 Amandine D. Marie
Amandine D. Marie Christophe Herbinger2
Christophe Herbinger2 Ciro Rico
Ciro Rico- 1School of Marine Studies, Molecular Analytics Laboratory, Faculty of Science Technology and Environment, The University of the South Pacific, Suva, Fiji
- 2Department of Biology, Dalhousie University, Halifax, NS, Canada
- 3Department of Mathematics and Statistics, Dalhousie University, Halifax, NS, Canada
- 4Instituto de Ciencias Marinas de Andalucía, Consejo Superior de Investigaciones Científicas, Cádiz, Spain
Sharks constitute one of the most threatened clades (Selachimorpha) of all marine fish, and substantial management efforts are required to help the recovery of their populations worldwide. Despite its significant impact on population dynamics and conservation, sharks’ reproductive and philopatric behavior has received little attention in fisheries management. The scalloped hammerhead shark (Sphyrna lewini), an endangered species listed on the IUCN’s Red List and on the CITES’ Appendix II, is an apex predator that potentially exhibits female philopatry to mating grounds. We reconstructed, for the first time in an open ocean species, the relationship among 166 juvenile individuals caught in a recently discovered aggregation of the Rewa Delta, Fiji, and determined the sample population’s mating system using 6,437 SNPs. Using two software packages, COLONY2 and SNP PEDIGREE, results revealed very high consistency in the identification of full and half sib. Moreover, COLONY2 allowed us to identify an equal breeding sex ratio for each cohort analyzed for this population (1.04:1; 1.02:1), as well as several cases of multiple paternity and numerous matings of the same male with different females suggesting polygynandry for this species. These findings reveal additional information about the complex life history of the scalloped hammerhead shark.
Introduction
Sharks, rays and chimeras (Chondrichthyes) are the most threatened fishes in the world due to the synergetic effects of overfishing, habitat destruction, pollution, climate change, slow growth, late maturity, low fecundity, and long life span (Davidson et al., 2016). It is estimated that at least 25% of shark and ray species are at high risk of extinction (Dulvy et al., 2014). Fishing mortality among Chondrichthyes has increased 227% since the end of World War II (Davidson et al., 2016). For sharks alone, the global human-induced mortality was estimated to be no less than 1.41 million tons in 2010 (Worm et al., 2013). Thus, effective management measures are urgently required to enforce sustainable harvest of sharks and rays.
While fisheries management is primarily concerned with estimating abundance in order to establish sustainable yields, the collapse of a fish stock provides scientists with the opportunity to study the causes and mechanisms behind the inability to predict the susceptibility to fishing of the overexploited species and its subsequent incapacity to recover (Rowe and Hutchings, 2003). Incorporating knowledge about the mating behavior of exploited species has been identified as an essential step to increase the effectiveness of fisheries management (Rowe and Hutchings, 2003) through improved understanding of associated population dynamics and conservation (Rowe and Hutchings, 2003; Lowerre-Barbieri et al., 2016). However, mating behavior has received little attention in fisheries management, probably due to the difficulties associated with studying the reproductive ecology of most large and commercially important marine fishes.
Determining key reproductive parameters such as mating patterns (e.g., number of parents involved in a cohort) using genetic markers in marine ecosystems has only been possible in confined environments harboring small or resident populations. For example, a recent study by Salles et al. (2016) presented the first resolved genealogical family tree of a reef fish, the orange clownfish (Amphiprion percula), by repeatedly genotyping, over a period of 10 years, all individuals of a local deme in Kimbe Island, Papua New-Guinea. Through their pedigree analysis, the authors showed a recurrent longitudinal philopatry over five generations and revealed the mating system of this iconic species. However, direct observations of the reproductive behavior of large marine predators such as sharks are inherently difficult and rare (Feldheim et al., 2004). A study by Dibattista et al. (2008) documented the family structure and reconstructed the genotypes of the adult population of lemon sharks (Negaprion brevirostris), but was again limited to a shallow and small inner lagoon in the Florida Keys, United States. The study also primarily used samples from neonates and juveniles. The lagoon serves as a nursery ground for approximately 75–100 juvenile lemon sharks in any one year and has been monitored for at least eight years by sampling most neonates every parturition season. The study revealed high levels of polyandry and strong female site fidelity, with at least 43 females and 163 males contributing to the litters engendered over 10 breeding seasons. Subsequently, Mourier et al. (2013) reconstructed the genetic relationships among individuals and determined the mating system of a naturally small island population of 40 adult sicklefin lemon sharks (Negaprion acutidens) in Moorea, French Polynesia. Results confirmed philopatry and estimated an average reproductive cycle of 2-year for most females, as well as fertilization by multiple males in up to 78% of litters showing the prevalence of a polyandrous mating system.
The scalloped hammerhead shark (SHS hereafter, Sphyrna lewini) is an apex predator species that has a circumglobal distribution in tropical and warm temperate latitudes over neritic and adjacent deep oceanic waters (Daly-Engel et al., 2012). Juveniles and adults occupy different habitats. Juveniles are demersal, gregarious, and primarily found in coastal areas, estuaries, and embayments, while adults are mainly solitary and inhabit pelagic waters. Viviparous mature females produce 13–31 neonates of 42–55 cm total length per litter after a gestation period of 9–10 months (Compagno, 1984). Current evidence from ecological, behavioral, and genetic data suggests female philopatry to mating grounds (Duncan and Holland, 2006; Daly-Engel et al., 2012). However, this hypothesis remains to be confirmed.
The SHS was declared an endangered species by the International Union for Conservation of Nature (IUCN) Red List in 2007 (Baum et al., 2007), and is currently listed on Appendix II of the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) (CITES, 2015). Several SHS populations have been heavily exploited worldwide by both inshore and offshore fisheries (Compagno, 1984). In Fiji waters, an important aggregation of neonates and young-of-the-year was recently reported in the Rewa Delta, on the island of Viti Levu (Brown et al., 2016; Marie et al., 2017).
In this study, a subsample of the pups captured by Marie et al. (2017) was genotyped to reconstruct the relationships among 166 individuals and to determine the population’s mating system. We were particularly interested in determining the minimum number of breeders that have contributed to two consecutive parturition seasons through kinship analyses. In addition, we also were interested in determining the breeding sex ratio and the effective population size (Ne). While the operational sex ratio (OSR), defined as the number of males to females within a population that are available to mate, is the common term used in behavioral ecology to study mating systems, it is impossible to identify every available mate in the open ocean. Therefore, we prefer to use the term breeding sex ratio (BSR), which refers to successfully breeding parents as a proxy of the OSR. This study constitutes a novel attempt to reconstruct the kin relationships among neonates and young-of-the-year captured in an estuarine environment without any parental information and born from live-bearing females that inhabit neritic and oceanic ecosystems, using the multilocus genotypes of a large battery of single nucleotide polymorphisms (SNPs).
Materials and Methods
Sampling Procedures
The study focused on the Rewa Delta (178.55°E, 18.15°S, Figure 1) on Viti Levu, Fiji. Sampling sites and field methods are detailed in Marie et al. (2017). Sampling was carried out using a stratified random design. In brief, after a pilot study, several locations were identified as sites of interest for long-term monitoring based on their accessibility, exposure to wind and swells, and strength of currents, which allowed for regular and consistently successful deployments. Surveys were carried out at least three times a month from September 2014 to March 2016 (see Marie et al., 2017). Sampling was conducted under a research permit provided by the Rewa Provincial Council and the Department of Fisheries. All handling procedures of live SHS specimens were approved under the Animal Ethics Committee section of the USP Research Committee, and performed in accordance with relevant guidelines and regulations. For each individual shark, the species was identified according to Last and Stevens (1994), then measured, sexed, and aged according to the umbilical scar status (Duncan and Holland, 2006). In addition, two biopsy tissue samples (0.5 cm2) were taken from the pelvic fins of each SHS and preserved in 95% ethanol until DNA extraction.
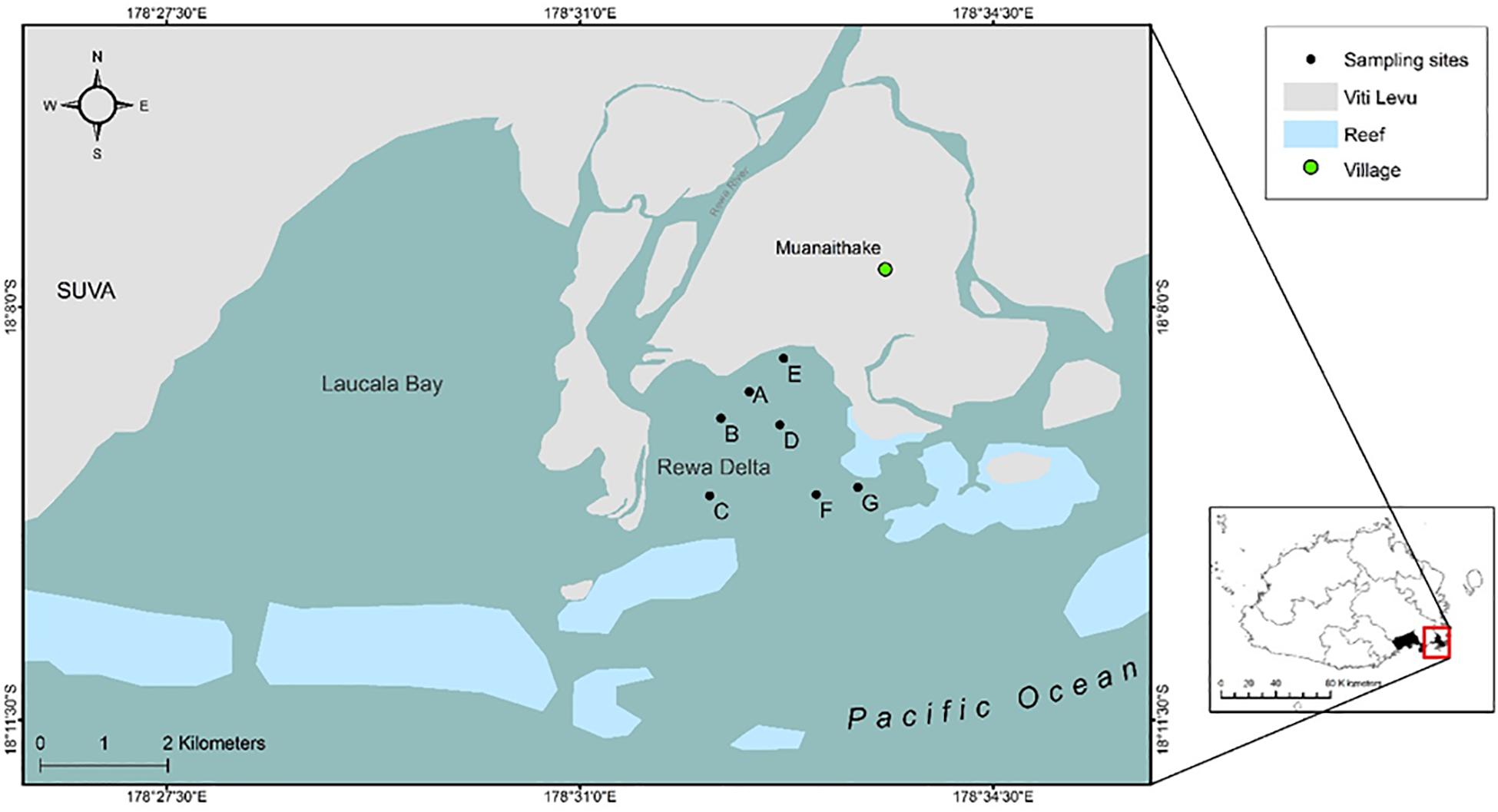
Figure 1. Geographical location of sampling sites (A–G) in the Rewa Delta, Viti Levu, Fiji. The two maps were created using ArcGis, version 10.2 (https://www.arcgis.com/features/index.html).
Because the breeding season at this site occurs during the austral summer, two different cohorts were included in the analyses (2014 – 2015 and 2015 – 2016, identified hereafter with the addition of the letters “A” and “B” presiding the sample number, respectively; e.g., ASHS#000). All individuals per parturition season were further grouped into cohorts based on size and umbilical scar status. Due to financial constraints, only a subsample of 166 SHS individuals out of the 1054 captures was genotyped and used for kinship analyses. In order to estimate the minimum number of parents that have contributed to each cohort within the given constraints, we selected samples caught within a week of each other and showing similar length (52.5 cm ± 5.6) and scar healing condition (mainly opened and semi-healed) as a method to improve the chances of getting some individuals from the same litter. This sampling strategy was chosen specifically to estimate the levels of polyandry and polygyny in the population, but it may bias estimates of breeding sex ratio. To assess the impact of the sampling strategy, we estimated the number of parents contributing to each cohort before and after removing all but one individual per family group. Likewise, for the estimates of effective population size, one individual per family group was included in the dataset. Supplementary Table 1 shows general information for each individual used in this study (e.g., total length, umbilical scar condition).
DArTseqTM Library Preparation and Sequencing
DNA was extracted from a small portion of pelvic fin (approximately 1 mm2) using a slightly modified version of Aljanabi and Martinez (1997) salting out method. In brief, after addition of the saline solution, the mixture was centrifuged for 30 min at 10,000 g. DNA precipitation and washing out of the pellet were achieved by incubation at −20°C with 600 μL isopropanol for 30 min and with 500 μL 70% ethanol for 10 min, respectively. Both steps were centrifuged after incubation at 10,000 g for 20 min. Diversity Arrays Technology Limited processed the extracted DNA using the DArTseqTM technique for reduced representation library construction, sequencing, and genotyping. We followed previously developed and tested complexity reduction protocols (Kilian et al., 2012). Genome complexity reduction was achieved with a double restriction digest using a PstI and SphI methylation-sensitive restriction enzyme combination. Libraries were sequenced on an Illumina HiSeq 2500 platform. For initial assessment of read quality and sequence representation, raw reads obtained were processed using Illumina CASAVA v.1.8.2 software. Then, DArTtoolbox was used to perform filtering and variant calling, and generate final genotypes (Kilian et al., 2012).
SNP Filtering
Following the generation of genotype data from DArTseq, the dataset was further filtered to retain only highly informative SNPs. This was achieved by filtering out duplicate SNPs (23.4% possessing identical Clone IDs), and retaining loci with a call rate of at least 95%, read depth > 7x, and minor allele frequency > 5%. We then searched for candidate loci under selection using BayeScan v.2.1 (Foll and Gaggiotti, 2008; Foll, 2012) software at a false discovery rate (FDR) = 0.1. The FDR is a statistical confidence measure, which allows us to assess how statistically true the results might be. Departure from Hardy-Weinberg Equilibrium (HWE) was then assessed for each locus using the software Arlequin v.3.5.2.2 (Schneider et al., 2000) using an exact test with 10,000 steps in the Markov Chain method and 100,000 dememorizations. Test for linkage disequilibrium was performed using the software PLINK v.1.07 (Chang et al., 2015). To assess the robustness of the reconstructed relationships to the number of SNPs used, a reduced data set was created using the same filtering but only retaining loci with a call rate at or greater than 98%.
Kinship Analyses
COLONY2 v.2.0.6.2 (Jones and Wang, 2010) was used to estimate sibship relationships as well as the most likely number of involved parents among the 166 neonates and young-of-the-year SHS. To perform this analysis, COLONY2 uses a maximum likelihood approach to assign individuals to clusters based on offspring relationship, categorized as full sibling, half sibling, or unrelated. This approach produced clusters of full sibling pups that share both parents, and half siblings that share at least one parent with at least one other individual in the cluster. To this end, we performed ten long runs to determine the consistency of the analyses using the following parameters: the full likelihood method, no updating of allele frequencies, weak siblingship prior and very high precision assuming polygamy of both sexes and outbreeding. In addition, as COLONY2 has been shown to overestimate the putative number of siblings (Sefc and Koblmuller, 2009), we also performed analyses using SNP PEDIGREE, a variant of PEDIGREE 2.2 that can handle SNP marker data. These additional analyses were performed in order to corroborate the COLONY2 results.
PEDIGREE 2.2 first uses the genetic marker information to construct pairwise likelihood ratios of being full sib vs. being unrelated for all individuals in the data set. These pairwise likelihood ratios are then used to construct an overall sibship partition score, which should be maximized under the true partition. The space of possible partitions is explored by a Markov Chain Monte Carlo (MCMC) algorithm (Smith et al., 2001; Herbinger et al., 2006). Full sib and half sib partitions are constructed by imposing constraints during the MCMC run on the observed microsatellite genotype patterns. Under this scenario, each group could be derived from a cross of a single male by a single female at all loci (full sib partition) or from a cross between 1 parent and several parents of the other sex at all loci (half sib partition) (Herbinger et al., 2006). SNP PEDIGREE follows the same overall logic with the following change: with bi-allelic markers such as SNPs, it is not possible to impose constraints on genotype patterns to differentiate full sib partitions from half sib partitions. Instead, the SNP marker information is used to construct a matrix of pairwise likelihood ratios of being full sib vs. being unrelated for all individuals in the data set, as well as a matrix of pairwise likelihood ratios of being half sib vs. being unrelated. The two different sets of pairwise likelihood ratios are then used to build full sib and half sib partition scores, which are maximized in separate MCMC runs. Finally, the full sib partition is nested within the half sib partition to identify which different full sib groups might be half sib to one another. The SNP PEDIGREE analyses were run on the subset of SNPs with a call rate of 98%. We also used the same set of SNPs using COLONY2 to validate robustness of results when reducing the number of SNPs.
Effective Population Size
The effective population size (Ne) was estimated using the full likelihood method assuming random mating among samples in COLONY2, as well as using the linkage disequilibrium method implemented in NeEstimator (version 2.0, Do et al., 2014). As stated above, only unrelated individuals (n = 104) were included in this analysis and only using the reduced dataset of SNPs.
Results
SNP Filtering
A total of 69,164 SNPs were genotyped using DArTseq. This number was reduced to 6,598 SNPs after quality filtering (call rate > 95%, read depth > 7x, MAF > 5%). BayeScan did not detect any loci as outliers. One hundred and fifty-one loci were found to significantly deviate from HWE expectations under random mating (P-value < 0.00001), and thus were removed from the dataset. We also identified and deleted ten SNPs (r2 > 75%) in linkage disequilibrium. The final, putatively neutral loci dataset contains 6,437 SNPs (large set).
The second filtering used more stringent quality thresholds (call rate > 98%, read depth > 7, MAF > 5%). No outliers were detected, while 13 loci were found to significantly deviate from HWE expectations (P-value < 0.00001), and were removed from the dataset. Two SNPs (r2 > 97%) were identified in linkage disequilibrium and also deleted from the database. The final dataset contains 1,361 SNPs (reduced set).
Kinship Analyses
COLONY2 runs were performed with both the large dataset (all 6,437 SNPs) and the reduced dataset (1,361 loci), while SNP PEDIGREE only used the reduced dataset. COLONY2 results with the large and reduced SNP datasets were identical. COLONY2 identified 92 putative full siblings, distributed in 33 groups ranging in size from two to seven individuals (Table 1). Out of the 72 remaining individuals, 37 had no relatives within the cohort (named singletons hereafter). Thirty-five individuals had no full siblings but did putatively have half-siblings. The results from SNP PEDIGREE were nearly identical. There was 93% concordance on the identification of full siblings between the two software programs (Table 1). Only seven individuals were not assigned to the same full sib groups by both software. For instance, individual ASHS#6 was assigned by SNP PEDIGREE as a member of the full sib family #12, but as a half-sib associated with the same group by COLONY2 (Table 1). In addition, three cases occurred in which SNP PEDIGREE identified pairs as full sib (families #34, #35 and #36), that COLONY2 independently labeled as half sibs (Table 1).
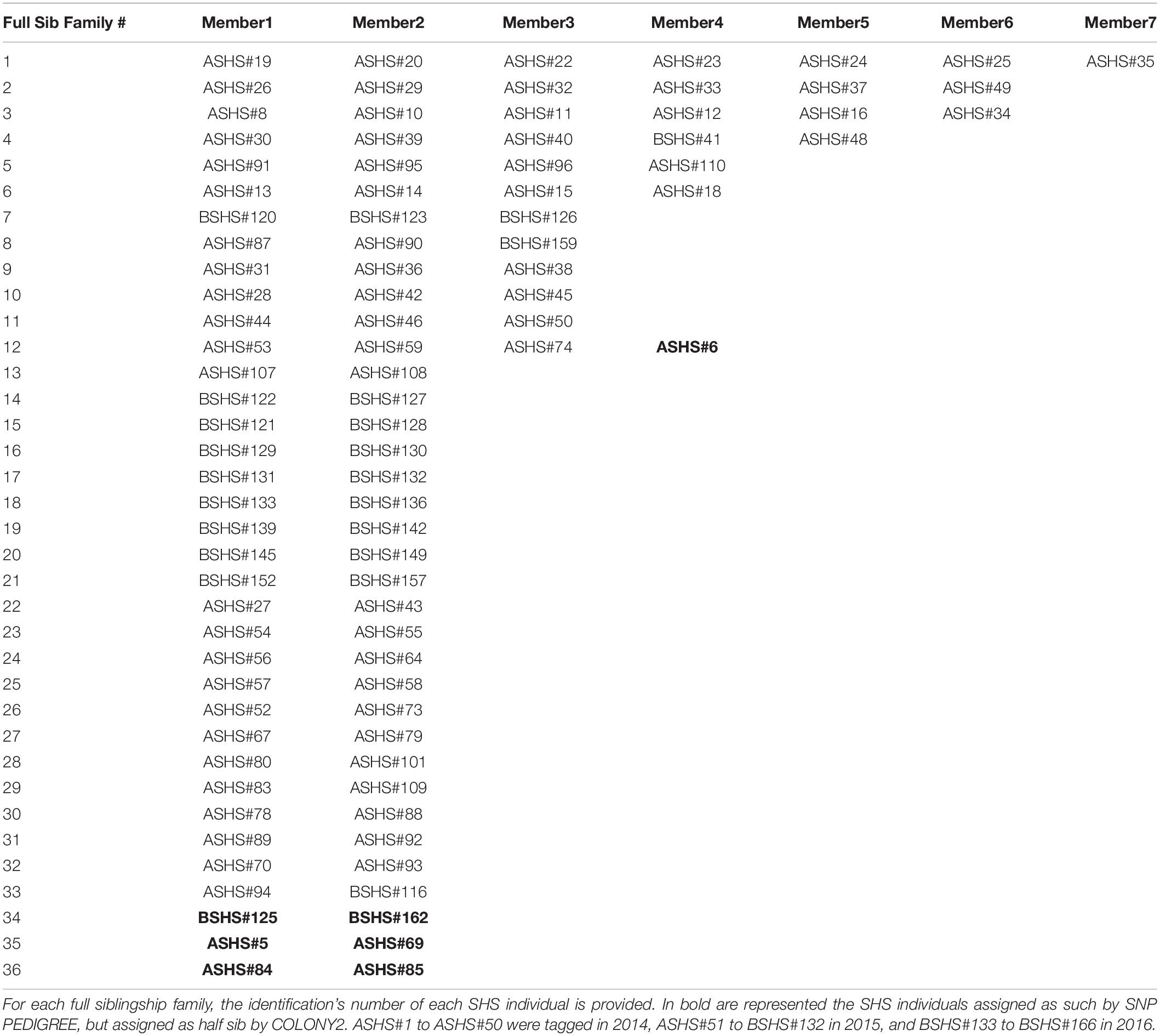
Table 1. Full sib assignments’ results obtained using COLONY2 and SNP PEDIGREE.
Using COLONY2, we were able to reconstruct the multilocus genotypes of the hypothetical parents of each sampled individual, which in turn allowed the identification of females that mated with a single male and the cases of multiple paternity, as well as the breeding sex ratio of this cohort (Table 2). To illustrate the results of this analysis, we labeled the parents of one sex with an asterisk (∗) plus a number (referred to as “Father” by COLONY2), and an asterisk plus a letter for the opposite sex (referred to as “Mother” by COLONY2) (Table 2). Note that, although COLONY2’s results showed the presence of multiple paternity, we cannot differentiate with certainty the maternal or paternal contribution, as none of the maker loci used can be associated to sex chromosomes.
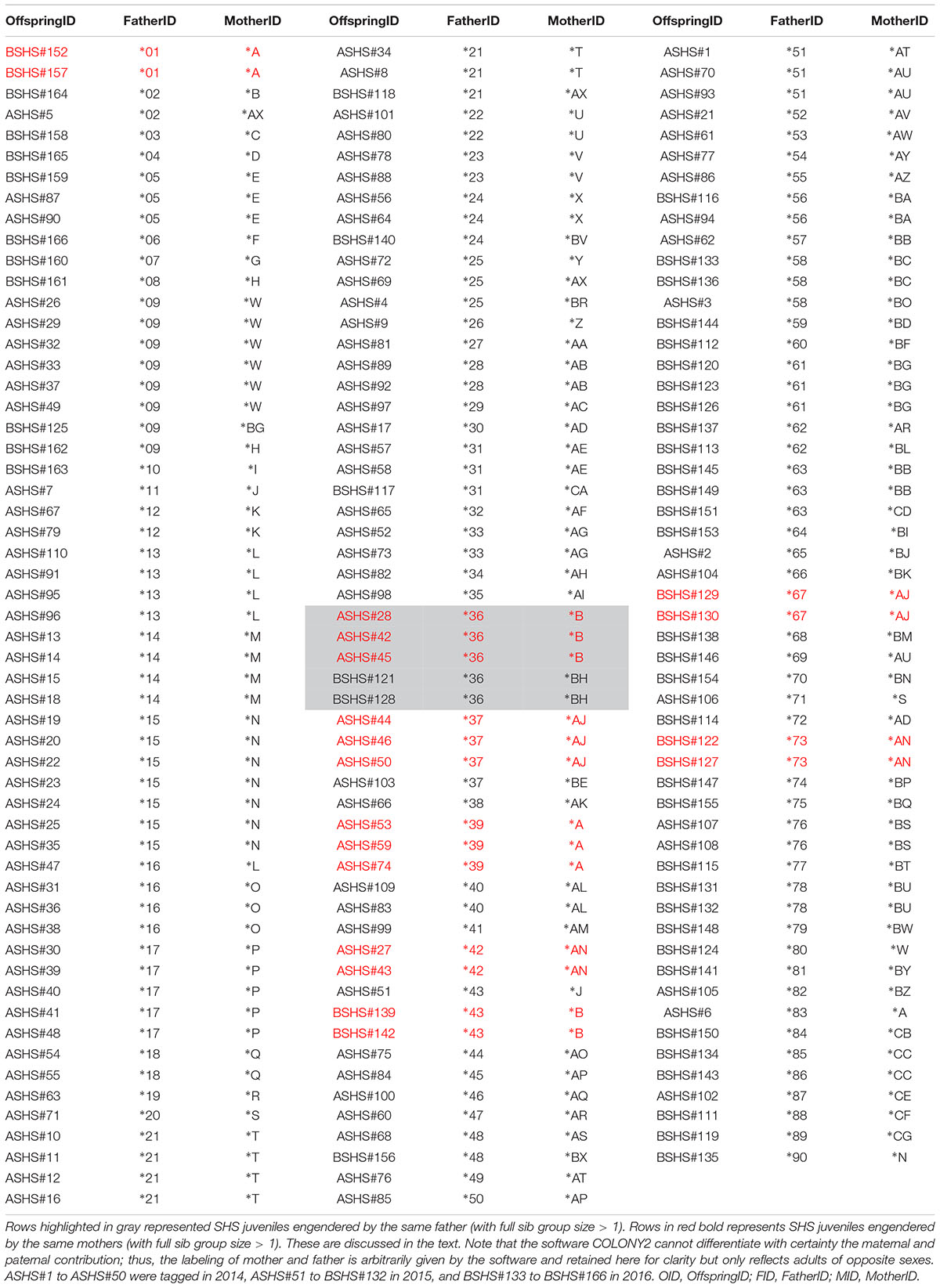
Table 2. Identification of the mother and the father of each SHS juvenile based on the reconstruction of the multilocus genotype of the two parents that contribute to each juvenile analyzed.
Agreement between COLONY2 and SNP PEDIGREE for identifying half sib relationships was also very good but harder to quantify, as both programs generate different types of half sib solutions. COLONY2 can reconstruct clusters of individuals who are related by common parents in complex patterns. For example, individuals ASHS#60, BSHS#137 and BSHS#113 belong to the same cluster 13 (Table 3), because ASHS#60 shares parent ∗AR with BSHS#137, while BSHS#113 shares parent ∗62 with BSHS#137. In contrast, SNP PEDIGREE generates a half sib partition where every individual in the half sib group should share at least one parent with every other member of the group. In the example above, not all three individuals ended up in the same SNP PEDIGREE half sib group because SHS#60 and SHS#113 do not share any common parent. COLONY2 and SNP PEDIGREE were in perfect agreement for 11 clusters (Cluster 4, 5, 8, 9, 10, 11, 12, 14, 15, 16, 17), which were pure half sib groups (all individuals sharing at least one parent, Table 3). The other COLONY2 clusters (1, 2, 3, 6, 7, and 13) were complex groupings originating from at least two male and two females parents. Not surprisingly, SNP PEDIGREE split them, generally into two or three half sib groupings, except for the large and very complex cluster 1, which was split into seven subgroups (Table 3). The only major discrepancy concerned individual ASHS#7, which was placed by COLONY2 in cluster 1 but was joined to a completely unrelated full sib family by SNP PEDIGREE.
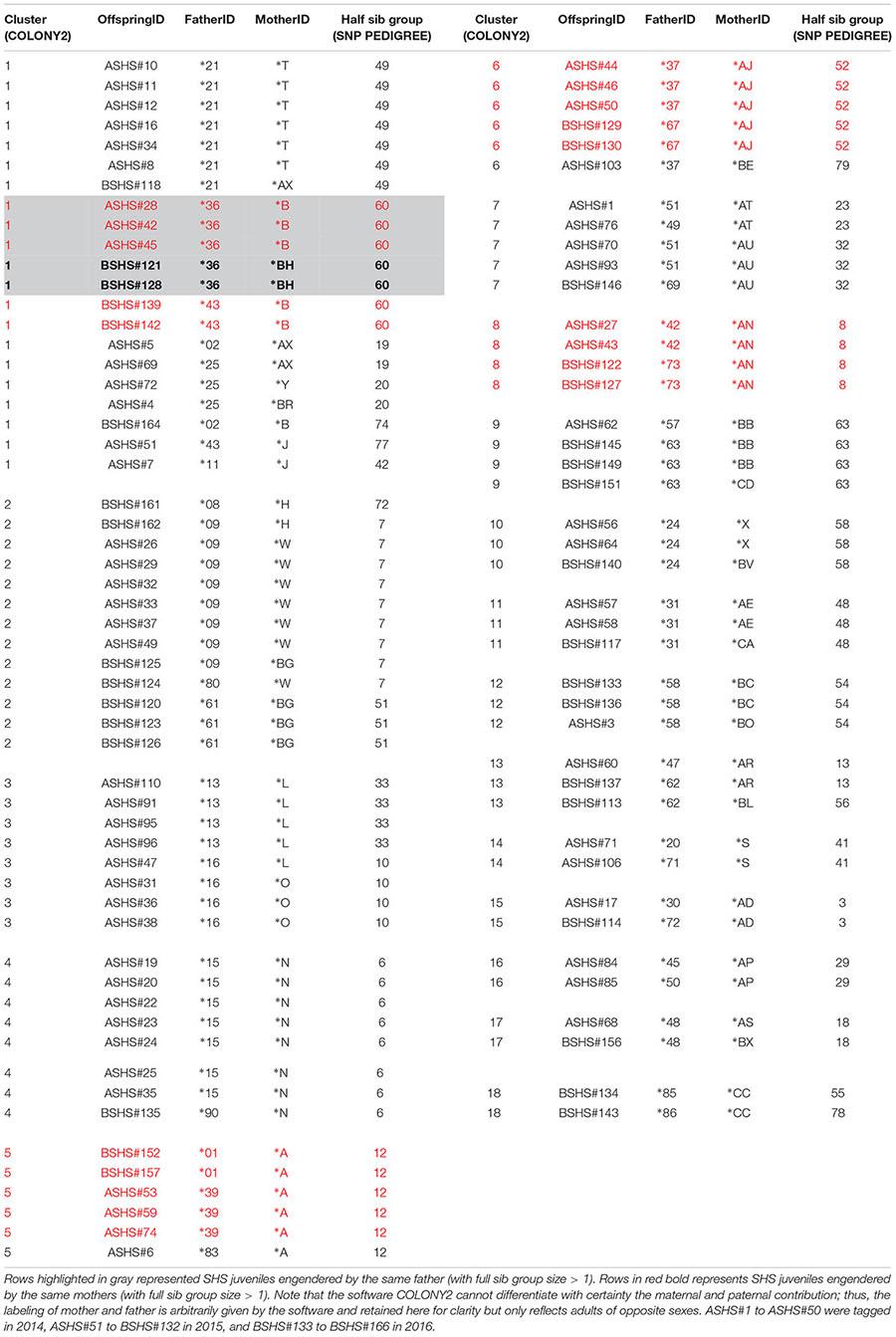
Table 3. Eighteen clusters of half sib and full sib identified by COLONY2 and agreement with SNP PEDIGREE half sib groups.
Irrespective of the putative sex of the breeder, COLONY2’s reconstruction of parental multilocus genotypes identified many instances where parents seemed to have had more than one partner in producing the 166 offspring that were analyzed. There were 13 instances where a “∗number” parent had two partners, and two instances with three partners; meanwhile, there were 17 instances where a “∗letter” parent had two partners, and two instances with three partners (Table 3). In most of these cases, the evidence of putative multiple paternity and/or more than one mating of a male with up to three females is based on a single offspring (Table 3). In five cases, however, the evidence is based on a larger full sib family size. In cluster 5, parent ∗A produced three and two offspring with two members of the opposite sex (∗39 and ∗1), respectively. Similarly, in cluster 6, parent ∗AJ produced three and two offspring respectively with ∗37 and ∗67; and in cluster 8, parent ∗AN produced two and two offspring respectively with ∗42 and ∗73 (Table 3). In cluster 1, Parent ∗B produced three and two offspring respectively with ∗36 and ∗43, while parent ∗36 produced three and two offspring with ∗B and ∗BH (Table 3).
A previous study documented seasonality of parturition from October to March (Marie et al., 2017), and thus the breeding ratio was estimated separately for the first cohort (ASHS#1 to ASHS#110 sampled from October 2014 to March 2015) and the second cohort (BSHS#111 to BSHS166, sampled from October 2015 to March 2016). COLONY2 identified the contribution of 58 and 56 parents of opposite sexes during the 2014-2015 season, and 43 and 42 in 2015-2016, suggesting an effectively equal breeding sex ratio of 1.04 to 1 and 1.02 to 1, respectively (Table 2).
Effective Population Size
Using 1,361 SNPs from 104 unrelated individuals, estimates of the effective population size yielded Ne = 191 (CI 95%: 149,0 – 241) and Ne = 343,6 (CI 95%: 343,0 – 344,3) using COLONY2 and NeEstimator, respectively.
Discussion
Analyses using COLONY2 revealed the presence of at least 33 full sib families ranging in size from two to seven and 72 unrelated individuals among 166 new born and young-of-the-year sharks. It is noteworthy that the pups within each full sib family were generally captured at the same date and location, as indicated by their consecutive identification numbers. Physical and temporal proximity support the expectation that these full siblings would be littermates. Sampled individuals were organized into 87 clusters of between 1 and 21 individuals. Cluster relationships ranged from singletons to isolated full- or half-sib families and complex, interlinked full and half-sib groups (Table 3).
Although methods used by COLONY2 and SNP PEDIGREE differ, results from both programs were highly consistent for both full sib and half sib determination. For the results that were not in perfect agreement, capture and ecological data can help determine, in some cases, the most parsimonious assignment. For instance, two pairs (ASHS#5 with ASHS#69; and BSHS#125 with BSHS#162) were assigned as full siblings by SNP PEDIGREE but as half siblings by COLONY2. Capture date, size, and status of the umbilical scar are not similar enough to suggest being littermates, therefore the COLONY2 half sib assignment is much more likely. In contrast, for individuals ASHS#84 and ASHS#85, capture and ecological data do not offer additional insights. These two individuals could well be full sibs as assigned by SNP PEDIGREE or could be half sibs as identified by COLONY2.
Interestingly, the individual ASHS#6 was identified as a member of the full sibling family 12 by SNP PEDIGREE, but was assigned as a half sib to the same group by COLONY2. This young-of-the-year was sampled in September 2014 (total length of 74.0 cm; healed umbilical scar; Supplementary Table 1) and the three others in February 2015 (each with total length of 49 cm and with semi-healed umbilical scar; Supplementary Table 1). It is therefore impossible that these are all littermates. However, it is not impossible that they are all still full siblings. The current data is insufficient to assess this claim. Mate fidelity over different breeding seasons is undocumented. However, some shark species are known to be able to store sperm (Pratt, 1993). From another perspective, the minimum time between gestations is known to be one year for other Carcharhiniformes such as bull sharks, which have a similar gestation period (Compagno, 1984). To the best of our knowledge, no study has documented gestation time specifically for hammerhead sharks. Furthermore, based on the estimated average monthly growth rate in this population determined through recapture in a previous study (Marie et al., 2017), the individual captured in 2014 is at least 1 year old (i.e., born before October 2013). Therefore, it is possible that the same female has two litters born at least 2 years apart but captured at least 17 months asunder. However, to be confident in this interesting observation, the probability that the same male contributes to both litters would need to be confirmed by a robust kinship assignment (i.e., both software concurrently identifying full siblingship).
In theory, the approach followed by COLONY2 is potentially more precise than the one followed SNP PEDIGREE (and by PEDIGREE 2.2), as the former maximizes the true full likelihood of the proposed configuration, while the latter maximizes an approximate partition score (Smith et al., 2001). However, based on microsatellite data, COLONY2 and PEDIGREE 2.2 yielded very similar results in a study of endangered salmon populations (Salmo salar), another species with complex mating system (Herbinger et al., 2006; de Mestral et al., 2012). The reconstructed half sib and full sib groups in these salmon studies seemed to result from a mating system mostly characterized by female polygamy and male monogamy, with a few instances of polygamy from both sexes. This proposed mating system was later confirmed using sex linked marker information (de Mestral et al., 2012). The present study demonstrates that COLONY2 and SNP PEDIGREE also generate nearly identical results when using SNP information.
Interestingly, COLONY2 generated the same solution with the full set of 6,437 SNPs and the reduced set of 1,361 loci, both of which agreed closely with SNP PEDIGREE results generated using the reduced set. This indicates that it is probably not necessary to use a very large SNP marker set to arrive at reliable results, which also allows a decreased computation time and associated genotyping cost. From these results, we suggest that in studies where a large number of individuals need to be genotyped, reliable results can be achieved with reduced monetary investment in sequencing. Furthermore, this study adds to the increasing literature on the effects of the number of SNP loci on population genetics and kinship analyses (e.g., Hauser et al., 2011; Willing et al., 2012; Mo et al., 2018). The empirical comparison of SNP analyses with full and reduced datasets performed in this study shows that reliable results can be obtained with a marker panel five times smaller than initially obtained using less stringent filtering conditions (95% call rate). While the need of approximately 1000 SNP markers in population genetics has already been validated with simulation studies (Willing et al., 2012), these results also provide an empirical test for the effectiveness of a reduced SNP panel in kinship assignments as previously shown in humans (Mo et al., 2018).
The siblingship reconstruction achieved using COLONY2 and confirmed with SNP PEDIGREE allowed us to identify many examples where putative fathers and putative mothers appeared to have progeny with multiple partners. Polyandry in Elasmobranchs has been documented in the majority of species studied to date using litters collected from the uteri of pregnant females captured on fishing boats (e.g., Daly-Engel et al., 2006; Boomer et al., 2013; Pirog et al., 2016; Rossouw et al., 2016; Green et al., 2017). However, the results of this study demonstrate polygyny in hammerhead sharks for the first time. Indeed, although the software COLONY2 cannot clearly differentiate true maternal or paternal contribution, polygynandry can be illustrated with the example of parent ∗B, which engendered offspring with ∗36 and ∗43; and, in addition to ∗B, parent ∗36 also engendered offspring with ∗BH. This result provides unequivocal evidence that at least two females and two males, engendered offspring with a minimum of two individuals of the opposite sex (Table 3) demonstrating that the SHS is a polygynandrous species. However, it should be noted that, in each of the five credible instances of polygyny and polyandry, the two full sib groups linked in a half sib relationship belonged to different cohorts (e.g., parents ∗B and ∗36 and ∗BH and 36∗engendered offspring during the first and second parturition period, respectively). This is the same pattern for each of the five instances of polygyny and multiple paternity.
As suggested from studies that have sampled litters, the low frequency of observed polygyny and multiple paternity in this study could simply be attributed to the low number of offspring analyzed here, leading to the identification of a few large full sib groups (Table 1). Indeed, in studies using litters, some authors suggested that the litter’s size, depending on the sampling effort, influences the level of reported multiple paternity results (Rossouw et al., 2016; Green et al., 2017). Most of these cases were based on a single offspring attributed to an additional partner.
In general, and especially in elasmobranch species, studying BSR or OSR can be challenging given how little information is available. For instance, the OSR reported from the resident population of manta rays (Manta alfredi) off Maui, Hawaii, which proposed 2.68 adult males per reproductively available female, was revealed to be heavily skewed (Deakos, 2012). The observation of an unequal OSR is thus common as it partially depends on adult sex ratio, which exhibits important variation in nature and causes implications that are not fully understood (Székely et al., 2014). Because we found an effectively equal BSR, we suggest that the OSR in this population is unlikely to deviate substantially from evenness. Furthermore, while both sexes may compete simultaneously for mates, the bias in OSR determines which one becomes the predominant competitor. Therefore, OSR is an important ecological factor that determines the mating system of the population with stronger levels of polygyny or polyandry depending on which sex is in excess (Emlen and Oring, 1977). Assuming our finding of an equal BSR is a good proxy of the overall OSR, one need to ask why do we see a high level of promiscuity? We suggest that the answer may lay in the effective population size. We found a comparatively small Ne for a migratory ocean dwelling species. It is possible that both sexes are mating with multiple partners as an inbreeding avoidance mechanism, rather than mate competition. It is well documented that polygamy may maintain genetic variation in a population, or increase effective population size (e.g., Sugg and Chesser, 1994).
In this study, the report of two sharks that mated with the same individual and then went to the Rewa Delta for parturition suggest the presence of at least one SHS mating site in the region. While this result can also be explained by sperm storage, we believe it is also pertinent to suggest the presence of a mating site in the region for the following reasons. First, Fiji as an oceanic island is isolated from the other suitable nursery grounds and provides adequate parturition areas given the number of large estuaries present in its topography. Consequently, it is possible that at least some of these females mate in the region. Second, SHS are documented to migrate up to 1,671 km (Bessudo et al., 2011) and other recorded juvenile aggregations are found at greater distances (e.g., Hawaiian Islands). Third, a follow up study recently provided evidence for the use of this parturition area by four genetically differentiated populations. One of them is represented by at least 80% of the samples while the other three are underrepresented, suggesting the existence of a local population (Marie et al., unpublished).
Numerous shark species have a complex life history, with high mobility and broad spatial ranges. Chapman et al. (2015) proposed a scheme called “triangle migrations,” which describes the spatial structure of coastal shark populations based on movements of individuals between nursery areas and habitats occupied by adults of different sexes, which have a tendency to exhibit spatial segregation most of the year. In this study, SHS juveniles come from the Rewa Delta and while adult movements, potential aggregations, and demographic and genetic connectivity in the region are unknown, the genetic differentiation uncovered in the above study suggest multiple populations and probably mating sites in the Pacific. This hypothesis, however, remains to be confirmed with additional studies.
Based on the multilocus genotypes obtained from SHS juveniles, we estimated the effective population size of a deme that could be segregated from other such demes or might be panmictic across the Pacific. The level of connectivity of this juvenile aggregation across the South Pacific remains to be determined. Ne is a parameter positively correlated with the ability of populations to persist in a changing environment and to evolve (Frankham et al., 2014). The values calculated in this study underscored possible reasons for concern. The 50/500 rule commonly used by conservation practitioners (Jamieson and Allendorf, 2012), but revised to >100/1000 by Frankham et al. (2014), shows that a minimum Ne of 100 is necessary to avoid immediate risk of extinction due to inbreeding depression. Our results show a potentially high long-term risk of extinction due to the loss of ecologically relevant genetic diversity. However, as suggested by a previous study on SHS in the Eastern Pacific (Nance et al., 2011), small Ne could also be associated with important evolutionary forces causing population divergence, rather than just restricted gene flow or inbreeding depression. Interestingly, the Ne estimates of the unrelated individuals collected in the Ba Estuary (Vierus et al., 2018), which belong to the same population of the Rewa Delta (Marie et al., unpublished), were of the same order of magnitude as current Ne values. In the case of the Ba Estuary samples, no subsampling was conducted to maximize the chances to obtain littermates, as all samples collected were genotyped, although some individuals were found to be siblings. These results suggest that removing all but one individual per family group produces unbiased estimates of the effective population size despite sample pruning for studying mating patterns in the Rewa Delta.
Data Availability Statement
The metadata on shark samples and the raw SNP loci generated for this study are available from Open Science Framework repository at https://osf.io/ykp4h/.
Ethics Statement
This study was reviewed and approved by The University of the South Pacific Animal Ethics Committee.
Author Contributions
CR and AM designed the project. CR wrote the grant proposals, which funded the project. AM organized and sampled all specimens. AM, CH, and PF performed the statistical analyses. All authors reviewed and contributed to the writing of the manuscript.
Funding
This work was primarily funded by The University of the South Pacific, Strategic Research Themes Program for Research and Innovation, and the Faculty of Science, Technology and Environment (Grant No. SRT/6175-3107-70766-617508 to CR). We thank Projects Abroad and Frontiers for additional funding for field work surveys in the Rewa Delta.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Celso Cawich, Semi Nagatalevu, Inoke Nagatalevu, Diego Cardeñosa, Gauthier Mescam, Ron Ronaivakulua, Sydney Shier, Kristian Miles, Brittany Waters, Niklas Komoss, Tom Vierus, Andrew Paris, Timoci Varinava, Ramesh Prasad, and Netani Tulele for their help during field work. We thank Tevita Soqo for producing Figure 1. We also acknowledge Aisake Batibasaga (Director of the Fisheries Department of the Ministry of Fisheries and Forest from 2014 to 2016), George Madden (Director of the Fisheries Department of the Ministry of Fisheries and Forest since 2016), Joji Kalounivalu (Roko Tui, Rewa Provincial Council, Rewa), Ian Campbell (Shark Conservation Programme, WWF-Fiji), and Mike Neuman (Beqa Adventure Divers) for their continued advice and/or support during the project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00676/full#supplementary-material
References
Aljanabi, S. M., and Martinez, I. (1997). Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25, 4692–4693. doi: 10.1093/nar/25.22.4692
Baum, J., Clarke, S., Domingo, A., Ducrocq, M., Lamónaca, A. F., Gaibor, N., et al. (2007). Sphyrna lewini (Northwest and Western Central Atlantic Subpopulation). Cambridge: The IUCN Red List of Threatened Species.
Bessudo, S., Soler, G. A., Klimley, A. P., Ketchum, J. T., Hearn, A., and Arauz, R. (2011). Residency of the scalloped hammerhead shark (Sphyrna lewini) at Malpelo Island and evidence of migration to other islands in the Eastern Tropical Pacific. Environ. Biol. Fish. 91, 165–176. doi: 10.1007/s10641-011-9769-3
Boomer, J. J., Harcourt, R. G., Francis, M. P., Walker, T. I., Braccini, J. M., and Stow, A. J. (2013). Frequency of multiple paternity in gummy shark, Mustelus antarcticus, and rig, Mustelus lenticulatus, and the implications of mate encounter rate, postcopulatory influences, and reproductive mode. J. Hered. 104, 371–379. doi: 10.1093/jhered/est010
Brown, K. T., Seeto, J., Lal, M. M., and Miller, C. E. (2016). Discovery of an important aggregation area for endangered scalloped hammerhead sharks, Sphyrna lewini, in the Rewa River estuary, Fiji Islands. Pac. Conserv. Biol. 22, 242–248.
Chang, C. C., Chow, C. C., Tellier, L., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Chapman, D. D., Feldheim, K. A., Papastamatiou, Y. P., and Hueter, R. E. (2015). There and back again: a review of residency and return migrations in sharks, with implications for population structure and management. Annu. Rev. Mar. Sci. 7, 547–570. doi: 10.1146/annurev-marine-010814-015730
CITES (2015). cites.org. Available at: https://cites.org/eng/app/appendices.php (accessed 04 February 16).
Compagno, L. J. V. (1984). FAO species catalogue. Volume4. Sharks of the world. An annotated and illustrated catalogue of shark species known to date. Part 2. Carcharhiniformes. Rome: Food and Agriculture Organization.
Daly-Engel, T. S., Grubbs, R. D., Holland, K. N., Toonen, R. J., and Bowen, B. W. (2006). Assessment of multiple paternity in single litters from three species of carcharhinid sharks in Hawaii. Environ. Biol. Fish. 76, 419–424. doi: 10.1007/s10641-006-9008-5
Daly-Engel, T. S., Seraphin, K. D., Holland, K. N., Coffey, J. P., Nance, H. A., Toonen, R. J., et al. (2012). Global phylogeography with mixed-marker analysis reveals male-mediated dispersal in the endangered scalloped hammerhead shark (Sphyrna lewini). PLoS One 7:e29986. doi: 10.1371/journal.pone.0029986
Davidson, L. N. K., Krawchuk, M. A., and Dulvy, N. K. (2016). Why have global shark and ray landings declined: improved management or overfishing? Fish Fish. 17, 438–458. doi: 10.1111/faf.12119
de Mestral, L. G., Herbinger, C. M., and O’Reilly, P. T. (2012). Mating structure of an endangered population of wild Atlantic salmon (Salmo salar) as determined using sibship reconstruction and a novel method of sex inference. Can. J. Fish. Aquat. Sci. 69, 1352–1361. doi: 10.1139/f2012-065
Deakos, M. H. (2012). The reproductive ecology of resident manta rays (Manta alfredi) off Maui, Hawaii, with an emphasis on body size. Environ. Biol. Fish. 94, 443–456. doi: 10.1007/s10641-011-9953-5
Dibattista, J. D., Feldheim, K. A., Thibert-Plante, X., Gruber, S. H., and Hendry, A. P. (2008). A genetic assessment of polyandry and breeding-site fidelity in lemon sharks. Mol. Ecol. 17, 3337–3351. doi: 10.1111/j.1365-294X.2008.03833.x
Do, C., Waples, R. S., Peel, D., Macbeth, G. M., Tillett, B. J., and Ovenden, J. R. (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14, 209–214. doi: 10.1111/1755-0998.12157
Dulvy, N. K., Fowler, S. L., Musick, J. A., Cavanagh, R. D., Kyne, P. M., Harrison, L. R., et al. (2014). Extinction risk and conservation of the world’s sharks and rays. eLife 3:e00590. doi: 10.7554/eLife.00590
Duncan, K., and Holland, K. (2006). Habitat use, growth rates and dispersal patterns of juvenile scalloped hammerhead sharks Sphyrna lewini in a nursery habitat. Mar. Ecol. Prog. Ser. 312, 211–221. doi: 10.3354/meps312211
Emlen, S. T., and Oring, L. W. (1977). Ecology, sexual selection, and the evolution of mating systems. Science 197, 215–223. doi: 10.1126/science.327542
Feldheim, K. A., Gruber, S. H., and Ashley, M. V. (2004). Reconstruction of parental microsatellite genotypes reveals female polyandry and philopatry in the lemon shark, Negaprion brevirostris. Evolution 58, 2332–2342. doi: 10.1111/j.0014-3820.2004.tb01607.x
Foll, M., and Gaggiotti, O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Frankham, R., Bradshaw, C. J. A., and Brook, B. W. (2014). Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 170, 56–63. doi: 10.1016/j.biocon.2013.12.036
Green, M. E., Appleyard, S. A., White, W., Tracey, S., and Ovenden, J. (2017). Variability in multiple paternity rates for grey reef sharks (Carcharhinus amblyrhynchos) and scalloped hammerheads (Sphyrna lewini). Sci. Rep. 7:1528. doi: 10.1038/s41598-017-01416-w
Hauser, L., Baird, M., Hilborn, R., Seeb, L. W., and Seeb, J. E. (2011). An empirical comparison of SNPs and microsatellites for parentage and kinship assignment in a wild sockeye salmon (Oncorhynchus nerka) population. Mol. Ecol. Resour. 11, 150–161. doi: 10.1111/j.1755-0998.2010.02961.x
Herbinger, C. M., O’Reilly, P. T., and Verspoor, E. (2006). Unravelling first-generation pedigrees in wild endangered salmon populations using molecular genetic markers. Mol. Ecol. 15, 2261–2275. doi: 10.1111/j.1365-294x.2006.02923.x
Jamieson, I. G., and Allendorf, F. W. (2012). How does the 50/500 rule apply to MVPs? Trends Ecol. Evol. 27, 578–584. doi: 10.1016/j.tree.2012.07.001
Jones, O. R., and Wang, J. (2010). COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resour. 10, 551–555. doi: 10.1111/j.1755-0998.2009.02787.x
Kilian, A., Wenzl, P., Huttner, E., Carling, J., Xia, L., Blois, H., et al. (2012). Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol. Biol. 888, 67–89. doi: 10.1007/978-1-61779-870-2_5
Lowerre-Barbieri, S. K., Burnsed, S. L. W., and Bickford, J. W. (2016). Assessing reproductive behavior important to fisheries management: a case study with red drum, Sciaenops ocellatus. Ecol. Appl. 26, 979–995. doi: 10.1890/15-0497
Marie, A. D., Miller, C., Cawich, C., Piovano, S., and Rico, C. (2017). Fisheries-independent surveys identify critical habitats for young scalloped hammerhead sharks (Sphyrna lewini) in the Rewa Delta, Fiji. Sci. Rep. 7:17273. doi: 10.1038/s41598-017-17152-0
Mo, S.-K., Ren, Z.-L., Yang, Y.-R., Liu, Y.-C., Zhang, J.-J., Wu, H.-J., et al. (2018). A 472-SNP panel for pairwise kinship testing of second-degree relatives. Forensic Sci. Int. Genet. 34, 178–185. doi: 10.1016/j.fsigen.2018.02.019
Mourier, J., Buray, N., Schultz, J. K., Clua, E., and Planes, S. (2013). Genetic network and breeding patterns of a Sicklefin lemon shark (Negaprion acutidens) population in the society Islands, French Polynesia. PLoS One 8:e73899. doi: 10.1371/journal.pone.0073899
Nance, H. A., Klimley, P., Galván-Magaña, F., Martínez-Ortíz, J., and Marko, P. B. (2011). Demographic processes underlying subtle patterns of population structure in the scalloped hammerhead shark, Sphyrna lewini. PLoS One 6:e21459. doi: 10.1371/journal.pone.0021459
Pirog, A., Jaquemet, S., Soria, M., and Magalon, H. (2016). First evidence of multiple paternity in the bull shark (Carcharhinus leucas). Mar. Freshw. Res. 68, 195–201.
Pratt, H. L. (1993). The storage of spermatozoa in the oviducal glands of western North Atlantic sharks. Environ. Biol. Fish. 38, 139–149. doi: 10.1007/978-94-017-3450-9_12
Rossouw, C., Wintner, S. P., and Bester-Van Der Merwe, A. E. (2016). Assessing multiple paternity in three commercially exploited shark species: Mustelus mustelus, Carcharhinus obscurus and Sphyrna lewini. J. Fish. Biol. 89, 1125–1141. doi: 10.1111/jfb.12996
Rowe, S., and Hutchings, J. A. (2003). Mating systems and the conservation of commercially exploited marine fish. Trends Ecol. Evol. 18, 567–572. doi: 10.1016/j.tree.2003.09.004
Salles, O. C., Pujol, B., Maynard, J. A., Almany, G. R., Berumen, M. L., Jones, G. P., et al. (2016). First genealogy for a wild marine fish population reveals multigenerational philopatry. Proc. Natl. Acad. Sci. U.S.A. 113, 13245–13250. doi: 10.1073/pnas.1611797113
Schneider, S., Roessli, D., and Excoffier, L. (2000). Arlequin Ver. 2.0: A Software for Population Genetics Data Analysis. Geneva: Genetics and Biometry Laboratory.
Sefc, K. M., and Koblmuller, S. (2009). Assessing parent numbers from offspring genotypes: the importance of marker polymorphism. J. Hered. 100, 197–205. doi: 10.1093/jhered/esn095
Smith, B. R., Herbinger, C. M., and Merry, H. R. (2001). Accurate partition of individuals into full-sib families from genetic data without parental information. Genetics 158, 1329–1338.
Sugg, D. W., and Chesser, R. K. (1994). Effective population sizes with multiple paternity. Genetics 137, 1147–1155.
Székely, T., Weissing, F. J., and Komdeur, J. (2014). Adult sex ratio variation: implications for breeding system evolution. J. Evol. Biol. 27, 1500–1512. doi: 10.1111/jeb.12415
Vierus, T., Gehrig, S., Brunnschweiler, J. M., Glaus, K., Zimmer, M., Marie, A. D., et al. (2018). Discovery of a multispecies shark aggregation and parturition area in the Ba Estuary, Fiji Islands. Ecol. Evol. 8, 7079–7093. doi: 10.1002/ece3.4230
Willing, E.-M., Dreyer, C., and Van Oosterhout, C. (2012). Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS One 7:e42649. doi: 10.1371/journal.pone.0042649
Keywords: scalloped hammerhead shark, mating system, sex ratio, kinship analyses, COLONY2, PEDIGREE
Citation: Marie AD, Herbinger C, Fullsack P and Rico C (2019) First Reconstruction of Kinship in a Scalloped Hammerhead Shark Aggregation Reveals the Mating Patterns and Breeding Sex Ratio. Front. Mar. Sci. 6:676. doi: 10.3389/fmars.2019.00676
Received: 29 May 2019; Accepted: 17 October 2019;
Published: 01 November 2019.
Edited by:
Lisa Marie Komoroske, National Oceanic and Atmospheric Administration (NOAA), United StatesReviewed by:
Kevin Feldheim, Field Museum of Natural History, United StatesDovi Kacev, University of California, San Diego, United States
Copyright © 2019 Marie, Herbinger, Fullsack and Rico. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amandine D. Marie, YW1zbWFyaWU4QGdtYWlsLmNvbQ==; Ciro Rico, Y2lyby5yaWNvQHVzcC5hYy5mag==