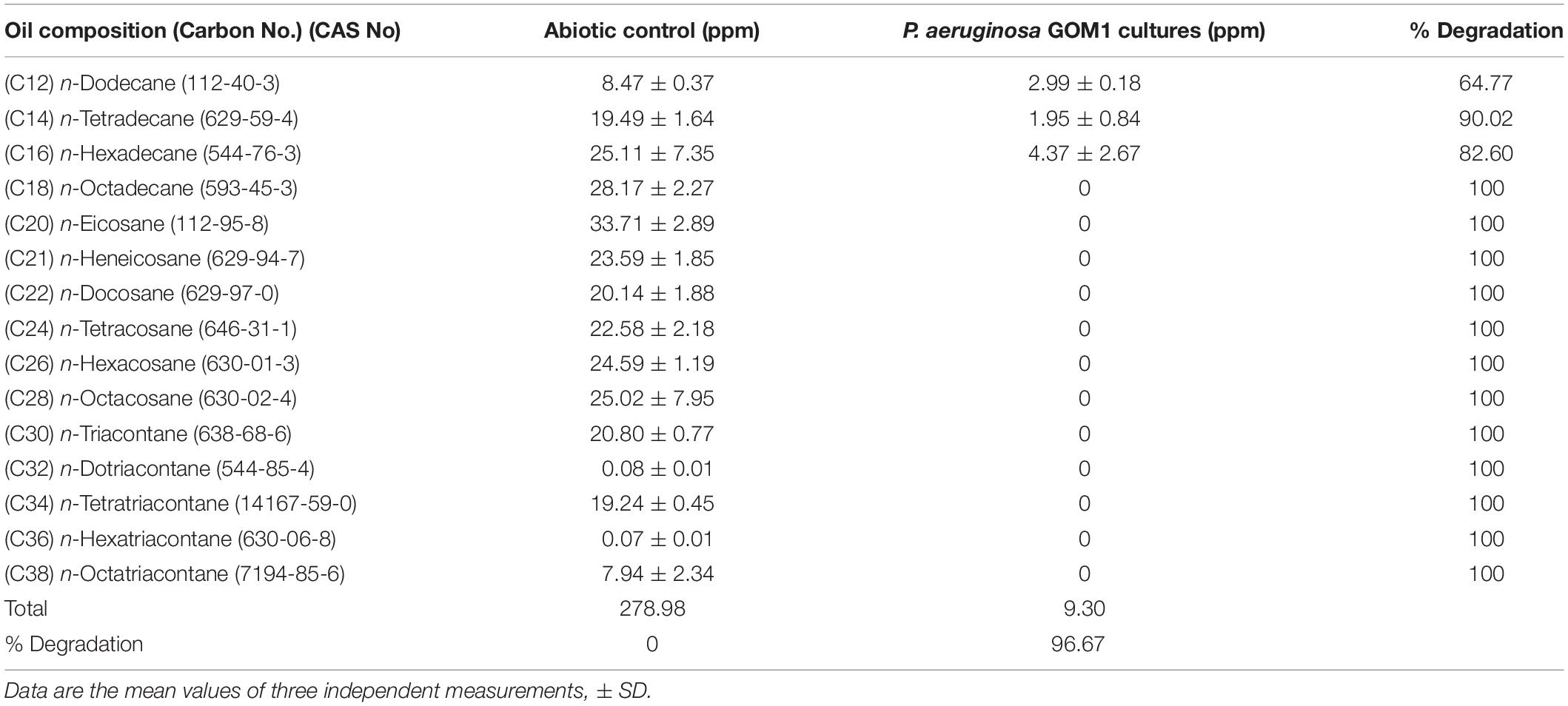
Table 1. Alkane degradation by P. aeruginosa GOM1 in BM medium with light crude oil (0.5%).
P. aeruginosa GOM1 Growth With Pure Alkanes as the Sole Carbon and Energy Source
The above results demonstrate that P. aeruginosa GOM1 can degrade alkanes and use them as its sole carbon and energy source. To further characterize such metabolic capability, we tested its growth on BM medium supplemented with a variety of pure alkanes (from C8 to C28). Bacterial growth was monitored by measuring the culture’s OD600nm. As seen in Figure 4A, this strain’s growth pattern differed depending on alkane chain-length. With C8, the lag phase time was 10 days, while in the presence of C11, growth had initiated by the second day of incubation. With medium- and long-chain alkanes (C12 to C28), bacterial growth was evident after the first day of incubation, displaying better growth of the strain overall when these alkanes were used as the sole carbon and energy source.
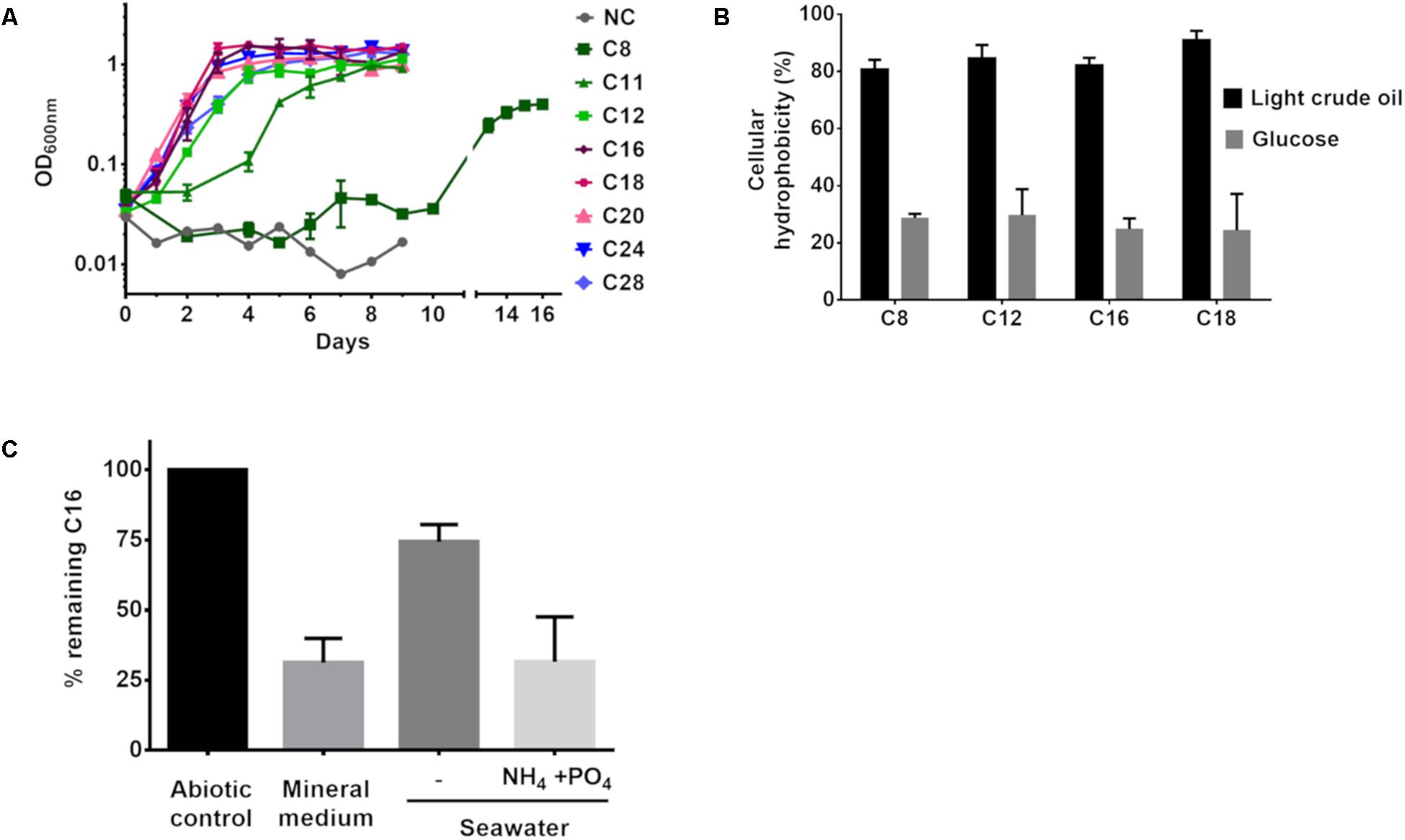
Figure 4. (A) GOM1 strain’s growth in BM medium with alkanes (0.5%) as sole carbon source. NC: Negative control (inoculated BM medium without any carbon source). (B) Cellular hydrophobicity of GOM1 strain previously grown in BM medium with light crude oil or glucose (0.5%) as sole carbon source. (C) Biodegradation of hexadecane in BM medium, sterile seawater with or without the addition of ammonium and phosphates at a C:N:P molar ratio of 100:10:1. For (A–C), data are mean values from three independent determinations. Error bars indicate the standard deviation.
Given that the ability to adhere to hydrocarbons is a trait of alkane-degrading bacteria (Rojo, 2009), we measured the capacity of P. aeruginosa GOM1 cells to adhere to various alkanes. Using the method reported by Rosenberg et al. (1980), we measured alkane adherence of GOM1 cells previously grown for 7 days with either glucose or light crude oil as the carbon source. As seen in Figure 4B, cells grown with glucose exhibited an adherence to alkanes (C8–C18) from 24 to 30%. In contrast, when cells had been previously adapted to growth with light crude oil, their alkane adherence was about 80–90%. These results indicate that growth in the presence of crude oil as the sole carbon source increased the cellular hydrophobicity of the GOM1 strain, which has been associated with a high capacity to degrade hydrocarbons (Obuekwe et al., 2009).
In order to confirm the alkane-degrading capacity of GOM1, we measured hexadecane degradation by this strain when grown in mineral medium supplemented with this alkane as the sole carbon source. By 15 days of incubation, P. aeruginosa GOM1 had degraded about 70% of the available C16 (Figure 4C). Given that GOM1 is a marine isolate, we also tested its capacity to degrade C16 in seawater. For this, the strain was grown in filter-sterilized seawater with C16 as the sole carbon source, and it was able to degrade about 25%. However, when the seawater medium was supplemented with nitrogen and phosphate at a C:N:P molar ratio of 100:10:1, GOM1 degraded about 70% of hexadecane after 15 days of incubation. Collectively, these results support that the P. aeruginosa GOM1 strain is able to degrade crude oil and alkanes both in mineral medium and in simulated marine conditions.
Expression of Alkane Hydroxylase Coding-Genes in P. aeruginosa GOM1
As previously described, alkB genes are differentially induced by alkanes in P. aeruginosa (Marín et al., 2003). We thus measured mRNA levels of alkB1 and alkB2 in P. aeruginosa GOM1 grown in mineral medium with different alkanes as the sole carbon and energy source. Cells grown in mineral medium with glucose were used as the reference control. Total RNA was isolated from cells at mid-exponential phase (OD600nm = 0.2–0.3) and used for qRT-PCR assays; the results are shown in Figure 5. Expression of alkB1 was induced (8- to 29-fold) when GOM1 was grown in the presence of all alkanes tested, with the highest transcript levels found with C11 (29.2-fold, compared to the control glucose condition). Expression of alkB2 increased 10- to 15-fold with alkanes C11-C28 compared to the glucose control, while induction was only 6-fold greater with C8. Additionally, we measured the expression of almA. As also shown in Figure 5, the highest induction levels of almA expression occurred with C20, C24, and C28 (about 6- to 7-fold compared to the glucose control), whereas short- and medium-chain alkanes (C8 to C16) only increased almA mRNA levels 3- to 5-fold.
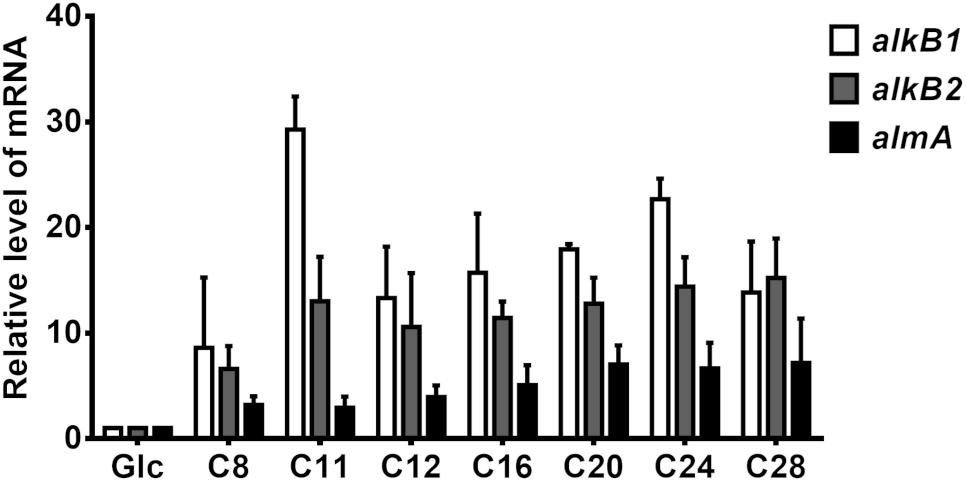
Figure 5. Expression of alkane-hydroxylase genes alkB1, alkB2, and almA in P. aeruginosa GOM1. The strain was grown in BM medium with pure alkanes or glucose (0.5%) at mid-exponential phase of growth (OD600nm: 0.2–0.3). The expression level of each gene in the glucose condition was considered as 1. The mRNA levels of 16s gene were used as internal control. Data are the mean values from three independent measurements. Error bars indicate the standard deviation.
Surfactant and Emulsifying Activities of GOM1 Culture’s Supernatants
Pseudomonas species can synthesize rhamnolipid surfactants which increase the solubility of hydrocarbons, promoting their biodegradation (Beal and Betts, 2000). The GOM1 strain contains the genes that encode for RhlA, RhlB, and RhlC enzymes (Supplementary Table S6), which are essential for rhamnolipid synthesis (Chong and Li, 2017). Given that the production of rhamnolipids depends on the carbon source available in the culture medium (Santos et al., 2002), we explored the capacity of the GOM1 strain to produce rhamnolipids in liquid PY medium supplemented with glucose, succinate, light crude oil or hexadecane (at 2% final concentration). After 5 days of incubation, the culture’s cell-free supernatants were analyzed on CTAB agar (Figure 6). Rhamnolipids were not detected in supernatants from cultures in PY medium without any additional carbon source added, while supernatants from GOM1 cultures in PY added with hexadecane gave a strong positive signal for rhamnolipid production (dark blue halo), and to a lesser extent, supernatants from cultures supplemented with succinate also exhibited a positive reaction. The addition of glucose or light crude oil to PY medium did not induce the production of rhamnolipids by the GOM1 strain.

Figure 6. Detection of rhamnolipids, surface and emulsifying activities of cell-free supernatants from P. aeruginosa GOM1 cultures. The strain was grown in either PY liquid medium without any additional carbon source, or PY medium supplemented with hexadecane, succinate, glucose or light crude oil (2% final concentration) for 5 days at 30°C. An aliquot (80 μl) of each cell-free supernatant was placed on CTAB agar plate wells and incubated for 24 h at laboratory temperature, followed by an additional incubation at 8°C for 12 h. A blue halo around the well indicates rhamnolipid production. Images are representative of three independent experiments. The readings of surface tension were validated with pure water (72.8 ± 0.2 mN/m). The Emulsification Index (%EI24) was obtained with kerosene. The data are the mean of three independent experiments (±standard deviation).
We also measured the ST of the culture supernatants. The PY culture medium without inoculum (negative control) reduced the ST of water from 73 to 64 mN m–1. Supernatants from cultures added with hexadecane showed the highest surface activity, reducing the ST of water up to 23 mN m–1, followed by supernatants from cultures added with glucose and succinate (32 and 34 mN m–1, respectively). Supernatants from PY cultures without any additional carbon source and with light crude oil showed a similar reduction of ST of water (42–43 mN m–1) (Figure 6). In addition, we measured the emulsifying activity with kerosene of all the supernatants, which showed an emulsification index (EI24) between 65 and 73%, except for those from cultures added with glucose (Figure 6). Collectively, these results demonstrate that the GOM1 strain produces biosurfactants.
Comparison of the Alkane-Degrading Capacity of P. aeruginosa Strains GOM1 and PAO1
As demonstrated above, the GOM1 strain is efficiently active on medium and long-chain alkanes. Given that hydrocarbon-degrading capacity is different among P. aeruginosa strains (Gunasekera et al., 2017), despite their high degree of conservation at the genomic level (Figure 1A), we thought it would be interesting to compare the marine GOM1 strain with another P. aeruginosa strain isolated from a different environment. For this, we used P. aeruginosa PAO1, a well-characterized strain isolated from a human wound (Holloway, 1955) which has been used as a reference laboratory strain, and yet it can grow using alkanes as the sole carbon source (Smits et al., 2003; Gunasekera et al., 2017).
First, we carried out comparative genomic analysis between P. aeruginosa GOM1 and PAO1. We found 5075 orthologous genes present in both strains (Supplementary Figure S5). We found 386 cluster genes that include 472 proteins in P. aeruginosa PAO1, and 1065 cluster genes that include 1246 proteins in P. aeruginosa GOM1, of which 50.4 and 58.5%, respectively, correspond to hypothetical proteins. These results demonstrate that the genomes of these two strains are very similar; however, they contain groups of unclassified proteins that could be responsible for functional differences between them.
Then, we compared the alkane-degrading capacity of P. aeruginosa strains GOM1 and PAO1. The strains were grown in mineral medium with C16 as the sole carbon source. GOM1 grew faster and to a greater density than PAO1 in the presence of C16, as measured both by culture OD600 and protein concentration (Figures 7A,B). Additionally, we compared expression levels of the alkane hydroxylase genes in these two strains at mid-exponential phase of growth (OD600nm: 0.2–0.3). Transcript levels of alkB1, alkB2 and almA were 2-, 16-, and 3-fold higher in GOM1, respectively, compared to PAO1 (Figure 7C). The C16 alkane degradation capacity of these strains was also compared. By 4 days of incubation, GOM1 had degraded 35% of C16, while PAO1 had only consumed 10%, as compared with the abiotic control. After 8 days of incubation, C16 degradation by GOM1 had reached 73%, while that of PAO1 had only reached 55% (Figure 7D). Collectively, these results indicate that the P. aeruginosa GOM1 environmental isolate has an enhanced adaptation for alkane degradation, compared to the PAO1 laboratory strain.
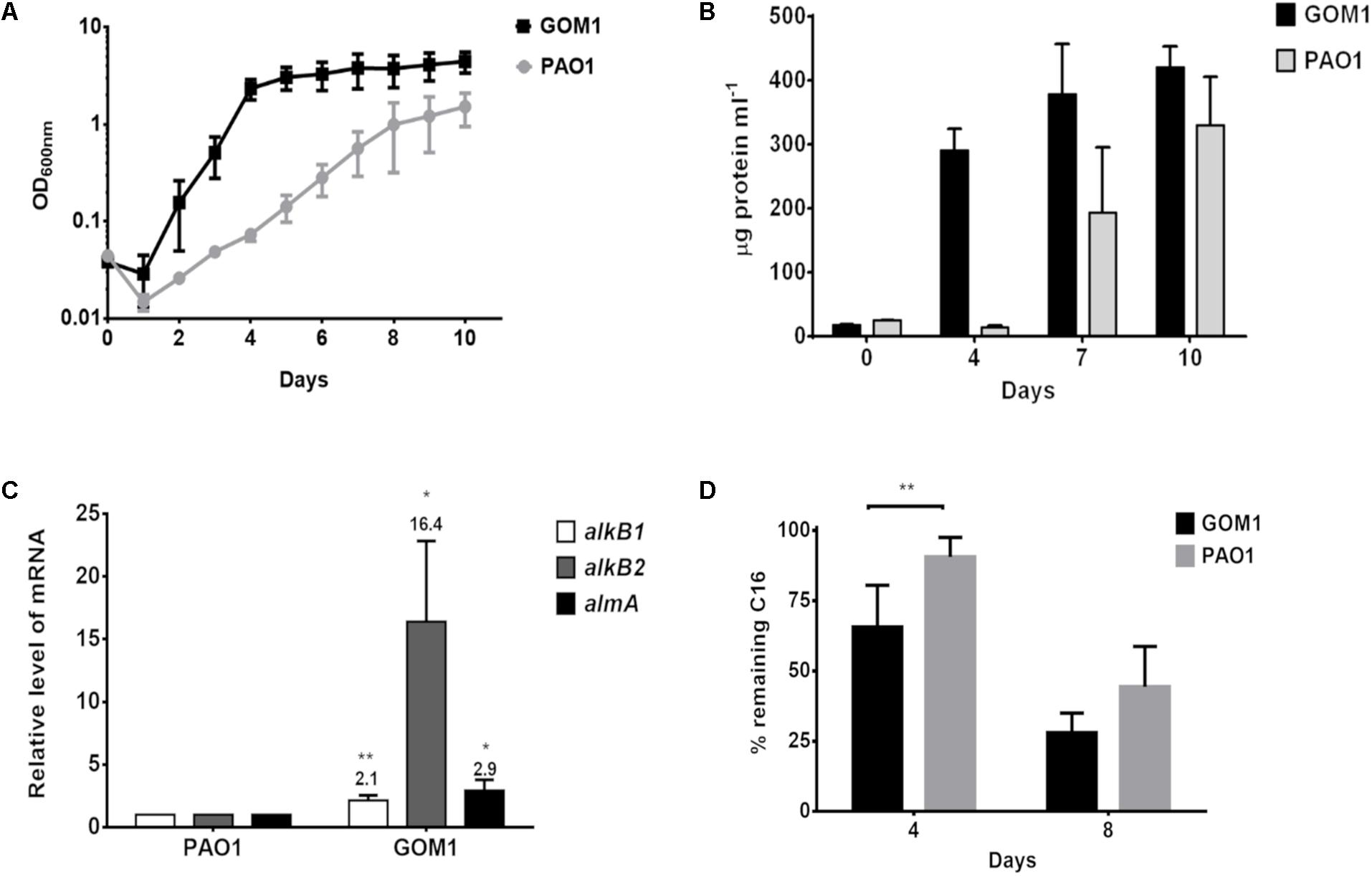
Figure 7. Pseudomonas aeruginosa GOM1 exhibits an enhanced adaptation for C16 degradation compared to the PAO1 strain. Growth in BM medium with C16 was monitored by measuring (A) optical density (600 nm) and (B) protein concentration (μg protein mL–1). (C) Comparison of the expression levels of alkB1, alkB2 and almA in strains PAO1 and GOM1 grown in BM medium with C16. The transcript level of each gene in PAO1 strain was considered as 1. The mRNA levels of 16s gene were used as internal control. Data are mean values of three independent measurements (D) Hexadecane degradation by PAO1 and GOM1 strains. The degradation percentage for each strain is relative to the abiotic control. Data are mean values from five independent determinations. Error bars indicate the standard deviation. Asterisks indicate statistical significance (unpaired t-test) between GOM1 and PAO1 data (∗p < 0.05, ∗∗p < 0.01).
Discussion
The Gulf of Mexico has been continuously exposed to hydrocarbons due to the presence of natural oil seeps, and discharges from anthropogenic activities such as extraction and transport of petroleum (MacDonald et al., 2015). Recently, we reported the bacterial diversity from different sediments in the swGoM, and identified several specialized hydrocarbon-degrading genera at a basal level, whose population size could potentially increase during an oil spill (Godoy-Lozano et al., 2018). We thus became interested in the isolation of bacterial consortia and species indigenous to the swGoM, to study their metabolic capabilities in hydrocarbon degradation. In this work, we isolated a strain from the swGoM and identified it as P. aeruginosa GOM1.
Phylogenetic analyses showed that GOM1 is closely related to P. aeruginosa WC55 (Figure 1B), another environmental strain isolated from the north Gulf of Mexico after the DWH spill, which degraded about 43% of DWH crude oil over 7 days (Thomas et al., 2014). The GOM1 strain is able to use different crude oils as carbon source, and it degraded about 96% of the aliphatic fraction in light crude oil (Figures 3A,B and Table 1). Gas Chromatography analyses and growth kinetics with pure n-alkanes revealed that P. aeruginosa GOM1 actively degraded medium and long-chain alkanes (Figures 3B, 4A). Other P. aeruginosa strains isolated from oil-contaminated soils have been shown to have a high degradation activity on aliphatic hydrocarbons. For example, P. aeruginosa NCIM 5514 degraded 60.63% of crude oil (3% v/v) with a removal of C19, C20, C21, C26, and C32 in a range of 95–97% at 60 days of incubation (Varjani and Upasani, 2016), while the SJTD-1 strain consumed 95–100% of C14, C16, and C18 at 36 h of incubation in cultures with 0.05% of n-alkanes (Liu et al., 2014).
Pseudomonas aeruginosa GOM1 demonstrated high surface and emulsifying activities. It is well known that P. aeruginosa species synthesize rhamnolipid biosurfactants which enhance alkane degradation (Zhang and Miller, 1995; Chong and Li, 2017). Rhamnolipid production by the GOM1 strain was stimulated by the presence of hexadecane (Figure 6), suggesting that the degradation of this alkane provided the raw material for the synthesis of rhamnolipids. Similar findings were previously reported for the Gram-positive bacterium Dietzia maris where hexadecane induced rhamnolipid production (Wang et al., 2014).
Marine ecosystems are usually oligotrophic, which limits bacterial growth. Addition of nutrients, mainly nitrogen and phosphate, has been reported to stimulate hydrocarbon degradation by indigenous bacterial populations (Nikolopoulou and Kalogerakis, 2010). The hexadecane degradation in seawater by the GOM1 strain increased from 25 to 70% with the addition of nitrogen and phosphate at molar ratio C:N:P 100:10:1 (Figure 4C), indicating that alkane biodegradation by P. aeruginosa GOM1 could be potentially stimulated in the marine environment. Furthermore, the addition of other carbon sources can stimulate the alkane degradation capacity of P. aeruginosa; Nie et al. (2017) reported that the addition of glutaric acid, a small organic acid produced in C16 metabolism, increased the hexadecane degradation rate of the P. aeruginosa strain NY3.
In GOM1 strain, the expression of alkB genes was induced in the presence of a broad range of alkanes (C8 to C28) (Figure 5), which correlates with its degradation activity on light crude oil’s aliphatic fraction (Figure 3B). The overlapping substrate ranges of AlkB enzymes have been previously reported in other bacteria with high alkane degradation capacity, such as P. aeruginosa strains SJTD-1 and RR1 (Yuste et al., 2000; Marín et al., 2003; Liu et al., 2014), Alcanivorax dieselolei B-5 (Liu et al., 2011), and Alcanivorax hongdengensis A-11-3 (Wang and Shao, 2012). The versatility of bacteria to metabolize a wide range of alkanes is relevant in the implementation of bioremediation strategies, as long chain alkanes are highly persistent in the marine environment (Thessen and North, 2017).
The analysis of the context of genes alkB1, alkB2, and almA revealed that some genomic regions are duplicated in GOM1 (Supplementary Figure S4). The role of these duplications is not well understood, but in the case of Pseudomonas, the role of duplicated operons has been identified. This is the case of P. aeruginosa PA14, for which the operons phzA1-G1 (Phz1) and phzA2-G2 (Phz2), encode nearly identical sets of proteins that catalyze the synthesis of phenazine-1-carboxylic acid, the precursor for several phenazine derivatives, which have diverse functions in this strain, including the infection process. Phz1 plays the major role in phenazine production during planktonic growth, while Phz2 participates almost exclusively during biofilm development and host infection. The differential expression of these duplicated genes could potentially be involved in the adaption of this bacterium to diverse environments (Recinos et al., 2012).
The almA gene, which is involved in the oxidation of long-chain alkanes (Liu et al., 2014), is also duplicated in P. aeruginosa GOM1. This could explain the high degradation activity of this strain on C18–C38 alkanes of light crude oil (Figure 3B), and the increase of almA mRNA levels in the presence of a wide range of alkanes (C8–C28) (Figure 5). Additional evidence that reinforces this hypothesis was reported by Liu et al. (2014) in P. aeruginosa SJTD-1, in which a copy of the almA gene is present, but was not induced with C12, C14, and C16, and was poorly activated with C18–C24 alkanes.
Pseudomonas aeruginosa GOM1 exhibited an enhanced adaptation to metabolize hexadecane compared to the PAO1 laboratory strain (Figure 7). Similar results were previously reported by Gunasekera et al. (2017); in this case, the P. aeruginosa ATCC 33988 strain, isolated from a fuel tank, showed higher expression of alkB1 and alkB2 than PAO1, along with greater alkane consumption. The authors suggested that the observed difference in alkane degradation between ATCC33988 and PAO1 might be related to differences in expression control, due to the alkB-genes promoters in ATCC 33988 strain having single nucleotide polymorphisms (SNPs). Interestingly, the alkB1 promoter (PalkB1) of the GOM1 strain has the same SNPs in ATCC 33988 PalkB1 compared to PAO1, consisting of a nucleotide substitution (cytosine replaced with guanine) and a deletion of a cytosine, at positions −55 and −60 (relative to the transcription start site), respectively. In the case of PalkB2, a cytosine at position +23 in both PAO1 and ATCC 33988, was replaced by a guanine in the GOM1 strain, and the −19 cytosine in both GOM1 and PAO1 was changed to thymine in ATCC 33988 (Supplementary Figure S6). Therefore, the differences in alkB expression observed among different P. aeruginosa strains could be associated, at least in part, to promoter polymorphisms.
In summary, a new P. aeruginosa strain indigenous to the swGoM was characterized, and it exhibited a high capacity to degrade crude oil. The GOM1 strain efficiently degraded medium and long-chain alkanes and was biostimulated for hexadecane degradation in seawater. These traits make P. aeruginosa GOM1 a strong potential candidate to be used in a consortium along with other PAH-degrading strains for the bioremediation of crude oil-polluted sites.
Data Availability
The datasets generated for this study can be found in DDBJ/ENA/GenBank, SDVY00000000.
Author Contributions
LM-M, JR-M, and LP-L conceived and designed the experiments. LM-M, JR-M, NR-G, EG-L, R-MG-R, DM-G, and AE-R performed the experiments and analyzed the data. All authors contributed to the writing of the manuscript and approved the final version. LP-L coordinated the IBt-L4-CIGoM group.
Funding
This research was funded by the National Council of Science and Technology of Mexico – Mexican Ministry of Energy-Hydrocarbon Trust, project 201441, and PAPIIT-DGAPA IN207019. This is a contribution of the Gulf of Mexico Research Consortium (CIGoM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sarahi Sánchez, Ileana Santos, and Humberto Sánchez for their technical support and Dr. Gloria Soberón for providing the P. aeruginosa PAO1 strain. We are grateful to Paul Gaytan, Jorge Yañez, Eugenio López, and Santiago Becerra for the oligonucleotide synthesis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00572/full#supplementary-material
Footnotes
- ^ http://trinotate.github.io
- ^ https://doi.org/10.6084/m9.figshare.8859428.v1
- ^ https://doi.org/10.6084/m9.figshare.8311487.v1
References
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Beal, R., and Betts, W. B. (2000). Role of rhamnolipid biosurfactants in the uptake and mineralization of hexadecane in Pseudomonas aeruginosa. J. Appl. Microbiol. 89, 158–168. doi: 10.1046/j.1365-2672.2000.01104.x
Borodovsky, M., and Lomsadze, A. (2014). Gene identification in prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr. Protoc. Microbiol. 32, .1–.1. doi: 10.1002/9780471729259.mc01e07s32
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Chaerun, S. K., Tazaki, K., Asada, R., and Kogure, K. (2004). Bioremediation of coastal areas 5 years after the Nakhodka oil spill in the Sea of Japan: isolation and characterization of hydrocarbon-degrading bacteria. Environ. Int. 30, 911–922. doi: 10.1016/j.envint.2004.02.007
Chong, H., and Li, Q. (2017). Microbial production of rhamnolipids: opportunities, challenges and strategies. Microb. Cell Fact. 16:137. doi: 10.1186/s12934-017-0753-2
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Cooper, D. G., and Goldenberg, B. G. (1987). Surface-active agents from two Bacillus species. Appl. Environ. Microbiol. 53, 224–229.
Dubinsky, E. A., Conrad, M. E., Chakraborty, R., Bill, M., Borglin, S. E., Hollibaugh, J. T., et al. (2013). Succession of hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ. Sci. Technol. 47, 10860–10867. doi: 10.1021/es401676y
Elango, V., Urbano, M., Lemelle, K. R., and Pardue, J. H. (2014). Biodegradation of MC252 oil in oil: sand aggregates in a coastal headland beach environment. Front. Microbiol. 5:161. doi: 10.3389/fmicb.2014.00161
George, K. W., and Hay, A. G. (2011). Bacterial strategies for growth on aromatic compounds. Adv. Appl. Microbiol. 74, 1–33. doi: 10.1016/B978-0-12-387022-3.00005-7
Ghosal, D., Ghosh, S., Dutta, T. K., and Ahn, Y. (2016). Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front. Microbiol. 7:1369. doi: 10.3389/fmicb.2016.01369
Godoy-Lozano, E. E., Escobar-Zepeda, A., Raggi, L., Merino, E., Gutierrez-Rios, R. M., Juarez, K., et al. (2018). Bacterial diversity and the geochemical landscape in the southwestern Gulf of Mexico. Front. Microbiol. 9:2528. doi: 10.3389/fmicb.2018.02528
Grady, S. L., Malfatti, S. A., Gunasekera, T. S., Dalley, B. K., Lyman, M. G., Striebich, R. C., et al. (2017). A comprehensive multi-omics approach uncovers adaptations for growth and survival of Pseudomonas aeruginosa on n-alkanes. BMC Genomics 18:334. doi: 10.1186/s12864-017-3708-4
Grosso-Becerra, M. V., Santos-Medellín, C., González-Valdez, A., Méndez, J. L., Delgado, G., Morales-Espinosa, R., et al. (2014). Pseudomonas aeruginosa clinical and environmental isolates constitute a single population with high phenotypic diversity. BMC Genomics 15:318. doi: 10.1186/1471-2164-15-318
Gunasekera, T. S., Bowen, L. L., Zhou, C. E., Howard-Byerly, S. C., Foley, W. S., Striebich, R. C., et al. (2017). Transcriptomic analyses elucidate adaptive differences of closely related strains of Pseudomonas aeruginosa in fuel. Appl. Environ. Microbiol. 83:e03249-16. doi: 10.1128/AEM.03249-16
Hassanshahian, M. (2014). Isolation and characterization of biosurfactant producing bacteria from Persian Gulf (Bushehr provenance). Mar. Pollut. Bull. 86, 361–366. doi: 10.1016/j.marpolbul.2014.06.043
Hollebone, B. (2015). “The oil properties data appendix,” in Handbook of Oil Spill Science and Technology, ed. M. Fingas, (New York, NY: John Wiley and Sons Inc), 557–681.
Holloway, B. W. (1955). Genetic recombination in Pseudomonas aeruginosa. Microbiology 13, 572–581. doi: 10.1099/00221287-13-3-572
Hunt, M., Kikuchi, T., Sanders, M., Newbold, C., Berriman, M., and Otto, T. D. (2013). REAPR: a universal tool for genome assembly evaluation. Genome. Biol. 14:R47. doi: 10.1186/gb-2013-14-5-r47
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Khan, N. H., Ahsan, M., Yoshizawa, S., Hosoya, S., Yokota, A., and Kogure, K. (2008). Multilocus sequence typing and phylogenetic analyses of Pseudomonas aeruginosa isolates from the ocean. Appl. Environ. Microbiol. 74, 6194–6205. doi: 10.1128/AEM.02322-07
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lamendella, R., Strutt, S., Borglin, S., Chakraborty, R., Tas, N., Mason, O., et al. (2014). Assessment of the deepwater horizon oil spill impact on Gulf coast microbial communities. Front. Microbiol. 5:130. doi: 10.3389/fmicb.2014.00130
Liu, C., Wang, W., Wu, Y., Zhou, Z., Lai, Q., and Shao, Z. (2011). Multiple alkane hydroxylase systems in a marine alkane degrader, Alcanivorax dieselolei B-5. Environ. Microbiol. 13, 1168–1178. doi: 10.1111/j.1462-2920.2010.02416.x
Liu, H., Liang, R., Tao, F., Ma, C., Liu, Y., Liu, X., et al. (2012). Genome sequence of Pseudomonas aeruginosa strain SJTD-1, a bacterium capable of degrading long-chain alkanes and crude oil. J. Bacteriol. 194, 4783–4784. doi: 10.1128/JB.01061-12
Liu, H., Sun, W. B., Liang, R. B., Huang, L., Hou, J. L., and Liu, J. H. (2015). iTRAQ-based quantitative proteomic analysis of Pseudomonas aeruginosa SJTD-1: a global response to n-octadecane induced stress. J. Proteom. 123, 14–28. doi: 10.1016/j.jprot.2015.03.034
Liu, H., Xu, J., Liang, R., and Liu, J. (2014). Characterization of the medium- and long-chain n-alkanes degrading Pseudomonas aeruginosa strain SJTD-1 and its alkane hydroxylase genes. PLoS One 9:e105506. doi: 10.1371/journal.pone.0105506
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275.
MacDonald, I. R., Garcia-Pineda, O., Beet, A., Daneshgar Asl, S., Feng, L., Graettinger, G., et al. (2015). Natural and unnatural oil slicks in the Gulf of Mexico. J. Geophys. Res. C Oceans. 120, 8364–8380. doi: 10.1002/2015JC011062
Mapelli, F., Scoma, A., Michoud, G., Aulenta, F., Boon, N., Borin, S., et al. (2017). Biotechnologies for marine oil spill cleanup: indissoluble ties with microorganisms. Trends Biotechnol. 35, 860–870. doi: 10.1016/j.tibtech.2017.04.003
Marín, M. M., Yuste, L., and Rojo, F. (2003). Differential expression of the components of the two alkane hydroxylases from Pseudomonas aeruginosa. J. Bacteriol. 185, 3232–3237. doi: 10.1128/JB.185.10.3232-3237.2003
Mnif, S., Chamkha, M., and Sayadi, S. (2009). Isolation and characterization of Halomonas sp. strain C2SS100, a hydrocarbon-degrading bacterium under hypersaline conditions. J. Appl. Microbiol. 107, 785–794. doi: 10.1111/j.1365-2672.2009.04251.x
Nadalin, F., Vezzi, F., and Policriti, A. (2012). GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 13:S8. doi: 10.1186/1471-2105-13-S14-S8
Nie, H., Nie, M., Xiao, T., Wang, Y., and Tian, X. (2017). Hexadecane degradation of Pseudomonas aeruginosa NY3 promoted by glutaric acid. Sci. Total Environ. 575, 1423–1428. doi: 10.1016/j.scitotenv.2016.09.223
Nie, Y., Chi, C. Q., Fang, H., Liang, J. L., Lu, S. L., Lai, G. L., et al. (2014). Diverse alkane hydroxylase genes in microorganisms and environments. Sci. Rep. 4:4968. doi: 10.1038/srep04968
Nikolopoulou, M., and Kalogerakis, N. (2010). “Biostimulation strategies for enhanced bioremediation of marine oil spills including chronic pollution,” in Handbook of Hydrocarbon and Lipid Microbiology, ed. K. Timmis, (Berlin: Springer), 2521–2529. doi: 10.1007/978-3-540-77587-4_187
Obuekwe, C. O., Al-Jadi, Z. K., and Al-Saleh, E. S. (2009). Hydrocarbon degradation in relation to cell-surface hydrophobicity among bacterial hydrocarbon degraders from petroleum-contaminated Kuwait desert environment. Int. Biodeterior. Biodegradation 63, 273–279. doi: 10.1016/j.ibiod.2008.10.004
Otto, T. D., Sanders, M., Berriman, M., and Newbold, C. (2010). Iterative correction of reference nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 261, 1704–1707. doi: 10.1093/bioinformatics/btq269
Poirier, M. C., Lair, S., Michaud, R., Hernández-Ramon, E. E., Divi, K. V., Dwyer, J. E., et al. (2019). Intestinal polycyclic aromatic hydrocarbon-DNA adducts in a population of beluga whales with high levels of gastrointestinal cancers. Environ. Mol. Mutagen. 60, 29–41. doi: 10.1002/em.22251
Ramos, J. L., Marqués, S., and Timmis, K. N. (1997). Transcriptional control of the Pseudomonas TOL plasmid catabolic operons is achieved through an interplay of host factors and plasmid-encoded regulators. Ann. Rev. Microbiol. 51, 341–373.
Recinos, D. A., Sekedat, M. D., Hernandez, A., Cohen, T. S., Sakhtah, H., Prince, A. S., et al. (2012). Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 109, 19420–19425. doi: 10.1073/pnas.1213901109
Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Rojo, F. (2009). Degradation of alkanes by bacteria. Environ. Microbiol. 11, 2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x
Rosenberg, M., Gutnick, D., and Rosenberg, E. (1980). Adherence of bacteria to hydrocarbons: a simple method for measuring cell-surface hydrophobicity. FEMS Microbiol. Lett. 9, 29–33.
Roy, A. S., Baruah, R., Gogoi, D., Borah, M., Singh, A. K., and Deka Boruah, H. P. (2013). Draft genome sequence of Pseudomonas aeruginosa strain N002, isolated from crude oil-contaminated Soil from Geleky, Assam, India. Genome Announc. 1, e00104–e00112. doi: 10.1128/genomeA.00104-12
Rzhetsky, A., and Nei, M. (1992). A simple method for estimating and testing minimum-evolution trees. Mol. Biol. Evol. 9:945.
Sahlin, K., Vezzi, F., Nystedt, B., Lundeberg, J., and Arvestad, L. (2014). BESST - Efficient scaffolding of large fragmented assemblies. BMC Bioinform. 15:281. doi: 10.1186/1471-2105-15-281
Santos, A. S., Sampaio, A. P. W., Vasquez, G. S., Santa Anna, L. M., Pereira, N., and Freire, D. M. G. (2002). Evaluation of different carbon and nitrogen sources in production of rhamnolipids by a strain of Pseudomonas aeruginosa. Appl. Biochem. Biotechnol. 98, 1025–1035.
Siegmund, I., and Wagner, F. (1991). New method for detecting rhamnolipids excreted by Pseudomonas species during growth on mineral agar. Biotechnol. Tech. 5, 265–268.
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J. M., and Birol, Ý (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Smits, T. H. M., Witholt, B., and van Beilen, J. B. (2003). Functional characterization of genes involved in alkane oxidation by Pseudomonas aeruginosa. Antonie van Leeuwenhoek 84, 193–200.
Sotirova, A., Spasova, D., Vasileva-Tonkova, E., and Galabova, D. (2009). Effects of rhamnolipid-biosurfactant on cell surface of Pseudomonas aeruginosa. Microbiol. Res. 164, 297–303. doi: 10.1016/j.micres.2007.01.005
Taboada, B., Estrada, K., Ciria, R., and Merino, E. (2018). Operon-mapper: a web server for precise operon identification in bacterial and archaeal genomes. Bioinformatics 34, 4118–4120. doi: 10.1093/bioinformatics/bty496
Thessen, A. E., and North, E. W. (2017). Calculating in situ degradation rates of hydrocarbon compounds in deep waters of the Gulf of Mexico. Mar. Pollut. Bull. 122, 77–84. doi: 10.1016/j.marpolbul.2017.06.004
Thomas, J. C., Wafula, D., Chauhan, A., Green, S. J., Gragg, R., and Jagoe, C. (2014). A survey of deepwater horizon (DWH) oil-degrading bacteria from the Eastern oyster biome and its surrounding environment. Front. Microbiol. 5:149. doi: 10.3389/fmicb.2014.00149
Tiwari, M., Sahu, S. K., and Pandit, G. G. (2017). Distribution of PAHs in different compartment of creek ecosystem: ecotoxicological concern and human health risk. Environ. Toxicol. Pharmacol. 50, 58–66. doi: 10.1016/j.etap.2017.01.008
Truong, D. T., Franzosa, E. A., Tickle, T. L., Scholz, M., Weingart, G., Pasolli, E., et al. (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. doi: 10.1038/nmeth.3589
Uzelac, G., Bertani, I., Kojic, M., Paszkiewicz, K. H., Studholme, D. J., Passos da Silva, D., et al. (2014). Draft genome sequence of beneficial rice rhizosphere isolate Pseudomonas aeruginosa PUPa3. Genome Announc. 2:e00654-14. doi: 10.1128/genomeA.00654-14
Valenzuela-González, F., Martínez-Porchas, M., Villalpando-Canchola, E., and Vargas-Albores, F. (2016). Studying long 16S rDNA sequences with ultrafast-metagenomic sequence classification using exact alignments (Kraken). J. Microbiol. Methods 122, 38–42. doi: 10.1016/j.mimet.2016.01.011
van Beilen, J. B., Smits, T. H. M., Roos, F. F., Brunner, T., Balada, S. B., Röthlisberger, M., et al. (2005). Identification of an amino acid position that determines the substrate range of integral membrane alkane hydroxylases. J. Bacteriol. 187, 85–91. doi: 10.1128/JB.187.1.85-91.2005
Varjani, S. J., and Upasani, V. N. (2016). Biodegradation of petroleum hydrocarbons by oleophilic strain of Pseudomonas aeruginosa NCIM 5514. Bioresour. Technol. 222, 195–201. doi: 10.1016/j.biortech.2016.10.006
Vives-Flórez, M., and Garnica, D. (2006). Comparison of virulence between clinical and environmental Pseudomonas aeruginosa isolates. Int. Microbiol. 9, 247–252.
Wang, W., Cai, B., and Shao, Z. (2014). Oil degradation and biosurfactant production by the deep sea bacterium Dietzia maris As-13-3. Front. Microbiol. 5:711. doi: 10.3389/fmicb.2014.00711
Wang, W., and Shao, Z. (2012). Genes involved in alkane degradation in the Alcanivorax hongdengensis strain A-11-3. Appl. Microbiol. Biotechnol. 94, 437–448. doi: 10.1007/s00253-011-3818-x
Wang, Z., and Wu, M. (2013). A phylum-level bacterial phylogenetic marker database. Mol. Biol. Evol. 30, 1258–1262. doi: 10.1093/molbev/mst059
Wentzel, A., Ellingsen, T. E., Kotlar, H. K., Zotchev, S. B., and Throne-Holst, M. (2007). Bacterial metabolism of long-chain n-alkanes. Appl. Microbiol. Biotechnol. 76, 1209–1221. doi: 10.1007/s00253-007-1119-1
Xue, J., Yu, Y., Bai, Y., Wang, L., and Wu, Y. (2015). Marine oil-degrading microorganisms and biodegradation process of petroleum hydrocarbon in marine environments: a review. Curr. Microbiol. 71, 220–228. doi: 10.1007/s00284-015-0825-7
Yuste, L., Corbella, M. E., Turiégano, M. J., Karlson, U., Puyet, A., and Rojo, F. (2000). Characterization of bacterial strains able to grow on high molecular mass residues from crude oil processing. FEMS Microbiol. Ecol. 32, 69–75. doi: 10.1016/s0168-6496(00)00015-5
Keywords: Gulf of Mexico, marine bacteria, hydrocarbon degradation, Pseudomonas aeruginosa, bioremediation
Citation: Muriel-Millán LF, Rodríguez-Mejía JL, Godoy-Lozano EE, Rivera-Gómez N, Gutierrez-Rios R-M, Morales-Guzmán D, Trejo-Hernández MR, Estradas-Romero A and Pardo-López L (2019) Functional and Genomic Characterization of a Pseudomonas aeruginosa Strain Isolated From the Southwestern Gulf of Mexico Reveals an Enhanced Adaptation for Long-Chain Alkane Degradation. Front. Mar. Sci. 6:572. doi: 10.3389/fmars.2019.00572
Received: 25 April 2019; Accepted: 29 August 2019;
Published: 18 September 2019.
Edited by:
Tony Gutierrez, Heriot-Watt University, United KingdomReviewed by:
Anahit Penesyan, Macquarie University, AustraliaStilianos Fodelianakis, École Polytechnique Fédérale de Lausanne, Switzerland
Copyright © 2019 Muriel-Millán, Rodríguez-Mejía, Godoy-Lozano, Rivera-Gómez, Gutierrez-Rios, Morales-Guzmán, Trejo-Hernández, Estradas-Romero and Pardo-López. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liliana Pardo-López, bGlsaWFuYUBpYnQudW5hbS5teA==
†Present address: Elizabeth Ernestina Godoy-Lozano, Centro de Investigación Sobre Enfermedades Infecciosas, Instituto Nacional de Salud Pública, Cuernavaca, Mexico Nancy Rivera-Gómez, Centro de Investigación en Salud Poblacional, Instituto Nacional de Salud Pública, Cuernavaca, Mexico Alejandro Estradas-Romero, Comisión Nacional para el Conocimiento y Uso de la Biodiversidad, Mexico City, Mexico