 Jiangtao Li
Jiangtao Li Jingyu Yang
Jingyu Yang Mingxue Sun
Mingxue Sun Lei Su
Lei Su Hu Wang
Hu Wang Jianqi Gao
Jianqi Gao Shijie Bai
Shijie Bai- 1State Key Laboratory of Marine Geology, Tongji University, Shanghai, China
- 2Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences, Sanya, China
The distribution and succession of microbial communities along the dispersion path of hydrothermal plumes has not been well investigated. In this study, we collected several types of samples from the Longqi hydrothermal field located on the Southwest Indian Ridge, including hydrothermal plumes at different stages of formation, a suite of water column samples across the non-buoyant hydrothermal plumes above this field, and a background seawater column approximately 350 km away from the hydrothermal field. Using CH4 concentration anomalies, three non-buoyant plume samples between 2,535 and 2,735 m were identified within the water column. Microbial community compositions within these plumes and background seawater samples were examined based on the 16S rRNA genes and revealed significant variations and successions in community composition between different portions of the hydrothermal plumes. Near the vent orifice, representing the initial stage of plume formation, microbial populations were characterized by abundant and diverse putative vent-associated communities including (hyper)thermophiles such as Aquificaceae and Hydrogenothermaceae within the phylum Aquificae, and some epsilonproteobacterial chemolithoautotrophs such as Sulfurovum, Sulfurimonas, and Caminibacter. By contrast, in the rising buoyant plumes and adjacent seawaters, most vent-associated microbial taxa were still present but made only minor contributions to community composition. Some microbial taxa that are common in seawater columns such as alphaproteobacterial Sphingomonadaceae and SAR11 clade, deltaproteobacterial SAR324 clade, and gammaproteobacterial Pseudomonas, together with Sulfurimonas and SUP05 clade, became predominant. Members within the Sulfurimonas and SUP05 clade flourished with considerable abundance in the non-buoyant plumes, although these plumes were mainly composed of alphaproteobacterial Rhodobacteraceae, gammaproteobacterial Alteromonadaceae and Saccharospirillaceae putatively derived from the surrounding ambient seawater. We also analyzed archaeal components in the initial discharge and rising buoyant plume stages, with both primarily consisting of thaumarchaeal Nitrosopumilales and euryarchaeal Marine Group II. Our results indicated that being characteristic microbial lineages within the hydrothermal plumes of the Southwest Indian Ridge, both Sulfurimonas and SUP05 clade display a common and abundant distribution across the plume path. However, out of these clades, Sulfurimonas is more abundant and widespread.
Introduction
Hydrothermal plumes, the result of mixing of hot, anoxic hydrothermal fluid and cold, oxygenated seawater, commonly occur at mid-ocean ridges worldwide and exert an important influence on ocean chemistry and microbiology on a global scale (Elderfield and Schultz, 1996; Jackson et al., 2010; Anantharaman et al., 2016). These plumes consist of two parts, or stages, with quite distinct and dynamic features: the buoyant plume, rising vertically hundreds of meters above the discharge orifices, and the non-buoyant plume, spreading out laterally over a few to thousands of kilometers away from originating vents (Baker et al., 1995; Helfrich and Speer, 1995; Fitzsimmons et al., 2017). Due to the entrainment of hydrothermal fluids, hydrothermal plumes are enriched in mineral particles, volatiles, and reduced metals compared with ambient seawater (Holden et al., 2012; Gartman et al., 2014). Theoretical models and biogeochemical measurements suggest that hydrothermal plumes can fuel vigorous microbial chemosynthetic activities (McCollom, 2000; Lam et al., 2004; Dick et al., 2009). In the past decade, cultivation-independent molecular approaches have been applied to characterize plume microbial communities and showed that they harbor a flourishing microbial biome (Orcutt et al., 2011; Dick et al., 2013, Dick, 2019). A wide range of key microbial lineages, such as the SUP05 clade, Alcanivorax, and methylotrophic groups in γ-Proteobacteria, SUP01 clade and Sulfurimonas in ε-Proteobacteria, SAR11 and SAR324 groups in α- and δ-Proteobacteria, respectively, and archaeal Marine Group I (MGI) and II (MGII) in Thaumarchaeota and Euryarchaeota, respectively, are often detected in hydrothermal plumes worldwide (Sunamura et al., 2004; Nakagawa et al., 2005; Dick and Tebo, 2010; German et al., 2010; Sylvan et al., 2012; Li et al., 2015; Sunamura and Yanagawa, 2015; Li J. et al., 2016). In addition to identifying the taxonomic composition of the microbial communities, several recent studies have attempted to link microbial function and metabolism to biogeochemical processes within hydrothermal plumes. For example, it has been suggested that methanotrophy, ammonia oxidation, cellular iron uptake and sulfur oxidation serve as the main energetic pathways fueling plume productivity and mediate the biogeochemical cycling of carbon, nitrogen, iron, and sulfur in the Guaymas Basin hydrothermal plumes (Baker et al., 2012; Lesniewski et al., 2012; Li et al., 2014b). Studies also show that bacterial and archaeal lineages in hydrothermal plumes have versatile metabolic potentials in diverse organic compounds such as carbohydrates (Li et al., 2014a; Li M. et al., 2016), lipids and proteins (Li et al., 2015). More recently, metagenomics analysis showed that oxidation of reduced sulfur species constitutes the most abundant and diverse chemolithotrophic energy metabolism in the Eastern Lau Spreading Center hydrothermal plumes (Anantharaman et al., 2016). Although these studies are insightful and microbiomes have received a great amount of attention in recent years, plume microbiology remains poorly understood. Where are plume microorganisms derived from (e.g., ambient seawater, seafloor vent, or subseafloor) (Lesniewski et al., 2012; Dick et al., 2013)? Once discharged from the orifices, hydrothermal fluids are progressively diluted by ambient seawater, and their chemical compositions continuously change with distance from the vents (Marbler et al., 2010). However, whether the spatial distribution and succession of microbial communities within hydrothermal plumes correspond to the chemical evolution of plumes is still unclear (Sheik et al., 2015; Anantharaman et al., 2016). In addition, although hydrothermal plumes are widely distributed throughout the global mid-ocean ridge system, many of them are still unexplored from the perspective of microbiology, especially at ultraslow spreading ridges (German et al., 2010; Li J. et al., 2016).
The Southwest Indian Ridge (SWIR) is a highly oblique ultraslow-spreading ridge, extending over 7,700 km between the Bouvet and Rodriguez triple junctions (Zhou and Dick, 2013). Studies in early 1997 detected anomalies of turbidity and chemical compositions in the seawater above the SWIR, suggesting the possible presence of hydrothermal venting (German et al., 1998). In subsequent years, addition hydrothermal plumes along the western SWIR were identified, and inactive sulfides and metalliferous sediments were also recovered from both the eastern and western SWIR (Münch et al., 2001; Bach et al., 2002; German, 2003). The first active hydrothermal field was discovered at 49°39′E in 2007 and subsequently, several active hydrothermal fields and hydrothermal anomalies were found between 49°E and 63°E along the SWIR (Tao et al., 2012, 2014). However, little is known about the local microbiology of these hydrothermal fields as only limited studies on the microbial populations of the SWIR plumes have been undertaken (Li J. et al., 2016; Djurhuus et al., 2017). During investigations by R/V Dayang Yihao in April 2014 and R/V Xiangyanghong 09 with HOV Jiaolong in December 2014, a series of water samples, including water columns through the non-buoyant plumes, the rising buoyant plumes and ambient near-bottom seawater near vents, were collected. In this study, we characterize the compositions of microbial community within these plumes at different formation stages and ambient seawater to explore the spatial distribution and variability of plume microorganisms. We found that microbial community composition within the different hydrothermal plume stages show significant variation and community succession.
Materials and Methods
Site Description and Sample Collection
The known hydrothermal fields on the SWIR are mainly centered on two ridge sections. One ridge section is located between the Indomed and Gallieni transform faults of the SWIR from 49°E to 53°E where six hydrothermal fields have been found, while the other section lies on the SWIR between the Melville transform fault and Rodriguez triple junction (RTJ) from 63°E to 64°E where two hydrothermal fields were discovered (Tao et al., 2012, 2014). The six hydrothermal fields on the section between 49°E and 53°E include Yuhuang (37°56′S, 49°16′E), Longqi (37°47′S, 49°39′E), Duanqiao (37°39′S, 50°24′E), Changbaishan carbonate field (37°37′S, 50°56′E), and two fields at 37°27′S, 51°19′E and 36°60′S, 53°15′E which are not yet named (Tao et al., 2014). Hydrothermal fields Tiancheng (27°51′S, 63°55′E) and Tianzuo (27°57′S, 63°33′E) are located on the 63°E to 64°E segment of the SWIR.
The Longqi vent field was the first confirmed active field on the SWIR (Figures 1A,B) (Tao et al., 2012). A series of water column anomalies (temperature, redox potential, methane, and turbidity) within this active field were reported in a previous study (Lin and Zhang, 2006). In 2007, an Autonomous Underwater Vehicle (AUV) obtained the images of active high-temperature venting, black smoker, sulfide deposits, and vent fauna at this field (Tao et al., 2012). The Longqi vent field is located on a high mound on the southeast wall of the ridge valley, at a water depth of ∼2,760 m, and consists of three active venting zones (S zone, M zone, and N zone) extending ∼1000 m laterally (Tao et al., 2012). In the Longqi vent field, there are at least eight active venting areas, each area displaying a variety of levels of hydrothermal discharge (Copley et al., 2016; Zhou et al., 2018). There are many active sulfide structures with variable sizes and morphologies. For example, a number of large sulfide edifices, some more than 15 m high, were observed, and the highest recorded hydrothermal fluids temperature was 379°C (Zhou et al., 2018). In addition to high-temperature vents, diffuse flows are also common in the Longqi field where low-temperature, clear fluids (∼<100°C) discharge, and yellow, flocculent Fe/Si precipitates and microbial mats are distributed (video observation, not published). In addition, inactive sulfide structures including some large chimneys are also distributed widely. Diverse and dense vent faunal assemblages such as shrimps, mussels, crabs, and gastropods around active or inactive hydrothermal structures were also commonly observed (Copley et al., 2016; Zhou et al., 2018).
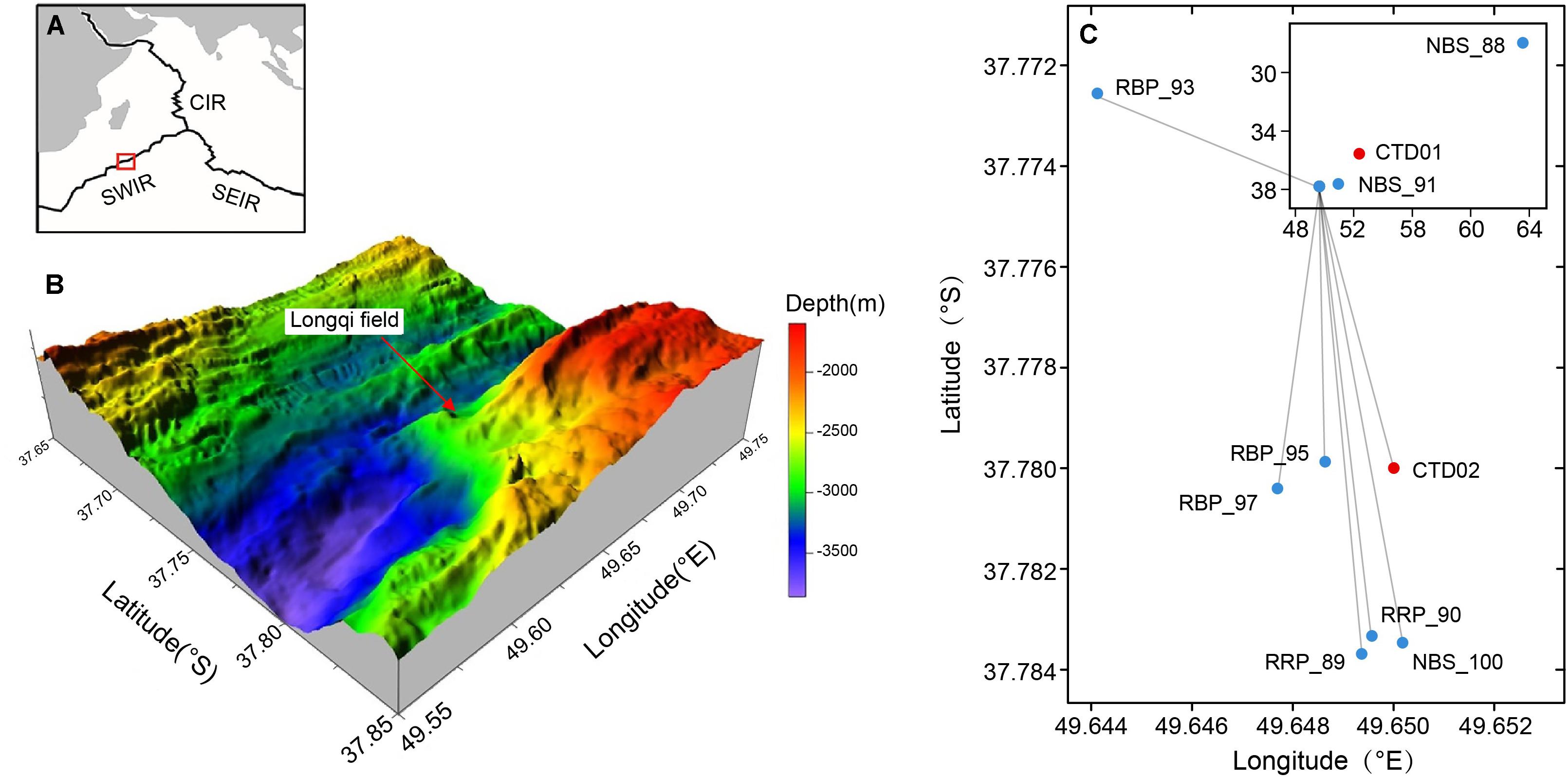
Figure 1. Map of the research area and locations of sampling sites from the Southwest Indian Ridge (SWIR). (A) Location of research area on the Southwest Indian Ridge. SEIR, Southeast Indian Ridge. CIR, Central Indian Ridge. (B) Bathymetry map and location of the Longqi vent field (Tao et al., 2014; Li J. et al., 2016). (C) Locations of sample sites in Longqi vent field and other areas (insert).
The Changbaishan carbonate area, located at 37.62°S, 50.95°E, is characterized by widespread carbonate deposits (Tao et al., 2014). To date, no temperature or turbidity anomalies have been detected in the water column and no sulfide deposits or other signs of hydrothermal activity were observed at this field. Geochemical data indicate that these carbonates belong to normal pelagic depositions, making it debatable whether it belongs to a potential hydrothermal field (Tao et al., 2014). Tianzuo field, located at 27°57′S, 63°33′E with a water depth of 2,949 m, is an inactive hydrothermal region. Black massive sulfides, red-brown iron-hydroxides, and polymetallic muds have been observed from this field by TV-guided grab (Tao et al., 2014).
Two water columns, designated as CTD01 and CTD02, were collected by a rosette sampler coupled with a CTD (SBE 911 plus, United States) and a MARP (Miniature Autonomous Plume Recorder) aboard the R/V Dayang Yihao during the DY115-30 cruise from April 1 to May 30, 2014. CTD01 water column was collected from the location of 35.54°S, 52.37°E with a max water depth of ∼ 3,410 m (Figure 1C). This sampling site was located approximately 350 km away from the Longqi field and did not host any known hydrothermal activity. Samples from this water column, considered as normal (background) pelagic environment, were taken at 12 different depths along a vertical profile between 100 and 3,365 m (Table 1). The CTD02 water column was collected above the active S zone (37.78°S, 49.65°E, ∼2,830 m water depth) of the Longqi field (Figures 1B,C), at 10 different depths, from 800 to 2,785 m (Table 1). Samples of potential non-buoyant plume were first identified by turbidity anomalies using a MARP with an optical backscatter sensor for turbidity measurement. During the CTD cast, a MAPR was attached to the CTD cable, about 50 m above the CTD rosette as described previously (Wang et al., 2011). Hydrothermal plume samples were confirmed by measurement of high concentrations of CH4, a commonly used tracer for hydrothermal plumes (Baker et al., 1995).
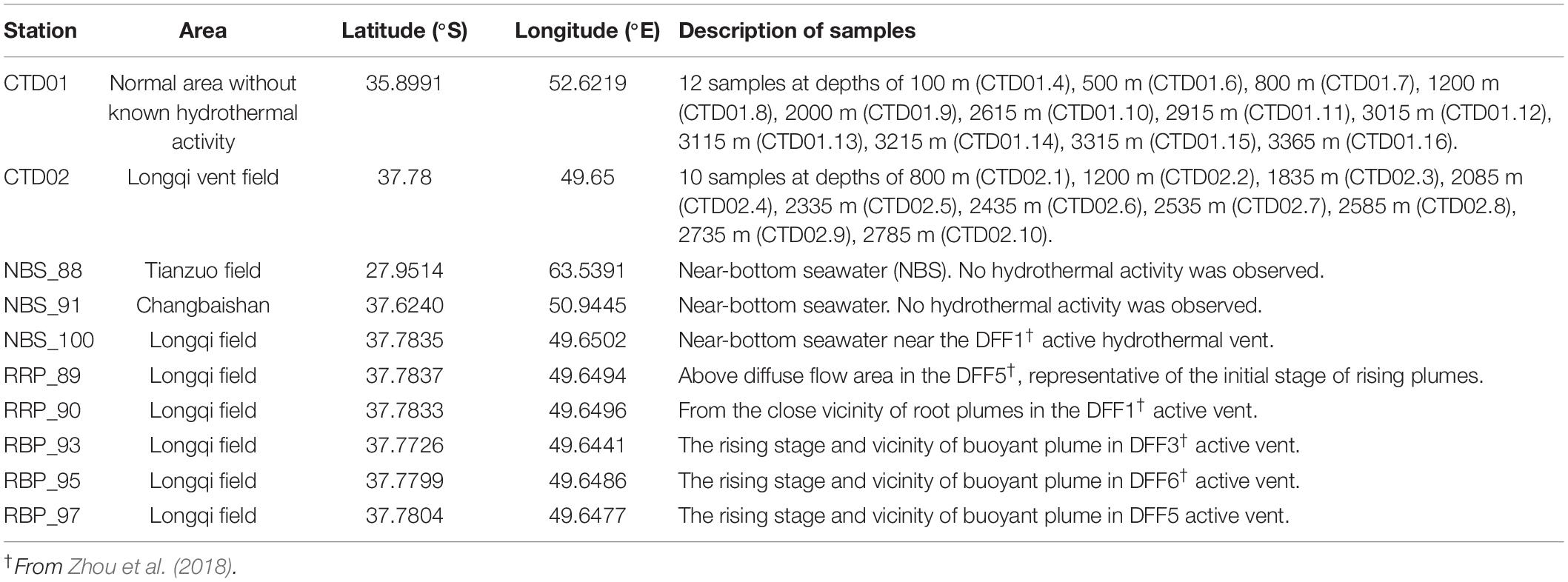
Table 1. The detailed information of samples collected for this study.
Samples of rising buoyant plumes and the surrounding ambient seawater were collected from the SWIR during the DY115-30 cruise of the R/V Xiangyanghong 09 with the HOV Jiaolong between November 20, 2014 to March 17, 2015. These samples were taken with Niskin bottles which were fixed to the front of HOV Jiaolong. Two near-bottom seawaters were collected from the inactive Tianzuo field (NBS_88) and Changbaishan carbonate area (NBS_91), respectively. The remaining samples were collected from the Longqi vent field (Figure 1C). Detailed information of these samples is listed in Table 1.
Determination of Dissolved CH4 Concentration
Gas chromatography (GC) was used onboard to determine the concentrations of dissolved CH4 in the water samples as described previously (Wang et al., 2015; Li J. et al., 2016). Generally, once the CTD rosette sampler came back to the deck, water samples for CH4 measurement were first taken by transferring water from each depth into different 100 ml extraction bottles. Then, CH4 was stripped from the water samples by continuous purging with helium gas and adsorbed with an activated charcoal trap which was dipped into liquid nitrogen. To release the adsorbed CH4, the charcoal trap was rapidly heated to 100°C by being submerged into boiling water. CH4 was then injected into an Agilent 6820 GC with a flame ionization detector. Chromatographic separation was achieved by a packed Porapak-Q stainless steel column (60/80 mesh, 3 mm i.d. × 4 m) at an oven temperature of 80°C.
DNA Extraction From Water Samples
DNA was extracted from all samples for analysis of microbial community structures. Approximately 2 liters of water for each sample was filtered through a polycarbonate (PC) membrane filter with 0.22 μm nominal pore size (47 mm, Millipore), and the filters were then frozen at −80°C until further processing. DNA was extracted from the membrane filter following the SDS-based extraction method as described in our previous study (Li J. et al., 2016). Briefly, 800 μL DNA extraction buffer (100 mM Tris-HCl, 100 mM sodium EDTA, 100 mM sodium phosphate, 1.5 M NaCl and 1% CTAB) was added into each centrifuge tube containing a PC membrane. The centrifuge tubes were frozen and thawed three times by alternating in liquid nitrogen and a 65°C water bath. Then, proteinase K was added to a final concentration of ∼ 0.2 mg mL–1. The centrifuge tubes were incubated at 65°C for 2 h. Afterward, 800 μL phenol/chloroform/isoamyl alcohol (25:24:1, v/v) was added and the tubes were centrifuged at 12,000 × g for 15 min. The top aqueous layer was carefully pipetted into new, clean 2 mL tubes and an equal volume of chloroform/isoamyl alcohol (24:1, v/v) was added. The tubes were centrifuged again at 12,000 × g for 10 min. The supernatant was transferred into new, clean tubes, and at the same time, 0.6 volume of cold isopropanol and 0.1 volume of sodium acetate (3M) were added. The tubes were incubated at −20°C for 1 h and centrifuged at 12,000 × g for 10 min. DNA pellets were carefully washed with 70% pre-cooled ethanol (−20°C) and resuspended into 50 μL sterile deionized H2O.
Pyrosequencing and Analysis of 16S rRNA Gene Sequences
DNA concentration was quantified with a Quant-iTTM PicoGreenTM dsDNA Assay Kit (Invitrogen, ThermoFisher Scientific, United States). Bacterial 16S rRNA genes were amplified with the primer set 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 533R (5′-TTA CCG CGG CTG CTG GCA C-3′) that included 10-nucleotide barcodes, while for the amplification of archaeal 16S rRNA gene, arch344F (5′-ACG GGG YGC AGC AGG CGC GA-3′) and arch915R (5′-GTG CTC CCC CGC CAA TTC CT-3′) containing 8-nucleotide barcodes. The PCR reaction conditions were: 94°C, 5 min; 94°C for 50 s, 53°C for 50 s, and 72°C for 50 s, 25 cycles; a final extension at 72°C for 5 min. PCR products were purified with QIAquick PCR Purification Kit (Qiagen, MD, United States) and then quantified with the NanoDrop 2000 (Thermo Scientific, United States). The pyrosequencing was performed with the 454 GS-FLX Titanium System (Roche, Basel, Switzerland).
We used QIIME 1.9.1 to perform analysis of the pyrosequenced amplicons. The following criteria were employed to remove the reads with low quality: (1) having ambiguous nucleotides; (2) shorter than 200 bp; (3) containing > 6 bp homopolymers; (4) the average flowgram score < 25 in a quality window of 50 bp. The Operational Taxonomic Units (OTUs) were generated based on 97% cutoff of sequence similarity, and the longest sequence of each OTU were used as the representative sequences. Taxonomy assignment was implemented using the RDP classifier against the SILVA 16S rRNA gene database (Version 132). Similarities among the different microbial communities were calculated using the similarity matrices generated according to the phylogenetic distance between reads (Unifrac distance) and beta diversity of NMDS was computed as part of the QIIME pipeline. To avoid the variation caused by an unequal sequence number across samples, resampling of sequences was performed with sing_rarefaction.py for each sample after OTU generation at a rarefication sequence level based on the sample with the fewest number of sequences. Based on the grouping results of NMDS ordination, statistical analyses including MRPP, ANOSIM and PERMANOVA were performed to test statistical significance in microbial compositions by R project (v 3.2.1) using the Vegan package1.
Nucleotide Sequence Accession Numbers
The nucleotide sequences obtained from the 454 sequencing of bacterial and archaeal 16S rRNA genes were deposited in SRA (Sequence Read Archive) under BioProject accession numbers PRJNA638507 and PRJNA638524.
Results
CH4 Concentration and Identification of Non-buoyant Plumes From Water Column
As a diagnostic tracer of hydrothermal plumes, CH4 concentration anomalies generally indicate the position of a plume in the water column (Baker et al., 1995; Wang et al., 2015). Therefore, CH4 concentrations of water samples from the CTD02 water column were measured to identify non-buoyant plume layers which are considered to have higher CH4 compositions than background seawater. Higher CH4 concentrations were observed at the depth interval between 2,535 and 2,735 m. As shown in Figure 2, CH4 concentrations in CTD02.7 (2,535 m), CTD02.8 (2,585 m), CTD02.9 (2,735 m) were 3.31, 4.41, and 4.23 nmol/L, respectively, while samples from other depths had lower CH4 concentrations ranging from 0.91 ∼ 1.96 nmol/L (Figure 2). Based on the CH4 anomalies, these three samples were identified as the non-buoyant hydrothermal plume at a height of about 100 ∼ 300 m above the hydrothermal orifices, with an anomaly thickness of around 200 m. Other samples of CTD02 water column belonged to pelagic seawater. Seawater CTD02.06, about 100 m above the upper layer of plumes, had a CH4 concentration ∼ 0.91 nmol/L, while seawater CTD02.10, the deepest sample (2,785 m) from CTD02 water column and collected from ∼ 50 m above the seafloor, was 1.76 nmol/L (Figure 2). Our results for CH4 concentrations of plume and background seawater, approximate height of the non-buoyant plume, and thickness of the plume layer are consistent with previous reports (Wang et al., 2015, 2019; Li J. et al., 2016).
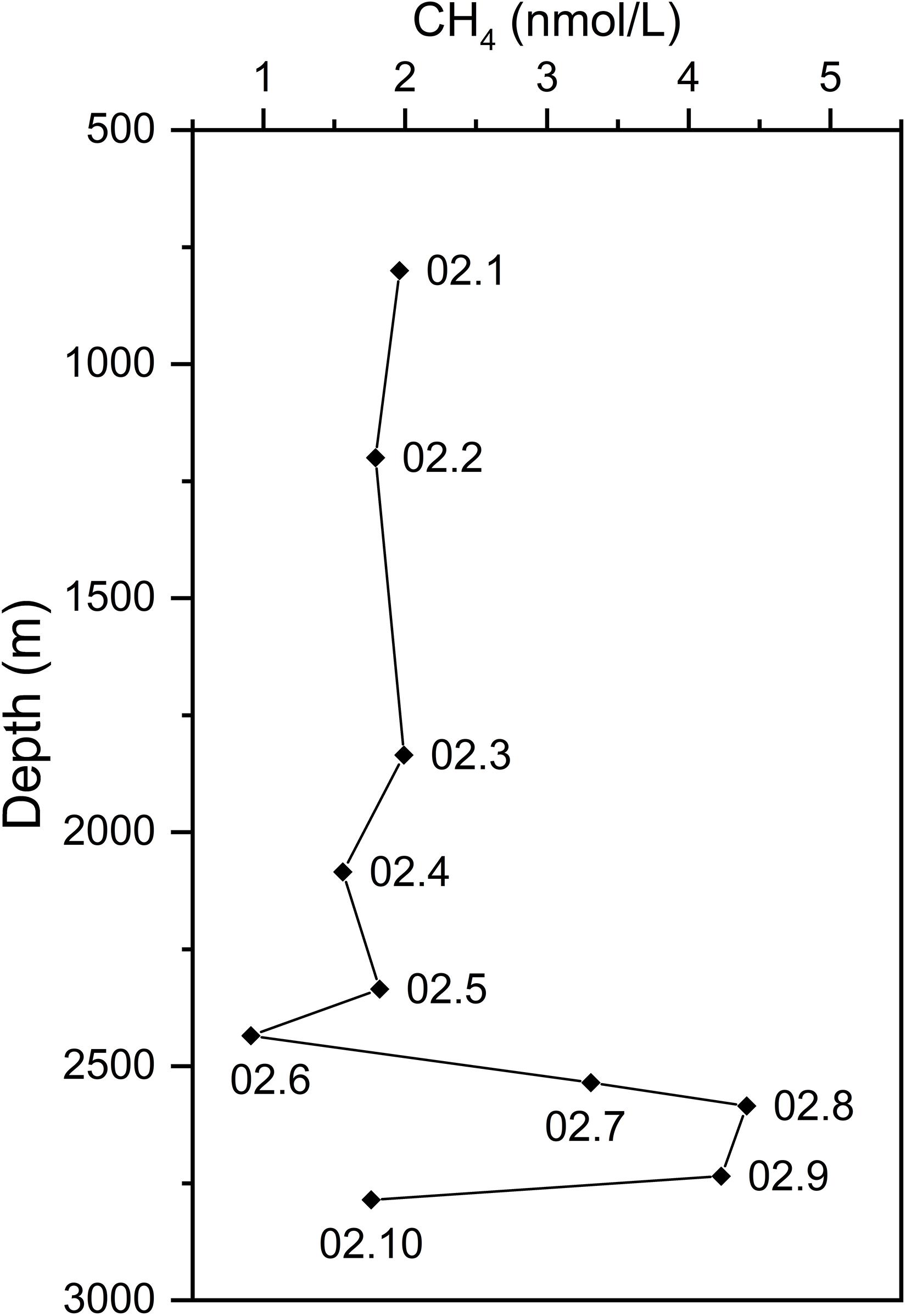
Figure 2. The profile of methane concentrations along the CTD02 water column above the Longqi vent field. Based on the anomalies of methane concentration, three samples, CTD02.7, 02.8, and 02.9, were identified as the non-buoyant plume samples.
The Diversity and Statistical Analysis of Microbial Communities
As shown in Table 1, there were a total of 30 samples, including 12 seawaters from CTD01 station, 10 samples from CTD02 station, and 8 samples collected by HOV Jiaolong of near-bottom seawater or rising buoyant plume near hydrothermal vents. All the samples were sequenced for their bacterial 16S rRNA genes (Supplementary Table S1), while only the 8 samples collected by HOV Jiaolong were sequenced for archaeal 16S rRNA genes (Supplementary Table S2) since the archaeal contribution is generally much smaller than that of bacteria in the seawater column (Sunamura and Yanagawa, 2015; Li J. et al., 2016). Detailed information on qualified reads and OTUs for each sample are listed in Supplementary Tables S1, S2. The α-diversity indices of these samples including Chao1, Shannon, and Simpson are also provided in Supplementary Tables S1, S2. For bacterial communities, the samples collected by HOV Jiaolong from the rising buoyant plumes and ambient seawaters generally had the highest Shannon and Chao1 values, while the seawater samples from the CTD01 station showed higher diversity than those of the CTD02 station. By contrast, seawaters of CTD01 water column displayed the best evenness as indicated by their higher Simpson values. The samples of CTD02 water column possessed lower Simpson values in general than those of the rising plume and surrounding seawater, indicative of relatively higher heterogeneity. Several samples such as CTD02.7, NBS_91 and RBP_93 showed relatively higher diversity indices values as compared with their surrounding samples (Supplementary Table S1). For archaeal communities, three samples including RBP_95, RBP_97, and NBS_100 which were collected from the rising portions of plumes and near-bottom seawater around an active vent (Table 1) showed the lowest values of Shannon and Chao1. By contrast, RRP_89 taken from the location adjacent to a diffuse flow and RBP_93 from a rising buoyant plume had the highest diversity indices values (Supplementary Table S2).
NMDS analysis of bacterial communities clearly separated these samples into two principal groups, one exclusively consisting of the rising buoyant plumes and ambient near-bottom seawater collected with HOV Jiaolong and another one composed completely of water column samples obtained by CTD sampler (Figure 3A). Although the rising buoyant plumes and ambient seawater samples grouped together, they were in a loose pattern except that RBP_95, RBP_97 and NBS_100 were closely enclosed in a subgroup (Figures 3A,B). Samples from the two water columns were further distinguished into two different subgroups, primarily based on their sampling locations (Figure 3A). To compare the bacterial communities within different hydrothermal plume stages and their surrounding environments, the rising buoyant plumes, the non-buoyant plume and its surrounding ambient seawater were picked out for further NMDS analysis. This revealed that samples of the non-buoyant plume and the surrounding seawater separate completely from those of the rising buoyant plumes and ambient near-bottom seawater samples (Figure 3B), indicative of a significant difference in their bacterial community compositions (P < 0.05) (Supplementary Table S4).
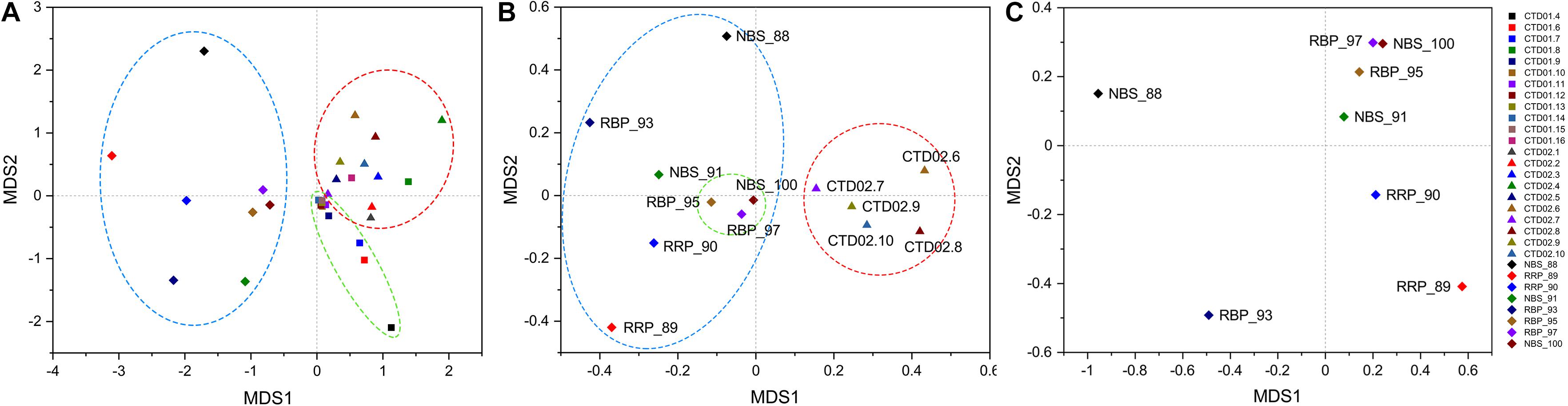
Figure 3. NMDS statistical analysis of bacterial and archaeal communities. (A) Bacterial results from all samples. Diamond: samples collected by HOV Jiaolong. Square: samples of CTD01 station. Triangle: samples of CTD02 water column including three non-buoyant plumes. (B) Bacterial results of those samples consisting of near-bottom seawater and rising buoyant plumes collected by HOV Jiaolong and of non-buoyant plumes and their surrounding seawater. (C) Archaeal results of samples collected by HOV Jiaolong from near vents and the seafloor.
Results of archaeal NMDS analysis revealed a loose clustering pattern within the rising buoyant plumes and ambient near-bottom seawater samples (Figure 3C). Three samples including RBP_95, RBP_97, and NBS_100 formed an independent, close group, clearly separated from other samples collected by HOV Jiaolong (Figure 3C). As shown in Figure 3, these three samples always clustered together in the statistical analyses of their bacterial and archaeal communities, indicating that these communities had similar compositions. Other samples were scattered in a loose pattern (Figure 3C).
Bacterial Community Structures Based on 16S rRNA Gene Analysis
Gammaproteobacteria was one of the dominant bacterial lineages, being found in most samples, but with dramatically variable abundances, ranging from 9 to 84%. As shown in Figure 4, γ-proteobacterial members generally accounted for 9% ∼ 19% of abundance in the CTD01 water column samples, but with three exceptions (CTD01.8, CTD01.15 and CTD01.16) whose γ-proteobacterial proportions were higher, 32% ∼ 62%. By contrast, γ-Proteobacteria dominated most of the samples from the CTD02 water column, accounting for 57% ∼ 84% of sequences with only two samples being exceptions, one seawater (CTD02.4) and one identified non-buoyant plume (CTD02.7), in which γ-Proteobacteria accounted for 23% ∼ 29% of the bacterial communities. Like the CTD01 water column samples, γ-proteobacterial members also accounted for a relatively lower proportion (12% ∼ 21%) in seven out of the eight rising buoyant plume and ambient seawater samples. Only within the bottom seawater NBS_88, taken from Tianzuo field, did γ-proteobacteria dominate, accounting for ∼47% percentage of the bacterial communities (Figure 4). At the family taxonomic level, the relatively abundant (>1%) groups of γ-Proteobacteria consisted of Alteromonadaceae, Alcanivoracaceae, Marinobacteriaceae, Shewanellaceae, and Saccharospirillaceae (Figure 5). It was further observed that these families, which are ubiquitous bacterial lineages in pelagic seawaters, almost exclusively dominated the seawater samples of the two CTD water columns. Another family, Thioglobaceae, exclusively composed of SUP05 clade members, which are putatively considered as the most universal bacterial lineage in hydrothermal plumes (Sunamura et al., 2004; Sunamura and Yanagawa, 2015), showed ubiquitous distribution within most samples in spite of relatively low abundance (Figure 5). In addition, a few γ-Proteobacteria families that were observed at only low relative abundances (<1%) in the rising buoyant plumes and the associated ambient near-bottom seawater, including Beggiatoaceae, Thioalkalispiraceae, Thiomicrospira, Thiohalorhabdus, Thioalkalispiraceae, and Thiomicrospirales, are all well known vent-associated organisms (Figure 5, Supplementary Figure S1, and Supplementary Table S3).
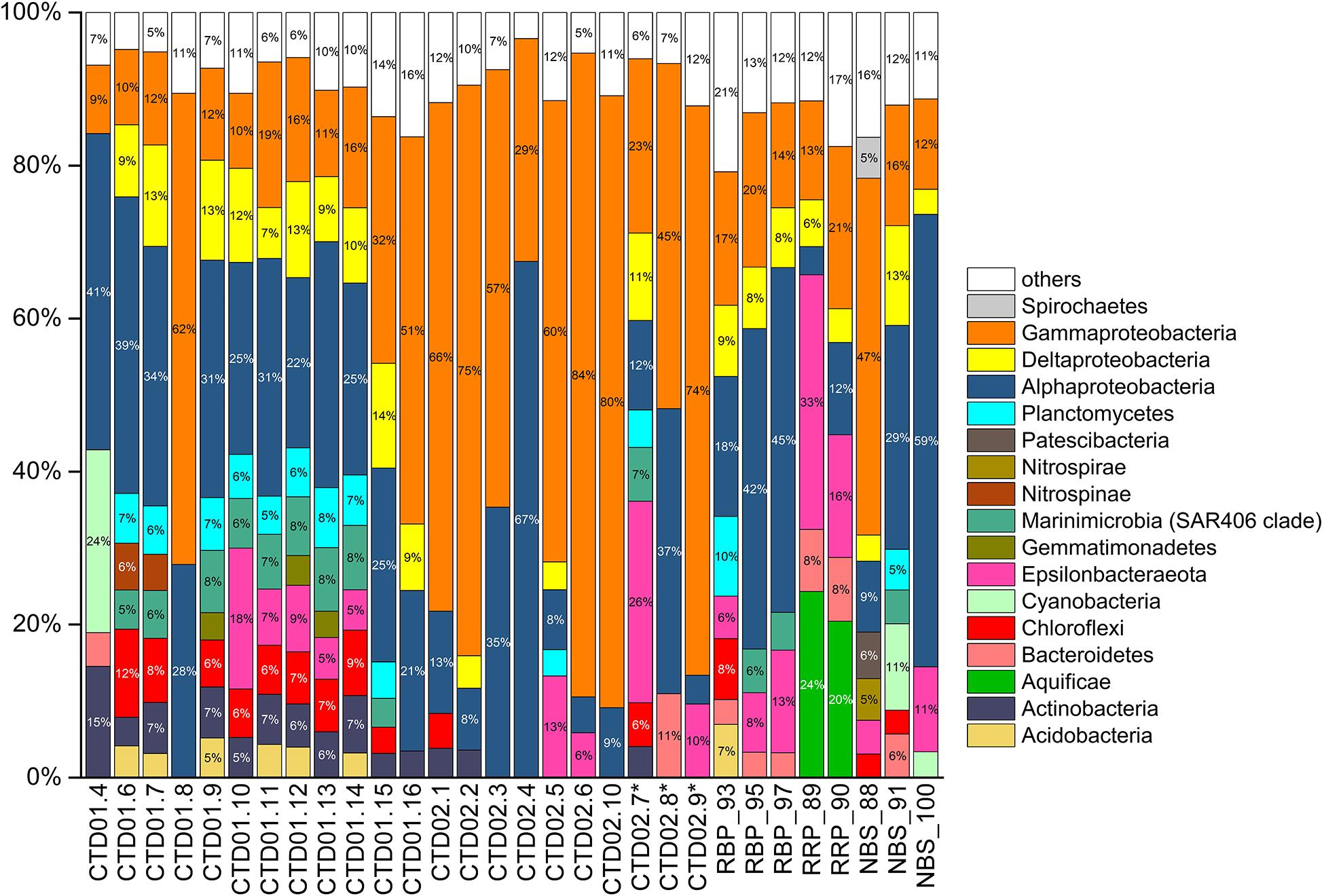
Figure 4. Histogram of bacterial communities from all samples at phylum and class levels (for Proteobacteria). Only bacterial lineages with abundances > 3% percentages are listed. The rest are classified into “others” group. ∗Indicates the three non-buoyant plume samples identified based on CH4 anomalies.
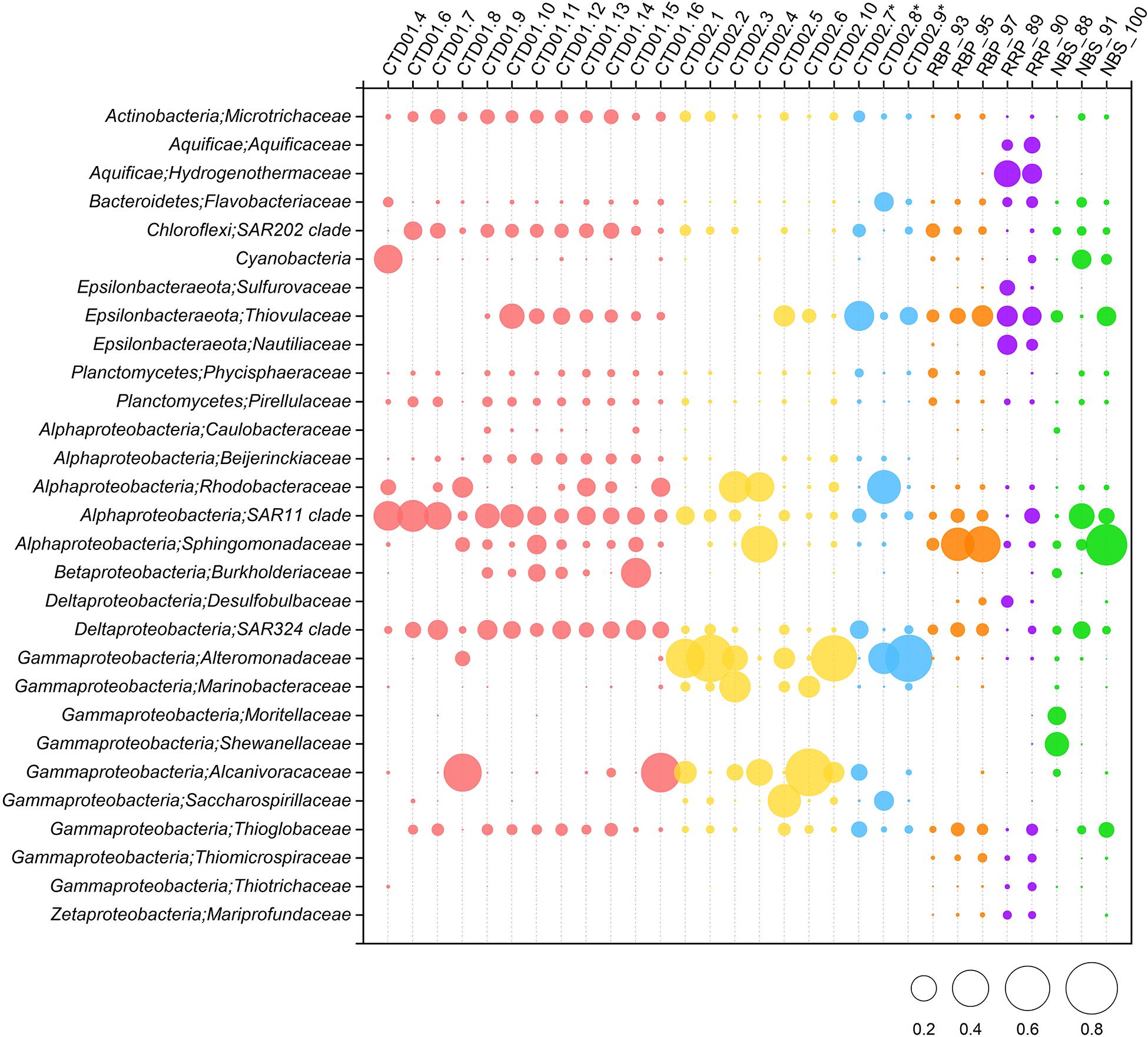
Figure 5. Relative abundances of dominant (>1% abundance) bacterial community members in the rising buoyant plumes and ambient near-bottom seawater, the non-buoyant plume and background seawater of the two water columns are at ∼family level. Some specific vent-associated bacterial groups despite of their low (<1%) abundances are shown. ∗ Indicates the three non-buoyant plume samples identified based on CH4 anomalies.
α-Proteobacteria also displayed a universal distribution, and high abundance, in the majority of seawater and plume samples (Figure 4). In the CTD01 station samples, α-proteobacteria generally accounted for 21% ∼ 41% of abundance, while CTD02 samples displayed a broad range of alphaproteobacterial abundance, varying between 4 and 67%. Meanwhile, the relative abundances of α-Proteobacteria within the rising plumes and nearby ambient seawaters were also heterogenous, with the highest proportion up to 59% in NBS_100 and the least, 4%, in RRP_89 (Figure 4 and Supplementary Table S3). The prevailing alphaproteobacterial groups were mainly derived from the SAR11 clade, Sphingomonadaceae, and Rhodobacteraceae (Figure 5), which are cosmopolitan members in seawater.
Members of ε-Proteobacteria were also frequently retrieved from both seawater and plume samples, although their relative abundances were significantly different. As shown in Figure 4, high abundances of ε-proteobacteria were mainly observed in the different plume samples and the adjacent seawater, accounting for approximately 4% ∼ 33% of each bacterial community. Within those samples representing the rising portions of plumes and the surrounding seawater, only one near-bottom seawater (NBS_88) had an ε-Proteobacteria abundance of <1% (Figure 4 and Supplementary Figure S1). The ε-proteobacterial sequences mainly classified as Campylobacterales and Nautiliales (Supplementary Table S3). They were further assigned into the families Arcobacteraceae, Nitratiruptoraceae, Sulfurovaceae, Sulfurospirillaceae, Thiovulaceae, and Nautiliaceae (Figure 5 and Supplementary Table S3) which are lineages often associated with hydrothermal vents. Among these, the family Thiovulaceae and its sole genus Sulfurimonas (Supplementary Table S3), were characterized by their high relative abundances (up to ∼ 26%) and universal distributions among the different plume stages and their adjacent environments. Moreover, ε-proteobacterial populations were also observed in the CTD01 water column (8 out of 12 samples), with five showing high abundance, 5%∼18%. By contrast, the Sulfurovaceae and Nautiliaceae were abundant only in RRP_89, accounting for 7% and 11% of sequences, respectively (Figure 5).
δ-Proteobacteria had an ubiquitous distribution among all our samples with relative abundances varying between 0.2 ∼ 14% (Figure 4 and Supplementary Figure S1). The SAR324 clade was the most abundant δ-proteobacterial lineage and was observed in most of the samples, especially in the seawater of the CTD01 water column (Figure 5). By contrast, another putative vent-associated lineage, the Desulfobacterales, was exclusively found in samples surrounding the rising buoyant plumes, but at low relative abundances (generally < 0.5%) (Figure 5).
Some bacterial lineages, generally belonging to putative vent-associated microorganisms, were exclusively distributed in the rising buoyant plumes and ambient near-bottom seawater. For example, sequences of Aquificae were only retrieved from samples collected by HOV Jiaolong. Two samples, RRP_89 and RRP_90, contained high proportions of Aquificae groups (20% ∼ 24%) primarily consisting of members from the Hydrogenothermaceae and Aquificaceae. Members of the Desulfurobacteriaceae (Aquificae) were found within both samples but at minor abundances (<1%). ζ-proteobacteria, that include well-known neutrophilic iron-oxidizers from hydrothermal vent areas (McAllister et al., 2019), were commonly detected from most rising buoyant plume and near-bottom seawater samples, with highest relative abundances in RRP_89 and RRP_90 of ∼2%, but less than 1% in other samples (Figures 4, 5 and Supplementary Table S3). Interestingly, other putative neutrophilic iron-oxidizing bacteria, belonging to the β-Proteobacteria family Gallionellaceae, were also exclusively found in the RRP_89 sample (Supplementary Table S3). The Gallionellaceae (their typical representatives, Gallionella spp.) are well-known Fe oxidizers in freshwater, but they have also been retrieved from hydrothermal vent environments such as low-temperature diffuse flows, microbial mats, and weathered hydrothermal chimneys (Kato et al., 2009; Sylvan et al., 2012; Li et al., 2017, 2020; Vander Roost et al., 2018). In addition, there were a number of groups with minor proportions (generally < 1%) such as Hydrogenedentes and Deinococcus-Thermus, exclusively retrieved in the rising plumes and ambient seawater. They were completely absent from all water column samples including the non-buoyant plumes (Supplementary Table S3).
In addition, a number of bacterial phyla including Acidobacteria, Actinobacteria, Chloroflexi, Marinimicrobia, Nitrospinae, Nitrospira, and Planctomycetes occurred universally among the different samples but generally at low relative abundances (usually < 5%) (Supplementary Figure S1). As putative photosynthetic bacteria, Cyanobacteria dominated the upper photic seawater CTD01.4 with >24% abundance (Figure 4). It was noted that cyanobacteria were commonly retrieved from different sample types at various depths, with an elevated abundance of cyanobacterial groups in two bottom seawater samples (NBS_91 and NBS_100) ranging between 3 and 11% (Figure 5).
Archaeal Community Compositions in the Rising Buoyant Plumes and Surrounding Seawaters
As shown in Figure 6, the archaeal compositions of the rising buoyant plumes and ambient near-bottom seawaters were dominated by members affiliated with the Nitrosopumilales of the Thaumarchaeota, Marine Group II and III (MGII and MGIII) of the Euryarchaeota, and the Woesearchaeota. Members of the Nitrosopumilales were observed in the rising buoyant plumes and surrounding near-bottom seawater with abundances varying from 43 to 71% (Figure 6). Euryarchaeal MGII clade, the second most abundant group, contributed significantly to archaeal populations to a varying degree, 17∼44% (Figure 6). Only in one near-bottom seawater (NBS_88) did MGII show a relatively lower abundance, ∼ 6%. By contrast, representatives of the MGIII clade accounted for only 2∼9% of abundance in all samples (Figure 6 and Supplementary Figure S2). These ubiquitous archaeal clades (Thaumarchaeota, MGII and MGIII) have been found to have versatile metabolic potentials, able to utilize diverse organic compounds in the hydrothermal plumes (Li et al., 2015). Woesearchaeota, was observed in all 8 samples but at generally low abundances (<5%) with the exception of NBS_88 where relative abundance was near 19%. As mentioned in previous studies (Castelle et al., 2015; Liu et al., 2018), these dominant archaeal lineages are universal members of archaeal communities in marine sediments and seawater. In addition, a number of taxa, such as thaumarchaeal Group 1.1c and Marine Benthic Group A (MBGA), Bathyarchaeota, Asgardaeota, Diapherotrites, Nanoarchaeaeota, were characterized by their common occurrence, but limited contributions to archaeal community compositions (Supplementary Figure S2 and Supplementary Table S3).
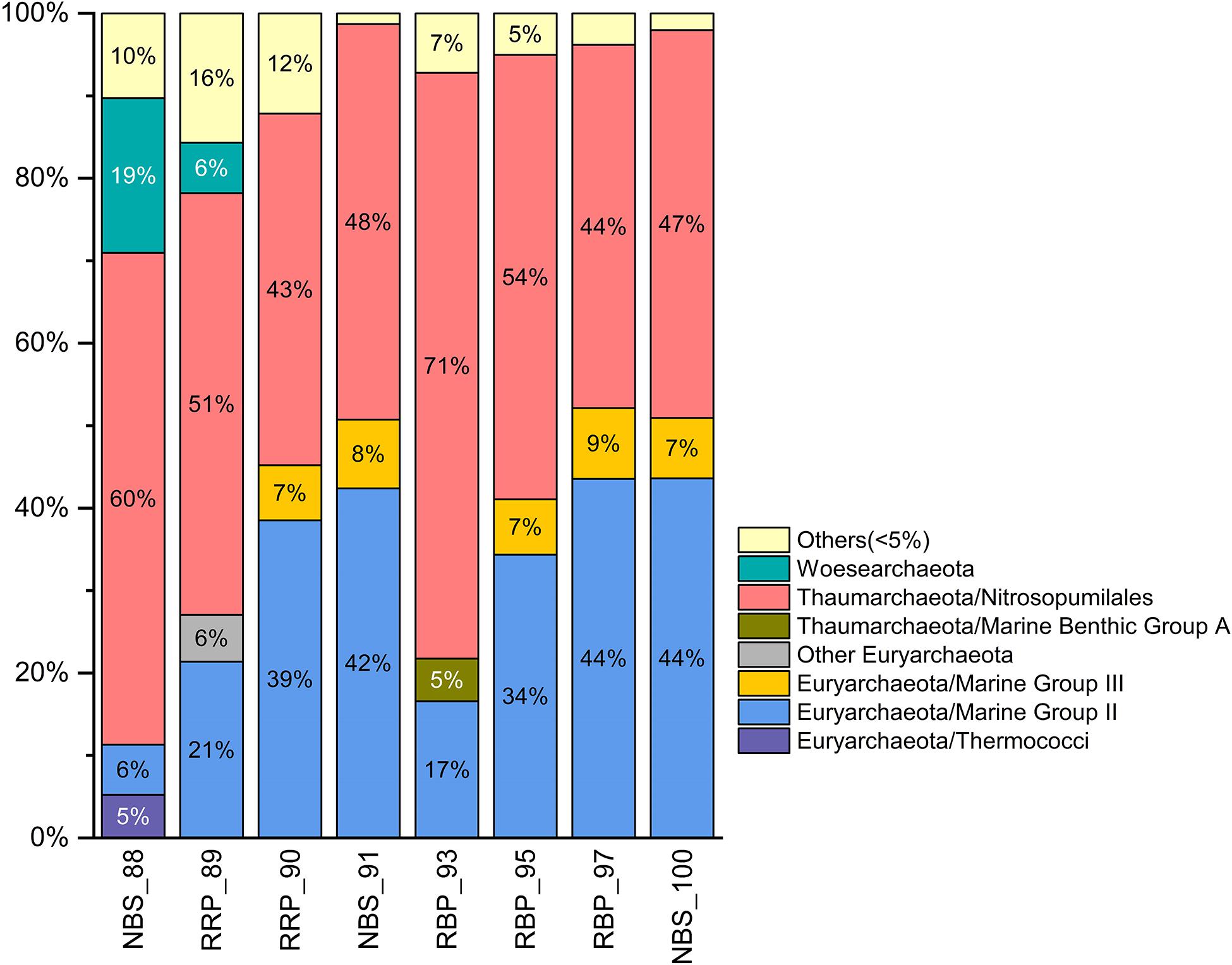
Figure 6. Histogram of archaeal communities in the rising buoyant plumes and ambient near-bottom seawater collected by HOV Jiaolong. Only archaeal lineages > 5% abundance are shown. The rest are classified into “others” group.
Additionally, two euryarchaeal clades, Thermococci and Archaeoglobi, were primarily retrieved from two near-bottom seawater samples taken around the hydrothermal vents (Supplementary Figure S2 and Supplementary Table S3). Their relative abundances were 3∼6%. Unlike the taxa with higher abundances, there were a number of vent-associated subclades which displayed localized distributions. For example, Methanomicrobia, Methanobacteria, Methanococci, which are known as typical methanogens, were detected mainly from RRP_89 and RRP_90 samples at 2∼3% abundance. Thermococci accounted for about 3% in RRP_89 sample. Other groups, such as JdfR-43, Hydrothermarchaeota, Thermplasmatales, Desulforococcales were only detected from RRP_89 and RRP_90 samples. In addition, Archaeoglobi and Thermococci within the Euryarchaeota were detected from a few samples with 3∼5% relative abundances (Supplementary Table S3).
Discussion
Representatives at Different Evolution Stages of Hydrothermal Plumes
In the present study, samples were collected from several locations to encompass the different evolutionary stages of hydrothermal plumes. As hot, low density hydrothermal fluids are discharged from seafloor orifices and encounter bottom seawater, the turbulent mixing of fluid and seawater initiate the formation of a buoyant hydrothermal plume (Sunamura and Yanagawa, 2015), which is primarily dispersed vertically. The buoyant plume rises with progressive dilution by ambient seawater and finally develops into a non-buoyant plume which spreads out only laterally. It is often difficult to collect spatially resolved plume samples along its dispersal path, from initial rising buoyant stages to final non-buoyant height (Takai et al., 2004; Sheik et al., 2015). Our sampling method had some technical limits in accurately accessing the rising buoyant plumes, especially for the low, narrow discharges near the orifices representing the early initial formation stage. For example, because the Niskin bottles were fixed horizontally, it was practically impossible to ensure that the bottle opening stayed completely within the narrow root of the rising plumes without contamination by ambient seawater. From this perspective, the root portions of plume, representing the earliest stage of plume development, was absent from our study. However, it has been revealed that intensive turbulences caused by hydrothermal fluid discharging out into seawater will constantly harvest microorganisms from the surroundings such as ambient near-bottom seawater, diffuse flows, and hydrothermal chimney structure, and entrain them into the root segment of rising buoyant plumes (Jiang and Breier, 2014; Sheik et al., 2015). Thereby, samples taken in close vicinity to the plume root (RRP_90) or from a position just above a diffuse flow (RRP_89) can, to some extent, reflect the microbial communities in the root portions of rising buoyant plumes. By contrast, three samples (RBP_93, RBP_95, and RBP_97) were collected from the rising regions of buoyant plumes at the heights of about 10 m above the vents, where plumes had diameters wide enough to surround the horizontal sampling opening completely. In addition to the buoyant plume samples, an additional three samples of near-bottom seawater were also taken. NBS_100, taken from the area near the Jabberwocky vent (or DFF1) (Copley et al., 2016; Zhou et al., 2018) of the Longqi vent field, but not adjacent to this active chimney, represented the near-bottom seawaters potentially affected by hydrothermal venting. NBS_88 and NBS_91 were collected from Tianzuo field and Changbaishan area, respectively, at which no active hydrothermal activities were observed, to represent background bottom seawater distant from hydrothermal activities. The non-buoyant hydrothermal plumes have a large lateral range, generally at heights of ∼ 200 m above the seafloor (Wang et al., 2015; Li J. et al., 2016; Djurhuus et al., 2017), making the CTD rosette sampler the most effective way to collect those samples. As described above, together with CH4 concentration anomalies, three non-buoyant plume samples (CTD02.7, CTD02.8, and CTD02.9) were successfully identified. Therefore, we collected multiple samples along the dispersal path of the hydrothermal plumes and their surrounding environments, thus able to represent the different developmental stages of a hydrothermal plume (initial, developing, and final) (Table 1).
Characterization of Microbial Communities Within Different Development Stages of Hydrothermal Plumes
RRP_89 and RRP_90, representing the initial roots of hydrothermal plumes, are characterized by significantly abundant (hyper)thermophilic microbial communities. It was shown that members of the families Aquificaceae, Hydrogenothermaceae, and Desulfurobacteriaceae within the phylum Aquificae, putative strict thermophilic and hyperthermophilic bacteria, were abundant totally accounting for ∼20% of sequences. Moreover, two additional thermophilic lineages, ε-proteobacterial Nautiliales (mainly affiliated with the genus Caminibacter) and Desulfobacterales, were also relatively abundant (4∼12% and 0.5∼5%, respectively) in both samples (Figure 5). In addition, archaeal (hyper)thermophiles such as Desulfurococcales, Thermoproteales, Archaeoglobales, Methanococcales and Thermococcales were also observed in these two samples (Supplementary Table S3). The high relative abundances and high diversity of (hyper)thermophilic microorganisms imply that hot habitats exerted a strong influence on the compositions of the communities in these two samples. As pointed out earlier, RRP_89 was collected from a venting site with diffuse hydrothermal flows. It has been previously demonstrated that low-temperature diffuse flows are important conduits to transfer subsurface microbiota onto the seafloor, and (hyper)thermophiles are good indicators for microbial life in the hot subsurface (Huber et al., 2002; Perner et al., 2010, 2013; Meier et al., 2016). Therefore, high abundance of thermophiles and hyperthermophiles strongly indicated the presence of a subsurface microbiome with hot habitats beneath the sampling location of RRP_89. Unlike RRP_89, near-bottom seawater RRP_90 was taken from a location near a plume root. The high temperature (>240°C) of hydrothermal fluids discharged from orifices ensures no microorganism could be conveyed from subsurface habitats into the rising buoyant plumes through fluid conduits within chimneys since the temperature is beyond the tolerance limit of life (122°C) (Takai et al., 2008). The intensive, consistent inward flows near the rising plume root (Jiang and Breier, 2014) would continuously entrain microorganisms from surrounding bottom seawaters and vent-associated habitats, such as chimneys and diffuse flows, into the rising plumes. Moreover, it has been simulated that turbulent kinetic energy and dissipation rate near the plume root are at maximum (Jiang and Breier, 2014), indicating strong turbulent mixing and entrainment of microorganisms in such regions. Owing to the short transport distance and low dilution with seawater, microbes derived from vent-associated habitats including various (hyper)thermophiles could remain at high relative abundances as found in our two samples (Figure 5).
The non-buoyant plumes show broadly similar microbial community structures to those of ambient seawater, but some significant differences in their microbial compositions were also observed. At high taxonomic level, the non-buoyant plumes and the surrounding seawater are primarily inhabited by γ-, α-, and ε-Proteobacteria, together with other relatively abundant bacterial lineages such as δ- Proteobacteria and Planctomycetes (Figure 4). The core bacterial lineages with high abundances in both non-buoyant plumes and ambient seawater consist of α-proteobacterial Rhodobacteraceae, δ-proteobacterial SAR324 clade, and γ-proteobacterial Alteromonadaceae, Alcanivoracaceae, and Saccharospirillaceae (Figure 5), all of which are ubiquitous in seawater (Meier et al., 2016). Statistical results of NMDS analysis on bacterial communities could barely distinguish non-buoyant hydrothermal plumes from the surrounding seawater column (Figure 3), supporting that microbial assemblages from the ambient seawater commonly populate non-buoyant plumes. This consistency of microbial compositions between seawater and non-buoyant plume is largely attributed to the extensive dilution by seawater as the estimated ratio of seawater to vent fluid in non-buoyant plumes is > 104:1 (Lupton et al., 1985; Dick et al., 2013; Jiang and Breier, 2014). This would indicate that non-buoyant plumes usually inherit the majority of their microbial assemblages from the surrounding seawater (for example, CTD02.8 and CTD02.9), while most vent-originated microbial signals are largely weak, and even completely depleted, because of their distance from the vents. Apart from the above mentioned water-column microorganisms, a characteristic of non-buoyant plumes is the high abundances of ε-proteobacterial Sulfurimonas and γ-proteobacterial SUP05 clade which are primarily niche-specific to areas that are hydrothermally active (Sheik et al., 2015; Sunamura and Yanagawa, 2015). One interesting thing to note is that although members of both Sulfurimonas and SUP05 clade were retrieved from all of three non-buoyant plumes, they dominated the bacterial communities exclusively in sample CTD02.7 which had the lowest CH4 concentration among the three non-buoyant plume samples. Moreover, it was also shown that microbial community structures among these three samples were not homogenous. It is likely due largely to the progressive variation of chemical compositions within non-buoyant plumes. In addition, seawater above the plume, such as CTD02.5 and CTD02.6, was also populated by members of Sulfurimonas and the SUP05 clade, indicating a potential plume-water column connection. There are some processes facilitating the exportation of non-buoyant plume microorganisms to contiguous seawaters (Dick et al., 2013), e.g., mesoscale eddies (Adams et al., 2011), transportation of ascending and descending plume particles (Cowen et al., 2001) and transparent exopolymer particles (TEP) (Shackelford and Cowen, 2006; Prieto and Cowen, 2007).
Sulfurimonas are prevalent in non-buoyant hydrothermal plumes. Although the SUP05 clade is usually detected as the most abundant and universal organismal group in deep-sea hydrothermal plumes worldwide (Sunamura and Yanagawa, 2015), Sulfurimonas became the dominant lineage populating the non-buoyant plumes of Longqi vent field on the SWIR (Figure 5). Members of the Sulfurimonas are putative chemolithoautotrophic, mesophilic sulfur and/or hydrogen oxidizers, and unlike SUP05 clade, they are ubiquitously distributed over diverse hydrothermal habitats including plumes, chimneys, sediments, and diffuse-flow fluids (Nakagawa et al., 2005; Campbell et al., 2006; Sheik et al., 2015; Mino et al., 2017). However, as seen in other hydrothermal fields including Guaymas Basin (Dick and Tebo, 2010), Lau Basin (Sheik et al., 2015; Anantharaman et al., 2016), Mid Cayman Rise (German et al., 2010), and Suiyo Seamount (Sunamura et al., 2004), Sulfurimonas are rarely detected with high abundances at distance from the vent orifices. Only few studies have determined the declining relative abundances of Sulfurimonas from vent-associated niches to the buoyant portions of the plumes (Nakagawa et al., 2005; Sheik et al., 2015). This is not surprised given the fact that there have been only limited investigations into non-buoyant hydrothermal plumes so far. Recently, the hydrothermal plumes of Gakkel Ridge were found to have high abundances of Sulfurimonas (Molari et al., 2017), indicating that the non-buoyant plumes of that location are excellent potential habitats for Sulfurimonas. The prevalence of Sulfurimonas was likely related to the high H2 concentrations (∼2.1–15.8 nM) in the non-buoyant plumes (Wang et al., 2015) being favorable to their growth as they are well known H2 oxidizers (Han and Perner, 2015). Members of the Sulfurimonas have demonstrated a strong ability to adapt to a range of environmental conditions, including cold seeps (Niemann et al., 2013), pelagic redoxcline (Grote et al., 2012) and terrestrial provinces (Lahme et al., 2019). In addition to the non-buoyant plumes above Longqi vent field, members of Sulfurimonas and SUP05 clade were also detected in the CTD01 water column samples (Figure 5) which we considered as normal background seawater, free of influence from hydrothermal activity. One possibility is that the non-buoyant plumes of CTD02 water column above the Longqi field spread horizontally out to the location of CTD01 station, and as a result, non-buoyant plume-associated microorganisms were transported there. Although there is an approximately 350 km distance between two sampling locations, it has been confirmed that non-buoyant hydrothermal plumes could spread out horizontally over more than 4,000 km from their hydrothermal vent source (Fitzsimmons et al., 2017). In such circumstance, membership of the Sulfurimonas and SUP05 clade, especially the latter which has a worldwide distribution (Sheik et al., 2015; Sunamura and Yanagawa, 2015; Dick, 2019), are good biosignatures to indicate non-buoyant hydrothermal plumes and hydrothermally impacted seawater.
The microbial communities of rising buoyant plumes are characterized by the biosignatures of both vent-associated communities and indicative populations of non-buoyant plume. Those putative seawater-derived microbial linages such as α-proteobacterial SAR11 clade and Sphingomonadaceae, δ-proteobacterial SAR324 clade and the most dominant archaeal lineages were consistently prevalent in our samples (Figures 5, 6), indicating a significant contribution by seawater dilution to the populations of the rising buoyant plumes. However, when compared with the root stage and non-buoyant plumes, the rising buoyant portion of the plumes showed an apparent divergence in microbial community structures (Figures 3A,B). On the one hand, putative colonizers of vent-associated niches occurred universally within the rising buoyant plumes. For example, a few sulfur-oxidizing populations such as ε-proteobacterial Sulfurovum, Sulfurospirillum, and γ-proteobacterial Thiotrichaceae and Thiomicrospiraceae, and typical iron-oxidizing bacteria affiliated with the ζ-Proteobacteria and the β-proteobacterial Gallionellaceae, which are ubiquitously distributed in chimneys, microbial mats, animals, and surrounding subsurface (Dick, 2019 and references therein), are commonly detected from rising plumes. However, their relative abundances were very low, usually less than 1%, compared with those of vent-associated habitats (Figure 5 and Supplementary Table S3). This is largely attributed to the relatively large distances to the vents, meaning greater dilution with seawater and less entrainment of vent inhabitants. Such inference is also supported by the similar distributions of the (hyper)thermophilic microorganisms mentioned above which are good indicators for hot, vent-associated habitats (Perner et al., 2010). On the other hand, Sulfurimonas and SUP05 clade, the principal indicator lineages in non-buoyant plumes, were also prevalent in the three rising buoyant plumes (Figure 5). As revealed by simulation modeling (Jiang and Breier, 2014), the rising plumes constantly entrain seawater from their vicinities by inward flows, simultaneously accompanied by rapid ascent and extensive seawater dilution. In such drastically dynamic plumes, the flourishing of the SUP05 clade and Sulfurimonas depends on persistent entrainment from nearby seawaters since it is impossible for them to grow and reproduce rapidly enough in situ to keep pace with quick and extensive seawater dilution (Sheik et al., 2015; Djurhuus et al., 2017). In other words, the seawater surrounding rising buoyant plumes necessarily harbor SUP05 clade and Sulfurimonas members with similar relative abundances. This is supported by the microbial community structure of the bottom seawater (NBS_100) collected from Longqi field, in which contain SUP05 clade and Sulfurimonas in proportions similar to the rising plumes (Figure 5 and Supplementary Table S3), thus clustering together into a tight statistical group (Figure 3). The Sulfurimonas and SUP05 clade members are conveyed into the non-buoyant plumes along these rising buoyant plumes serving as the linkage between vent-associated environments (seawater included) and the non-buoyant plumes.
Succession of Microbial Communities Along the Dispersal Pathway of Hydrothermal Plumes
Microbial community evolution and succession along a dispersing hydrothermal plume can be depicted based on the characterization of microbial communities at different plume developmental stages, indicating a spatially significant variation in microbial community compositions. Results of NMDS statistical analysis reveal that the samples collected relatively close to the discharge origins of hydrothermal vents are distinct from those of non-buoyant plumes and the ambient seawater column taken by CTD sampler (Figure 3). A clear shift in microbial communities was observed along the dispersal path. In the root segments and surrounding near-bottom seawater, microbial populations were characterized by particularly abundant and diverse putative vent-associated microbial communities consisting of (hyper)thermophiles, and ε- and γ-proteobacterial chemolithoautotrophs. By sharp contrast, the relative abundances of such vent-associated microbial lineages significantly decreased in the rising buoyant plumes and were barely detectable in the non-buoyant plumes (Supplementary Table S3). Meanwhile, although dominant hydrothermal microbial inhabitants are mainly replaced by universal seawater microorganisms, several putative plume-specialized bacterial lineages such as γ-proteobacterial SUP05 clade and ε-proteobacterial Sulfurimonas were greatly increased. Microbial succession of hydrothermal plumes along the dispersal pathway is likely due to progressive dilution by the surrounding seawater (Sheik et al., 2015; Sunamura and Yanagawa, 2015; Djurhuus et al., 2017). From the initial discharge out of seafloor orifices, the rising and development of hydrothermal plumes are constantly accompanied by dilution with ambient seawater until the final non-buoyant stage (Jiang and Breier, 2014). At the root of the plumes, intense turbulence and vortices or eddies induced by the mixing between the discharged fluids and seawater act to transfer and/or entrain microorganisms from vent-associated environments such as subsurface, seafloor, and chimney into the rising plumes (Dick et al., 2013; Jiang and Breier, 2014; Sheik et al., 2015). However, due to extensive physical mixing and dilution with the surrounding ambient seawater, niches within hydrothermal plumes are immeasurably distinct from those of the surrounding vents. That means plumes are not habitable for most vent-associated microorganisms which are unable to survive, grow and reproduce due to the rigorously metabolic and physiological challenges (Sheik et al., 2015; Djurhuus et al., 2017). In such cases, known vent-associated microbial taxa conservatively function as tracers to indicate the dispersal path and dilution degree of hydrothermal fluids as they are carried into plumes, and are quickly reduced in abundances as plumes rise and dilute. Here we plotted the relative abundances of typical vent-associated microbial lineages according to plume dispersal positions (Figure 7). Thermophilic and hyperthermophilic microbial lineages, chemolithoautotrophs (mainly from ε- and γ-Proteobacteria) and vent fauna-related bacterial symbionts present an approximate distance-decay relationship in relative abundances with distance to the vent orifices, supporting the indication that dilution by seawater exerts an important role in regulating microbial distribution and succession. In addition, in view of the distribution of these vent-associated microorganisms, it is also indicated that most vent-associated microbial populations are only dispersed for a limited distance along the rising segment of the hydrothermal plumes. The rising plume acts as a conveyor to deliver SUP05 clade and Sulfurimonas members from vent-associated environments and bottom seawaters to the final non-buoyant plume which favors their growth.
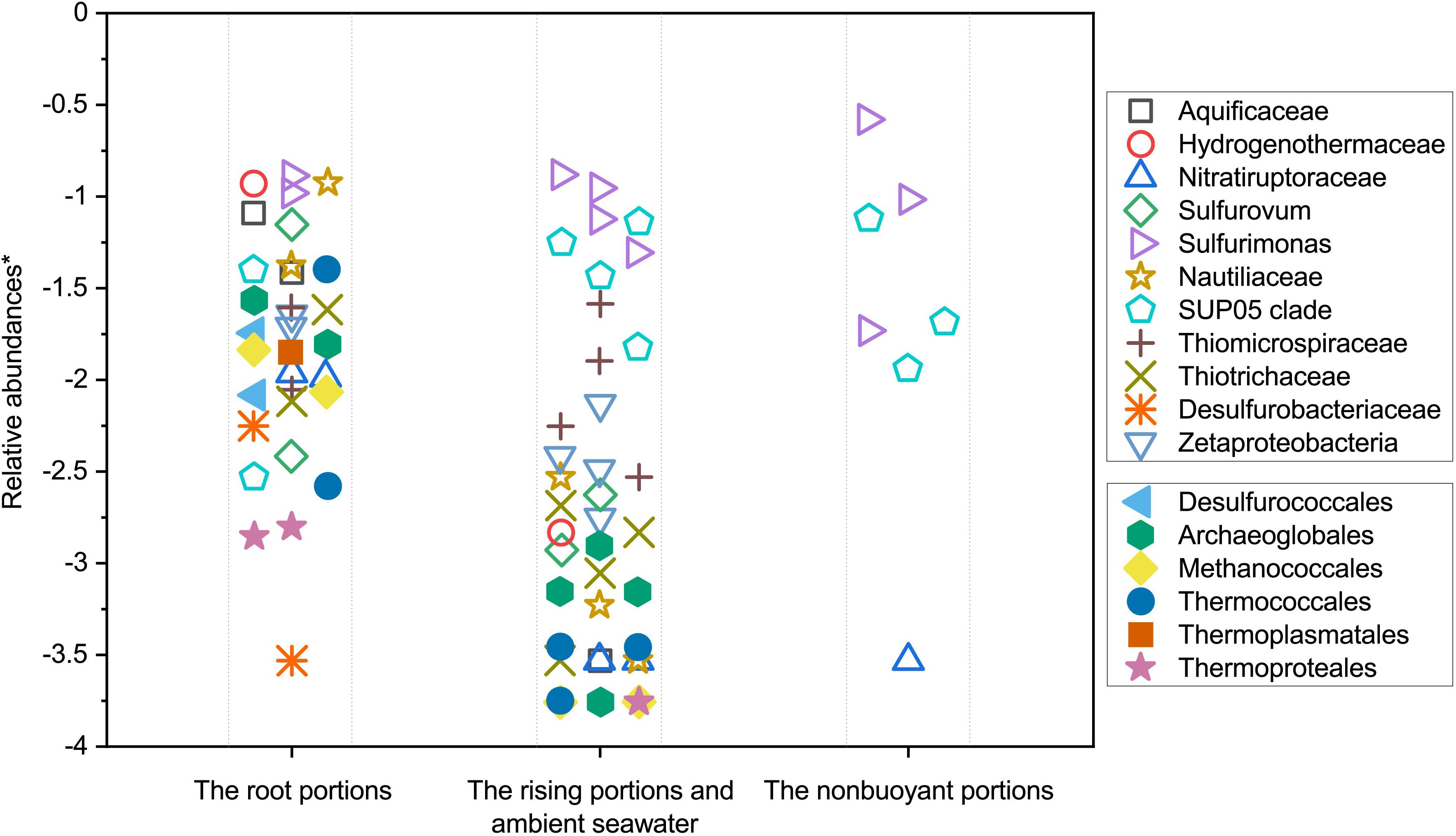
Figure 7. Variation in relative abundances of typical vent-derived and plume-specialized lineages among the different stages (root, buoyant, and non-buoyant) of hydrothermal plumes roughly corresponding to the increasing distances from the vent origin. ∗ The values were calculated by taking the log of the relative abundances.
Conclusion
In this study, we collected samples from ambient seawater and from distinct hydrothermal plume stages from the Southwest Indian Ridge. Hydrothermal plumes were sampled from the Longqi hydrothermal field and represented the different formation stages including the initial root segment, the rising buoyant portion, and the final non-buoyant plume. Our results show that there is a significant shift in microbial community compositions along the dispersal path of hydrothermal plumes. As hydrothermal plumes ascend away from the discharge orifices, vent-associated microbial populations are continually diluted and removed, while seawater-derived microorganisms increase with constant input from ambient seawater, suggesting that along the development path of a hydrothermal plume, dilution with seawater exerts an important role in regulating microbial composition, distribution, and succession. The Sulfurimonas and SUP05 clade are signal lineages of plumes at all different formation stages, and comparatively, the Sulfurimonas is more abundant and prevailing. We also found that the non-buoyant plumes of Longqi hydrothermal field can spread out horizontally for a great distance, as supported by the detection of Sulfurimonas and SUP05 clade members from a water column ∼350 km away where no hydrothermal activity is hosted.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject (PRJNA638507 and PRJNA638524).
Author Contributions
JL designed the experiments and took the samples. JY, MS, LS, and JG were responsible for the microbiological experiments. HW measured the concentrations of methane from water column. JL and SB treated and analyzed the sequence data. JL wrote the manuscript with contributions from all coauthors. All the authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (NSFC, Nos. 41373071 and 42072333).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Qunhui Yang for providing the water samples of 2 CTD water columns for this study. We thank all the crew and scientists on the DY115-30 cruise and their supports were essential.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.581381/full#supplementary-material
Footnotes
References
Adams, D. K., Mcgillicuddy, D. J., Zamudio, L., Thurnherr, A. M., Liang, X., Rouxel, O., et al. (2011). Surface-generated mesoscale eddies transport deep-sea products from hydrothermal vents. Science 332, 580–583. doi: 10.1126/science.1201066
Anantharaman, K., Breier, J. A., and Dick, G. J. (2016). Metagenomic resolution of microbial function in deep-sea hydrothermal plumes across the Eastern Lau Spreading Center. ISME J. 10, 225–239. doi: 10.1038/ismej.2015.81
Bach, W., Banerjee, N. R., Dick, H. J. B., and Baker, E. T. (2002). Discovery of ancient and active hydrothermal systems along the ultra-slow spreading Southwest Indian Ridge 10°–16°E. Geochem. Geophys. Geosyst. 3, 1–14. doi: 10.1029/2001GC000279
Baker, B. J., Lesniewski, R. A., and Dick, G. J. (2012). Genome-enabled transcriptomics reveals archaeal populations that drive nitrification in a deep-sea hydrothermal plume. ISME J. 6, 2269–2279. doi: 10.1038/ismej.2012.64
Baker, E. R., German, C. R., and Elderfield, H. (1995). “Hydrothermal plumes over spreading-center axes: global distributions and geological inferences,” in Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions, Geophys. Monogr. Ser, Vol. 91, eds S. Humphris, R. A. Zierenberg, L. S. Mullineaux, and R. E. Thomson, (Washington, DC: AGU), 47–71. doi: 10.1029/gm091p0047
Campbell, B. J., Summers Engel, A., Porter, M. L., and Takai, K. (2006). The versatile ε-proteobacteria. Nat. Rev. Microbiol. 4, 458–468.
Castelle, C., Wrighton, K., Thomas, B., Hug, L. A., Brown, C. T., Wilkins, M. J., et al. (2015). Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701. doi: 10.1016/j.cub.2015.01.014
Copley, J. T., Marsh, L., Glover, A. G., Hühnerbach, V., Nye, V. E., Reid, W. D. K., et al. (2016). Ecology and biogeography of megafauna and macrofauna at the first known deep-sea hydrothermal vents on the ultraslow-spreading Southwest Indian Ridge. Sci. Rep. 6:39158. doi: 10.1038/sre39158
Cowen, J. P., Bertram, M. A., Wakeham, S. G., Thomson, R. E., Lavelle, J. W., Baker, E. T., et al. (2001). Ascending and descending particle flux from hydrothermal plumes at Endeavour Segment, Juan de Fuca Ridge. Deep Sea Res. Part I: Oceanogr. Res. Pap. 48, 1093–1120. doi: 10.1016/s0967-0637(00)00070-4
Dick, G. J. (2019). The microbiomes of deep-sea hydrothermal vents: distributed globally, shaped locally. Nat. Rev. Microbiol. 17, 271–283. doi: 10.1038/s41579-019-0160-2
Dick, G. J., Anantharaman, K., Baker, B. J., Li, M., Reed, D. C., and Sheik, C. S. (2013). The microbiology of deep-sea hydrothermal vent plumes: ecological and biogeographic linkages to seafloor and water column habitats. Front. Microbiol. 4:124. doi: 10.3389/fmicb.2013.00124
Dick, G. J., Clement, B. G., Fodrie, F. J., Webb, S. M., Bargar, J. R., and Tebo, B. M. (2009). Enzymatic microbial Mn(II) oxidation and Mn biooxide production in the Guaymas Basin hydrothermal plume. Geochim. Cosmochim. Acta 73, 6517–6530. doi: 10.1016/j.gca.2009.07.039
Dick, G. J., and Tebo, B. M. (2010). Microbial diversity and biogeochemistry of the Guaymas Basin deepsea hydrothermal plume. Environ. Microbiol. 12, 1334–1347. doi: 10.1111/j.1462-2920.2010.02177.x
Djurhuus, A., Mikalsen, S.-O., Giebel, H.-A., and Rogers, A. D. (2017). Cutting through the smoke: the diversity of microorganisms in deep-sea hydrothermal plumes. R. Soc. Open Sci. 4:160829. doi: 10.1098/rsos.160829
Elderfield, H., and Schultz, A. (1996). Mid-ocean ridge hydrothermal fluxes and the chemical composition of the ocean. Annu. Rev. Earth Planet. Sci. 24, 191–224. doi: 10.1146/annurev.earth.24.1.191
Fitzsimmons, J. N., John, S. G., Marsay, C. M., Hoffman, C. L., Nicholas, S. L., Toner, B. M., et al. (2017). Iron persistence in a distal hydrothermal plume supported by dissolved–particulate exchange. Nat.Geosci. 10, 195–201. doi: 10.1038/ngeo2900
Gartman, A., Findlay, A. J., and Luther, G. W. I. I. I. (2014). Nanoparticulate pyrite and other nanoparticles are a widespread component of hydrothermal vent black smoker emissions. Chem. Geol. 366, 32–41. doi: 10.1016/j.chemgeo.2013.12.013
German, C. R., Baker, E. T., Mevel, C., and Tamaki, K. the Fuji Scientific Team (1998). Hydrothermal activity along the Southwest Indian Ridge. Nature 395, 490–493. doi: 10.1038/26730
German, C. R., Bowen, A., Coleman, M. L., Honig, D. L., Huber, J. A., Jakuba, M. V., et al. (2010). Diverse styles of submarine venting on the ultraslow spreading Mid-Cayman Rise. Proc. Natl. Acad. Sci. U.S.A. 107, 14020–14025. doi: 10.1073/pnas.1009205107
German, C. R. (2003). Hydrothermal activity on the eastern Southwest Indian Ridge (50°-70°E): Evidence from core-top geochemistry, 1887 and 1998. Geochem. Geophys. Geosyst. 4:9102. doi: 10.1029/2003GC000522
Grote, J., Schott, T., Bruckner, C. G., Glöckner, F. O., Jost, G., Teeling, H., et al. (2012). Genome and physiology of a model ε-proteobacterium responsible for sulfide detoxification in marine oxygen depletion zones. Proc. Natl. Acad. Sci. U.S.A. 109, 506–510. doi: 10.1073/pnas.1111262109
Han, Y., and Perner, M. (2015). The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 6:989. doi: 10.3389/fmicb.2015.00989
Helfrich, K. R., and Speer, K. G. (1995). Ocean hydrothermal circulation: mesoscale and basin-scale flow. Geophys. Monogr. 91, 347–356. doi: 10.1029/gm091p0347
Holden, J., Breier, J., Rogers, K., Schulte, M., and Toner, B. (2012). Biogeochemical processes at hydrothermal vents: microbes and minerals, bioenergetics, and carbon fluxes. Oceanography 25, 196–208. doi: 10.5670/oceanog.2012.18
Huber, J. A., Butterfield, D. A., and Baross, J. A. (2002). Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat. Appl. Environ. Microbiol. 68, 1585–1594. doi: 10.1128/aem.68.4.1585-1594.2002
Jackson, P. R., Ledwell, J. R., and Thurnherr, A. M. (2010). Dispersion of a tracer on the East Pacific Rise (9°N to 10°N), including the influence of hydrothermal plumes. Deep Sea Res. Part I Oceanogr. Res. Pap. 57, 37–52. doi: 10.1016/j.dsr.2009.10.011
Jiang, H., and Breier, J. A. (2014). Physical controls on mixing and transport within rising submarine hydrothermal plumes: a numerical simulation study. Deep Sea Res. Part I Oceanogr. Res. Pap. 92, 41–45. doi: 10.1016/j.dsr.2014.06.006
Kato, S., Kobayashi, C., Kakegawa, T., and Yamagishi, A. (2009). Microbial communities in iron-silica-rich microbial mats at deep-sea hydrothermal fields of the southern Mariana Trough. Environ. Microbiol. 11, 2094–2111. doi: 10.1111/j.1462-2920.2009.01930.x
Lahme, S., Enning, D., Callbeck, C. M., Menendez Vega, D., Curtis, T. P., Head, I. M., et al. (2019). Metabolites of an oil field sulfide-oxidizing, nitrate-reducing Sulfurimonas sp. cause severe corrosion. Appl. Env. Microbiol. 85:e01891-18.
Lam, P., Cowen, J. P., and Jones, R. D. (2004). Autotrophic ammonia oxidation in a deep-sea hydrothermal plume. FEMS Microbiol. Ecol. 47, 191–206. doi: 10.1016/s0168-6496(03)00256-3
Lesniewski, R. A., Jain, S., Anantharaman, K., Schloss, P. D., and Dick, G. J. (2012). The metatranscriptome of a deep-sea hydrothermal plume is dominated by water column methanotrophs and lithotrophs. ISME J. 6, 2257–2268. doi: 10.1038/ismej.2012.63
Li, J., Cui, J., Yang, Q., Cui, G., Wei, B., Wu, Z., et al. (2017). Oxidative weathering and microbial diversity of an inactive seafloor hydrothermal sulfide chimney. Front. Microbiol. 8:1378. doi: 10.3389/fmicb.2017.01378
Li, J., Su, L., Wang, F., Yang, J., Gu, L., Sun, M., et al. (2020). Elucidating the biomineralization of low-temperature hydrothermal precipitates with varying Fe, Si contents: indication from ultrastructure and microbiological analyses. Deep Sea Res. Part I. 157:103208. doi: 10.1016/j.dsr.2019.103208
Li, J., Zhou, H., Fang, J., Wu, Z., and Peng, X. (2016). Microbial distribution in a hydrothermal plume of the Southwest Indian Ridge. Geomicrobiol. J. 33, 401–415. doi: 10.1080/01490451.2015.1048393
Li, M., Jain, S., and Dick, G. J. (2016). Genomic and transcriptomic resolution of organic matter utilization among deep-sea bacteria in guaymas basin hydrothermal plumes. Front. Microbiol. 7:1125. doi: 10.3389/fmicb.2016.01125
Li, M., Baker, B. J., Anantharaman, K., Jain, S., Breier, J. A., and Dick, G. J. (2015). Genomic and transcriptomic evidence for scavenging of diverse organic compounds by widespread Deep-Sea Archaea. Nat. Commun. 6:8933. doi: 10.1038/ncomms9933
Li, M., Jain, S., Baker, B. J., Taylor, C., and Dick, G. J. (2014a). Novel hydrocarbon monooxygenase genes in the metatranscriptome of a natural deep-sea hydrocarbon plume. Environ. Microbiol. 16, 60–71. doi: 10.1111/1462-2920.12182
Li, M., Toner, B. M., Baker, B. J., Breier, J. A., Sheik, C. S., and Dick, G. J. (2014b). Microbial Iron Uptake as a mechanism for dispersing iron from deep-sea hydrothermal vents. Nat. Commun. 5:3192. doi: 10.1038/ncomms4192
Lin, J., and Zhang, C. (2006). The first collaborative China-international cruises to investigate mid-ocean ridge hydrothermal vents. InterRidge News 15, 33–34.
Liu, X., Li, M., Castelle, C. J., Probst, A. J., Zhou, Z., and Pan, J. (2018). Insights into the ecology, evolution, and metabolism of the widespread woesearchaeotal lineages. Microbiome 6:102.
Lupton, J. E., Delaney, J. R., Johnson, H. P., and Tivey, M. K. (1985). Entrainment and vertical transport of deep-ocean water by buoyant hydrothermal plumes. Nature 316, 621–623. doi: 10.1038/316621a0
Marbler, H., Koschinsky, A., Pape, T., Seifert, R., Weber, S., Baker, E. T., et al. (2010). Geochemical and physical structure of the hydrothermal plume at the ultramafic-hosted Logatchev hydrothermal field at 14°45’N on the Mid-Atlantic Ridge. Mar. Geol. 271, 187–197. doi: 10.1016/j.margeo.2010.01.012
McAllister, S. M., Moore, R. M., Gartman, A., Luther, G. W., Emerson, D., and Chan, C. S. (2019). The Fe(II)-oxidizing Zetaproteobacteria: historical, ecological and genomic perspectives. FEMS Microbiol. Ecol. 95:fiz015. doi: 10.1093/femsec/fiz015
McCollom, T. M. (2000). Geochemical constraints on primary productivity in submarine hydrothermal vent plumes. Deep Sea Res. Part IOceanogr. Res. Pap. 47, 85–101. doi: 10.1016/s0967-0637(99)00048-5
Meier, D. V., Bach, W., Girguis, P. R., Gruber-Vodicka, H. R., Reeves, E. P., Richter, M., et al. (2016). Heterotrophic Proteobacteria in the vicinity of diffuse hydrothermal venting. Environ. Microbiol. 18, 4348–4368. doi: 10.1111/1462-2920.13304
Mino, S., Nakagawa, S., Makita, H., Toki, T., Miyazaki, J., Sievert, S. M., et al. (2017). Endemicity of the cosmopolitan mesophilic chemolithoautotroph Sulfurimonas at deep-sea hydrothermal vents. ISME J. 11, 909–919. doi: 10.1038/ismej.2016.178
Molari, M., Wegener, G., McDermott, J., Bach, W., and Boetius, A. the Shipboard Parties of Rv Polarstern (2017). Microbial dynamics in deep-sea hydrothermal plumes of the ultraslow spreading Gakkel Ridge. Goldschmidt2017 Abstract
Münch, U., Lalou, C., Halbach, P., and Fujimoto, H. (2001). Relict hydrothermal events along the super-slow Southwest Indian spreading ridge near 63°56’E: mineralogy, chemistry and chronology of sulfide samples. Chem. Geol. 177, 341–349. doi: 10.1016/s0009-2541(00)00418-6
Nakagawa, S., Takai, K., Inagaki, F., Hirayama, H., Nunoura, T., Horikoshi, K., et al. (2005). Distribution, phylogenetic diversity and physiological characteristics of epsilon-Proteobacteria in a deep-sea hydrothermal field. Environ. Microbiol. 7, 1619–1632. doi: 10.1111/j.1462-2920.2005.00856.x
Niemann, H., Linke, P., Knittel, K., MacPherson, E., Boetius, A., Brückmann, W., et al. (2013). Methane-carbon flow into the benthic food web at cold seeps: a case study from the Costa Rica subduction zone. PLoS One 8:e74894. doi: 10.1371/journal.pone.0074894
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422. doi: 10.1128/mmbr.00039-10
Perner, M., Gonnella, G., Hourdez, S., Böhnke, S., Kurtz, S., and Girguis, P. (2013). In situ chemistry and microbial community compositions in five deep-sea hydrothermal fluid samples from Irina II in the Logatchev field. Environ. Microbiol. 15, 1551–1560. doi: 10.1111/1462-2920.12038
Perner, M., Petersen, J. M., Zielinski, F., Gennerich, H. H., and Seifert, R. (2010). Geochemical constraints on the diversity and activity of H2-oxidizing microorganisms in diffuse hydrothermal fluids from a basalt- and an ultramafic-hosted vent. FEMS Microbiol. Ecol. 74, 55–71. doi: 10.1111/j.1574-6941.2010.00940.x
Prieto, L., and Cowen, J. P. (2007). Transparent exopolymer particles in a deep-sea hydrothermal system: guaymas Basin. Gulf of California. Mar. Biol. 150, 1093–1101. doi: 10.1007/s00227-006-0430-1
Shackelford, R., and Cowen, J. P. (2006). Transparent exopolymer particles (TEP) as a component of hydrothermal plume particle dynamics. Deep Sea Res. Part I Oceanogr. Res. Pap. 53, 1677–1694. doi: 10.1016/j.dsr.2006.08.001
Sheik, C., Anantharaman, K., Breier, J., Sylvan, J. B., Edwards, K. J., and Dick, G. J. (2015). Spatially resolved sampling reveals dynamic microbial communities in rising hydrothermal plumes across a back-arc basin. ISME J. 9, 1434–1445. doi: 10.1038/ismej.2014.228
Sunamura, M., Higashi, Y., Miyako, C., Ishibashi, J., and Maruyama, A. (2004). Two bacteria phylotypes are predominant in the Suiyo Seamount hydrothermal plume. Appl. Environ. Microbiol. 70, 1190–1198. doi: 10.1128/aem.70.2.1190-1198.2004
Sunamura, M., and Yanagawa, K. (2015). “Microbial cell densities, community structures and growth in the hydrothermal plumes of subduction hydrothermal systems,” in Subseafloor Biosphere Linked to Hydrothermal System: TAIGA Concept, eds J.-I. Ishibashi, K. Okino, and M. Sunamura, (Berlin: Springer).
Sylvan, J. B., Pyenson, B. C., Rouxel, O., German, C. R., and Edwards, K. J. (2012). Time-series analysis of two hydrothermal plumes at 9 degrees 50°N East Pacific Rise reveals distinct, heterogeneous bacterial populations. Geobiology 10, 178–192. doi: 10.1111/j.1472-4669.2011.00315.x
Takai, K., Nakamura, K., Toki, T., Tsunogai, U., Miyazaki, M., Miyazaki, J., et al. (2008). Cell proliferation at 122°C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation. Proc. Natl Acad. Sci. U.S.A. 105, 10949–10954. doi: 10.1073/pnas.0712334105
Takai, K., Oida, H., Suzuki, Y., Hirayama, H., Nakagawa, S., Nunoura, T., et al. (2004). Spatial distribution of marine crenarchaeota group I in the vicinity of deep-sea hydrothermal systems. Appl. Environ. Microbiol. 70, 2404–2413. doi: 10.1128/aem.70.4.2404-2413.2004
Tao, C., Li, H., Deng, X., Lei, J., Wang, Y., Zhang, K., et al. (2014). “Hydrothermal Activity on ultraslow spreading ridge: new hydrothermal fields found on the Southwest Indian ridge,” in 2014 AGU Fall Meeting, (Washington, DC: American Geophysical Union).
Tao, C., Lin, J., Guo, S., Chen, Y., Wu, G., Han, X., et al. (2012). First active hydrothermal vents on an ultraslow-spreading center: southwest Indian Ridge. Geology 40, 47–50. doi: 10.1130/g32389.1
Vander Roost, J., Daae, F. L., Steen, I. H., Thorseth, I. H., and Dahle, H. (2018). Distribution patterns of iron-oxidizing Zeta- and Beta-Proteobacteria from different environmental settings at the Jan Mayen Vent Fields. Front. Microbiol. 9:3008. doi: 10.3389/fmicb.2018.03008
Wang, H., Yan, Q., Yang, Q., Ji, F., Wonh, K. H., and Zhou, H. (2019). The size fractionation and speciation of iron in the Longqi hydrothermal plumes on the Southwest Indian Ridge. J.Geophys. Res. Oceans 124, 4029–4043. doi: 10.1029/2018JC014713
Wang, H., Zhou, H., Yang, Q., Lilley, M. D., Wu, J., and Ji, F. (2015). Development and application of a gas chromatography method for simultaneously measuring H2 and CH4 in hydrothermal plume samples. Limnol. Oceanogr. Methods 13, 722–730. doi: 10.1002/lom3.10061
Wang, T., Chen, Y. J., and Tao, C. (2011). Revisit the K-segment of the Southeast Indian Ridge for new evidence of hydrothermal plumes. Chin. Sci. Bull. 56, 3605–3609. doi: 10.1007/s11434-011-4723-5
Zhou, H., and Dick, H. (2013). Thin crust as evidence for depleted mantle supporting the marion rise. Nature 494, 195–200. doi: 10.1038/nature11842
Zhou, Y., Zhang, D., Zhang, R., Liu, Z., Tao, C., Lu, B., et al. (2018). Characterization of vent fauna at three hydrothermal vent fields on the Southwest Indian Ridge: implications for biogeography and interannual dynamics on ultraslow-spreading ridges. Deep Sea Rese. Part I Oceanogr. Res. Pap. 137, 1–12. doi: 10.1016/j.dsr.2018.05.001
Keywords: hydrothermal plumes, the rising buoyant and non-buoyant plume, microbial spatial distribution and succession, Sulfurimonas, SUP05 clade, the Southwest Indian Ridge
Citation: Li J, Yang J, Sun M, Su L, Wang H, Gao J and Bai S (2020) Distribution and Succession of Microbial Communities Along the Dispersal Pathway of Hydrothermal Plumes on the Southwest Indian Ridge. Front. Mar. Sci. 7:581381. doi: 10.3389/fmars.2020.581381
Received: 08 July 2020; Accepted: 15 October 2020;
Published: 05 November 2020.
Edited by:
Cinzia Corinaldesi, Marche Polytechnic University, ItalyReviewed by:
Meng Li, Shenzhen University, ChinaMarianne Quéméneur, UMR 7294 Institut Méditerranéen d’Océanographie (MIO), France
Copyright © 2020 Li, Yang, Sun, Su, Wang, Gao and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijie Bai, YmFpc2hpamllQGlkc3NlLmFjLmNu