 Xue Zhao1,2Anne Flore Deton Cabanillas3Alaguraj Veluchamy4
Xue Zhao1,2Anne Flore Deton Cabanillas3Alaguraj Veluchamy4 Chris Bowler3Fabio Rocha Jimenez Vieira3*
Chris Bowler3Fabio Rocha Jimenez Vieira3* Leila Tirichine1*
Leila Tirichine1*- 1Department of Biology, Université de Nantes, CNRS, UFIP, UMR 6286, Nantes, France
- 2Universit Paris-Saclay, CNRS, INRAE, Univ Evry, Institute of Plant Sciences Paris-Saclay (IPS2), Orsay, France
- 3Institut de Biologie de l’Ecolenormale Supérieure (IBENS), Ecole Normale Supérieure, CNRS, INSERM, PSL Université Paris, Paris, France
- 4Biological and Environmental Science and Engineering Division, Laboratory of Chromatin Biochemistry, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Polycomb (PcG) and Trithorax (TrxG) complexes are two evolutionarily conserved epigenetic regulatory components that act antagonistically to regulate the expression of genes involved in cell differentiation and development in multicellular organisms. The absence of PcG in both yeast models Saccharomyces cerevisiae and Schizosaccharomyces pombe suggested that polycomb proteins might have evolved together with the emergence of multicellular organisms. However, high throughput sequencing of several microalgal genomes and transcriptomes reveals an unprecedented abundance and diversity of genes encoding the components of these complexes. We report here the diversity of genes encoding PcG and TrxG proteins in microalgae from the Marine Microbial Eukaryote Transcriptome Sequencing Project database (MMETSP) and detected at broad scale in Tara Oceans genomics datasets using a highly sensitive method called eDAF (enhanced Domain Architecture Filtering). Further, we explored the correlation between environmental factors measured during the Tara Oceans expedition and transcript levels of PcG and TrxG components. PcG and TrxG are responsible for the deposition of a number of histone marks among which a TrxG associated mark, H3K4me3 which we profiled genome wide in the model diatom Phaeodactylum tricornutum to understand its role in microalgae and revisited the previously published histone code and co-occurrence with other histone marks including the antagonizing Polycomb deposited mark H3K27me3.
Introduction
Polycomb (PcG) and trithorax (TrxG) protein complexes were initially isolated in Drosophila as factors responsible for the maintenance of expression of HOX genes, important determinants of body patterning (Schuettengruber et al., 2007). Several decades of research has revealed that these two complexes are involved in a plethora of biological processes including X chromosome inactivation, genomic imprinting, cell cycle control, stem cell biology, and cancer (Schuettengruber et al., 2017). PcG and TrxG complexes have been shown to act antagonistically to modify chromatin via histone-modifying or chromatin-remodeling activities that repress or activate their target genes, respectively (Geisler and Paro, 2015). Both complexes are highly conserved among eukaryotes and their recent discovery in single celled species raises important questions about their function and role in unicellular organisms and in the evolution of multicellularity. Of note, PcG complexes are not found in two widely studied yeast species Saccharomyces cerevisiae and Schizosaccharomyces pombe and their recent discovery and conservation in several unicellular microalgae points to their ancient origin and importance (Shaver et al., 2010).
In animals and plants, polycomb proteins form at least two distinct complexes, PRC1 and PRC2. In mammals, PRC1 regulates gene expression through controlling chromatin compaction and catalyzing mono-ubiquitylation of histone H2A, whereas PRC2 is responsible for chromatin structure and methylation of lysine 27 of histone H3 (H3K27) (Gil and O’Loghlen, 2014). PRC1 contains four core subunits: polycomb (Pc), posterior sex combs (Psc), polyhomeotic (Ph; absent in plants), sex combs extra (Sce) or dRING (Barrero and Izpisua Belmonte, 2013). The PRC2 complex comprises four core components: a histone methyltransferase, enhancer of zeste E(z), a WD40 domain protein, extra sex combs (ESC), a zinc finger protein, suppressor of zeste 12 (Su(z)12), and another WD40 domain protein, Nurf-55 (Margueron and Reinberg, 2011).
TrxG complex has been classified into three groups based on the function of its members (1) SET domain containing proteins which methylate histones including COMPASS and COMPASS-like involved in the methylation of Lysine 4 of histone H3 for general gene activation as well as methylation of specific genes and methylation of H3K36me3 (2) ATP dependent chromatin remodeling factors with few proteins that can read the histone methylation marks deposited by the SET domain proteins, and (3) TrxG proteins known to bind directly to specific DNA sequences. Our focus here is on the first group of TrxG complex proteins considering their role in antagonizing PcG mediated repression.
Both complexes have been studied intensively in multicellular species but only scarcely investigated in unicellular organisms such as microalgae or yeast model species in which PRC1 and PRC2 complexes do not exist. Here, we draw a comprehensive picture of the diversity and abundance of both complexes in the Marine Microbial Eukaryote Transcriptome Sequencing Project database (MMETSP) and metagenome/metatranscriptome samples from Tara Oceans using the enhanced Domain Architecture Framework, eDAF, which is an ensemble of algorithms to process, enhance and expand the output of DAMA (Bernardes J.S. et al., 2016) or any other domain architecture algorithm (Terrapon et al., 2009; Yeats et al., 2010; Ochoa et al., 2011). Since eDAF can improve domain annotations, it is complementary to more prominent tools, such as InterPro (Mitchell et al., 2019). It also strongly improves the abilities of CLADE (Bernardes J. et al., 2016) by allowing it to analyze nucleotide sequences and propose the most probable translation of a given sequence (Figure 1).
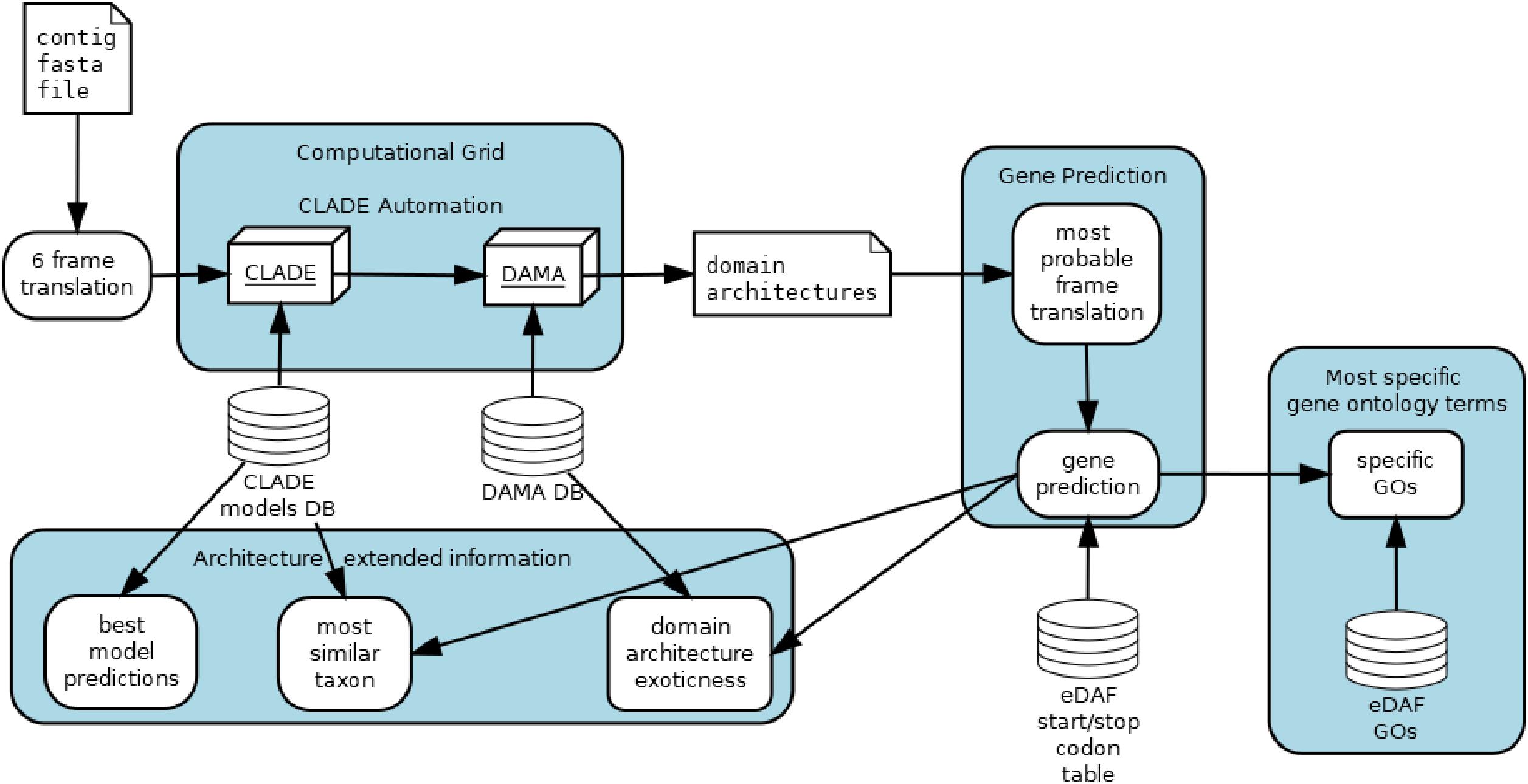
Figure 1. eDAF flow chart.
Both PcG and TrxG complexes are histone writers involved in post translational modifications of histones (PTMs), namely H3K27me3, H2AK119 Ub and H3K4me3/K36me3 and probably more as some recent work points to the involvement of enhancer of zeste in tri-methylation of lysine 9 of histone H3 (Frapporti et al., 2019). Therefore, these complexes play an important role in genome regulation providing a plasticity in mediating genome responses to environmental factors and developmental triggers. In this study, we illustrate the importance of these PTMs in particular a trithorax deposited mark, H3K4me3 using the model diatom Phaeodactylum tricornutum where chromatin immunoprecipitation was used with deep sequencing to investigate the pattern of its distribution genome wide and its role in microalgae. Further, we revisited the epigenetic code in P. tricornutum (Veluchamy et al., 2015) and compared H3K4me3 mapping profile with the previously investigated Polycomb associated mark H3K27me3 (Veluchamy et al., 2015) to address for the first time their co-occurrence in a unicellular species.
Materials and Methods
Material and Growth Conditions
Cells of P. tricornutum reference strain Pt1 8.6 CCAP 1055/1 (CCMP2561) were harvested after a week of growth in artificial sea water (Vartanian et al., 2009) at 19°C, under 12/12 light dark period with a light intensity of 75 μmol photons m-2 s-1.
Enhanced Domain Architecture Framework Analysis
eDAF is composed of four different modules: gene prediction (GP), most specific gene ontology terms (SGO), architecture extended information (AEI) and CLADE automation (CA) (Figure 1). Each of these modules is detailed below.
Gene Prediction
The gene prediction module allows CLADE to analyze all possible six-frame translations of each uniGene (Carradec et al., 2018). Once CLADE/DAMA predicts a domain architecture for a frame, we use a quadratic regression (Gergonne, 1974) to select the frame with the highest score. The variables of quadratic regression are the coverage, the e-value, and the average length of the PFAM domain (El-Gebali et al., 2019). The regression might select the frame with the best domain architecture, that is, with the highest number of conserved regions, the best coverages and e-values. Note that we call domain architecture an arrangement of conserved regions (domains) typically found in protein sequences. Since the selected frame can contain more than one gene, we trim it to where start/stop codons are detected; however, we did not consider start and stop codons within domains. We have considered the standard list of start/stop codons and users can change it by adding/removing entries in the eDAF start/stop codon table.
Most Specific Gene Ontology Terms
After gene predictions, search is performed for their functional annotations. “PFAM2GO” that provides the Gene Ontology terms (Mitchell et al., 2015) for each domain detected by CLADE is used. Domains are represented by a set of GO terms organized in a hierarchical representation. To look for the most specific gene ontology terms of each domain, the postgresql database is used to execute a depth first search (Even, 2011) over the gene ontology tree terms (Carbon and Mungall, 2018). This search algorithm retains only the terms located on the leaves by avoiding generic terms, such as “cellular activity,” that do not provide conclusive knowledge of protein function. However, generic terms can also be found on the leaves; thus, only terms whose depth is higher than the three levels are retained. Also, terms that are in the downward path of more profound terms are not retained. To produce the most enriched terms for a gene, all specific domain terms are considered, and only those with a frequency higher than three standard deviations above the mean of all identified terms (0.1% most enriched/frequent terms) are retained. Finally, repetitive/similar terms are grouped and collapsed by an approximate string matching algorithm (Sellers, 1980).
Architecture Extended Information (AEI)
Architecture Extended Information displays some DAMA and CLADE outputs not explored before. Each CLADE prediction is associated with one or more models and each model is associated to a specific taxonomy (species level). AEI recovers the corresponding taxa of each gene prediction and displays them to the user. The frequency of observed domain co-occurrence (internally part of DAMA database) are shown to highlight exotic (rarely observed) composition of conserved regions in the predictions.
CLADE Automation
CLADE is a set of powerful tools that need some expertise to be properly used. To make it user friendly, we implemented an automation. Basically, the user should only call one script and inform the gene fasta file; CLADE automation module will produce the submission files and the corresponding jobs to be sent to the computational grid. Currently, CLADE automation only works with Condor Computational Grid (Thain et al., 2005), but it can be easily adapted to other schedulers.
eDAF Analysis
We conducted an extensive set of scans over more than 11,720 transcriptomes distributed among 414 species (the MMETSP dataset) using eDAF. First, we used eDAF to interrogate the MMETSP transcripts by first translating each into the 6 possible frame translations. Next, eDAF fed the translations into CLADE and DAMA to identify the conserved domains. Then, eDAF selected the most probable translation of each transcript (as described in the previous sections). eDAF outputs the resulting proteins and the domain architecture of the corresponding MMETSP transcripts. Finally, to identify the true positives among the MMETSP sequences, we used eDAF to identify the domain architecture of both Polycomb and Thrithorax reference proteins (sequences reported as true positives in the literature). We retained only those MMETSP sequences with the same domain architecture of the references.
Phylogenetic Analysis
Both reference sequences and MMETSP true positives were aligned with Clustal Omega (Sievers et al., 2011). The corresponding alignments were used to produce the phylogenetic trees, which were built with MrBayes (Harmanci et al., 2014) using the following parameters (fixed rate model Blosum; mcmc nruns = 1; ngen = 1000000; samplefreq = 100; sump burnin = 250; sumt burnin = 250).
ChIP-Seq Analysis
ChIP-Seq was performed on the reference strain of P. tricornutum Pt1 8.6 as described previously (Veluchamy et al., 2015) using a monoclonal antibody anti-trimethyl histone H3 (Lys4) (H3K4me3, Cell signaling Technology 9751S). Library preparation was performed at Fasteris next generation sequencing facilities (Switzerland), using the Illumina TruSeq kit and sequencing was performed on a HiSeq platform (V4 chemistry) with 1 × 50 bp single-reads. Two replicates with input each around 10 million reads were sequenced. Quality check on reads was done using FASTQC1 with a cut-off Phred score of 20. Reads with minimum length of 36 bp were retained after trimming using Trimmomatic. Parameters for Trimmomatic were set as follows: Minimum length of 36 bp; Mean Phred quality score greater than 30; leading and trailing bases removal with base quality below 3; sliding window of 4:15. The reads were then mapped onto Phatr3 using Bowtie with unique read mapping parameter (Langmead and Salzberg, 2012).
For peak calling, we used three different peak detection methods such as MUSIC (Harmanci et al., 2014), BCP (Xing et al., 2012) and MACS2 (Zhang et al., 2008). We found a significant overlap between them and took consensus peaks from the three methods. Genome-wide analysis of tag density profile on TSS (transcriptional start sites) was performed using deepTools (Ramirez et al., 2014). Mean and standard deviations of the coverage depth were calculated and plotted using Qualimap and samtools. Analysis and visualization of the data were performed using Integrative genomics viewer from Broad Institute (IGV) (Robinson et al., 2011) and R module ChIP peak annotation.
Co-occurrence and Correlation Analysis
ChIP-seq peaks were annotated with gene annotation from Phatr3 (Rastogi et al., 2018). The intersection of different sets of genes overlapping with individual ChIP-seq peaks was performed using UpSetR (Conway et al., 2017). All combinations of overlap are derived and ordered from most to least overlapping categories. Non-overlapping peak sets are removed from the plot.
The aligned histone modification ChIP-seq dataset was binned for coverage with a window size of 1 kb. Replicate correlation was performed using multiBigwigSummary tools from deepTools. Pearson pairwise correlation coefficients were calculated from tag density. Scatter plot for correlation depicts the tag density for a bin of 1 kb each.
Sequence data were deposited in NCBI Gene Expression Omnibus database (accession number GSE139676).
Results
Unlike in mammals and plants, Polycomb and Trithorax complexes are not well studied in unicellular species. Only a few recent studies of PcG proteins have been reported in microalgae (Shaver et al., 2010). To further investigate the diversity of Polycomb and Trithorax complexes in single-cell species, we first made an extensive scan of the MMETSP database using eDAF (see section “Materials and Methods”) and reference sequences (Supplementary Table S1). We retained only MMETSP sequences whose corresponding domain architectures were identical to at least one of the reference sequences (see methods for details). Next, we aligned both reference and MMETSP sequences with Clustal Omega (Sievers et al., 2011) and built phylogenetic trees. In total, 203 homologs were identified from the MMESTP database that possess at least one of the components of the PRC1 or PRC2 complexes (Supplementary Table S2).
Phylogenetic Distribution of Polycomb Complexes in Marine Unicellular Species
Three core components of PRC2 are discussed here, E(z), Esc and Su(z)12. Nurf55 was not considered in our study because it is ubiquitous and is related to a high number of different biological processes that do not involve PRC2. Phylogenies of E(z), Esc and Suz(12) show similarity to human and plant genes with very conserved domains displaying high support values (above 85%) (Figure 2). PRC2 components are in principle very conserved, however, the phylogenetic tree of E(z), Esc and Su(z)12 polypeptides all fail to reconstruct or represent the consensus tree of eukaryotic monophyletic tree, not only because of the normal weakness of individual gene phylogeny, but might also be due to insufficient information in the database. Despite this, diatom and green algae E(z) homologs are well-clustered by their domain architecture (Figure 2A), although there is one exception, Thalassiosira oceanica, a centric diatom, surprisingly clustered with Alveolata and Rhizaria polypeptides. About 90 E(z) homologs share two conserved regions, CXC domain known as pre-SET domain and the SET domain itself (Supplementary Table S2). Esc homologs have two WD-40 repeat domains which can form a platform for protein-protein interactions (Supplementary Table S2). Esc transcripts are not as widely distributed as E(z) and Su(z)12 in diatom species. Consequently, the phylogenetic tree of Esc cannot well resolve the relationship among the species for which Esc sequences were detected (Figure 2B). Using the VEFS-Box of the polycomb domain to construct the phylogenetic tree of Su(z)12, we found that Chlorophytes and diatom Su(z)12 sequence formed a well-supported cluster like E(z), but with a few exceptions such as some pennate diatoms, Pseudo-nitzschia delicatissima, P. arenysensis and Fragilariopsis kerguelensis which were found to cluster with Rhizaria and Fungi (Figure 2C). The high conservation of domain architecture suggests the ancient origin of PRC2 proteins and the important function of this complex during evolution, while the branching of E(z) and Su(z)12 suggests the early divergence of these two genes.
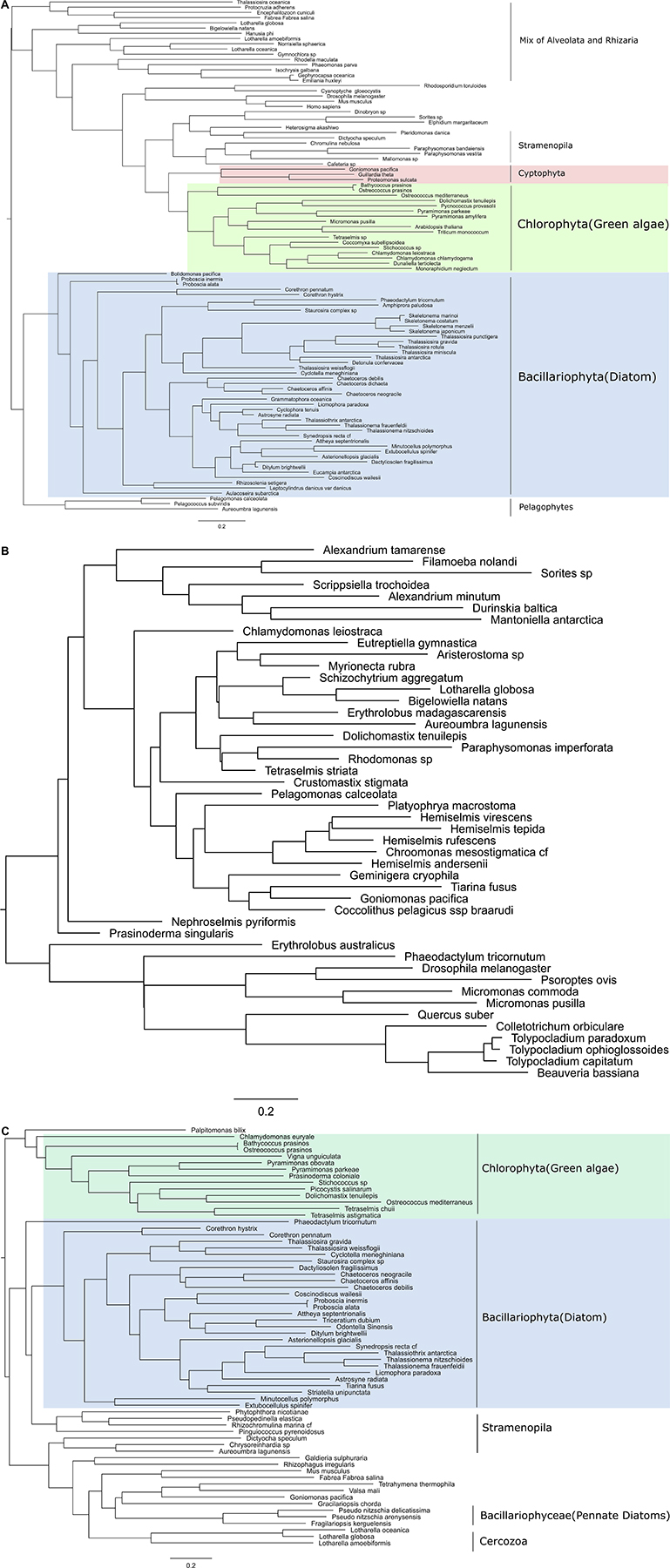
Figure 2C. Phylogenetic trees of PRC1 components. (A) RING, (B) Psc, (C) Pc.
Like in PRC2, PRC1 components show strict conservation in unicellular species (Figure 3 and Supplementary Table S3). RING1 is the catalytic enzyme in the PRC1 complex which deposits H2AK119Ubi. C3HC4 type RING finger domain was the only conserved domain found and the proteins containing this domain typically play a key role in the ubiquitination pathway (Joazeiro and Weissman, 2000). Psc homologs which consist of two conserved domains, C3HC4 type RING finger and RAWUL domains, can function alone or together with RING1 to act as E3 ligase (Vidal, 2019). Phylogenetic tree of Psc was found to cluster well by species. Clusters of diatoms, green algae, Rhizaria and hacrobia species were found (Figure 3B). Similar to E(z), diatom homologs of Psc are closer to other Stramenopile species such as Ochrophyta, while green algae are clustered with Hacrobia species. Further, our study shows that Psc and E(z) proteins in Haptophyta are closer to green lineages while Cryptophyta homologs are closer to SAR lineages. Although Haptophyta and Cryptophyta both belong to Hacrobia, several studies support that they do not form a monophyletic group (Baurain et al., 2010; Burki et al., 2012) which is supported by our finding. Psc and RING1 are known to share RING finger and RAWUL domains from plants to mammals, but in our study only Psc have RAWUL domains whereas RING1 homologs do not. Interestingly, our results indicate that Psc and RING1 homologs are found almost mutually exclusively (Figure 4), except in four species, Chrysochromulina brevifilum, Emiliania huxleyi, Isochrysis galbana and Neoparamoeba aestuarina, suggesting the simplicity of PRC1 complexes or the existence of two types of PRC1 complexes in unicellular species, RING1-Type and Psc-Type. This composition found only in Haptophytes might be an indication of regulation mechanisms more similar to plants and animals compared to the rest of investigated species from the SAR lineage.
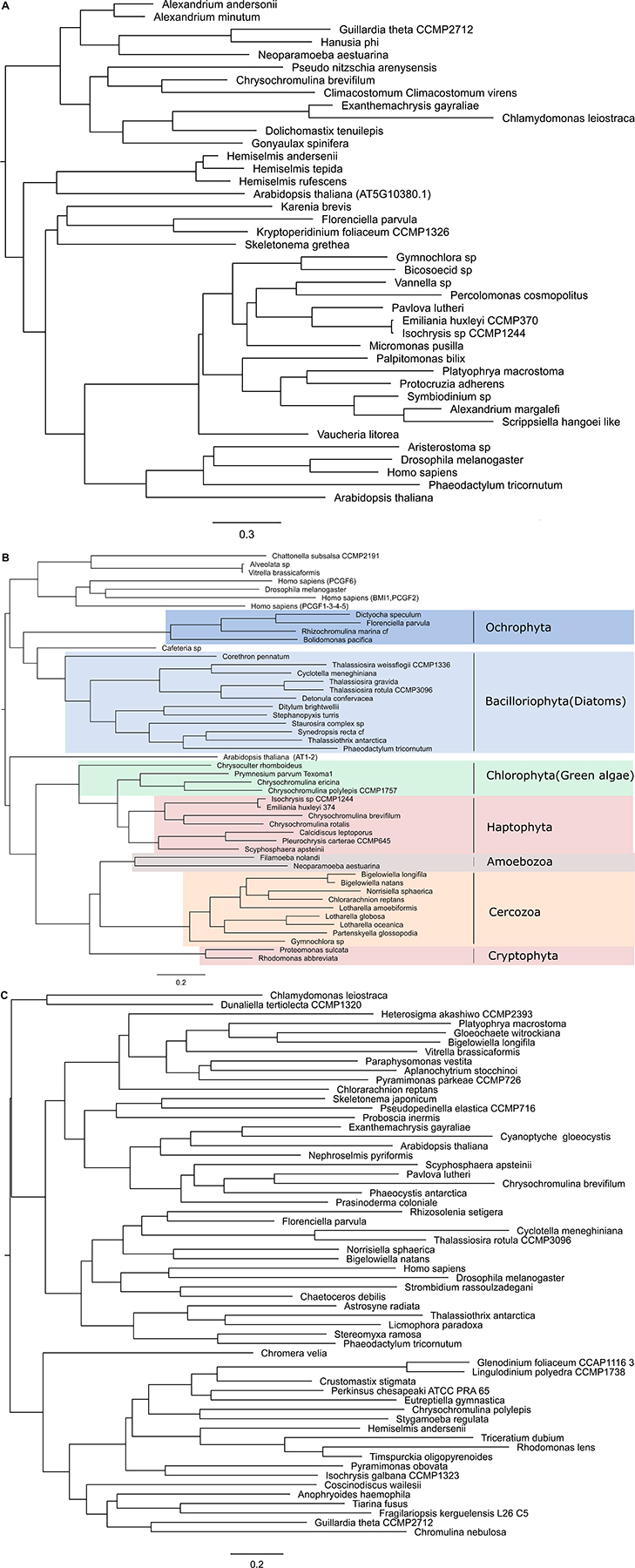
Figure 3C. Phylogenetic trees of PRC2 components. (A) Enhancer of zeste, (B) ESC, (C) Su(z)12.
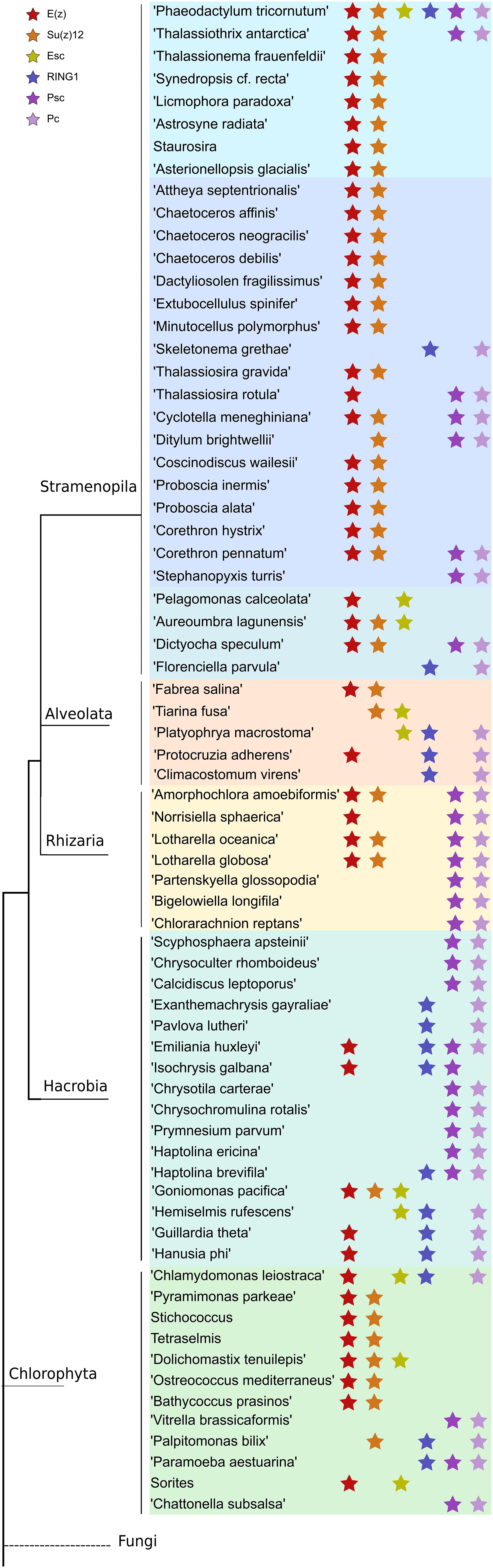
Figure 4. Distribution of PRC1 and PRC2 components in a phylogenetic tree among microalgae species in MMETSP. Each star marks the presence of one component of either PRC1 or PRC2. Different color backgrounds represent species belongs to the same phylum or group.
Pc homologs have a chromodomain and act as reader of H3K27me3. Yeast two-hybrid experiments show that Psc can directly interact with Pc and Ph through the RING finger domain (Kyba and Brock, 1998) which is in line with our finding that Pc expression always co-occurs with Psc and RING1 (Figure 4), whereas Ph was not detected in our study.
To further investigate the coexistence of polycomb complexes within species, we looked at all the ones that have at least one homolog in either PRC1 or/and PRC2 complex (Figure 4). It seems that PRC1 proteins are more present in Hacrobia exclusively without PRC2 components. The only exception is Emiliania huxleyi and Isochrysis galbana which have E(z) in PRC2. While PRC2 components are more abundant in Stramenopila and Chlorophyta, which exclusively lack PRC1 subunits, 24 species have both PRC1 and PRC2 core components with two catalytic enzymes from each complex (Supplementary Table S2 and Figure 4). P. tricornutum is the only species that contains all the subunits of both complexes PRC1 and PRC2 (Supplementary Table S3 and Figure 4). It is important to note that MMETSP is based on transcripts and most of its species have poorly assembled genomes which means that undetected components might be due to a poor or lack of expression. On the other hand, P. tricornutum is one of the rare microalgae species with a fully sequenced high quality genome.
Diversity of Domain Features in the Trithorax Subfamily
TrxG proteins can be divided into 3 classes: histone-modifying SET domain proteins, ATP-dependent chromatin-remodeling factors which can recognize methylated sites by the SET domain, and a third class that includes proteins with specific DNA sequences (Schuettengruber et al., 2011). We chose references from the SET domain TrxG proteins and found that TrxG homologs are more diverse compared to PcG protein homologs, with 54 sequences that can be sorted into 4 clades according to phylogeny and domain structures (Figures 5A,B). Clade 1 contains five domains, Bromodomain, PHD finger domain, PHD-like zinc-binding domain, F/Y-rich N-terminus domain and SET domain. Clade 2 is composed of three domains: PHD finger domain, F/Y-rich N-terminus domain and SET domain. Clade 3 is small and can be considered to be a sub-set of either clade 1 without the Bromodomain or clade 2 with extra F/Y-rich N-terminal domains. Clade 4 has the simplest combinations with only PHD finger and SET domains.
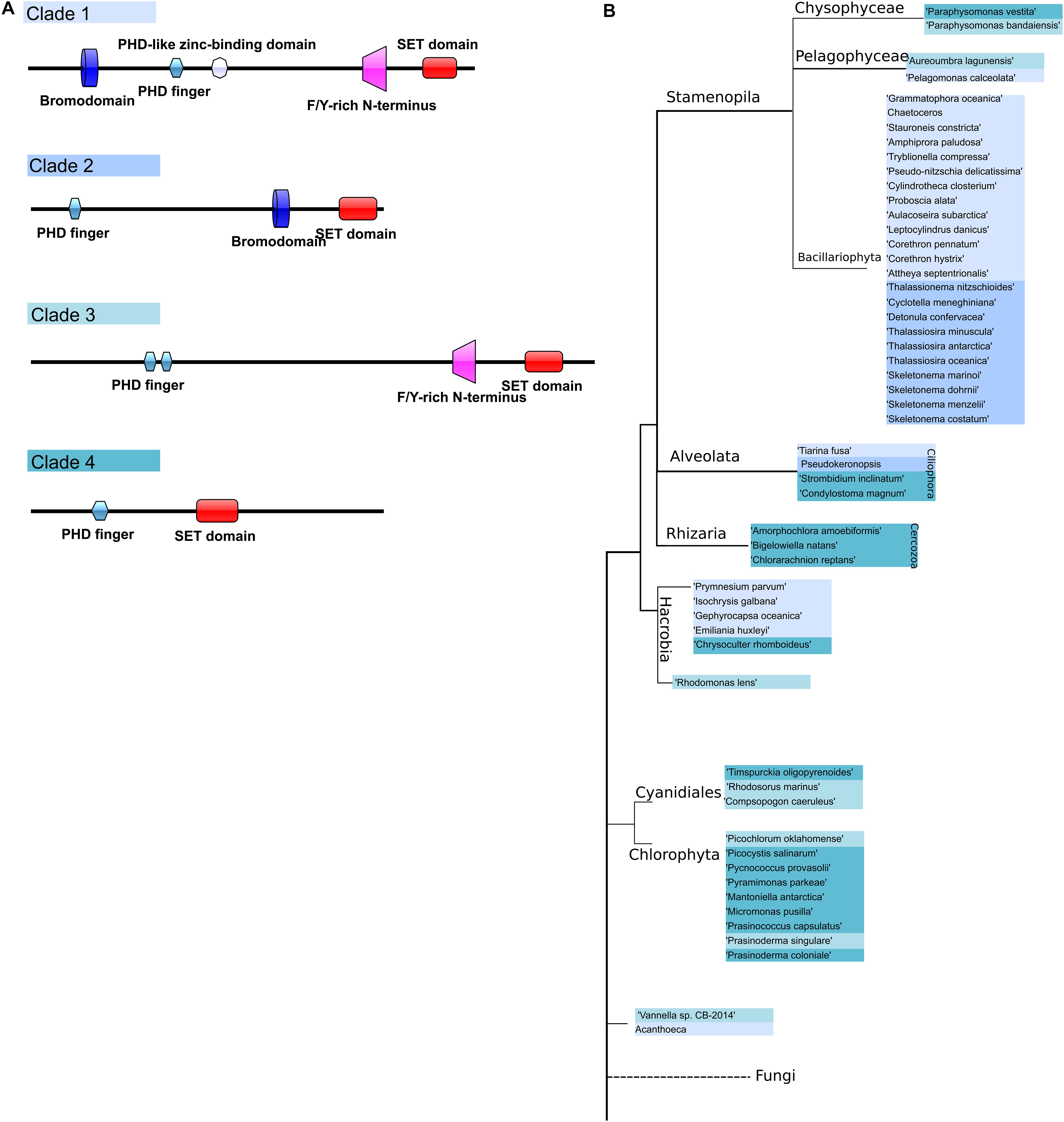
Figure 5. Domain architecture of Trithorax homologs in microalgae and their phylogenetic distribution. (A) Scheme representing the different groups of TrxG protein domain structures, (B) TrxG group distribution in a phylogenetic tree.
To explore the distribution and evolution of TrxG protein homologs, phylogenetic analysis was performed as described above. Interestingly, all diatom species were found to possess a Bromodomain which belongs to Clade 1 and Clade 2, with a clear distinction between pennate diatoms, which are all in Clade 1, while centric diatoms are restricted to Clade 2. Some Stramenopile groups such as Chysophyceae and Pelagophyceae might have lost the Bromodomain during evolution (Figure 5). Three cercozoan species were found to have Clade 4-type Trithorax homologs in Rhizaria that lack the Bromodomain. On the other hand, in the Alveolata we found Clade 4-type trithorax as well as Clades 1 and 2.
Compared to the complicated situation in SAR group organisms, green lineage organisms show simpler combinations of domains. All 12 green algae have either Clade 3 or Clade 4 type Trithorax with the following combination: PHD finger domain and SET domain, sometimes with the F/Y-rich N-terminal domain. As expected, a phylogenetic tree of Trithorax homologs shows that sequences from green lineage organisms cluster with the reference sequences ATX1 and ATX2 from the plant model species Arabidopsis thaliana (Figure 6). Among the five species found in Haptophyta, except for Chrysoculter rhomboideus, four have Clade 1-type Trithorax homologs.
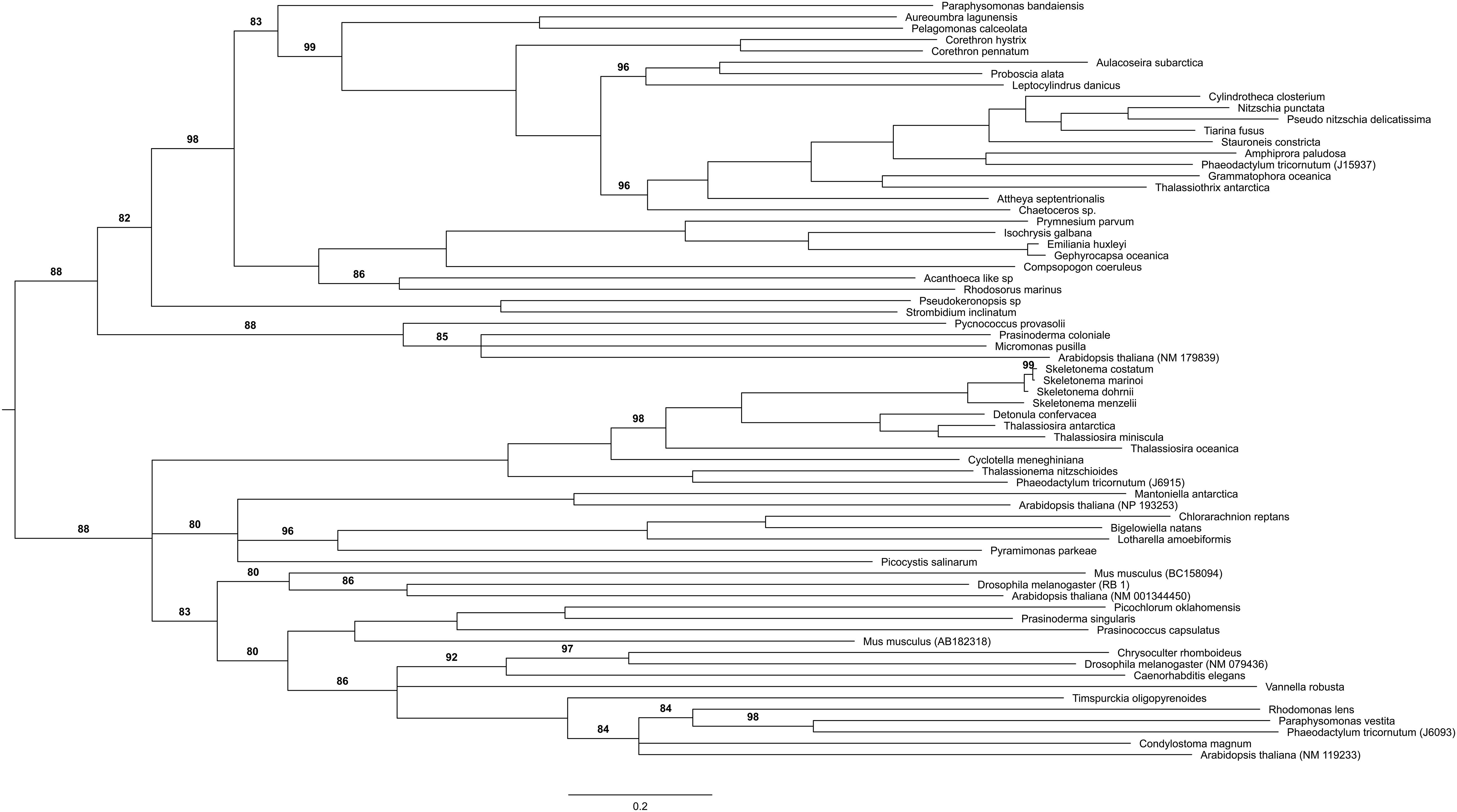
Figure 6. Phylogenetic tree of TrxG components.
Assessment of Polycomb and Trithorax Complexes in Environmentally Sampled Microalgae
To investigate the presence of Polycomb and Trithorax members in the environment, we used metagenomes and metatranscriptomes from Tara Oceans and applied eDAF (same procedure described in the Materials and Methods section) to detect all the unigenes. We retained only sequences with the exact same arrangements of conserved regions as the reference sequences (Supplementary Table S3), and considered only surface samples in 0.8 to 5, 5 to 20, 20 to 180 and 180 to 2000 μm size fractions.
Both RING and enhancer of zeste genomic sequences were weakly detected in 28 stations (9 and 25, respectively) out of 68 where diatoms were found (Carradec et al., 2018). However, their expression is significant in response to nitrate and phosphate although to a lesser extent than what was observed for the Trithorax complex (Figure 7B). This anti-correlation can be explained by the quality of DNA sequence reads, as reported previously in a much wider study of Tara Oceans samples (Terrapon et al., 2009). The majority of transcripts from genes encoding Trithorax components belong to Stramenopiles and only a few are shared between Chlorophyta and Haptophyceae. On the other hand, transcripts from genes encoding EZ and RING components are dominated by Dinophyceae (Supplementary Figure S1). The proportions of genomic DNA and transcripts for each class of microalgae can be accessed using Krona interactive charts in the following links: (https://ndownloader.figshare.com/files/20019587?private_link=302f866ec48cc9a2e1ed, https:// ndownloader.figshare.com/files/20019590?private_link=302f866 ec48cc9a2e1ed).
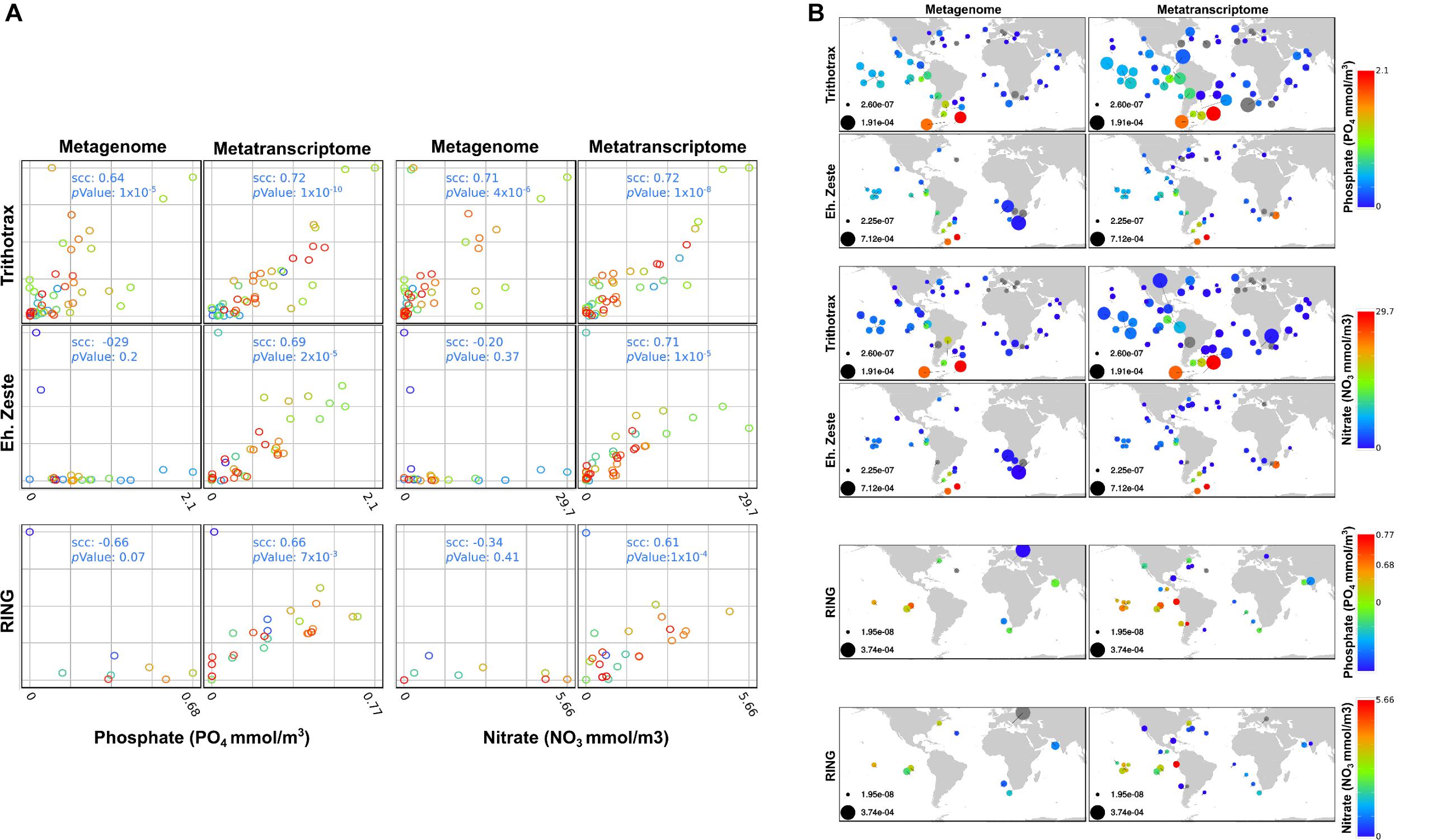
Figure 7. Correlation analysis of Metagenome and metatranscriptome and distribution of PcG and TrxG in TARA stations. (A) Correlation of the relative levels of gene presence (Metagenome) and gene expression (Metatranscriptome) detected by TARA station for bacillariophyta sequences when compared with 3 different environmental levels. The relative levels correspond to the abundance in RPKM of each gene/complex at each station normalized by the total abundance in RPKM of bacillariophyta sequences detected at the same station, and then re-scaled to the unit interval (minimum and maximum levels are presented in figure B). Abundances at each station were computed by pooling 4 size fractions, namely: 0.8 to 5, 5 to 20, 20 to 180 and 180 to 2000 μm. Spearman correlations coefficients (scc) and p values represent statistical association of the environment levels of Phosphate, Nitrate and bioavailable Nitrogen with the relative levels of RING genes, Enhancer of zeste genes and the Trithorax complex. We note that correlations between each respective Metagenome and Metatranscriptome pair were not significant enough to be shown (scc < 0.22) and colors were used to distinguish different stations, for visual clarity. (B) Distribution of RING genes, Enhancer of zeste genes and the Trithorax complex among TARA stations. Disk sizes represent the relative levels of gene presence (Metagenome) and gene expression (Metatranscriptome) at each TARA station in RPKM. Color intensities shows the different environment levels of Phosphate, Nitrate and bioavailable Nitrogen according to the corresponding color bars, and gray disks represent relative levels of genes where environmental factors were not available.
Several environmental factors were measured during the Tara Oceans expedition, so we examined whether there was any correlation with the expression of genes encoding Polycomb and Trithorax components. Only phosphate and nitrate showed significant correlations with enhancer of zeste, RING and Trithorax. Trithorax members were found to be highly expressed in response to increasing levels of nitrate and phosphate. Enhancer of zeste shows the same trend although to a lesser extent. RING was also found to correlate moderately with increasing levels of both nutrient except in TARA Stations 84 and 85 in the South Atlantic Ocean where they respond to lower levels of nitrate and phosphate (Figures 7A,B).
Among the other environmental factors measured during the Tara Oceans expedition, we observed a weak correlation between salinity and the expression of RING genes in 9 stations (scc: 0.56, p value: 0.04). The low number of stations is due to the absence of genes encoding RING proteins in most of the stations. We also obtained a weak correlation between the expression of genes encoding Trithorax components in 46 stations with measured oxygen levels (scc: 0.54 p value: 0.47). However, the observed high p values prevent us from drawing any conclusions.
Trithorax Deposited H3K4me3 Is an Active Mark Exclusive to Genes in P. tricornutum
Phaeodactylum tricornutum is the first and only Stramenopile so far for which a chromatin landscape has been drawn, with the mapping of five histone marks including H3K4me2, H3K9/14Ac, H3K9me2/me3, and H3K27me3 (Veluchamy et al., 2015). To explore the role of H3K4me3, known to be deposited by the Trithorax complex described above, and investigate its relationship to the repressive mark H3K27me3 reported to antagonize H3K4me3 (Aach et al., 2014), we performed chromatin immunoprecipitation with deep sequencing on two independent replicates of cultures of the reference strain Pt1 8.6 (Supplementary Figure S2).
H3K4me3 was found to cover around 42% of the genome targeting only genes (Supplementary Table S4). A total of 8,431 genes out of 12,152 were marked by H3K4me3, which was found mainly on exons (2,109 on exons out of 2,168 total peaks). H3K4me3 localizes at the 5′-end of transcribed regions (TSSs) and shows a similar profile to H3K9 acetylation and H3K4me2 but is different from the broader pattern of repressive marks such as H3K9me3 and H3K27me3 (Supplementary Figure S3).
We subsequently investigated the correlation between H3K4me3 distribution and transcript levels using ChIP-Seq from this study and RNA-Seq data generated previously in the same growth conditions (Bernardes J.S. et al., 2016). Analysis using a t-test shows significant differences in expression levels between marked and unmarked genes (t-test p-value:4.6e-08). Analysis of categorization of expression quantiles (10 quantiles) shows that highly expressed quantiles have 956 genes marked and 255 genes unmarked. In the low expression quantiles, 343 genes are marked and 988 genes are unmarked by H3K4me3 (Supplementary Figure S3). Furthermore, the genes that are uniquely marked by H3K4me3 show similar levels of gene expression than those marked uniquely by either H3K4me2 or H3K9Ac, which are widely recognized as active marks. Overall, H3K4me3 marked genes show increased levels of expression (FPKM), compared to the H3K4me3 unmarked genes, further confirming that H3K4me3 is an active mark in P. tricornutum.
Combinatorial Analysis of Histone Marks in P. tricornutum
To investigate the relationship between H3K4me3 and previously characterized histone marks (Schuettengruber et al., 2007), we analyzed the co-marking patterns including five histone marks, namely H3K4me2, H3K9/14Ac, H3K9me2, H3K9me3, H3K27me3, and DNA methylation (Supplementary Figure S4). Histone marks and DNA methylation can occur in 40 different combinations (Figure 8). Correlation analysis with transcript data defines principally three chromatin states (CS), active, repressive and intermediate. The three active marks of the study localize together on the highest number of genes compared to the rest of the combinations. They predominantly appear together with one repressive mark, H3K9me2. The lowest number of genes are co-marked by H3K9me3, H3K27me3 and DNA methylation, leading to a repressive CS. While co-occurrence of DNA methylation and the three repressive histone marks clearly define a repressive chromatin state, their association with one or more active marks switches to an intermediate or active CS with a particular signature of H3K4me3 which tends to have a significant effect by itself on the increase of global transcript levels (Figure 8). Mapping of an additional active mark does not change the pattern of co-occurrence of four repressive histone marks, which is unique to P. tricornutum, suggesting an interdependence or cooperation for transcriptional regulation of genes and TEs.
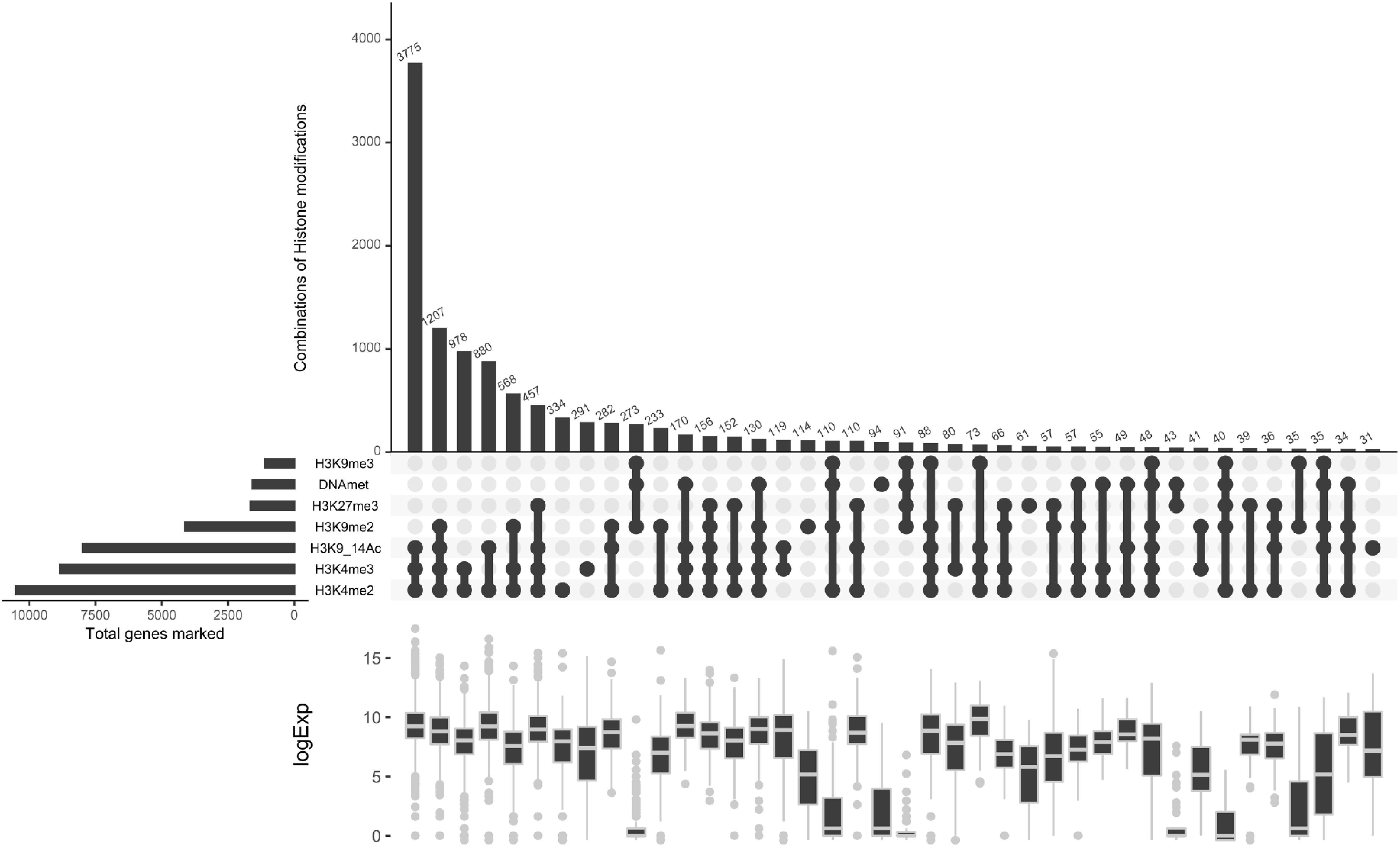
Figure 8. Co-occurrence analysis. A co-occurrence plot for the ChIP-seq peaks of different histone modification. For each set of intersection, a black filled circle is drawn. The size of the intersections is depicted in vertical bar. The vertical black line connects the different dataset that intersect. The number of genes for each chromatin state is shown on the top of the bars.
Considering the antagonistic relationship of both PcG and TrxG complexes in the regulation of several biological processes of the cell reported in plants and animals (Geisler and Paro, 2015; Poynter and Kadoch, 2016), we investigated the expression output when genes are co-marked with H3K27/K4me3. This analysis revealed two scenarios: first, 80 genes were exclusively co-marked with an intermediate level of expression and, second, a total of 814 co-marked genes with higher or lower expression are shared with either acetylation and H3K4m2 (CC6 with 457 genes), acetylation, H3K4m2 and H3K9me2 (CC 13 with 156 genes), only H3K4me2 (CC14 with 152) or H3K4me2 and H3K9me2 (CC25 with 66 genes) (Figure 8). Exclusive loci co-marking with both H3K4me3 and H3K27me3 suggests a bivalency that likely maintains genes in a poised state ready for activation or repression in response to relevant signals. These distinct readouts define a new histone code with the co-occurrence of active and repressive marks which likely cooperate or antagonize each other for gene regulation, with a distinct signature of balanced expression when H3K4me3 co-occurs alone with H3K27me3 and higher or lower expression when combined with active or repressive marks, respectively.
GO Functional Categories of H3K4me3 Marked Genes
To gain insights into the functional categories enriched in H3K4me3 marked genes, we performed a GO classification based on DAMA and CLADE and refined by eDAF results. We found an enrichment mostly in categories such as: (1) tetratricopeptide repeat containing proteins which act as a protein-protein interaction module involved in regulation of different cellular functions including cell cycle, hormone signaling, neurogenesis, protein folding and transport, and transcriptional control (Schapire et al., 2006); (2) mitochondrial carrier proteins the mediate the transport of ions, nucleotides and metabolites across mitochondrial membranes (Palmieri et al., 2011); (3) WD domain, G-beta repeat involved in a variety of functions ranging from signal transduction and transcription regulation to cell cycle control and apoptosis; (4) Aldo/Keto reductase families of proteins which are important intermediates in many metabolic pathways, including sugar metabolism, steroid biosynthesis, amino acid metabolism, and biosynthesis of secondary metabolites (Ellis, 2002); and (5) Kelch motif involved in fundamental cellular activities in multiple cellular compartments such as actin-binding activity, organization of cytoskeletal, plasma membrane and organelle structures (Adams et al., 2000). Overall, functional categories of H3K4me3 marked genes are mostly related to general biological processes involved in house-keeping functions.
The cooperation of two antagonistic marks, H3K4me3 and H3K27me3, for regulation of developmental processes in plants and animals compelled us to analyze the GO categories inherent to these cases. Interestingly, we see developmental and cell differentiation related categories when genes are exclusively co-marked with both H3K27me3/K4me3. When shared with other marks (CC6, CC13, CC14, and CC27), broader GO terms emerge (p-value of 2.52383E-73 for genes only enriched with H3K4me3 versus H3K4me3/K27me3 and no matter the others, p-value 4.54346E-98 for genes only co-marked by H3K4me3/K27me3 versus genes marked only by H3K27me3 or H3K4me3, p-value of 1.094673-123 for genes only marked by H3K4me3 or only H3K27me3 versus genes marked only by H3K4me3, Supplementary Table S4).
Discussion
In eukaryotes, the life cycle goes through growth/developmental phases characterized by specific spatial and temporal regulation of genome expression. This regulation is not only defined by their DNA sequence which remains constant between cell types but also by their gene expression pattern controlled by epigenetic mechanisms including dynamic chromatin states. Two groups of evolutionary conserved protein complexes known as Polycomb (PcG) and trithorax (TrxG) play a crucial role in such regulation by preventing or promoting gene expression, respectively (Schuettengruber et al., 2017). While TrxG are scarcely documented in only a few microalgae, PcG proteins are reported to be likely present in the last common ancestor of eukaryotes and to subsequently have been lost during evolution in certain single celled lineages such as both yeast model species S. pombe and S. cerevisiae (Shaver et al., 2010; Margueron and Reinberg, 2011). In the present study, using eDAF, which is an ensemble of modules for gene prediction, ontology, architecture extended information and automation, we interrogated MMETSP, currently the largest transcriptome reference database for eukaryotic marine microbes, and found that although the coexistence of PcGs and TrxGs is not found frequently in unicellular species (39 out of 203; 19.21%), there are several species that have both PcG catalytic enzymes and TrxGs, including Bigelowiella natans, Corethron hystrix, Attheya septentrionalis, Pelagomonas calceolate, and Proboscia alata (Supplementary Table S2). This number of species might be limited by the nature of the MMETSP database, which contains only transcriptomics sequences, and sequencing more unicellular genomes might reveal additional species with PcG and TrxG complexes.
Recently, studies on PRC2 and its associated epigenetic mark H3K27me3 in microalgae revealed interesting insights on their diversity and role in silencing of genes and transposable elements, which are their main targets (Shaver et al., 2010; Veluchamy et al., 2015; Mikulski et al., 2017; Frapporti et al., 2019). In our study, we found that both PcG and TrxG proteins are not well represented in dinoflagellates. This might reflect the unorthodox nature of histone proteins in dinoflagellates (Marinov and Lynch, 2016) which do not play a major role in genome packaging and heterochromatinization (Gornik et al., 2012). Although previous study show that dinoflagellates have most of the chromatin reader, writer and eraser protein families, including a strikingly large number of SET-domain proteins (Marinov and Lynch, 2016), our search of MMETSP did not yield such examples, except for a few species including Alexandrium and Symbiodinium which were found to have Esc and/or RING homologs, although no transcripts of other PcGs and TrxGs complex subunits could be found (Supplementary Table S3). Possible explanations for the poor representation in dinoflagellates is either the silencing of histone writers in the dinoflagellates contained in MMETSP or it may be a distinctive feature like other epigenetic modifying enzymes such as DNA methyltransferases (DNMTs) that are peculiar and unlike classical DNMTs (de Mendoza et al., 2018).
Trithorax family is a diverse group of proteins involved primarily in gene activation either by nucleosome positioning, histone modifications such as methylating lysine 4 of histone H3 or direct interactions with transcription machineries (Kingston and Tamkun, 2014). Here we focus on MLL/COMPASS complex which has histone methyltransferase activity. MLL family is made up of three pairs of structurally similar proteins, namely MLL1-MLL2, MLL3-MLL4, and SET1A-SET1B (Ruthenburg et al., 2007; Crump and Milne, 2019). Surprisingly, TrxG protein homologs distribution in diatoms is more complicated than expected. They can be divided into two groups: centric and pennates with structure differences that might reflect their evolutionary history with 90 million years of divergence. Green algae TrxG homologs are as simple as the first H3K4 methyltransferase Set1, identified in S. cerevisiae (Briggs et al., 2001), only have SET and PHD finger domains, which is similar to SET1A-SET1B group in MLL family. Compared with green algae which possess only clade 3 and 4, diatoms TrxGs are more close to human TrxG genes which reflects the close relationship between stramenopiles and animals.
Using the same reference sequences, we probed the presence of both TrxG and PcG complexes in environmental samples from small size fractions of Tara Oceans. Our results point to the presence of TrxG complex including the four constituent proteins, all with SET and PHD finger domains in combination with either Bromodomain or F/Y motifs, which correlate with high concentrations of nitrate and phosphate. Although less important, this correlation is significant for both EZ and RING components of PRC2 and PRC1, respectively. These results suggest a Polycomb and Trithorax mediated regulation in response to nutrient availability. A weak correlation was observed between transcript levels of RING, TrxG and low salinity and oxygen, respectively. To the best of our knowledge, this is the first report of expression of Polycomb and Trithorax genes in environmentally sampled marine eukaryotic microalgae and tehir correlation to macronutrient availability. This may suggest a role of chromatin master regulators in nutrient uptake which is important in nutrient cycling and ultimately primary productivity. Beyond the conservation of Trithorax proteins in unicellular marine microalgae sharing conserved domains with animal and plant homologs, the TrxG deposited histone mark H3K4me3 shows similar patterns of distribution in P. tricornutum compared to what is described in multicellular species (Barski et al., 2007; Zhang et al., 2009). It is found over genes spanning transcriptional start sites and 5′ promoter regions and correlates with active gene expression, suggesting a ubiquitous role of H3K4me3 as a transcriptionally activating mark.
Analysis of the combinatorial readout of histone PTMs in P. tricornutum identified principally three chromatin states that likely reflect a coordinated regulation of genes. Although not exhaustive, this histone code is overall conserved pointing to key PTMs that seem to be important for defining each of the chromatin states confirming previously published study (Veluchamy et al., 2015). Despite the overall conservation of the histone code, unique patterns emerge from the co-occurrences of repressive marks that are not documented in animals and plants, in particular those that combine several repressive marks (e.g., C21 with 91 genes) with a dramatic effect on gene repression compared to the other CS. Our previous work (Veluchamy et al., 2015) has identified such patterns, and profiling a new active histone mark which targets a high number of genes uncovers genes that are uniquely marked with several repressive histone marks and DNA methylation, suggesting a stable pattern of gene regulation that likely involves a crosstalk. This suggests several scenarios of regulation including a cumulative effect or recruitment loop as described in other studies (Strahl and Allis, 2000; Roudier et al., 2011). Genetic studies in model species such as P. tricornutum are an important step in elucidating these mechanisms. These chromatin states are an indication of the overall transcriptional output of several marks when they co-occur. However, it needs to be extended to additional key marks which will undoubtedly refine the histone code which is a useful proxy for understanding how chromatin states mediate the regulation of genes in response to developmental and environmental triggers.
Functional annotation of exclusively H3K4me3 marked genes revealed an enrichment in categories such as protein degradation and peptide processing into amino acids, RING finger containing proteins known for their role in diverse cellular processes such as regulation of transcription, cell cycle, signaling and secretory pathway (Deshaies, 1999), FYVE domain containing proteins associated to vacuolar protein sorting and endosome function (Leevers et al., 1999; Jensen et al., 2001), proteins with DNA ligase domains with a role in a wide range of DNA transactions (Martin and MacNeill, 2002), all of which are indicative of rather general house-keeping functions. However, gene ontology annotation of H3K4me3/K27me3 loci show enrichment in categories related to cellular architecture with a majority of GO terms referring to regulation of actin polymerization or depolymerization, regulation of cell shape, microtubule cytoskeleton, myosin complex, positive regulation of cellular components organization and peptidyl tyrosine phosphorylation important for cell proliferation, cell cycle progression, metabolic homeostasis, differentiation and development (Hunter, 2009). Although less important, these GO categories are present when genes are marked by H3K3me4/K27me3 and other marks. It is tempting to postulate that co-marking by H3K4me3 and H3K37me3 is a combinatorial action of both marks in transcriptional regulation of genes related to specific functions such as cell differentiation. This analysis is based on the intersection of ChIP-Seq tracks and so it will be important in future studies to perform sequential ChIP to monitor the coexistence of these two marks or the unique co-occurrence of several repressive histone marks within a single nucleosome.
The existence and diversity of PcG and TrxG complexes in eukaryotic unicellular species throughout the tree of life opens a whole range of questions on the molecular mechanisms of their interactions; their role in cell differentiation and other biological processes, how they are recruited to their targets, and understanding their contribution to a fundamental mechanism, the emergence of multicellularity. One particular aspect is understanding the cross talk between these two complexes for gene regulation, especially in a bivalency context where a timely activation is required as well as the maintenance of repression. Future studies in P. tricornutum and other emerging unicellular models will undoubtedly bring new insights and deepen our understanding of the evolutionary history of PcG and TrxG complexes.
Data Availability Statement
The datasets generated for this study can be found in the GSE139676.
Author Contributions
LT, XZ, and FV conceived and designed the study. AD performed the experimental work. LT, XZ, AV, and FV performed the bioinformatic analysis, analyzed and interpreted the results. LT, XZ, and FV wrote the manuscript with input from all authors. All authors read and approved the manuscript.
Funding
LT acknowledges funds from the CNRS and the region of Pays de la Loire (ConnecTalent EPIALG project). CB acknowledges the European Research Council Advanced Award Diatomite. XZ was supported by a Ph.D. fellowship from the Chinese Scholarship Council (CSC-201604910722).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00189/full#supplementary-material
FIGURE S1 | Krona Pie charts. (A–C) represent metagenomics krona charts of TrxG, EZ and RING. (D–F) show meta-transcriptomic Krona charts of TrxG, EZ and RING respectively.
FIGURE S2 | Profile plot of histone marks including H3K4me3 and gene expression levels of H3K4me3 marked versus unmarked genes. (A) Averaged tag-density profile for 6 histone modifications are plotted for TSS of 12k genes with 3 kb upstream and 10 kb downstream regions. Regions are ordered and scaled to TSS as reference point. (B) Boxplot showing the differences in expression levels of genes with and without histone modifications. Box represents 10–90% of data and the outliers are shown as whiskers. Median of the data is shown in a small dot in the center of the box (t-test: p-value = 1.581e-08).
FIGURE S3 | H3K4me3 Correlation plot. Pairwise scatterplots showing correlation of the two H3K4me3 ChIP-seq replicates. Genome is split into bins and enrichment scores per bin were calculated. Pearson correlation coefficient for the comparison of enrichment scores is marked.
FIGURE S4 | PTMs distribution. Upper panel shows average ChIPseq profile (read density) over TSS of genes. Lower panel shows heatmaps of ChIPseq data of individual histone modifications and input is shown over TSS.
TABLE S1 | Reference sequences used in this study.
TABLE S2 | List of species found in MMETSP with PRC1 or/and PRC2 components and Trithorax components. The table contains species have at least one PcG proteins with present or absent of TrxG protein which located at the last column. TrxG column also specify the trithorax homologs clade classification which is discussed in this paper, species with both PcG catalytic enzyme (E(z) and RING/Psc) and TrxG were highlight in yellow background.
TABLE S3 | Protein sequences of PRC1, PRC2 and TrxG components. The table shows the PFAM domains, their coordinates within the sequences and score.
TABLE S4 | Gene annotations and functional categories of histone marked genes in P. tricornutum. Genes annotated for each histone modification are summarized and tabulated. Histone marked genes are given value 1 and unmarked genes in the genome are given value 0.
Footnotes
References
Aach, J., Prashant, M., and George, M. C. (2014). CasFinder flexible algorithm for identifying specific Cas9 targetsin genomes. bioRxiv [Preprint]. doi: 10.1101/005074
Adams, J., Kelso, R., and Cooley, L. (2000). The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell. Biol. 10, 17–24. doi: 10.1016/s0962-8924(99)01673-6
Barrero, M. J., and Izpisua Belmonte, J. C. (2013). Polycomb complex recruitment in pluripotent stem cells. Nat. Cell Biol. 15, 348–350. doi: 10.1038/ncb2723
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Baurain, D., Brinkmann, H., Petersen, J., Rodríguez-Ezpeleta, N., Stechmann, A., Demoulin, V., et al. (2010). Phylogenomic evidence for separate acquisition of plastids in cryptophytes, haptophytes, and stramenopiles. Mol. Biol. Evol. 27, 1698–1709. doi: 10.1093/molbev/msq059
Bernardes, J., Zaverucha, G., Vaquero, C., and Carbone, A. (2016). Improvement in protein domain identification is reached by breaking consensus, with the agreement of many profiles and domain co-occurrence. PLoS Comput. Biol. 12:e1005038. doi: 10.1371/journal.pcbi.1005038
Bernardes, J. S., Vieira, F. R., Zaverucha, G., and Carbone, A. (2016). A multi-objective optimization approach accurately resolves protein domain architectures. Bioinformatics 32, 345–353. doi: 10.1093/bioinformatics/btv582
Briggs, S., Bryk, M., Strahl, B. D., Cheung, W. L., Davie, J. K., Dent, S. Y. D., et al. (2001). Histone H3 lysine 4 methylation is mediated by set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 15, 3286–3295. doi: 10.1101/gad.940201
Burki, F., Okamoto, N., Pombert, J. F., and Keeling, P. J. (2012). The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc. Biol. Sci. 279, 2246–2254. doi: 10.1098/rspb.2011.2301
Carradec, Q., Pelletier, E., Da Silva, C., Alberti, A., Seeleuthner, Y., Blanc-Mathieu, R., et al. (2018). A global ocean atlas of eukaryotic genes. Nat. Commun. 9:373. doi: 10.1038/s41467-017-02342-2341
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Crump, N. T., and Milne, T. A. (2019). Why are so many MLL lysine methyltransferases required for normal mammalian development? Cell Mol. Life Sci. 76, 2885–2898. doi: 10.1007/s00018-019-03143-z
de Mendoza, A., Bonnet, A., Vargas-Landin, D. B., Ji, N., Li, H., Yang, F., et al. (2018). Recurrent acquisition of cytosine methyltransferases into eukaryotic retrotransposons. Nat. Commun. 9:1341. doi: 10.1038/s41467-018-03724-3729
Deshaies, R. J. (1999). SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 15, 435–467. doi: 10.1146/annurev.cellbio.15.1.435
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Ellis, E. M. (2002). Microbial aldo-keto reductases. FEMS Microbiol. Lett. 216, 123–131. doi: 10.1111/j.1574-6968.2002.tb11425.x
Frapporti, A., Miró, P. C., Arnaiz, O., Holoch, D., Kawaguchi, T., Humbert, A., et al. (2019). The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat. Commun. 10:2710. doi: 10.1038/s41467-019-10648-10645
Geisler, S. J., and Paro, R. (2015). Trithorax and Polycomb group-dependent regulation: a tale of opposing activities. Development 142, 2876–2887. doi: 10.1242/dev.120030
Gergonne, J. D. (1974). The application of the method of least squares to the interpolation of sequences. Hist. Math. Hist. Math. 1, 439–447. doi: 10.1016/0315-0860(74)90034-2
Gil, J., and O’Loghlen, A. (2014). PRC1 complex diversity: where is it taking us? Trends Cell Biol. 24, 632–641. doi: 10.1016/j.tcb.2014.06.005
Gornik, S. G., Ford, K. L., Mulhern, T. D., Bacic, A., McFadden, G. I., Waller, R. F., et al. (2012). Loss of nucleosomal DNA condensation coincides with appearance of a novel nuclear protein in dinoflagellates. Curr. Biol. 22, 2303–2312. doi: 10.1016/j.cub.2012.10.036
Harmanci, A., Rozowsky, J., and Gerstein, M. (2014). MUSIC: identification of enriched regions in ChIP-Seq experiments using a mappability-corrected multiscale signal processing framework. Genome Biol. 15:474. doi: 10.1186/s13059-014-0474-473
Hunter, T. (2009). Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146. doi: 10.1016/j.ceb.2009.01.028
Jensen, R. B., La Cour, T., Albrethsen, J., Nielsen, M., and Skriver, K. (2001). FYVE zinc-finger proteins in the plant model Arabidopsis thaliana: identification of PtdIns3P-binding residues by comparison of classic and variant FYVE domains. Biochem. J. 359, 165–173. doi: 10.1042/bj3590165
Joazeiro, C. A., and Weissman, A. M. (2000). RING finger proteins: mediators of ubiquitin ligase activity. Cell 102, 549–552. doi: 10.1016/s0092-8674(00)00077-75
Kingston, R. E., and Tamkun, J. W. (2014). Transcriptional regulation by trithorax-group proteins. Cold Spring Harb. Perspect. Biol. 6:a019349. doi: 10.1101/cshperspect.a019349
Kyba, M., and Brock, H. W. (1998). The drosophila polycomb group protein Psc contacts ph and Pc through specific conserved domains. Mol. Cell Biol. 18, 2712–2720. doi: 10.1128/mcb.18.5.2712
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leevers, S. J., Vanhaesebroeck, B., and Waterfield, M. D. (1999). Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr. Opin. Cell Biol. 11, 219–225. doi: 10.1016/s0955-0674(99)80029-80025
Margueron, R., and Reinberg, D. (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. doi: 10.1038/nature09784
Marinov, G. K., and Lynch, M. (2016). Conservation and divergence of the histone code in nucleomorphs. Biol. Direct. 11:18. doi: 10.1186/s13062-016-0119-114
Martin, I. V., and MacNeill, S. A. (2002). ATP-dependent DNA ligases. Genome Biol. 3:REVIEWS3005. doi: 10.1186/gb-2002-3-4-reviews3005
Mikulski, P., Komarynets, O., Fachinelli, F., Weber, A. P. M., and Schubert, D. (2017). Characterization of the polycomb-group mark H3K27me3 in unicellular algae. Front. Plant Sci. 8:607. doi: 10.3389/fpls.2017.00607
Mitchell, A., Chang, H. Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2019). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360. doi: 10.1093/nar/gky1100
Ochoa, A., Llinas, M., and Singh, M. (2011). Using context to improve protein domain identification. BMC Bioinform. 12:90. doi: 10.1186/1471-2105-12-90
Palmieri, F., Pierri, C. L., De Grassi, A., Nunes-Nesi, A., and Fernie, A. R. (2011). Evolution, structure and function of mitochondrial carriers: a review with new insights. Plant J. 66, 161–181. doi: 10.1111/j.1365-313X.2011.04516.x
Poynter, S. T., and Kadoch, C. (2016). Polycomb and trithorax opposition in development and disease. Wiley Interdiscip. Rev. Dev. Biol. 5, 659–688. doi: 10.1002/wdev.244
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A., and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. doi: 10.1093/nar/gku365
Rastogi, A., Maheswari, U., Dorrell, R. G., Vieira, F. R. J., Maumus, F., Kustka, A., et al. (2018). Integrative analysis of large scale transcriptome data draws a comprehensive landscape of Phaeodactylum tricornutum genome and evolutionary origin of diatoms. Sci. Rep. 8:4834. doi: 10.1038/s41598-018-23106-x
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Roudier, F., Ahmed, I., Bérard, C., Sarazin, A., Mary-Huard, T., Cortijo, S., et al. (2011). Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 30, 1928–1938. doi: 10.1038/emboj.2011.103
Ruthenburg, A. J., Allis, C. D., and Wysocka, J. (2007). Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell 25, 15–30. doi: 10.1016/j.molcel.2006.12.014
Schapire, A. L., Valpuesta, V., and Botella, M. A. (2006). TPR proteins in plant hormone signaling. Plant Signal. Behav. 1, 229–230. doi: 10.4161/psb.1.5.3491
Schuettengruber, B., Bourbon, H. M., Di Croce, L., and Cavalli, G. (2017). Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171, 34–57. doi: 10.1016/j.cell.2017.08.002
Schuettengruber, B., Chourrout, D., Vervoort, M., Leblanc, B., and Cavalli, G. (2007). Genome regulation by polycomb and trithorax proteins. Cell 128, 735–745. doi: 10.1016/j.cell.2007.02.009
Schuettengruber, B., Martinez, A. M., Iovino, N., and Cavalli, G. (2011). Trithorax group proteins: switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 12, 799–814. doi: 10.1038/nrm3230
Sellers, P. (1980). The theory and computation of evolutionary distances: pattern recognition. J. Algorith. 1, 359–373. doi: 10.1016/0196-6774(80)90016-4
Shaver, S., Casas-Mollano, J. A., Cerny, R. L., and Cerutti, H. (2010). Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics 5, 301–312. doi: 10.4161/epi.5.4.11608
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Terrapon, N., Gascuel, O., Marechal, E., and Breehelin, L. (2009). Detection of new protein domains using co-occurrence: application to Plasmodium falciparum. Bioinformatics 25, 3077–3083. doi: 10.1093/bioinformatics/btp560
Thain, D., Tannenbaum, T., and Livny, M. (2005). Distributed computing in practice: the condor experience. Concurr. Comput. Pract. Exp. 17, 323–356. doi: 10.1002/cpe.938
Vartanian, M., Descles, J., Quinet, M., Douady, S., and Lopez, P. J. (2009). Plasticity and robustness of pattern formation in the model diatom Phaeodactylum tricornutum. New Phytol. 182, 429–442. doi: 10.1111/j.1469-8137.2009.02769.x
Veluchamy, A., Rastogi, A., Lin, X., Lombard, B., Murik, O., Thomas, Y., et al. (2015). An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol. 16:102. doi: 10.1186/s13059-015-0671-678
Vidal, M. (2019). Polycomb assemblies multitask to regulate transcription. Epigenomes 3:12. doi: 10.3390/epigenomes3020012
Xing, H., Mo, Y., Liao, W., and Zhang, M. Q. (2012). Genome-wide localization of protein-DNA binding and histone modification by a Bayesian change-point method with ChIP-seq data. PLoS Comput. Biol. 8:e1002613. doi: 10.1371/journal.pcbi.1002613
Yeats, C., Redfern, O. C., and Orengo, C. (2010). A fast and automated solution for accurately resolving protein domain architectures. Bioinformatics 26, 745–751. doi: 10.1093/bioinformatics/btq034
Zhang, X., Bernatavichute, Y. V., Cokus, S., Pellegrini, M., and Jacobsen, S. E. (2009). Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 10:R62. doi: 10.1186/gb-2009-10-6-r62
Keywords: epigenetics, environment, microalgae, diatoms, bioinformatics, Phaeodactylum tricornutum, polycomb, trithorax
Citation: Zhao X, Deton Cabanillas AF, Veluchamy A, Bowler C, Vieira FRJ and Tirichine L (2020) Probing the Diversity of Polycomb and Trithorax Proteins in Cultured and Environmentally Sampled Microalgae. Front. Mar. Sci. 7:189. doi: 10.3389/fmars.2020.00189
Received: 09 December 2019; Accepted: 11 March 2020;
Published: 31 March 2020.
Edited by:
Jose M. Eirin-Lopez, Florida International University, United StatesReviewed by:
Daniel Garcia-Souto, University of Vigo, SpainChiara Lanzuolo, Institute of Cell Biology and Neurobiology (CNR), Italy
Copyright © 2020 Zhao, Deton Cabanillas, Veluchamy, Bowler, Vieira and Tirichine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Rocha Jimenez Vieira, cm9jaGFAYmlvbG9naWUuZW5zLmZy; ZmFiaW9yanZpZWlyYUBnbWFpbC5jb20=; Leila Tirichine, dGlyaWNoaW5lLWxAdW5pdi1uYW50ZXMuZnI=; TGVpbGEuVGlyaWNoaW5lQHVuaXYtbmFudGVzLmZy