 Kai-Le Zhong1,2,3
Kai-Le Zhong1,2,3 Xiao-Han Song1,2,3Han-Gil Choi4Shimada Satoshi5
Xiao-Han Song1,2,3Han-Gil Choi4Shimada Satoshi5 Florian Weinberger6
Florian Weinberger6 Stefano G. A. Draisma7
Stefano G. A. Draisma7 De-Lin Duan1,2
De-Lin Duan1,2 Zi-Min Hu1,2*
Zi-Min Hu1,2*- 1Key Laboratory of Experimental Marine Biology, Center for Ocean Mega-Science, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3College of Earth and Planetary Sciences, University of Chinese Academy Sciences, Beijing, China
- 4Faculty of Biological Science, Wonkwang University, Iksan, South Korea
- 5Faculty of Core Research, Natural Science Division, Ochanomizu University, Tokyo, Japan
- 6Department of Benthic Ecology, GEOMAR Helmholtz-Zentrum für Ozeanforschung Kiel, Kiel, Germany
- 7Excellence Center for Biodiversity of Peninsular Thailand, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
The repeated transgression and regression of coastlines mediated by the late Quaternary glacial–interglacial cycles make the northwest Pacific a hot spot to study marine speciation and population diversity. The red alga Agarophyton vermiculophyllum is an ecologically important species native to the northwest Pacific, capturing considerable research interest due to its wide-range invasiveness in Europe and North America. However, the knowledge of phylogeographic structure and intraspecific genetic diversity across the entire native range was still scarce. Here, we used 1,214-bp of mitochondrial cytochrome c oxidase subunit 1 (cox1) to explore phylogeographic patterns, lineage structure, and population genetic differentiation of 48 A. vermiculophyllum populations in the northwest Pacific. Our DNA data revealed overall high haplotype diversity and low nucleotide diversity and five phylogeographically structured genetic lineages that diverged significantly from each other. S-DIVA analysis showed the ancestors of A. vermiculophyllum originating from multiple areas encompassing the Japan–Pacific coast, East and South China Seas. This combined evidence indicates that A. vermiculophyllum might have survived in multiple scattered glacial refugia during the late Quaternary climate oscillations in the northwest Pacific. Such knowledge may help to better understand how palaeoclimate interacted with contemporary environments to contribute to intraspecific genetic variation and provide a new perspective for conserving natural resource of A. vermiculophyllum in the northwest Pacific.
Introduction
The northwest Pacific spans three major marginal seas, the Sea of Japan, the East China Sea (including Yellow–Bohai Sea), and the South China Sea (Figure 1). In the late Quaternary, global sea level fell 120–140 m during glacial maxima and thus led to the exposure of the marginal seas (Lambeck et al., 2002). Consequently, the Yellow–Bohai Sea was completely exposed, the East China Sea shrank eastward to the Okinawa trough, the South China Sea became a semi-closed sea, and the Sea of Japan with a shallower bedrock became an almost enclosed sea (Figure 1; Wang, 1999; Oba and Irino, 2012). During the interglacial periods, the three marginal seas were inundated by rising sea levels. These repeated transgression–regression cycles, together with long-distance dispersal mediated by ocean currents and recent anthropogenic activities, imposed great impacts on phylogeographic patterns and population genetic differentiation in this area (Cheang et al., 2010; Wang et al., 2016). The northwest Pacific has therefore been proposed as an ideal model for studying how sea level oscillations, caused by repeated glaciations, structured distribution range and spatial population genetic diversity and marine phylogeography (Ni et al., 2014). In the northwest Pacific, the reduction of marginal seas during the late Quaternary is thought to have led to a large decrease of marine species, while some ancestral relict populations may have survived in isolated refugia. The discontinuity of these refugia made an essential contribution to population-level isolation and differentiation (Ni et al., 2014). Phylogeographic studies of a variety of marine organisms have proposed several marine glacial refugia in the northwest Pacific, which scattered from the South China Sea (Hu et al., 2018a) to Taiwan island (Lee et al., 2012), the Okinawa trough (Hu et al., 2015), the Sea of Japan (Wang, 1999), Jeju island (Lee et al., 2012), and the Japanese Pacific coast (Hu et al., 2011, 2015, 2017). When sea levels rose after the Last Glacial Maximum (LGM), the surviving populations in marine refugia expanded to colonize new habitats, eliminating population differentiation caused by isolation in different refugia (Xue et al., 2014). Ocean currents form another important driving force of post-glacial population colonization. The dominant ocean current systems in the northwest Pacific constitute the Kuroshio Current and its branches (the Yellow Sea Warm Current and the Tsushima Warm Current), the China Coastal Current, the South China Sea Warm Current, and the Oyashio Current (Figure 1). In the Yellow–Bohai Sea, for example, the Yellow Sea Warm Current originating from the Okinawa trough has been reported to drive genetic homogeneity in some marine organisms (Hu et al., 2015; Li et al., 2017; Liu et al., 2018).
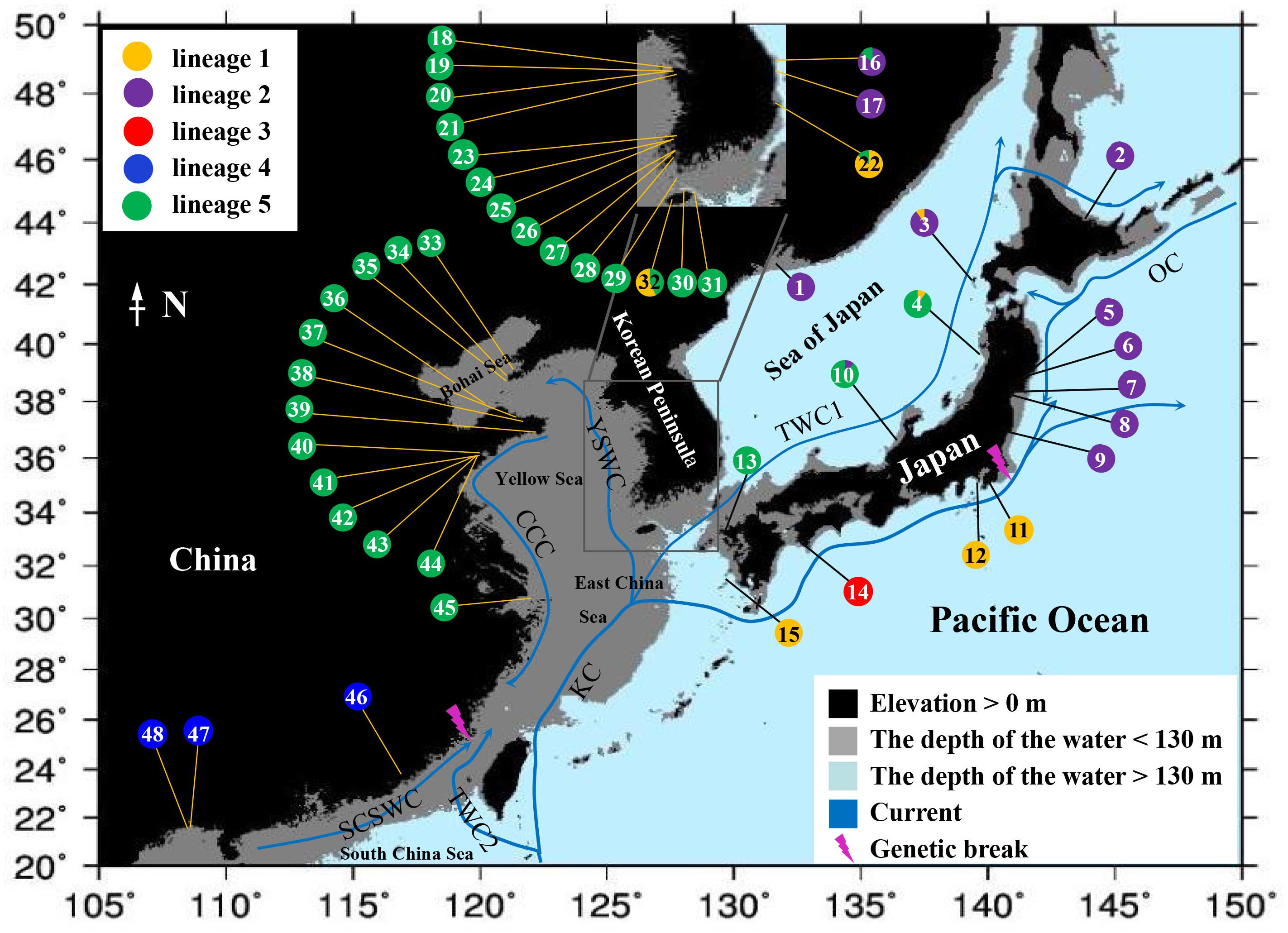
Figure 1. Geographical distribution of mtDNA cox1 haplotypes in the northwest Pacific. Numbers in pie charts correspond to sampling localities in Supplementary Table S1. Colors corresponds to the lineages indicated in the phylogenetic tree (Figure 2) and haplotype network (Figure 3). The black areas represent the elevation >0 m, the gray areas represent the depth of water <130 m, and the light blue areas represent the depth of water >130 m (sea levels during the last glacial maximum). KC, Kuroshio Current; TWC1, Tsushima Warm Current; YSWC, Yellow Sea Warm Current; CCC, China Coastal Current; TWC2, Taiwan Warm Current; SCSWC, South China Sea Warm Current; OC, Oyashio Current.
The red alga Agarophyton vermiculophyllum (Ohmi) Gurgel, J. N. Norris et Fredericq, is native to the northwest Pacific (Gurgel et al., 2018). It was originally described as Gracilariopsis vermiculophylla Ohmi from the Akkeshi Bay, Hokkaido, Japan, but was transferred a decade later to the genus Gracilaria Greville and only recently was transferred to a new genus Agarophyton Gurgel, J. N. Norris et Fredericq, together with two other former Gracilaria species (Gurgel et al., 2018). Its life cycle consists of an alternation of isomorphic haploid and diploid stages. It is considered an important economic and ecological species because it is one of the main raw materials for extracting agar (Armisen, 1995) and used as a biofilter in seawater eutrophication restoration (Abreu et al., 2011).
The past few decades witnessed an invasion of A. vermiculophyllum in Europe and North America (Krueger-Hadfield et al., 2017). The invasion of A. vermiculophyllum has captured widespread research interest, particularly its impacts on coastal ecosystem and adaptation to environmental variables (Davoult et al., 2017; Sotka et al., 2018). Some studies indicate that strong asexual reproduction, high tolerance to abiotic stresses, low palatability, and high resistance to epiphytes account for the rapid invasion, establishment, and expansion in new habitats (Hu and Lopez-Bautista, 2014). Kim et al. (2010) and Krueger-Hadfield et al. (2017) speculated the Sea of Japan and northeastern Japan as the donor areas of invasive A. vermiculophyllum populations in Europe and North America, possibly mediated by the exported oyster Crassostrea gigas Thunberg, 1793 from Japan, but these studies lacked adequate sampling from its native range.
Molecular studies of A. vermiculophyllum in its native range are still scarce. Kim et al. (2010) and Gulbransen et al. (2012) compared genetic diversity based on mitochondrial cox1 (cytochrome c oxidase subunit 1) and found that A. vermiculophyllum in the native northwest Pacific had substantially higher genetic diversity and haplotype richness compared to the invasive populations in Europe and North America. Krueger-Hadfield et al. (2017) detected three mitochondrial lineages of A. vermiculophyllum in the northwest Pacific and high genetic differentiation among proximal geographic populations using microsatellite genotyping. They found two cox1 genetic groups that differed by two C/T substitutions which showed a segregated distribution pattern north and south of ∼35°N. Recently, Hu et al. (2018b) reported a significant genetic break in A. vermiculophyllum along the coast of China using Amplified Fragment Length Polymorphism data. Nevertheless, they did not yet thoroughly understand phylogeographic structure and potential cryptic lineage diversity in A. vermiculophyllum across its native rang in the northwest Pacific.
In the northwest Pacific, A. vermiculophyllum is distributed from the far east of Russia in the north to tropical Guangxi, China, and Vietnam in the south (Xia, 1999; Skriptsova and Choi, 2009). In this study, we expanded the sampling area of A. vermiculophyllum in its native range in the northwest Pacific compared to previous studies to (i) quantify phylogeographic structure and genetic variation of A. vermiculophyllum in the northwest Pacific and (ii) detect the diverged genetic lineages and reconstruct their putative phylogeographic histories. We are especially interested in testing the hypothesis proposed by recent phylogeographical studies (Hu et al., 2011; Dong et al., 2012; Ni et al., 2014; Li et al., 2017) in which sea level drops during the Quaternary ice ages and oceanic currents interactively contributed to geographical distribution and genetic differentiation of coastal marine organisms in the northwest Pacific. This knowledge may enhance our understanding of the distribution shifts and genetic exchange patterns of marine species in this area and provide action guidelines for conserving natural resources of A. vermiculophyllum.
Materials and Methods
Sample Collection
Agarophyton vermiculophyllum populations were collected during 2014–2018 from Hokkaido, Japan (44.05°N) to Beihai, China (21.45°N). In total, 546 specimens were sampled from 39 sites (14 from Japan, nine from Korea, and 16 from China) (Supplementary Table S1). At each site, 5–33 individuals were collected randomly with an interval distance >10 m to minimize the chance of collecting genetically identical samples. All samples were stored in silica gel. Due to the scarcity of Korean samples, molecular data of eight additional sites from Korea (62 specimens) and one site from Russia (three specimens) were retrieved from Kim et al. (2010) [marked with asterisk (∗) in Supplementary Table S1]. In total, we analyzed 611 samples of A. vermiculophyllum from 48 populations.
DNA Extraction, PCR Amplification and Sequencing
Genomic DNA was extracted from 5–20-mg dried samples using the Onestep-LysisTM Plant DNA Isolation Kit (PosPolysaccharides & Polyphenolics-rich, Nobelab, Beijing, China) following the manufacturer’s instructions. We amplified partial mitochondrial DNA (mtDNA) cox1 region with primers 43F (5′-TCA ACA AAT CAT AAA GAT ATT GGW ACT-3′) and 1549R (5′-AGG CAT TTC TTC AAA NGT ATG ATA-3′) (Geraldino et al., 2006). PCR amplification was performed in 50 μl of reaction volume, containing 25 μl 2 × EasyTaq Master Mix (Cwbio, Beijing, China), 1 μl of each forward and reverse primer (10 μmol L–1), 1 μl DNA template (15 ng μl–1) and 22 μl RNase-free water. Amplification was run in a PCR Thermal Cycler (LongGene, China) with an initial denaturation at 94°C for 10 min followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 72°C for 2 min, with a final extension at 72°C for 10 min (Yang et al., 2008). PCR products were purified using a Gel Extraction Kit (Omega, Norcross, GA, United States), and bidirectional sequencing was performed on the ABI3730 sequencing platform (Applied Biosystems, Foster City, CA, United States) using another primer set 622F (5′-CCT GTN TTA GCA GGW GCT ATT ACA ATG C-3′) and 880R (5′-ACA GTA TAC ATA TGA TGN GCT CAA AC-3′) (Yang et al., 2008).
Molecular Diversity, Phylogeny, and Genetic Structure
BioEdit v.7.2.5 (Hall, 1999) was used to align newly obtained sequences with previously published eight mtDNA cox1 (GenBank: EF434932–EF434339) (Yang et al., 2008). In total, 619 mtDNA cox1 sequences including eight reference sequences were imported into MEGA v.7.0.21 (Kumar et al., 2016) for a comprehensive alignment and manual editing. Four genetic diversity indices, the number of haplotype (Nh), the number of polymorphic sites (S), haplotype diversity (Hd) and nucleotide diversity (π), and neutrality tests (Tajima’s D and Fu’s Fs) were estimated in Arlequin v.3.5.1.3 (Excoffier et al., 2005). The distribution of characteristic nucleotide mutations in each cox1 haplotype was calculated in MEGA.
Bayesian inference (BI) and maximum likelihood (ML) trees were reconstructed using BEAST v.1.8.0 (Drummond et al., 2012) and PhyML v.3.1 (Guindon and Gascuel, 2003), respectively. The best-fit nucleotide substitution model of mtDNA cox1 (GTR + G, G = 0.1490) was determined based on the Akaike Information Criterion in jModelTest v.0.1.1 (Posada, 2008). For BI analysis, a Yule process tree prior with the UPGMA tree was constructed in BEAST. An uncorrected lognormal relaxed clock with default estimated rate of 1.0 was set. BEAST was run with 5 × 107 Markov Chain Monte Carlo (MCMC), sampling every 1,000 steps under the GTR + G model. The MCMC output was the tree data set and checked for convergence [effective sample sizes (ESS) >200] in Tracer v.1.7.1 (Rambaut et al., 2018). To obtain the condensed tree, the MCMC output was summarized in TreeAnnotator v.1.7.5 (Drummond et al., 2012), the first 10% was discarded as “burn-in”. The ML tree was constructed based on nearest neighbor interchange (NNI). Phylogenetic support for each node was assessed by bootstrapping with heuristic analysis of 1,000 replicates. For both the BI and ML analysis, the other two members of the genus Agarophyton were chosen as outgroup (Gurgel et al., 2018), i.e., A. tenuistipitatum (C. F. Chang et B.- M. Xia) Gurgel, J. N. Norris et Fredericq (GenBank: EF434924), and A. chilense (C. J. Bird, McLachlan et E. C. Oliveira) Gurgel, J. N.Norris et Fredericq (GenBank: EF434915).
Genetic clustering analysis was inferred based on an admixture model using rmaverick v.1.0.5 (Verity, 2019) in the R package. ClustalX v.1.83 (Larkin et al., 2007), xmfaconvertor, and xmfa2struct were used to convert cox1 sequence files to the rmaverick input .str files. The values of K were set from 1 to 10, and 10 replicates were run for each K using 104 burn-in iterations and 2,000 sampling iterations.
Genealogic relationships of mtDNA cox1 haplotypes were inferred using the median-joining algorithm implemented in Network v.5.0.1.1 (Bandelt et al., 1999). Intraspecific relationships of cox1 haplotypes were also assessed by minimum spanning network (MSN) in Arlequin.
Analyses of molecular variance (AMOVA) were conducted in Arlequin to detect spatial partitioning of genetic variance among sampling sites and regions. We used four criteria for population grouping: (i) Country, populations were divided into three groups: Japan, Korea, and China, and the PRI population was not included due to the single locality from Russia; (ii) Longitude, populations were divided into two groups (West and East) by the central line of the Tsushima Strait (130°E); (iii) Biogeographic basins (Wang, 1999), populations were divided into four groups: Japan–Pacific coast (JPC), Sea of Japan (SOJ), East China Sea including Yellow–Bohai Sea (ECS), and South China Sea (SCS); (iv) Marine provinces (Longhurst, 2007), populations were divided into two groups: the Kuroshio Current province (KURO) and China Sea Coastal province (CHIN). The significance (p = 0.05) of the fixation indices was calculated based on 104 permutations. Pairwise genetic differentiations (Fst) and Nei’s genetic distance among lineages were calculated using Arlequin and POPGENE v.1.32 (Yeh et al., 1999). In addition, pairwise genetic differentiations (Fst) and gene flow (Nm) among populations were also calculated in Arlequin. Based on the estimated substitution rate of mtDNA cox1 in the class Florideophyceae (Le Gall and Saunders, 2010; Bringloe and Saunders, 2019), we set a range of 0.7–1.0%/Ma to broadly calculate divergence times between cox1 lineages in A. vermiculophyllum in the native northwest Pacific.
Historical Phylogeography
S-DIVA (Statistical Dispersal-Vicariance Analysis) implemented in RASP v.3.1 (Yu et al., 2015) was used to infer the phylogeographic history of A. vermiculophyllum through statistically evaluating the alternative ancestral ranges at each node in a phylogenetic tree. The tree data set and condensed tree produced by BEAST and TreeAnnotator were loaded in RASP with the outgroup removed and then saved as “trees file.” Four main biogeographic regions (Wang, 1999) were defined based on geographic distribution of cox1 haplotypes: (A) Japan–Pacific coast, (B) Sea of Japan, (C) East China Sea including Yellow–Bohai Sea, and (D) South China Sea. The “Distribution file” was produced based on the distribution of cox1 haplotypes through all defined biogeographic regions. “Trees file” and “distribution file” were loaded in RASP to perform the S-DIVA analysis. The topology and Most Likely States (MLS) functions were used to infer the probability of ancestor distribution at each node of the phylogeny.
Results
Genetic Diversity
The length of the cox1 alignment was 1,214 nucleotides (nt) and consisted of 611 sequences. A total of 45 polymorphic sites (3.71%) were detected (Supplementary Table S2), including 28 singletons (2.31%) and 17 parsimony informative sites (1.40%). Thirty-three haplotypes were defined (GenBank accession numbers: MN817547-MN817579), of which 26 were restricted to a single population and seven were found in two or more populations (Supplementary Table S1). The two dominant haplotypes H1 (90 specimens, 15%) and H3 (339 specimens, 55%) were found in 11 (23%) and 28 (58%) of the populations, respectively (Supplementary Table S1). There were 28 populations harboring a single haplotype and six populations had more than two haplotypes (maximum four) (Supplementary Table S1). Haplotype diversity (total Hd = 0.6632) ranged from 0.0000 to 0.7905, and nucleotide diversity (total π = 0.2473 × 10–2) varied from 0.0000 to 0.3766 × 10–2 (Supplementary Table S1). The east of Jeju Island population (JEE, 31 in Figure 1), Korea had the highest haplotype diversity (Hd = 0.7905), and the south of Jeju Island population (JES, 32 in Figure 1), Korea had the highest nucleotide diversity (π = 0.3766 × 10–2) (Supplementary Table S1). At a regional scale, populations from Japan showed much higher genetic diversity (133 specimens, Hd = 0.7236, π = 0.3275 × 10–2) than those from Korea (213 specimens, Hd = 0.5282, π = 0.1361 × 10–2), China (262 specimens, Hd = 0.5265, π = 0.2100 × 10–2) and Russia (three specimens, Hd = 0.0000, π = 0.0000) (Supplementary Table S1). The value of neutrality test of the A. vermiculophyllum populations in the northwest Pacific is negative (Tajima’s D = −1.4414, p < 0.05; Fu’s Fs = -10.1928, p < 0.05) (Supplementary Table S1).
Phylogenetic and Phylogeographic Structure
Phylogenetic analyses of the 33 A. vermiculophyllum cox1 haplotypes with two outgroup taxa resulted in five strongly supported A. vermiculophyllum clades (lineages 1–5 in Figure 2) in both the BI and ML. BI and ML trees presented highly concordant phylogenetic structure. Haplotype 10 differed from haplotype 32/33 by eight bases, so haplotype 10 was regarded as an independent genetic lineage (Figure 3). Another four diverged lineages were supported with high values for main nodes (BI: posterior probabilities = 0.71∼0.99, ML: bootstrap values = 78∼96) except for lineage 5 in the ML (Figure 2). The haplotype network (Figure 3) revealed the same five distinct lineages in A. vermiculophyllum which were basically in accordance with phylogenetic trees. Both lineages 2 and 5 showed significant star-like structure, and the neutrality test of them was negative (Figure 3 and Table 1). In addition, there were 3–11 substitutions detected among the five lineages (e.g., lineage 2 differed by three bases from lineage 5, and lineage 1 differed by 11 bases from lineage 4) (Figure 3). Positions 227, 329, 377, 398, 443, 485, 722, 767, 860, 945, 989, 1040, 1106, and 1154 of the cox1 alignment showed nucleotide substitutions unique to certain lineages (Supplementary Table S2). For example, at the 398th site, nucleotide A corresponded to lineage 5, while nucleotide G corresponded to the other four lineages. Similar distribution patterns of nucleotides were found at other sites. In addition, the optimal K value (K = 5) was detected by genetic clustering analysis (Supplementary Figure S1), which was consistent with phylogenetic results and biogeographical distribution of haplotypes (Figures 1, 2).
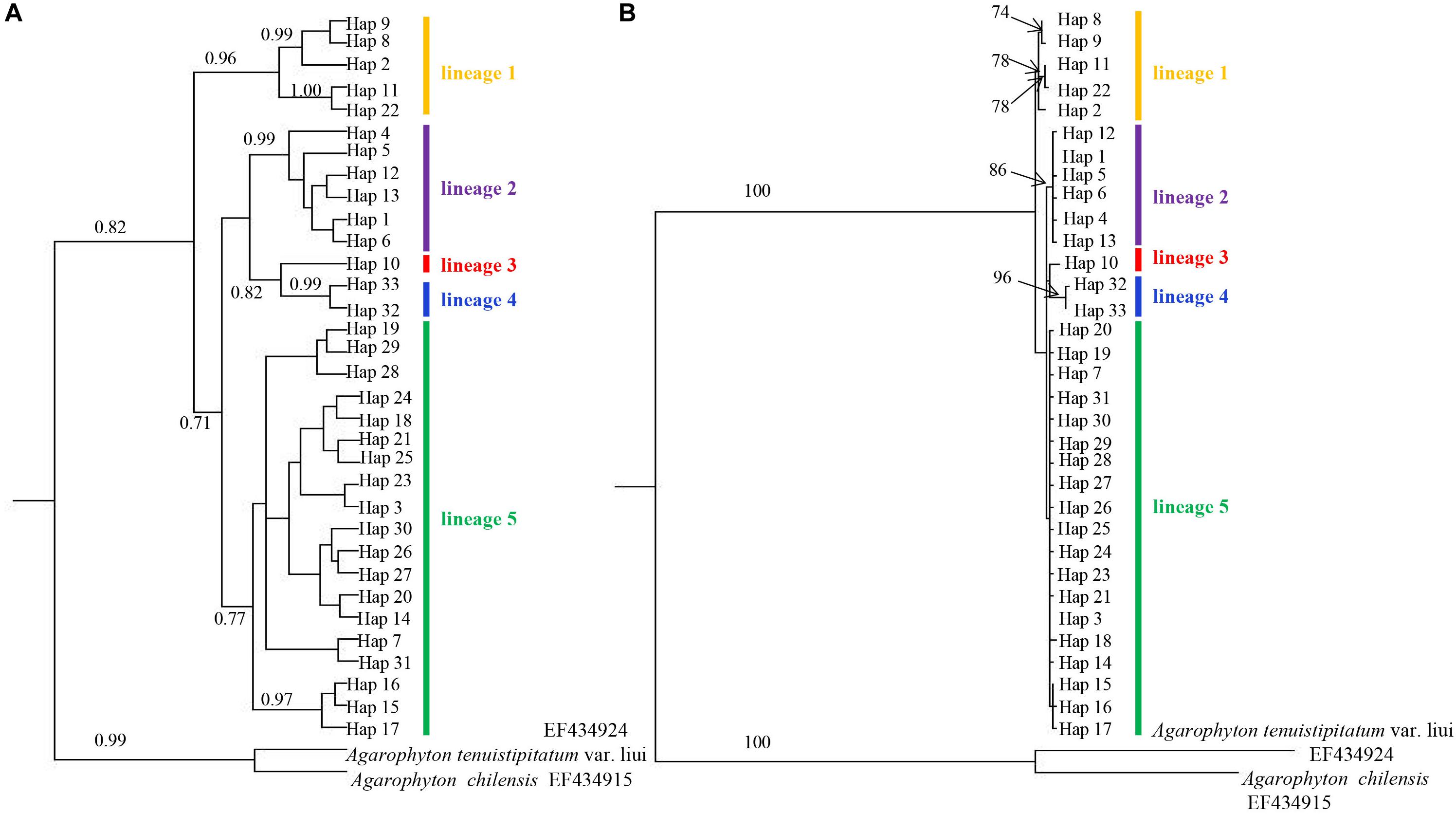
Figure 2. Bayesian inference (BI) tree (A) and maximum likelihood tree (ML) (B) of Agarophyton vermiculophyllum in the northwest Pacific inferred from mitochondrial DNA (mtDNA) cox1. Numbers near branches are posterior probabilities (BI) and bootstrap percentages (ML). Values <0.5 or <50 not shown.
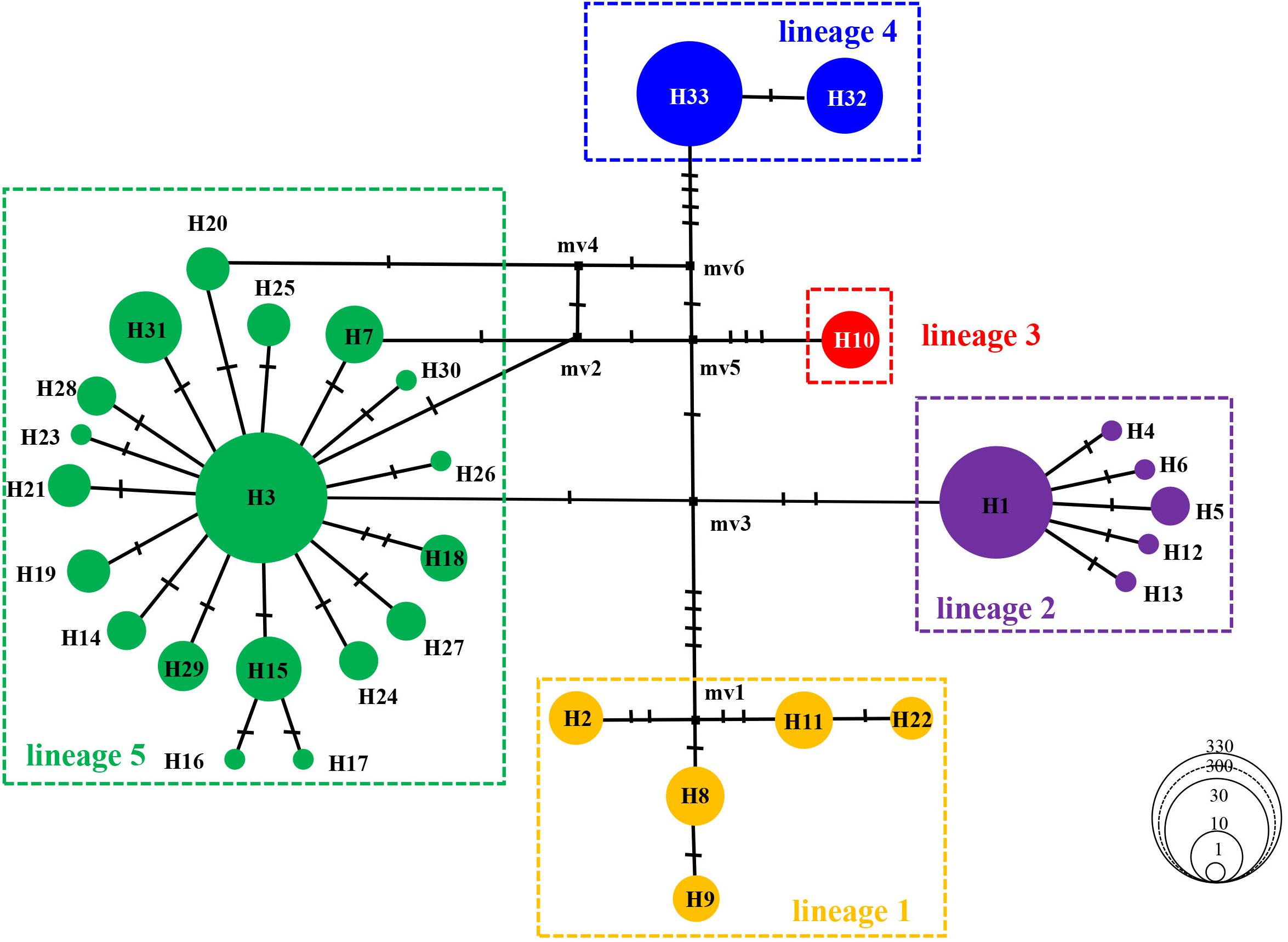
Figure 3. Median joining network of mitochondrial DNA (mtDNA) cox1 haplotypes in the northwest Pacific. The size of each circle is proportional to the frequency of haplotypes. Color coding of lineages as in Figure 1. The small black squares represent extinct or unsampled haplotypes.
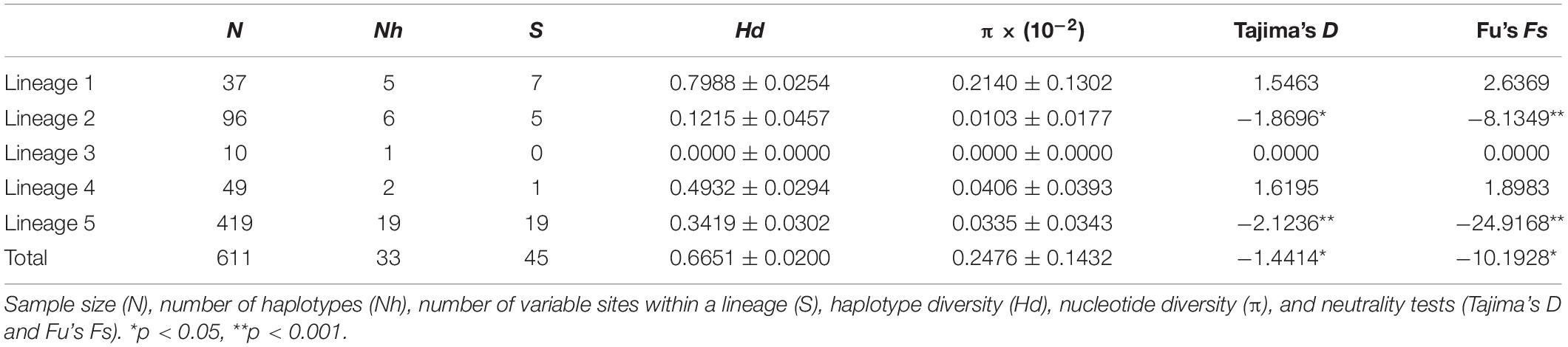
Table 1. Molecular diversity for four lineages of Agarophyton vermiculophyllum in the northwest Pacific.
Mapping the distribution of the five lineages showed a basic biogeographic pattern (Figure 1): Lineage 1 consisting of five haplotypes was mainly distributed in the south of Japan–Pacific coast and the Sea of Japan. Lineage 2 consisting of six haplotypes was dominated by H1 and mainly distributed in the north of Japan–Pacific coast and the Sea of Japan. Lineage 3 was only distributed in one population in the south of Japan–Pacific. Lineage 4 consisting of two haplotypes was distributed in three southern Chinese populations. The most widely distributed Lineage 5 consisting of 19 haplotypes was dominated by H3 and mainly distributed in the East China Sea, the Yellow–Bohai Sea, and the Sea of Japan. Most populations (42/48) only harbored one lineage (Figure 1). Lineage 1 exhibited higher levels of genetic diversity (Hd = 0.7988, π = 0.2140 × 10–2) than the others (Table 1).
AMOVA revealed that most genetic variations occurred among populations within groups (67–75%) except the grouping criterion of biogeographic basins (61% of the variation occurred among groups), and all measures were statistically significant (p < 0.001) (Table 2). Pairwise Fst estimates exhibited high levels of genetic differentiation among lineages (Fst > 0.8000, p < 0.001), and Nei’s genetic distance varied between 0.0726 and 0.2716 (Table 3). Population-level Fst values also revealed high levels of genetic differentiation and limited connection among populations (70.8% of Fst > 0.25, p < 0.05; 11.3% of Nm > 1) (Supplementary Table S3). In particular, the geographically proximate populations (13, 14, and 15) in the south of Japan–Pacific were highly differentiated (Fst = 1.00, p < 0.001; Nm = 0.0000). The populations 11, 12, 14, and 15 from Japan, the populations 22 and 29 from Korea, and the populations SS and ZP from China were highly differentiated from the others (Fst = 0.4209–1.0000, p < 0.05) (Supplementary Table S3). Sequence divergence among five lineages varied from 0.41 to 1.03%, and the estimated divergence time between them dating to 0.21–0.74 Ma.
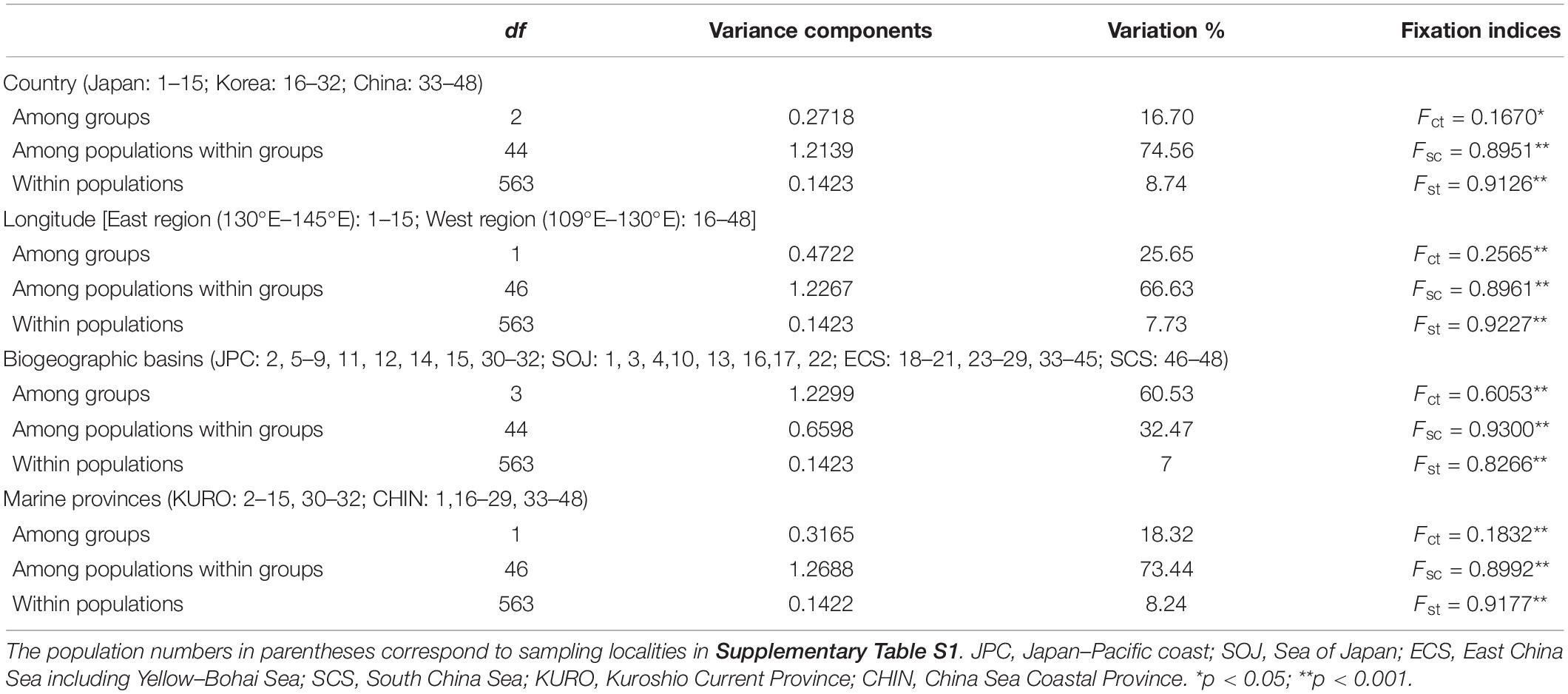
Table 2. Analysis of molecular variance (AMOVA) to partition genetic variance under different grouping criteria.
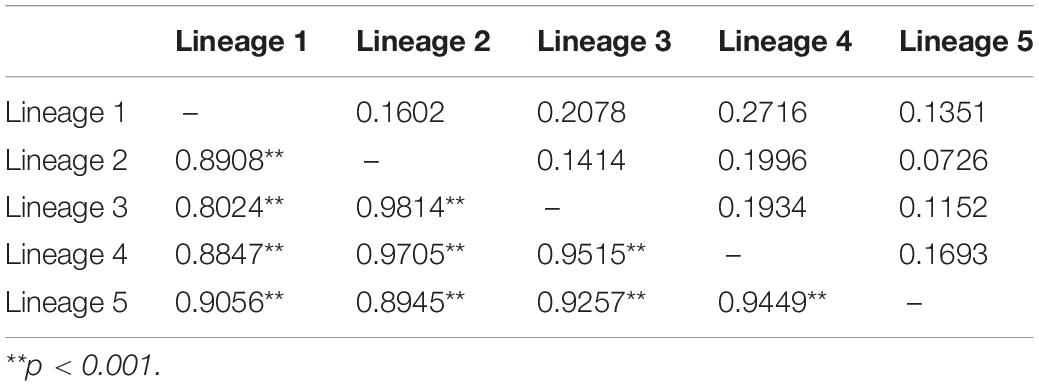
Table 3. Genetic differentiation (Fst, below diagonal) and Nei’s genetic distance (above diagonal) among five genetic lineages of Agarophyton vermiculophyllum in the native northwest Pacific.
Historical Phylogeography
S-DIVA analysis of mtDNA cox1 haplotypes suggested that current A. vermiculophyllum populations originated from a wide ancestral region encompassing the Japan–Pacific coast, East and South China Sea (node 65, ACD in Figure 4), associated with multiple dispersal and vicariance events (Figure 4). Lineage 1 originated in the Japan–Pacific coast and the Sea of Japan (node37, AB); lineage 2 originated along the Japan–Pacific coast (node 42, A); Lineages 3 and 4 were isolated due to vicariance events from the common ancestor distributed along Japan–Pacific coast and the South China Sea (node 44, AD); lineage 5 originated from the East China Sea (node 63, C). A total of 12 dispersal events and eight vicariance events were detected (Figure 4).
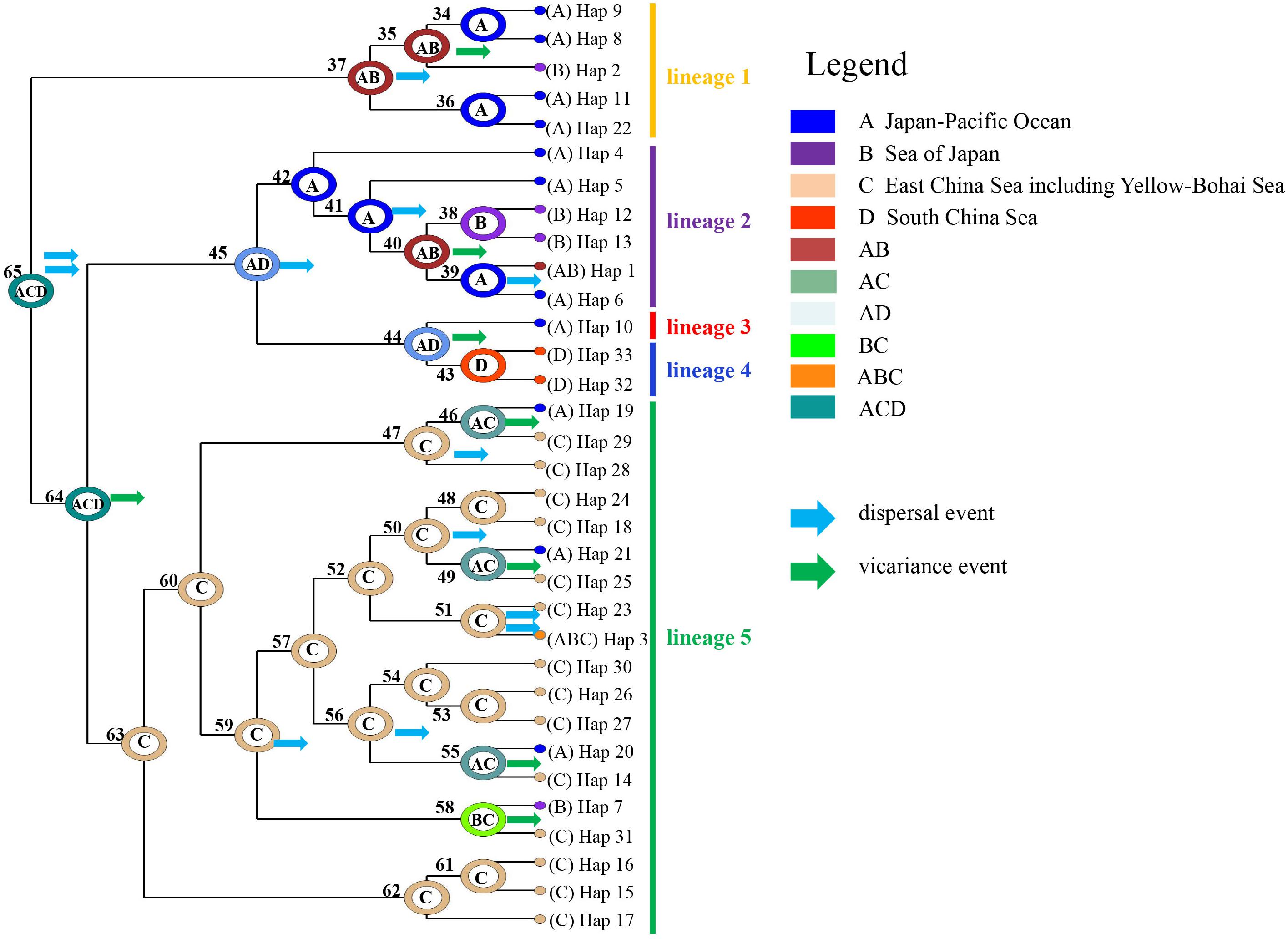
Figure 4. Ancestral phylogeographic reconstruction of Agarophyton vermiculophyllum based on mitochondrial DNA (mtDNA) cox1. Colored circles on each node indicate the likelihood of occurrence of each ancestral haplotype at an inferred ancestral biogeographic region. Blue and green arrows indicate potential dispersal and vicariance events, respectively.
Discussion
Genetic Diversity of A. vermiculophyllum in the Northwest Pacific
In this study, the detected overall haplotype (Hd = 0.6632) and nucleotide diversity (π = 0.3766 × 10–2) are similar to previous results reported in A. vermiculophyllum in the northwest Pacific by Kim et al. (2010) (Hd = 0.731, π = 0.329 × 10–2) and Liu et al. (2016) (Hd = 0.589, π = 0.318 × 10–2), respectively. Such high haplotype diversity and low nucleotide diversity patterns suggest that A. vermiculophyllum in the native northwest Pacific may have experienced rapid population growth over a short period because sudden population expansion cannot support this species to have sufficient time to accumulate enough nucleotide mutations (Grant and Bowen, 1998). The haplotype network of A. vermiuclophyllum (e.g., lineages 2 and 5 in Figure 3) hints to recent population expansion as well because new haplotypes derived from ancestral haplotypes (H3 and H1) usually formed a star-like topology during population expansion (Posada and Crandall, 2001). This conjecture of population expansion is also concordant with the neutrality test (Tajima’s D = −1.4414, p < 0.05; Fu’s Fs = −10.1928, p < 0.05; Supplementary Table S1).
The high haplotype diversity of A. vermivulophyllum in the northwest Pacific may be ascribed to high environmental heterogeneity (Nei, 1987). Discontinuous habitat (e.g., Fucus vesiculosus, Zardi et al., 2013) and variation of salinity (e.g., Cladophora, Hayakawa et al., 2012) have been reported to impact genetic diversity and differentiation of intertidal seaweeds. A. vermiculophyllum is distributed in both estuaries and marine coast in the northwest Pacific. Regional habitat discontinuity and heterogeneity (e.g., sandy beach, rocky reef, gravel and mud beach, Supplementary Table S1) may have caused population-level genetic differentiation. In addition, A. vermiculophyllum is well adapted to salinity variation of 5–60‰ (Yokoya et al., 1999). Such salinity gradients may have imposed strong selection on allele frequencies (e.g., Furcellaria lumbricalis, Kostamo et al., 2012) and apomictic reproduction and phenotypic variation (e.g., Ulva prolifera, Ogawa et al., 2014), ultimately leading to genotypic differentiation.
We detected five genetic lineages in A. vermiculophyllum in the native northwest Pacific. Three of them (lineages 1, 2, and 5) showed similar distribution ranges as reported by Krueger-Hadfield et al. (2017). However, the ∼35°N segregation based on nucleotides at sites 945 and 1040 is not as clear as in Krueger-Hadfield et al. (2017). Although haplotypes with a T at these positions all occur north of ∼35°N (e.g., lineage 2), haplotypes with a C were found on both sides of this latitudinal divide. Twelve additional unique nucleotide substitutions also correspond to lineage differentiation, respectively (Supplementary Table S2). Our data suggest that a gradual nucleotide change in lineage distribution happened at 35°N, and that nucleotide substitutions at a few sites are not sufficient to delineate genetic lineages and their geographic extent as described by Krueger-Hadfield et al. (2017).
Phylogeographic History of A. vermiculophyllum in the Northwest Pacific
Sea levels in the northwest Pacific dropped by 120–140 m during the glacial maxima in the late Quaternary (e.g., the Last Glacial Maximum, Lambeck et al., 2002), leading to the isolation of the Sea of Japan and a shift of the coastline of the South China Sea and the East China Sea (and the presumed disappearance of the Yellow–Bohai Sea) (Wang, 1999). This coastline configuration imposed significant impacts on distribution range and genetic diversity of marine species (Cheang et al., 2010; Ni et al., 2014; Hu et al., 2015). Our S-DIVA analysis illustrated that the current distribution range of A. vermiculophyllum stem from the ancestral relics that persisted historical glaciation periods and experienced multiple dispersal events (Figure 4). It should be noted that the sister relationships between the five lineages have not been resolved phylogenetically. Sequencing multiple genes or complete mitochondria of specimens from each lineage may help to address this issue. Phylogeographic studies have proposed Japan–Pacific coast (Hu et al., 2011, 2015, 2017), Okinawa trough (Wang, 1999; Hu et al., 2015), and northern South China Sea (Hu et al., 2018a) as glacial refugia in the northwest Pacific. The locations of these glacial refugia broadly coincide with the distribution of five lineages in A. vermiculophyllum in the native northwest Pacific. For example, lineage 2 originated from the north of Japan–Pacific coast and spread southward into the Sea of Japan driven by the Oyashio Current and the Tsushima Warm Current. Similarly, lineages 4 and 5 originated from the South China Sea and the Okinawa trough, respectively, and colonized the South China Sea coast (lineage 4) and the Yellow–Bohai Sea (lineage 5) driven by the South China Sea Warm Current, the Yellow Sea Warm Current and the China Coastal Current (Figures 1, 4). Mitochondrial lineages 2/3/4, despite their geographic distance in the northwest Pacific, exhibit close genetic relationships, which likely resulted from the persistence of the most recent common ancestor in isolated refugia during the ice age and subsequently multiple dispersal and vicariance events (node 45 in Figure 4). Therefore, ancestral persistence in multiple scattered glacial refugia during the late Quaternary ice ages is likely the major phylogeographic process contributing to extant lineage differentiation and distribution.
Geographically, we found two distinct genetic breaks (Figure 1). The first break occurred in the south-central coast of China, which exactly corresponds to the boundary between the distribution range of lineages 4 and 5 (Figure 1). As illustrated by Hu et al. (2018b), the genetic break may have resulted from the physical barrier of the Yangtze river diluted water. The diluted water is characterized by profoundly decreased seawater salinity and pH, which can affect the trans-regional dispersal of intertidal marine organisms (Wang et al., 2003) and hence population-level genetic connectivity. It has been reported that the growth of A. vermiculophyllum is inhibited by salinity fluctuation, and the greater the salinity fluctuation, the more obvious the inhibition (Nejrupa and Pedersena, 2012). In northern China, spore release of A. vermiculophyllum occurs mainly in summer, especially from June to July (Li and Li, 1986). The Yangtze river diluted water, which is influenced by southwestward monsoon in summer, deflected to the northeast paralleling with the Taiwan Warm Current, consequently caused the deflection of southward China Coastal Current and prevented the northern populations spreading to the south (Dong et al., 2012). Alternatively, the genetic break may have occurred near the Taiwan Strait as the reported genetic split between the brown algae Colpomenia peregrina and Colpomenia sinuosa (Song et al., 2019). Such a break may be the result of the isolation of the Taiwan Strait land bridge caused by the sea level drop during glaciation periods (Ota, 1998), leading to the isolation of species/populations on both sides of the land bridge which have also long been defined as marine ecoregions 52 and 113 (Spalding et al., 2007). To test this hypothesis, we need to sample more populations from the region between populations SS (45 in Figure 1) and ZP (46 in Figure 1). It should be noted that lineage 4 has close relatives (lineages 2 and 3) that exist further north than lineage 5. Therefore, gene flow probably had occurred in the past and the Yangtze river was not the primary vicariance event that separated lineages 4 and 5.
Another genetic break was detected between lineages 1 and 2 in the central Japan–Pacific coast (Figure 1) which corresponds to the boundary of the marine ecoregions 48 and 51 (Spalding et al., 2007), suggesting a long-term genetic isolation despite their geographic proximity. This differentiation pattern is similar to results found in Sargassum horneri (Turner) C. Agardh and Sargassum fusiforme (Harv.) Setchel in the northwest Pacific (Hu et al., 2011, 2017). The central Japan–Pacific coast is characterized by the convergence of the cold Oyashio Current and the warm Kuroshio Current (Figure 1). The interplaying current systems, especially the coastal upwelling, can create distinct physical barrier for genetic exchange, particularly for the passive drifting spores and fragments of A. vermiculophyllum (Yu and Kim, 2018). However, the current and upwelling regime may not have served as the primary vicariance separating lineages 1 and 2 because they are only distantly related. Lineage 2 is specifically restricted to the north of the Japanese Archipelago and east coast Korean Peninsula, and the underlying phylogeographic processes probably correlates with contemporary ocean currents, but more evidence is needed with wide-range sampling.
Conservation Insights for A. vermiculophyllum
Agarophyton vermiculophyllum is an important economic and ecological species that deserves to be protected in the situation of increasing biodiversity destruction in the northwest Pacific (Armisen, 1995; Abreu et al., 2011; Tanaka et al., 2012). Our mtDNA-based phylogeographical study provides a basis for how to conserve A. vermiculophyllum populations in the wild. We detected the highest population genetic diversity and number of genetic lineages in the Japanese Archipelago (Figure 1 and Supplementary Table S1). In particular, this area is rich in unique and endemic haplotypes, and the Fst values between adjacent populations are much higher than those between the distant Korean and Japanese populations in the south of Japan–Pacific coast, reflecting a potential habitat fragmentation (Rossiter et al., 2007; Hu et al., 2017). In addition, gene flow between populations in this area is extremely low (Nm = 0.0000), indicating very low population connectivity. In order to prevent the loss of local genetic diversity caused by a genetic drift, the Japanese Archipelago can be considered as a special conservation priority. In addition, the five genetic lineages with significant genetic differentiation were isolated since the late Quaternary ice ages with significant genetic differentiation. Particularly, lineage 5 has the highest genetic diversity and is reciprocally monophyletic with the other four lineages; it thus should be taken as an independent Evolutionary significant unit (ESU) to be preserved to maintain commercial access and ecological sustainability (Moritz, 1994; Hu et al., 2017).
Data Availability Statement
The datasets generated for this study can be found in NCBI GenBank, NCBI Accession No.’s MN817547–MN817579.
Author Contributions
Z-MH conceived the project. K-LZ conducted the molecular experiments, data collection, analysis, and writing. X-HS assisted in data analysis and writing. H-GC, SS, and FW provided the partial samples and assisted data interpretation. SD, D-LD, and Z-MH commented the results and modified the manuscript to final version.
Funding
This study was supported by the National Natural Science Foundation of China (41761144057 and 31971395), the Sino-German Centre for Research Promotion (GZ1375), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB42000000), and the Thailand Research Fund (RDG6130002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Zhong-Min Sun and Mr. Ruo-Yu Liu for providing assistance in field collections.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00366/full#supplementary-material
References
Abreu, M. H., Pereira, R., Yaris, C., Buschmann, A. H., and Sousa-Pinto, I. (2011). IMTA with Gracilaria vermiculophylla: productivity and nutrient removal performance of the seaweed in a land-based pilot scale system. Aquaculture 312, 0–87. doi: 10.1016/j.aquaculture.2010.12.036
Armisen, R. (1995). World-wide use and importance of Gracilaria. J. Appl. Phycol. 7, 231–243. doi: 10.1007/BF00003998
Bandelt, H. J., Forster, P., and Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1002/ece3.2381
Bringloe, T. T., and Saunders, G. W. (2019). Trans-arctic speciation of Florideophyceae (Rhodophyta) since the opening of the Bering Strait, with consideration of the “species pump” hypothesis. J. Biogeogr. 46, 694–705. doi: 10.1111/jbi.13504
Cheang, C. C., Chu, K. H., and Ang, P. O. (2010). Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Mol. Ecol. 19, 2933–2948. doi: 10.1111/j.1365-294X.2010.04685.x
Davoult, D., Surget, G., Stiger-Pouvreau, V., Noisette, F., Riera, P., Stagnol, D., et al. (2017). Multiple effects of a Gracilaria vermiculophylla invasion on estuarine mudflat functioning and diversity. Mar. Environ. Res. 131, 227–235. doi: 10.1016/j.marenvres.2017.09.020
Dong, Y. W., Wang, H. S., Han, G. D., Ke, C. H., Zhan, X., Nakano, T., et al. (2012). The impact of Yangtze River discharge, ocean currents and historical events on the biogeographic pattern of Cellana toreuma along the China coast. PLoS One 7:e36178. doi: 10.1371/journal.pone.0036178
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Excoffier, L., Laval, G., and Schneider, S. (2005). Arlequin (ver. 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. 1, 47–50. doi: 10.1177/117693430500100003
Geraldino, P. J. L., Yang, E. C., and Boo, S. M. (2006). Morphology and molecular phylogeny of Hypnea flexicaulis (Gigartinales, Rhodophyta) from Korea. Algae 21, 417–423. doi: 10.4490/ALGAE.2006.21.4.417
Grant, W. S., and Bowen, B. W. (1998). Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J. Hered. 89, 415–426. doi: 10.1093/jhered/89.5.415
Guindon, S., and Gascuel, O. (2003). A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Gulbransen, D. J., Mcglathery, K. J., Marklund, M., Norris, J. N., and Gurgel, C. F. D. (2012). Gracilaria vermiculophylla (Rhodophyta, Gracilariales) in the Virginia coastal bays, USA: cox1 analysis reveals high genetic richness of an introduced macroalga. J. Phycol. 48, 1278–1283. doi: 10.1111/j.1529-8817.2012.01218.x
Gurgel, C. F. D., Norris, J. N., Schmidt, W. E., Le, H. N., and Fredericq, S. (2018). Systematics of the Gracilariales (Rhodophyta) including new subfamilies, tribes, subgenera, and two new genera, Agarophyton gen. nov. and Crassa gen. nov. Phytotaxa 374, 1–23. doi: 10.11646/phytotaxa.374.1.1
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 41, 95–98. doi: 10.1371/journal.pone.0079975
Hayakawa, Y. I., Ogawa, T., Yoshikawa, S., Ohki, K., and Kamiya, M. (2012). Genetic and ecophysiological diversity of Cladophora (Cladophorales. Ulvophyceae) in various salinity regimes. Phycol. Res. 60, 86–97. doi: 10.1111/j.1440-1835.2012.00641.x
Hu, Z. M., Kantachumpoo, A., Liu, R. Y., Sun, Z. M., Yao, J. T., Komatsu, T., et al. (2018a). A late Pleistocene marine glacial refugium in the south-west of Hainan Island, China: phylogeographical insights from the brown alga Sargassum polycystum. J. Biogeogr. 45, 355–366. doi: 10.1111/jbi.13130
Hu, Z. M., Liu, R. Y., Zhang, J., Duan, D. L., Wang, G. G., and Li, W. H. (2018b). A unique genetic lineage at the southern coast of China in the agar-producing Gracilaria vermiculophylla (Gracilariales. Florideophyceae). Algae 33, 269–278. doi: 10.4490/algae.2018.33.8.30
Hu, Z. M., Li, J. J., Sun, Z. M., Gao, X., Yao, J. T., Choi, H. G., et al. (2017). Hidden diversity and phylogeographic history provide conservation insights for the edible seaweed Sargassum fusiforme in the northwest Pacific. Evol. Appl. 10, 366–378. doi: 10.1111/eva.12455
Hu, Z. M., Li, J. J., Sun, Z. M., Oak, J. H., Zhang, J., Fresia, P., et al. (2015). Phylogeographic structure and deep lineage diversication of the red alga Chondrus ocellatus Holmes in the northwest Pacific. Mol. Ecol. 24, 5020–5033. doi: 10.1111/mec.13367
Hu, Z. M., and Lopez-Bautista, J. (2014). Adaptation mechanisms and ecological consequences of seaweed invasions: a review case of agarophyte Gracilaria vermiculophylla. Biol. Invas. 16, 967–976. doi: 10.1007/s10530-013-0558-0
Hu, Z. M., Uwai, S., Yu, S. H., Komatsu, T., Ajisaka, T., and Duan, D. L. (2011). Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol. Ecol. 20, 3894–3909. doi: 10.1111/j.1365-294X.2011.05220.x
Kim, S. Y., Weinberger, F., and Boo, S. M. (2010). Genetic data hint at a common donor region for invasive Atlantic and Pacific populations of Gracilaria vermiculophylla (Gracilariales. Rhodophyta). J. Phycol. 46, 1346–1349. doi: 10.1111/j.1529-8817.2010.00905.x
Kostamo, K., Korpelainen, H., and Olsson, S. (2012). Comparative study on the population genetics of the red algae Furcellaria lumbricalis occupying different salinity conditions. Mar. Biol. 159, 561–571. doi: 10.1007/s00227-011-1835-z
Krueger-Hadfield, S. A., Kollars, N. M., Strand, A. E., Byers, J. E., Shainker, S. J., Terada, R., et al. (2017). Genetic identification of source and likely vector of a widespread marine invader. Ecol. Evol. 7, 4432–4447. doi: 10.1002/ece3.3001
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lambeck, K., Esat, T. M., and Potter, E. K. (2002). Links between climate and sea levels for the past three million years. Nature 419, 199–206. doi: 10.1038/nature01089
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Le Gall, L., and Saunders, G. W. (2010). DNA barcoding is a powerful tool to uncover algal diversity: a case study of the phyllophoraceae (Gigartinales, Rhodophyta) in the Canadian flora. J. Phycol. 46, 374–389. doi: 10.1111/j.1529-8817.2010.00807.x
Lee, K. M., Yang, E. C., Coyer, J. A., Zuccarello, G. C., Wang, W. L., Choi, C. G., et al. (2012). Phylogeography of the seaweed Ishige okamurae (Phaeophyceae): evidence for glacial refugia in the northwest Pacific region. Mar. Biol. 159, 1021–1028. doi: 10.1007/s00227-012-1882-0
Li, J. J., Hu, Z. M., Gao, X., Sun, Z. M., Choi, H. G., Duan, D. L., et al. (2017). Oceanic currents drove population genetic connectivity of the brown alga Sargassum thunbergii in the north-west Pacific. J. Biogeogr. 44, 230–242. doi: 10.1111/jbi.12856
Li, X. L., and Li, M. Z. (1986). Growth comparison between sporophyte and female gametophyte of Gracilaria verrucosa. Trans. Oceanol. Limnol. 3, 63–68. doi: 10.13984/j.cnki.cn37-1141
Liu, J., Zhan, D., Zhang, R., Zeng, L., Wang, G. G., and Hu, Z. M. (2018). AFLP analysis revealed a north to south genetic break in the brown alga Sargassum thunbergii along the coast of China. J. Appl. Phycol. 30, 1–9. doi: 10.1007/s10811-018-1417-2
Liu, R. Y., Sun, Z. M., Yao, J. T., Hu, Z. M., and Duan, D. L. (2016). Genetic diversity of the habitat-forming red alga Gracilaria vermiculophylla along Chinese coasts. Biodiv. Sci. 24, 781–790. doi: 10.17520/biods.2016038
Moritz, C. (1994). Defining ‘Evolutionary Significant Units’ for conservation. Trends Ecol. Evol. 9, 373–375. doi: 10.1016/0169-5347(94)90057-4
Nejrupa, L. B., and Pedersena, M. F. (2012). The effect of temporal variability in salinity on the invasive red alga Gracilaria vermiculophylla. Eur. J. Phycol. 47, 254–263. doi: 10.1080/09670262.2012.702225
Ni, G., Li, Q., Kong, L. F., and Yu, H. (2014). Comparative phylogeography in marginal seas of the northwestern Pacific. Mol. Ecol. 23, 534–548. doi: 10.1111/mec.12620
Oba, T., and Irino, T. (2012). Sea level at the Last Glacial Maximum, constrained by oxygen isotopic curves of planktonic foraminifera in the Japan Sea. J. Q. Sci. 27, 941–947. doi: 10.1002/jqs.2585
Ogawa, T., Ohki, K., and Kamiya, M. (2014). High heterozygosity and phenotypic variation of zoids in apomictic Ulva prolifera (Ulvophyceae) from brackish environments. Aquat. Bot. 120, 185–192. doi: 10.1016/j.aquabot.2014.05.015
Ota, H. (1998). Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their paleogeographical implications. Popul. Ecol. 40, 189–204. doi: 10.1007/bf02763404
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Posada, D., and Crandall, K. A. (2001). Intraspecific gene genealogies: trees grafting into networks. Trends Ecol. Evol. 16, 37–45. doi: 10.1016/s0169-5347(00)02026-7
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rossiter, S. J., Benda, P., Dietz, C., Zhang, S. Y., and Jones, G. (2007). Rangewide phylogeography in the greater horseshoe bat inferred from microsatellites: implications for population history, taxonomy and conservation. Mol. Ecol. 16, 4699–4714. doi: 10.1111/j.1365-294X.2007.03546.x
Skriptsova, A. V., and Choi, H. G. (2009). Taxonomic revision of Gracilaria “verrucosa” from the Russian Far East based on morphological and molecular data. Bot. Mar. 52, 331–340. doi: 10.1515/BOT.2009.008
Song, X. H., Hu, Z. M., Sun, Z. M., Draisma, S. G. A., Fresia, P., and Duan, D. L. (2019). Species diversity and distribution of the genus Colpomenia (Scytosiphonaceae. Phaeophyceae) along the coast of China. Algae 34, 217–228. doi: 10.4490/algae.2019.34.7.22
Sotka, E. E., Baumgardner, A. W., Bippus, P. M., Destombe, C., Duermit, E. A., Endo, H., et al. (2018). Combining niche-shift and population genetic analyses predicts rapid phenotypic evolution during invasion. Evol. Appl. 11, 781–793. doi: 10.1111/eva.12592
Spalding, M. D., Fox, H. E., Allen, G. R., Davidson, N., and Robertson, J. (2007). Marine ecoregions of the world: a bioregionalization of coastal and shelf areas. Bioscience 57, 573–583. doi: 10.1641/B570707
Tanaka, K., Taino, S., Haraguchi, H., Prendergast, G., and Kiraoka, M. (2012). Warming off southwestern Japan linked to distributional shifts of subtidal canopy-forming seaweeds. Ecol. Evol. 2, 2854–2865. doi: 10.1002/ece3.391
Verity, B. (2019). Rmaverick. Available at: https://bobverity.github.io/rmaverick/articles/basic_tutorial.html (accessed March 10, 2020).
Wang, B. D., Wang, X. L., and Zhan, R. (2003). Nutrient conditions in the Yellow Sea and the East China Sea. Estuar. Coast. Shelf Sci. 58, 127–136. doi: 10.1016/S0272-7714(03)00067-2
Wang, P. X. (1999). Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Mar. Geol. 156, 5–39. doi: 10.1016/S0025-3227(98)00172-8
Wang, Z. Y., Zhang, Y., Zhao, L. L., Song, N., Han, Z. Q., and Gao, T. X. (2016). Shallow mitochondrial phylogeographical pattern and high levels of genetic connectivity of Thamnaconus hypargyreus in the South China Sea and the East China Sea. Biochem. Syst. Ecol. 67, 110–118. doi: 10.1016/j.bse.2016.05.013
Xia, B. M. (1999). Flora Algarum Marinarum Sinicarum Tomus II, Rhodophyta no. V Ahnfeltiales, Gigartinales, Rhodymeniales. Beijing: Science Press.
Xue, D. X., Wang, H. Y., Zhang, T., and Liu, J. X. (2014). Population genetic structure and demographic history of Atrina pectinata based on mitochondrial DNA and microsatellite markers. PLoS One 9:e95436. doi: 10.1371/journal.pone.0095436
Yang, E. C., Kim, M. S., Geraldino, P. J. L., Sahoo, D., Shin, J. A., and Boo, S. M. (2008). Mitochondrial cox1 and plastid rbcL genes of Gracilaria vermiculophylla (Gracilariaceae, Rhodophyta). J. Appl. Phycol. 20, 161–168. doi: 10.1007/s10811-007-9201-8
Yeh, F. C., Yang, R. C., and Boile, T. (1999). Popgene Version 1.31 Microsoft Window-Based Freeware for Population Genetic Analysis. Edmonton: University of Alberta and Centre for International Forestry Research.
Yokoya, N. S., Kakita, H., Obika, H., and Kitamura, T. (1999). Effects of environmental factors and plant growth regulators on growth of the red alga Gracilaria vermiculophylla from Shikoku island, Japan. Hydrobiologia 399, 339–347. doi: 10.1023/a:1017072508583
Yu, H. J., and Kim, J. K. (2018). Upwelling and eddies affect connectivity among local populations of the goldeye rockfish, Sebastes thompsoni (Pisces, Scorpaenoidei). Ecol. Evol. 8, 4387–4402. doi: 10.1002/ece3.3993
Yu, Y., Harris, A. J., Blair, C., and He, X. J. (2015). RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. doi: 10.1016/j.ympev.2015.03.008
Keywords: Agarophyton vermiculophyllum, cox1, genetic lineages, marginal sea, phylogeography, red seaweed
Citation: Zhong K-L, Song X-H, Choi H-G, Satoshi S, Weinberger F, Draisma SGA, Duan D-L and Hu Z-M (2020) MtDNA-Based Phylogeography of the Red Alga Agarophyton vermiculophyllum (Gigartinales, Rhodophyta) in the Native Northwest Pacific. Front. Mar. Sci. 7:366. doi: 10.3389/fmars.2020.00366
Received: 11 December 2019; Accepted: 30 April 2020;
Published: 04 June 2020.
Edited by:
Zhijun Dong, Yantai Institute of Coastal Zone Research (CAS), ChinaReviewed by:
Anja Schulze, Texas A&M University at Galveston, United StatesMarie-Laure Guillemin, Austral University of Chile, Chile
Copyright © 2020 Zhong, Song, Choi, Satoshi, Weinberger, Draisma, Duan and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zi-Min Hu, aHV6aW1pbjk3MTJAMTYzLmNvbQ==; aHV6bUBxZGlvLmFjLmNu