 Wirulda Pootakham1†
Wirulda Pootakham1† Tanaporn Uengwetwanit2†
Tanaporn Uengwetwanit2† Chutima Sonthirod1Kanchana Sittikankaew2Nitsara Karoonuthaisiri2*
Chutima Sonthirod1Kanchana Sittikankaew2Nitsara Karoonuthaisiri2*- 1National Omics Center, National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency, Pathum Thani, Thailand
- 2National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency, Pathum Thani, Thailand
Background
The black tiger shrimp, Penaeus monodon, is one of the major cultured penaeid shrimp species, contributing 9% of total crustacean production (FAO, 2018). With its economic and nutritional importance, extensive research and development programs have been initiated to gain better understanding of various biological processes in black tiger shrimp and to apply this basic knowledge to improve farming practices and advance selective breeding programs with an ultimate goal of sustainable shrimp farming industry. Although several transcriptome studies in shrimp have been performed using expressed sequence tags (ESTs) (Lehnert et al., 1999; Supungul et al., 2002; Tassanakajon et al., 2006), microarray technology (Karoonuthaisiri et al., 2009; Wongsurawat et al., 2010; Leelatanawit et al., 2011; Uawisetwathana et al., 2011), and second-generation sequencing (SGS) technologies (Rotllant et al., 2015; Nguyen et al., 2016; Huerlimann et al., 2018; Uengwetwanit et al., 2018), the efforts were significantly impeded by the lack of high-quality reference genome assembly in this organism. It is also extremely challenging to obtain a complete, full-length transcriptome assembly from short-read RNA sequencing (RNAseq) data given the high contents of repetitive elements, which have been estimated to be 46.68–51.18% (Huang et al., 2011; Yuan et al., 2018).
In this study, we applied long-read Pacific Biosciences (PacBio) isoform sequencing (Iso-seq) to generate the first full-length transcriptome assembly for black tiger shrimp. Full-length mRNA sequences from nine major organs and hemocytes were obtained using PacBio's circular consensus sequencing (CCS) technology. PacBio sequencing platform enables full-length transcripts from their 5′ ends to poly(A) tails to be captured in single long reads, making this an ideal approach for constructing a reference transcriptome assembly without reference genome sequences (Dong et al., 2015; Kuo et al., 2017; Workman et al., 2018). The ability to capture full-length transcripts also facilitates the discovery of novel isoforms and alternative transcripts that vary with developmental stages (Thatcher et al., 2016), cell types (Swarup et al., 2016) and stress conditions (Yan et al., 2012; Liu et al., 2016). Without a high-quality genome sequence draft, we believe that this high-quality reference transcriptome assembly will be a valuable resource for transcriptome profiling studies under various conditions in black tiger shrimp.
Data Description
Sample Collection
A total of nine organs and hemocytes were harvested from two male and two female 4-month-old juvenile black tiger shrimps (Yui Gung Oog, Prachuap Khiri Khan, Thailand). The shrimps were specific pathogen free: they were tested for White Spot Syndrome Virus (WSSV), Yellowhead disease, Taura Syndrome (TS) virus and acute hepatopancreatic necrosis disease (AHPND). All tissue samples (except ovary and testis) were pooled from four individuals while the ovary and testis tissues were pooled from two individuals. This study was performed in accordance with the recommendations of Animal Research Ethics Guidelines, and the protocol was approved by the National Center of Genetic Engineering and Biotechnology Animal Research Ethics Committee (approval number BT-IACUC-RF01-10-01). The following organs were dissected and immediately frozen in liquid nitrogen: gill, heart, hepatopancreas, intestine, ovary, pleopod, stomach, testis and thoracic ganglia. Haemolymph was centrifuged at 3,000 rpm at 4°C for 5 min to pellet the hemocytes, which were then frozen in liquid nitrogen. All samples were stored at −80°C until RNA extraction.
RNA Extraction and PacBio Sequencing
Total RNA was extracted from the frozen tissues using TRI REAGENT according to the manufacturer's instruction (Molecular Research Center, USA). Contaminated genomic DNA was removed by DNase I treatment at 0.5 U/μg total RNA at 37°C for 30 min (Promega, USA). One ug of DNase-treated RNA sample from each tissue was used for cDNA synthesis. SMRTbell library construction was carried out using the SMRTbell Template Prep Kit 1.0-SPv3 protocol, following the manufacturer's instruction (Pacific Biosciences, Menlo Park, USA). Barcodes were used to keep track of the tissue origin of the samples. The libraries were run on the PacBio Sequel System using the sequencing chemistry version 3.0 and 10-h movie times (the sequencing was outsourced to NovogenAIT, Singapore and Macrogen, Korea).
Transcriptome Assembly and Annotation
SMRTlink version 6.0 (Pacific Biosciences, Menlo Park, CA, USA) was used to filter and process raw subreads, using the Iso-seq3 pipeline to obtain highly accurate long reads. Subreads shorter than 50 nt (default parameter) or with the ReadScore lower than 0.8 were removed prior to generating the circular consensus sequence (CCS) reads using the following parameters: minPasses = 1 and minPredictedAccuracy = 0.9. Full-length non-chimeric (FLNC) or non full-length (NFL) reads were classified based on the presence or absence of 5′ primer, 3′ primer and the poly(A) tail. FLNC reads were clustered using the Iso-seq3 cluster module in the SMRTlink to generate consensus sequences, which were subsequently polished with the Arrow software (https://www.pacb.com/wp-content/uploads/SMRT_Tools_Reference_Guide_v600.pdf) using NFL reads. We obtained a total of 1,604,098 reads of insert (from all tissues combined), ranging from 50 to 14,973 nt with an average read length of 2,942 and an N50 of 3,242 nt. Of those reads of insert, 82,441 were high-quality FLNC transcripts (Table 1). To exclude contaminating sequences from other organisms, we aligned FLNC transcripts to virus, bacteria, fungi and human sequences (from the NCBI RefSeq database release number 97) using BLASTN and identified 22 FLNC transcripts that exhibited over 90% identity to those sequences with at least 90% sequence coverage. Those were removed prior to downstream analyses. We subsequently used CPAT version 1.2.4 (Wang et al., 2013) to filter out 5,627 long non-coding RNA (lncRNA) and clustered the remaining transcript sequences at 99% identity using UCLUST (Edgar, 2010) to obtain a set of 22,418 non-redundant sequences in the final transcriptome assembly. Compared to our full-length transcriptome assembly, the previously reported assembly derived from Illumina short reads contained a greater number of transcripts (236,085 sequences; TRA: GGLH00000000) with considerably shorter average read length (956 nt) and N50 length (1,431 nt) (Huerlimann et al., 2018).
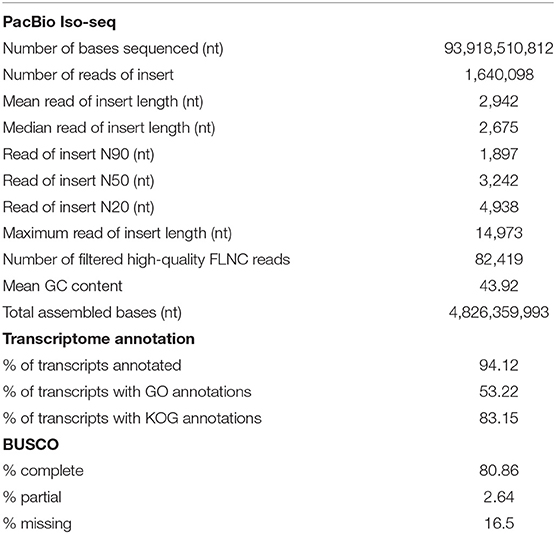
Table 1. Assembly statistics for P. monodon transcriptome.
The annotations of this reference assembly, including gene ontology (GO) and eukaryotic orthologous groups (KOG) assignments, were carried out using Blast2GO version 5.2 (Götz et al., 2008) (BioBam, Valencia, Spain; Supplementary Table 1). Approximately 94% of sequences in the assembly were annotated, and 53% and 83% had been assigned GO and KOG annotations, respectively (Table 1). The highest number of transcripts sequenced was affiliated with cellular metabolic process, organic substance metabolic process, primary metabolic process and nitrogen compound metabolic process (Figure 1A). We also investigated the completeness of the transcriptome assembly by assessing the coverage of the benchmarking universal single-copy orthologs (BUSCO) using the eukaryotic gene set (Simão et al., 2015). Of the 429 conserved eukaryotic genes, 80.86% were identified as “complete” in the assembly and an additional 2.64% were partially assembled. Our assembly had a much higher proportion of “missing” orthologs (16.5%) compared to the assembly reported by Huerlimann et al. (2018), which was 98% complete based on BUSCO analysis. The fact that our shrimp samples were from a single stage (4-month old) while those used in Huerlimann et al. (2018) were from multiple stages and that some of the tissues sequenced in Huerlimann et al. (2018) were not included in our study could attribute to the missing orthologs in the BUSCO assessment. Most of the conserved orthologs that were missing in our assembly were those involved in the DNA replication processes.
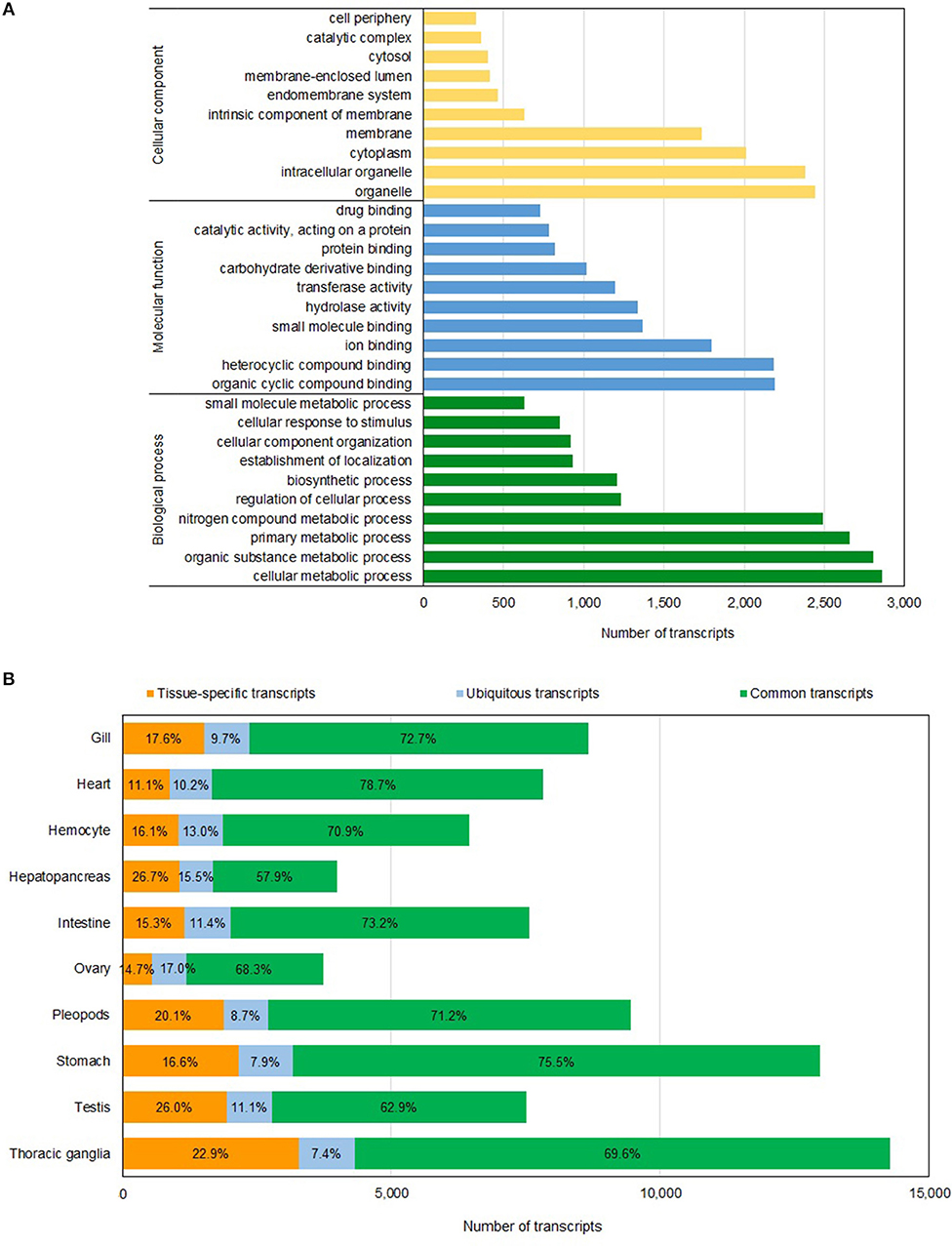
Figure 1. Gene annotation and transcript distribution. (A) Distribution of functional categories based on GO terms in P. monodon transcriptome assembly. (B) Distribution of transcripts in each tissue type based on their presence in the tissues studied. Tissue-specific transcripts (orange bar) are present in only one tissue type; ubiquitous transcripts (blue bar) are present in all tissue types, and common transcripts (green bar) are present in more than one tissue type but not detected in all tissues examined. The number represents the percentage of each transcript category in each tissue type.
RNA samples from nine different tissue types and hemocytes were sequenced to generate the reference transcriptome assembly. We kept track of the origin of individual full-length transcript sequence and classified each of them as follows: “tissue-specific” when it is present in only one tissue type, “ubiquitous” when it is present in all tissue types investigated and “common” when it is present in between two to nine tissue types studied. Of the 22,418 non-redundant transcript sequences in the assembly, 462 transcripts were present in all tissues examined (Supplementary Table 2). The number of non-redundant transcripts detected in each tissue type is displayed in Figure 1B and Supplementary Table 2. Hepatopancreas (26.7%) and testis (26.0%) tissues had the highest proportions of tissue-specific transcripts while heart tissues had the lowest proportion of tissue-specific transcripts (11.1%; Figure 1B and Supplementary Table 2). Interestingly, between 54 and 65% of the transcripts detected in each individual tissue type were also present in the stomach. On contrary, only 17-30% of the transcripts detected in each individual tissue type were also present in either hepatopancreas or ovary tissues (Supplementary Table 2).
The ability to capture full-length transcripts using Iso-seq technology provided a unique opportunity to detect alternative splicing events in different tissue types. To identify candidate transcripts with multiple isoforms, we examined clusters of FLNC transcripts containing sequences of multiple lengths. Examples of transcripts with different isoforms are shown in Supplementary Figure 1. A transcript annotated as T-cell immunomodulatory protein had two alternative 5′ splice sites and displayed a hemocyte-specific isoform with a truncated 3′ end, which likely represented an exon-skipping event. Another example was the thoracic ganglia-specific isoform (annotated as protein lines-like) appeared to skip an exon near the 5′ end of the transcript. Complementary information from genome sequences is required to map the exon-intron junctions and determine if the alternative splicing events observed are exon skipping or intron retention. PacBio technology eliminates the need to assemble transcripts from short-read data and allows (long) full-length transcripts to be sequenced in single-molecule reads, providing direct evidence for alternatively spliced isoforms.
Re-use Potential
This study reports the first full-length transcriptome assembly for P. mondon, utilizing PacBio long-read single molecule real-time (SMRT) sequencing technology to obtain full-length transcript sequences. Future transcriptome profiling studies and genome sequencing projects in P. monodon and closely related species will greatly benefit from the full-length Iso-seq based transcriptome assembly reported here and the previously reported Illumina-based transcriptome assembly.
Data Availability Statement
The transcriptome assembly is available from NCBI GenBank database under BioProject ID PRJNA602748, and the transcriptome annotations are provided in Supplementary Table 1.
Author Contributions
NK, TU, and WP conceived and designed the experiment. KS collected shrimp samples and carried out the RNA extraction. CS and TU performed bioinformatics analyses. WP and NK wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Center for Genetic Engineering and Biotechnology (RI grant number P1851724 and Platform grant number P1651718), National Science and Technology Development Agency (grant number P1950419), and partially supported by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 734486.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00172/full#supplementary-material
References
Dong, L., Liu, H., Zhang, J., Yang, S., Kong, G., Chu, J. S. C., et al. (2015). Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics 16:1039. doi: 10.1186/s12864-015-2257-y
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
FAO (2018). The State of Fisheries and Aquaqulture 2018. Meeting the Sustainable Development Goals. Rome: FAO.
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Huang, S.-W., Lin, Y.-Y., You, E.-M., Liu, T.-T., Shu, H.-Y., Wu, K.-M., et al. (2011). Fosmid library end sequencing reveals a rarely known genome structure of marine shrimp Penaeus monodon. BMC Genomics 12:242. doi: 10.1186/1471-2164-12-242
Huerlimann, R., Wade, N. M., Gordon, L., Montenegro, J. D., Goodall, J., Mcwilliam, S., et al. (2018). De novo assembly, characterization, functional annotation and expression patterns of the black tiger shrimp (Penaeus monodon) transcriptome. Sci. Rep. 8:13553. doi: 10.1038/s41598-018-31148-4
Karoonuthaisiri, N., Sittikankeaw, K., Preechaphol, R., Kalachikov, S., Wongsurawat, T., Uawisetwathana, U., et al. (2009). ReproArray(GTS): a cDNA microarray for identification of reproduction-related genes in the giant tiger shrimp Penaeus monodon and characterization of a novel nuclear autoantigenic sperm protein (NASP) gene. Comp. Biochem. Physiol. Part D Genomics Proteomics 4, 90–99. doi: 10.1016/j.cbd.2008.11.003
Kuo, R. I., Tseng, E., Eory, L., Paton, I. R., Archibald, A. L., and Burt, D. W. (2017). Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genomics 18:323. doi: 10.1186/s12864-017-3691-9
Leelatanawit, R., Uawisetwathana, U., Klinbunga, S., and Karoonuthaisiri, N. (2011). A cDNA microarray, UniShrimpChip, for identification of genes relevant to testicular development in the black tiger shrimp (Penaeus monodon). BMC Mol. Biol. 12:15. doi: 10.1186/1471-2199-12-15
Lehnert, S. A., Wilson, K. J., Byrne, K., and Moore, S. S. (1999). Tissue-specific expressed sequence tags from the black tiger shrimp Penaeus monodon. Mar. Biotechnol. 1, 465–0476. doi: 10.1007/PL00011803
Liu, J., Chen, X., Liang, X., Zhou, X., Yang, F., Liu, J., et al. (2016). Alternative splicing of rice WRKY62 and WRKY76 transcription factor genes in pathogen defense. Plant Physiol. 171, 1427–1442. doi: 10.1104/pp.15.01921
Nguyen, C., Nguyen, T. G., Nguyen, L. V., Pham, H. Q., Nguyen, T. H., Pham, H. T., et al. (2016). De novo assembly and transcriptome characterization of major growth-related genes in various tissues of Penaeus monodon. Aquaculture 464, 545–553. doi: 10.1016/j.aquaculture.2016.08.003
Rotllant, G., Wade, N. M., Arnold, S. J., Coman, G. J., Preston, N. P., and Glencross, B. D. (2015). Identification of genes involved in reproduction and lipid pathway metabolism in wild and domesticated shrimps. Mar. Genomics 22, 55–61. doi: 10.1016/j.margen.2015.04.001
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Supungul, P., Klinbunga, S., Pichyangkura, R., Jitrapakdee, S., Hirono, I., Aoki, T., et al. (2002). Identification of immune-related genes in hemocytes of black tiger shrimp (Penaeus monodon). Mar. Biotechnol. 4, 487–494. doi: 10.1007/s10126-002-0043-8
Swarup, R., Crespi, M., and Bennett, M. J. (2016). One gene, many proteins: mapping cell-specific alternative splicing in plants. Dev. Cell 39, 383–385. doi: 10.1016/j.devcel.2016.11.002
Tassanakajon, A., Klinbunga, S., Paunglarp, N., Rimphanitchayakit, V., Udomkit, A., Jitrapakdee, S., et al. (2006). Penaeus monodon gene discovery project: the generation of an EST collection and establishment of a database. Gene 384, 104–112. doi: 10.1016/j.gene.2006.07.012
Thatcher, S. R., Danilevskaya, O. N., Meng, X., Beatty, M., Zastrow-Hayes, G., Harris, C., et al. (2016). Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 170, 586–599. doi: 10.1104/pp.15.01267
Uawisetwathana, U., Leelatanawit, R., Klanchui, A., Prommoon, J., Klinbunga, S., and Karoonuthaisiri, N. (2011). Insights into eyestalk ablation mechanism to induce ovarian maturation in the black tiger shrimp. PLoS ONE 6:e24427. doi: 10.1371/journal.pone.0024427
Uengwetwanit, T., Ponza, P., Sangsrakru, D., Wichadakul, D., Ingsriswang, S., Leelatanawit, R., et al. (2018). Transcriptome-based discovery of pathways and genes related to reproduction of the black tiger shrimp (Penaeus monodon). Mar. Genomics 37, 69–73. doi: 10.1016/j.margen.2017.08.007
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J. P., and Li, W. (2013). CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41:e74. doi: 10.1093/nar/gkt006
Wongsurawat, T., Leelatanawit, R., Thamniemdee, N., Uawisetwathana, U., Karoonuthaisiri, N., Menasveta, P., et al. (2010). Identification of testis-relevant genes using in silico analysis from testis ESTs and cDNA microarray in the black tiger shrimp (Penaeus monodon). BMC Mol. Biol. 11:55. doi: 10.1186/1471-2199-11-55
Workman, R. E., Myrka, A. M., Wong, G. W., Tseng, E., Welch, K. C. Jr., and Timp, W. (2018). Single-molecule, full-length transcript sequencing provides insight into the extreme metabolism of the ruby-throated hummingbird Archilochus colubris. GigaScience 7:giy009. doi: 10.1093/gigascience/giy009
Yan, K., Liu, P., Wu, C.-A., Yang, G.-D., Xu, R., Guo, Q.-H., et al. (2012). Stress-induced alternative splicing provides a mechanism for the regulation of microRNA processing in Arabidopsis thaliana. Mol. Cell 48, 521–531. doi: 10.1016/j.molcel.2012.08.032
Keywords: black tiger shrimp, Penaeus monodon, transcriptome, Iso-seq, PacBio sequencing
Citation: Pootakham W, Uengwetwanit T, Sonthirod C, Sittikankaew K and Karoonuthaisiri N (2020) A Novel Full-Length Transcriptome Resource for Black Tiger Shrimp (Penaeus monodon) Developed Using Isoform Sequencing (Iso-Seq). Front. Mar. Sci. 7:172. doi: 10.3389/fmars.2020.00172
Received: 22 January 2020; Accepted: 05 March 2020;
Published: 24 March 2020.
Edited by:
Pei-Yuan Qian, Hong Kong University of Science and Technology, Hong KongReviewed by:
Ka Yan Ma, The Chinese University of Hong Kong, ChinaMarta Gomez-Chiarri, University of Rhode Island, United States
Adam Michael Reitzel, University of North Carolina at Charlotte, United States
Copyright © 2020 Pootakham, Uengwetwanit, Sonthirod, Sittikankaew and Karoonuthaisiri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nitsara Karoonuthaisiri, bml0c2FyYUBhbHVtbmkuc3RhbmZvcmQuZWR1
†These authors have contributed equally to this work