 Kerstin B. J. Glaus1*
Kerstin B. J. Glaus1* Sharon A. Appleyard2†
Sharon A. Appleyard2† Brian Stockwell1†
Brian Stockwell1† Juerg M. Brunnschweiler3
Juerg M. Brunnschweiler3 Mahmood Shivji4
Mahmood Shivji4 Eric Clua5
Eric Clua5 Amandine D. Marie1,6
Amandine D. Marie1,6 Ciro Rico1,7
Ciro Rico1,7- 1School of Marine Studies, Faculty of Science, Technology and Environment, The University of the South Pacific, Suva, Fiji
- 2CSIRO National Research Collections Australia, Australian National Fish Collection, Hobart, TAS, Australia
- 3Independent Researcher, Zurich, Switzerland
- 4Save Our Seas Foundation Shark Research Center, Nova Southeastern University, Fort Lauderdale, FL, United States
- 5PSL Research University, Labex CORAIL, CRIOBE USR 3278 CNRS-EPHE-UPVD, Université de Perpignan, Perpignan, France
- 6ESE, Ecology and Ecosystems Health, Agrocampus Ouest, INRAE, Rennes, France
- 7Instituto de Ciencias Marinas de Andalucía (ICMAN), Consejo Superior de Investigaciones Científicas, Puerto Real, Spain
The bull shark (Carcharhinus leucas) is a large, mobile, circumglobally distributed high trophic level predator that inhabits a variety of remote islands and continental coastal habitats, including freshwater environments. Here, we hypothesize that the barriers to dispersal created by large oceanic expanses and deep-water trenches result in a heterogeneous distribution of the neutral genetic diversity between island bull shark populations compared to populations sampled in continental locations connected through continuous coastlines of continental shelves. We analyzed 1,494 high-quality neutral single nucleotide polymorphism (SNP) markers in 215 individual bull sharks from widespread locations across the Indian and Pacific Oceans (South Africa, Indonesia, Western Australia, Papua New Guinea, eastern Australia, New Caledonia, and Fiji). Genomic analyses revealed partitioning between remote insular and continental populations, with the Fiji population being genetically different from all other locations sampled (FST = 0.034–0.044, P < 0.001), and New Caledonia showing marginal isolation (FST = 0.016–0.024, P < 0.001; albeit based on a small sample size) from most sampled sites. Discriminant analysis of principal components (DAPC) identified samples from Fiji as a distinct cluster with all other sites clustering together. Genetic structure analyses (Admixture, fastStructure and AssignPOP) further supported the genetic isolation of bull sharks from Fiji, with the analyses in agreement. The observed differentiation in bull sharks from Fiji makes this site of special interest, as it indicates a lack of migration through dispersal across deep-water trenches and large ocean expanses.
Introduction
Reduced genetic variation, population isolation and genetic drift may lead to genetic divergence and speciation among terrestrial organisms inhabiting islands (Grant, 1985). Assessing genetic structure among island demes can help in identifying small, genetically differentiated, and spatially isolated populations (Allendorf et al., 2008). While terrestrial organisms inhabiting oceanic islands generally show significant levels of genetic differentiation when compared to their continental conspecifics, marine taxa with large dispersal capabilities are expected to show little divergence as their habitats appear to be continuous (Roberts, 1997). However, growing evidence suggests that biogeographic barriers, produced for example by ocean currents (Hare et al., 2005), changes in sea surface temperature (Kelly and Eernisse, 2007), physical barriers such as oceanographic fronts (Galarza et al., 2009), and resource availability (Palumbi, 1994) can impede gene flow (Cowman and Bellwood, 2013) in some marine taxa (Cowen et al., 2000; Leis, 2002; Hauser and Carvalho, 2008). Spatial distribution of the genetic variation to establish connectivity patterns among marine taxa has been subject of extensive research in recent years (Cowen et al., 2000; Cowen and Sponaugle, 2009; Crandall et al., 2019), including sharks (e.g., Momigliano et al., 2017). However, data is limited when it comes to the quantification of population structure in coastal shark species inhabiting both island and continental coastal waters. Such studies can be vital for the identification of management units and metapopulations. For example, an analysis examining the population structure of the tiger shark on a global scale using a suite of mitochondrial and nuclear (microsatellites) genetic markers revealed population isolation for the Hawaiian Islands (Bernard et al., 2016).
Overexploitation has led to drastic declines in shark populations worldwide (Dulvy et al., 2014). In general, various studies indicate that many commercially exploited shark species have a low intrinsic rebound potential due to their low fecundity and slow growth rates, and have further been found to have low genetic diversity and smaller population sizes compared with other marine taxa (Martin et al., 1992; Daly-Engel et al., 2010; Karl et al., 2011; Vignaud et al., 2013). Additionally, coastal sharks are vulnerable to increased mortality rates due to their proximity to human populations, where fishing pressure is typically higher and habitat degradation is more pervasive. Therefore, identifying the genetic structuring of populations is fundamental for determining whether, and to what degree, population differentiation exists. Furthermore, this information is essential for understanding the state of subpopulations that need to be treated as separate management units (Palsbøll et al., 2007).
While global surveys examining broad scale patterns of genetic differentiation confirmed connectivity within and between ocean basins in some highly migratory pelagic sharks (Hoelzel et al., 2006; Castro et al., 2007; Sequeira et al., 2013), oceanic expanses can cause genetic population structure in coastal sharks (Schultz et al., 2008; Ahonen et al., 2009; Ovenden et al., 2009; Portnoy et al., 2010; Momigliano et al., 2017). For example, sicklefin lemon shark populations, Negaprion acutidens, in Australia and French Polynesia, separated by oceanic distances of at least 750 km across the Tonga trench with a maximum depth of over 10,000 m (van der Hilst, 1995), show moderate but statistically significant genetic differentiation (FST = 0.070–0.087, p < 0.001), with South Pacific archipelagos probably serving as stepping stones for rare dispersal events (Schultz et al., 2008). In addition, to oceanic expanses, physical barriers such as fronts, eddies, gyres and other oceanographic factors can prevent dispersal. In some tropical and subtropical shark species, restricted gene flow is associated with cool thermal barriers such as the Benguela upwelling (Duncan et al., 2006; Keeney and Heist, 2006). For example, in the blacktip shark, Carcharhinus limbatus, the cold Benguela upwelling along the south-West coast of Africa restricts contemporary female-mediated gene flow between Atlantic and Indo-Pacific populations (Keeney et al., 2005; Keeney and Heist, 2006; Sodré et al., 2012). Contrastingly, the Benguela upwelling appears not to be a barrier to the temperate copper shark, C. brachyurus, as evidenced by a lack of genetic differentiation between Namibia and South Africa (Benavides et al., 2011). Additionally, coastal sharks may be limited by their habitat, exhibiting fidelity to discrete locations for feeding, mating, parturition, maturation and migratory routes (Castro, 1983; Simpfendorfer and Milward, 1993; Heupel et al., 2007; Speed et al., 2010; Brunnschweiler and Baensch, 2011). For example, increasing evidence suggests that many coastal sharks have strong philopatric behavior (Karl et al., 2011; Chapman et al., 2015). The degree of segregation of these sites, and site fidelity (philopatry) can directly affect the level of population subdivision and genetic divergence among geographic regions (Keeney et al., 2005).
Reproductive philopatry has been described in scalloped hammerheads, Sphyrna lewini (Duncan et al., 2006), blacktips (Keeney et al., 2005), and bull sharks, Carcharhinus leucas (Karl et al., 2011; Tillett et al., 2012). Furthermore, natal philopatry and long-term fidelity to parturition sites has been documented, for the first time in any shark species at Bimini, Bahamas (Feldheim et al., 2014). Restricted maternal gene flow between populations caused by female site fidelity may result in small local effective population size and thus increased vulnerability to exploitation (Duncan and Holland, 2006; Duncan et al., 2006; Nance et al., 2011). Thus, overfishing may have detrimental effects on large coastal shark species due to limited exchange with other regional populations.
One of the most iconic coastal apex predators is the bull shark (C. leucas; Müller and Henle, 1839). This large shark is globally distributed in warm, temperate, tropical and subtropical waters (Compagno, 2001). Evidence based on highly restricted maternal gene flow suggests site-fidelity and reproductive philopatry (Tillett et al., 2012), while tracking studies documented large-scale movements of up to 1,500 km in individual bull sharks (Carlson et al., 2010; Daly et al., 2014; Heupel et al., 2015; Lea et al., 2015). For example, a recent study documented that a substantial segment of a Great Barrier Reef bull shark population displayed long migrations of up to 1,400 km to other coral reefs and/or inshore coastal habitats along the Australian east coast (Espinoza et al., 2016). Microsatellite markers revealed, for the first time, genetic structuring in West Atlantic bull shark populations (Karl et al., 2011) and strong genetic differentiation between bull sharks from the West North Atlantic and Indo-Pacific, with significant genetic structuring in specimens from Fiji (Testerman, 2014). Similarly, a more recent study by Pirog et al. (2019) utilized both mitochondrial and microsatellites DNA markers in 11 sites across the Western Atlantic, Western Pacific and Western Indian Ocean and found clustering between the Western Atlantic and those from the Indian and Pacific Oceans. However, given that the bull shark is a coastal species (Brunnschweiler et al., 2010), it seems likely that barriers to gene flow exist within its range, which includes distant oceanic islands separated by hundreds of kilometers of open ocean.
New methodological advances such as co-dominant, genome-wide single nucleotide polymorphism (SNP) loci, have the potential to improve resolution in the estimation of population structuring, migration rates, dispersal, and population assignment (Morin et al., 2004; Benestan et al., 2015; Gagnaire et al., 2015; Selkoe et al., 2016). Here, we utilize SNP markers to test the hypothesis that barriers to dispersal created by large oceanic expanses and deep-water trenches result in a heterogeneous distribution of neutral genetic diversity between island bull shark populations compared to demes sampled in continental locations and connected through continuous coastline of continental shelves.
To provide a broad geographic framework for our study, and to place the findings in the context of existing knowledge on bull shark population structure, we obtained samples from South Africa, Indonesia, Western Australia, Papua New Guinea, eastern Australia, New Caledonia and Fiji. Specific aims of the study were: i) to compare inbreeding coefficients and levels of heterozygosity of bull sharks within the sampled range, and ii) to assess the population structure in bull sharks on a genomic scale using SNPs. This study contributes to the understanding of genetic patterns of insular – coastal shark populations that inhabit historically understudied regions.
Materials and Methods
Ethics Statement
All shipping procedures of shark tissue samples were conducted under the relevant import and export permits issued by The Australian Government, Department of Agriculture and Water Resources to Diversity Arrays Technology Pty Ltd., Canberra, Australia and the Ministry of Fisheries and Forests in Fiji. Bull shark tissues from Papua New Guinea came from the CSIRO/ACIAR/PNG-NFA FIS/2012/102 “Sustainable management of the shark resources of Papua New Guinea: socioeconomic and biological characteristics of the fishery.” Handling of live shark specimens were approved under the Research Permit issued by the Fiji Ministry of Education, Heritage and Arts to Beqa Adventure Divers and performed in accordance with relevant guidelines and regulations.
Sampling Procedures
Bull shark samples (white muscle and fin clips, 1 cm2) were obtained from fisheries independent surveys, observers on board commercial fleets, and recreational and artisanal fishers from 2004 to 2017. The samples of 215 individual bull sharks came from seven locations (defined as a priori populations, hereafter) and six countries across the Indian and Pacific Oceans (Figure 1, Table 1). All samples were stored in 95% ethanol until DNA extraction, library preparation and sequencing.
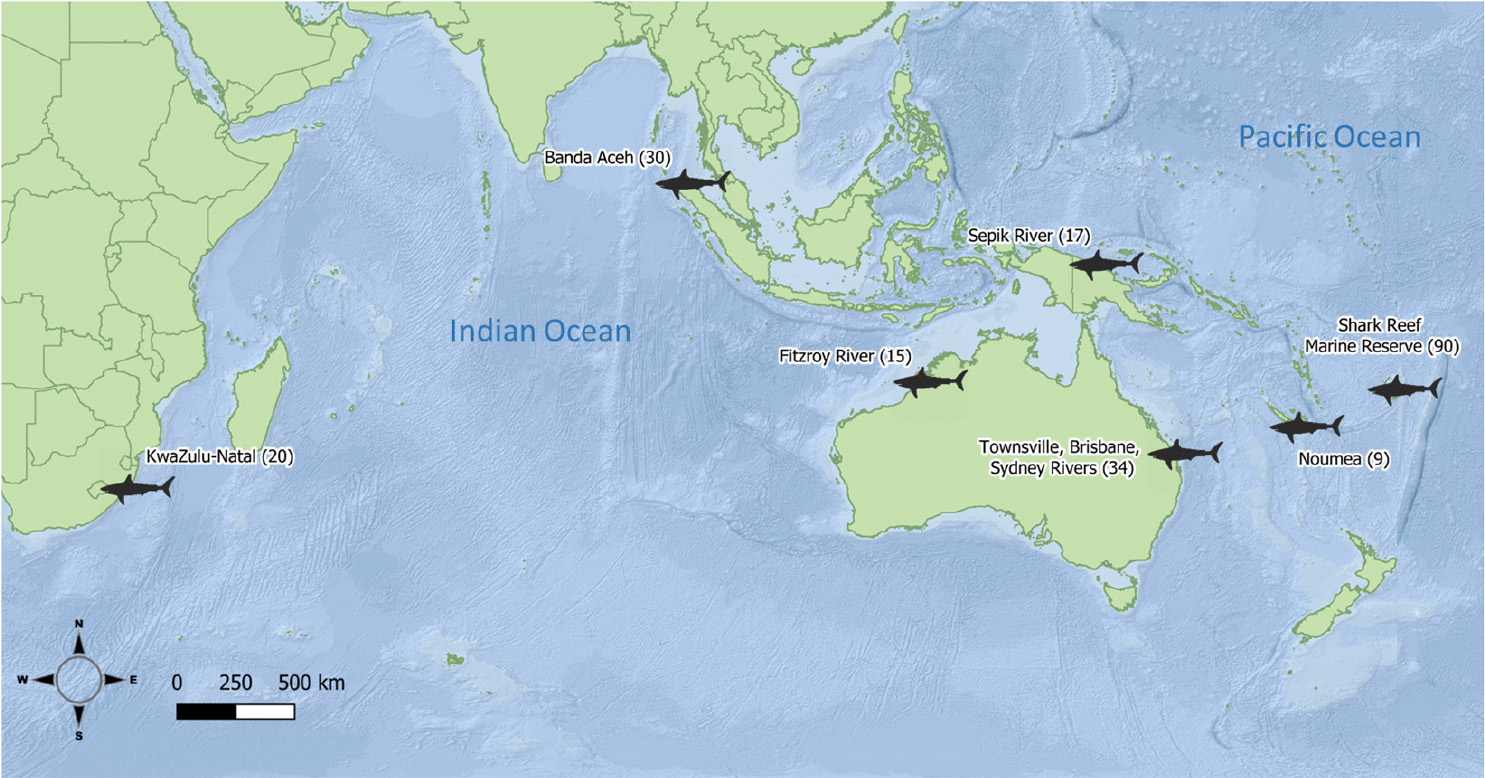
Figure 1. Locations of sample sites with numbers of samples in parentheses.
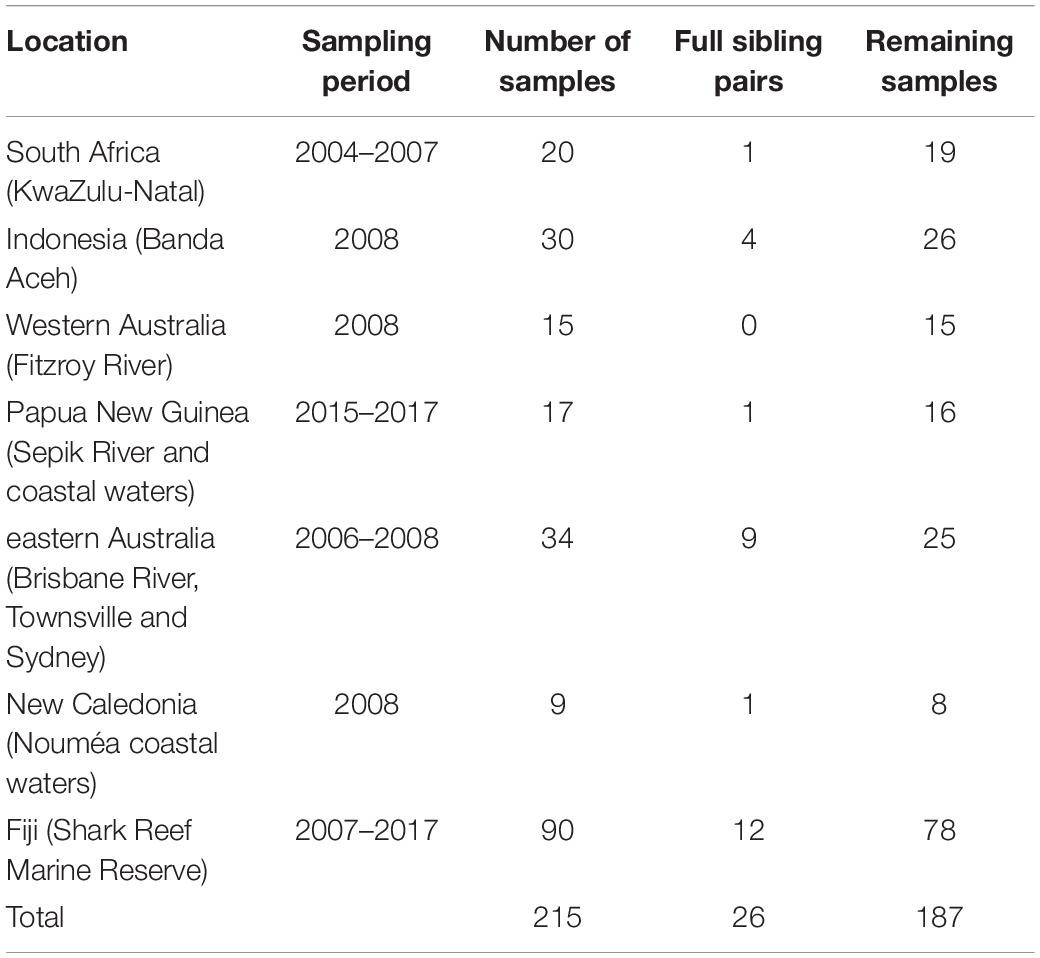
Table 1. Number of original bull shark samples per location, full sibling pairs detected with COANCESTRY, and remaining samples after one individual for each pair was removed.
Extraction and Sequencing
Tissue samples were sent to Diversity Arrays Technology (DArT-SeqTM) in Canberra Australia where genomic DNA was extracted from the tissues using standard robotic methods. DNA was processed for reduced representation library construction, sequenced and genotyped by DArT-SeqTM following previously developed and tested complexity reduction protocols for scalloped hammerhead sharks (Sphyrna lewini) (Marie et al., 2019). Briefly, genome complexity reduction was achieved with a double restriction digest using a PstI and SphI methylation-sensitive restriction enzyme combination. Libraries were sequenced on an Illumina HiSeq 2500 platform, and raw reads obtained following sequencing were processed using Illumina CASAVA v.1.8.2 software for initial assessment of read quality and sequence representation. The DArT PL proprietary software pipeline, DArTtoolbox was used for further filtering and variant calling to generate the final genotypes set.
SNP Filtering
The dataset was filtered by excluding duplicate SNPs possessing identical Clone IDs and by removing loci with a call rate (proportion of individuals scored for a locus) <99%, read depth >7 and minor allele frequencies (MAF) <2%. Detection of loci under selection was implemented in BayeScan v.2.1 (Foll and Gaggiotti, 2008). The most conservative neutral model in BayeScan was used to minimize falsely detected loci under selection (Lotterhos and Whitlock, 2014). Runs consisted of 100,000 iterations with a burn-in length of 50,000 iterations (Foll and Gaggiotti, 2008; Foll, 2012). Once probabilities had been calculated for each locus, the BayeScan function plot_R was used in the R v.3.2.0 statistical package (Venables et al., 2003) to identify putative outlier loci. A range of false discovery rate (FDR) values from 0.01 to 0.20 were evaluated based on preliminary testing and recommendations by Gondro et al. (2013). Departure from Hardy-Weinberg Equilibrium was tested for each locus using the software Arlequin v.3.5.2.2 (Schneider et al., 2000) using an exact test with 10,000 steps in the Markov Chain method and 100,000 dememorizations. Tests for linkage disequilibrium was performed using the software PLINK v.1.07 (Chang et al., 2015). Related individuals were detected using TrioML with COANCESTRY v. 1.0.1.7 (Wang, 2011). To determine cut-off r values for categorizing relatedness coefficients produced by the various algorithms into full-sib and less-related brackets, COANCESTRY was again used to simulate 200 dyads in each group based on the same allele frequencies as the original population samples. These values became the cut-offs for delineating full siblings in the empirical dataset. One individual from each of the full sibling pairs was removed for the final neutral data set. This new data set was used in all the subsequent analyses.
Allelic Diversity and Population Structuring
Allelic diversity indices including average observed (Ho) and unbiased expected heterozygosities corrected for population sample size (Hnb) were computed in Genetix v.4.05.2 (Belkhir et al., 1996), together with the inbreeding coefficient (FIS) values using GENEPOP v.4.6 (Rousset, 2008). Pairwise FST estimates were calculated using Arlequin v.3.5.1.3 followed by correction of significance levels for multiple pairwise tests (Excoffier and Lischer, 2010). An analysis of molecular variance (AMOVA) was performed in GenoDive v.3.0 using 10,000 permutations to estimate F-statistics in order to detect population genetic partitioning between sampling locations (Mengoni and Bazzicalupo, 2002). P values were adjusted by using the Bonferroni correction (Rice, 1989). Multiple AMOVAs were performed: i) one overall analysis including all sampling locations as separate populations, ii) Fiji versus all other sites grouped as one population, iii) insular (New Caledonia, Fiji) versus continental locations, and iv) Pacific Ocean versus Indian Ocean. Visualization of broad-scale population structure and population assignment was carried out by performing a Discriminant Analysis of Principal Components (DAPC) in the R package adegenet 1.4.2 (Jombart, 2008; Jombart and Ahmed, 2011). DAPC was carried out for all neutral loci. An α-score optimization was used to determine the number of principal components to retain. Alpha-score optimization indicated that the first nine PCs (42.7% of total variation) should be retained for analysis. Given the wide temporal range for sampling in Fiji (2007–2017), separate DAPC analyses were run to determine the temporal stability within Fiji samples. For this, the sampled years that have a sufficiently large number of individuals within each year were split as a separate populations [2010 (n = 28), 2011 (n = 22), and 2017 (n = 18)]. The samples collected in 2007 (n = 6), 2008 (n = 1), and 2009 (n = 3) were considered inadequate for this analysis given the small sample sizes. Furthermore, to determine if the pattern of differentiation obtained when using all Fijian samples simultaneously was stable, the DAPC analyses were also run comparing all Indo-Pacific populations against individual years (2010, 2011, and 2017) from Fiji.
The determination of the optimal number of clusters (k) was tested using both fastStructure (Raj et al., 2014) and Admixture (Alexander et al., 2009). Both software packages were written for large autosomal SNP data sets and unlike other programs can test the likelihood of a k of one. Admixture utilizes a cross-validation procedure when determining the optimal k. While, for fastStructure the full model was selected with a seed of 100. For both programs, k ranging from 1 to 10 was run. Additionally, fastStructure included the python script chooseK.py which was run after the ten k scenarios were finished and the optimal k was selected. Once the optimal k was determined the probability of individual population assignments was calculated and visualized using both Admixture and AssignPOP (Chen et al., 2018).
Results
Filtering and Genotypic Diversity
Genotyping by sequencing using DArT-SeqTM resulted in 59,601 SNPs prior to quality control filtering (Carson et al., 2014), which in turn resulted in 1,494 loci using a 99% call rate, read depth >7, and 2% MAF. Among the 1,494 SNPs that passed all quality control filters for the 215 individuals, zero SNPs were identified as outlier loci putatively under positive selection (FDR 1%). No loci were detected to be out of Hardy-Weinberg Equilibrium and in addition no loci were determined to be linked.
A total of 28 full sibling pairs were identified in COANCESTRY (Table 1), with Fiji having the most pairs (12) followed by eastern Australia (9). Most of the other sites only had one or two pairs, while no full sibling pairs were identified within the West Australian samples. After removal of one individual from each of the pairs, 187 samples were retained.
Overall, population-level indices of genetic diversity, including Ho, Hnb, and FIS, were variable across a priori populations (Table 2). The Ho were lowest within Papua New Guinea (0.128 ± 0.162) and South Africa (0.157 ± 0.168) and highest in bull sharks from New Caledonia (0.182 ± 0.188) and Indonesia (0.214 ± 0.166) (Table 2). Inbreeding estimates were highest in specimens collected in eastern Australia (FIS = 0.355) and lowest in bull sharks from New Caledonia (FIS = −0.064) and Western Australia (FIS = −0.049).
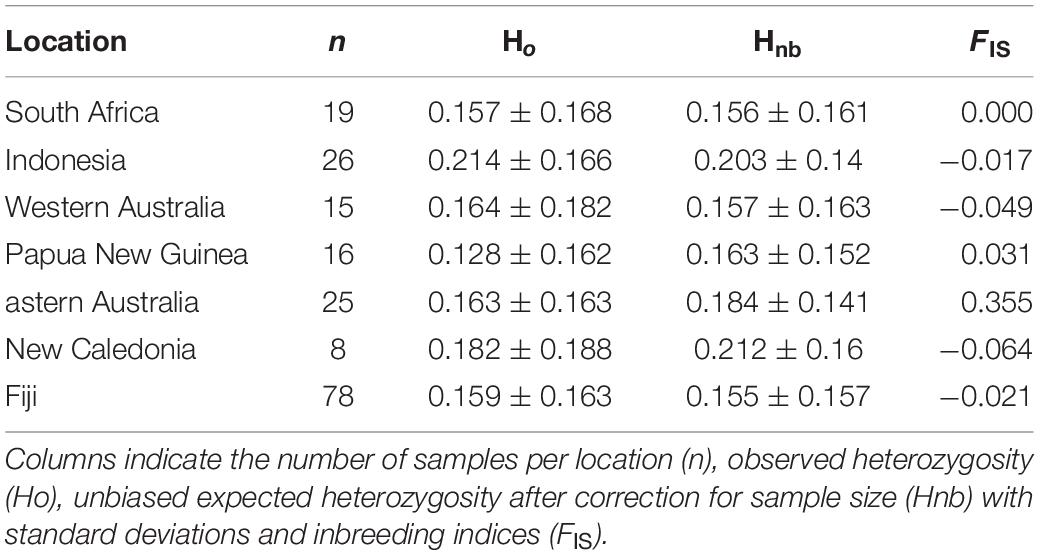
Table 2. Summary of average genetic diversity statistics per population.
Population Structuring
Pairwise FST values were highest (0.034–0.044) between Fiji and all other sites, while values were moderate (0.016–0.024) for New Caledonia and all other continental sites (Table 3). The AMOVA results allocated the vast majority of the variation within individuals and when sampling sites were treated separately without groupings, roughly 2.7% of the variation was found among a priori populations (Table 4). The analysis showed significant variation among groups, when Fiji and New Caledonia were grouped as one versus a second group consisting of all continental sites (Table 4). This reflects the pairwise FST values, in that Fiji and New Caledonia were significantly different from all other sites.
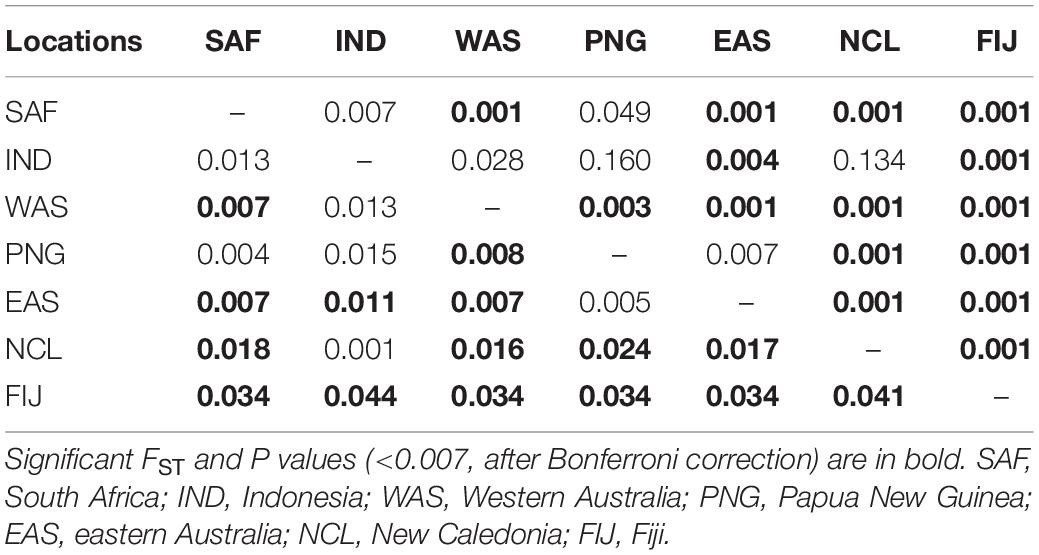
Table 3. Pairwise FST (lower diagonals) and P values (upper diagonal) between the seven locations sampled.
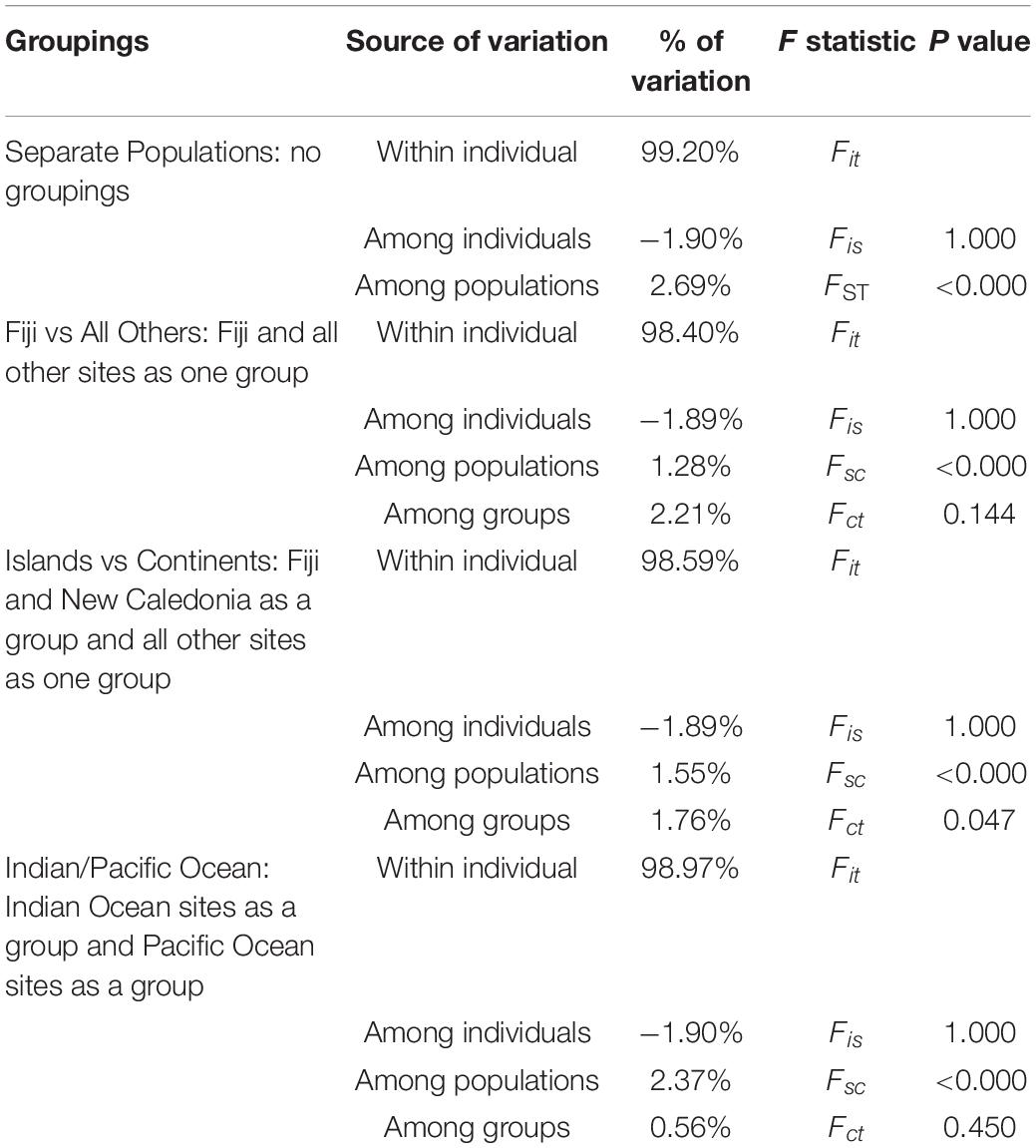
Table 4. Summary of AMOVA results for the various groupings.
The DAPC scatterplot showed a clear distinction between Fiji and the other locations. New Caledonia was not differentiated from the continental locations, which may be a result of the small sample size (Figure 2). The analysis of temporal samples within Fiji resulted in scatter plots that placed the individuals compared, from three different years, overlapping to an extent that supports stability in the distribution of the neutral genetic diversity within this insular population. Furthermore, when sampled years were analyzed separately with the rest of populations, again, sharks strongly overlapped within the Indo-Pacific and within Fiji. This confirmed the patterns of differentiation observed irrespective of the sampling year in Fiji and ruled out any temporal substructure (Supplementary Materials; Supplementary Figure S1). Both fastStructure and Admixture reported an optimal k of two, and we arbitrarily labeled them population A and B. For Admixture the cross-validation inflection point was at k = 2, while for fastStructure the python script chooseK.py selected a k of 2 based on marginal likelihood. The population assignment analyses for both Admixture and AssignPOP assigned the same individuals to the same populations, with all continental locations and New Caledonia belonging to population A, and Fiji belonging to population B (Figure 3). The best performing assignment model in AssignPOP was Support Vector Machine (SVM) with an assignment accuracy of 0.999 (s.d. = 0.001) for population A and 1.0 for population B.
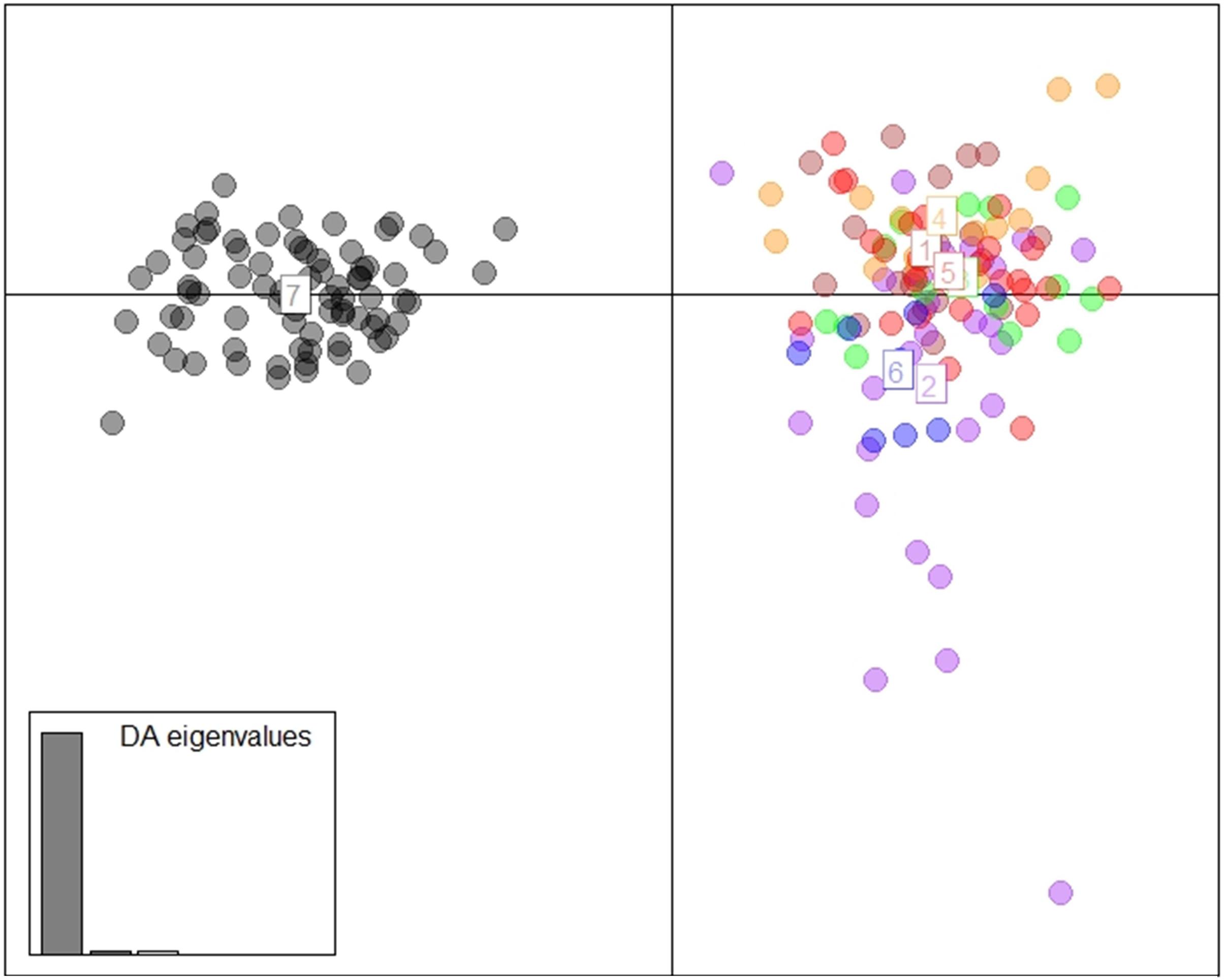
Figure 2. DAPC using 1,494 neutral loci and 9 principle components. (1 = South Africa, 2 = Indonesia, 3 = Western Australia, 4 = Papua New Guinea, 5 = eastern Australia, 6 = New Caledonia, and 7 = Fiji).
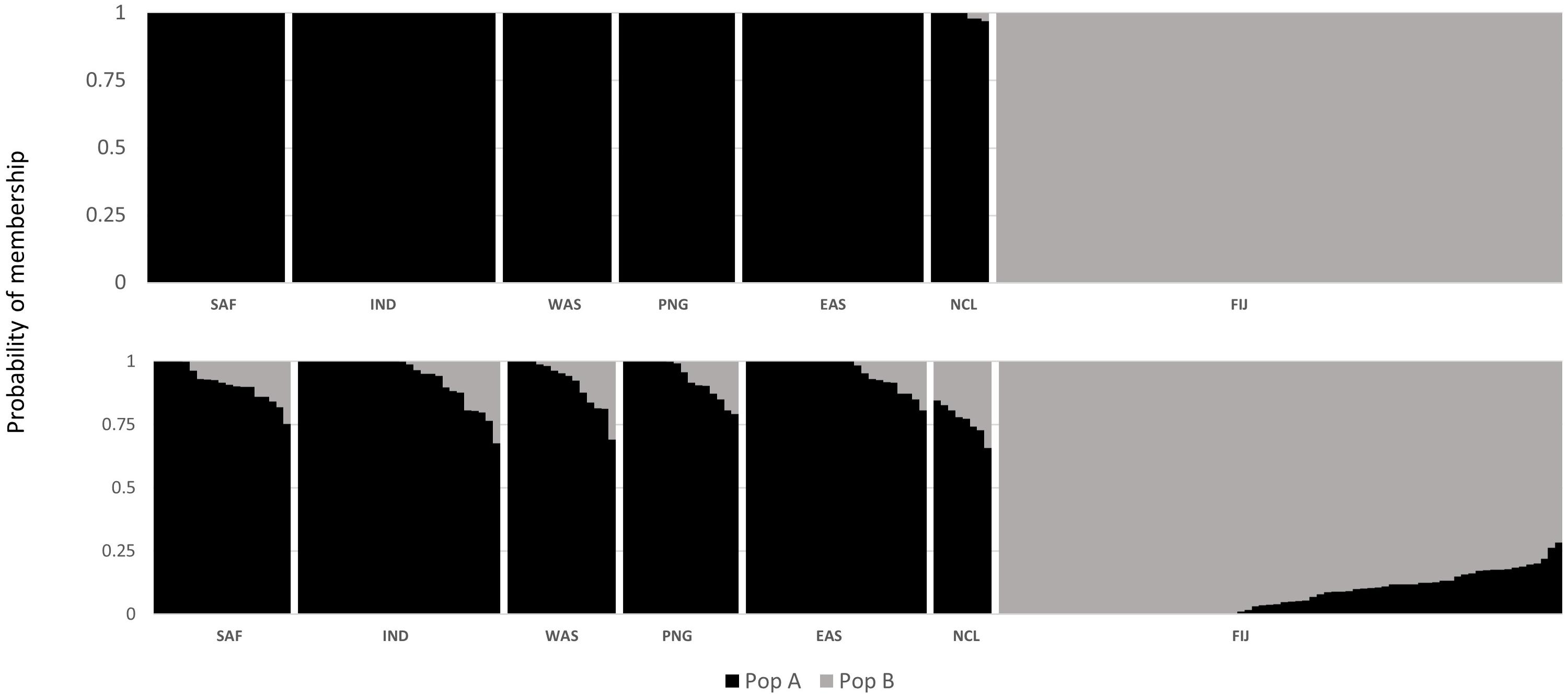
Figure 3. Probability of population placement based on an optimal k of PopAssign (top) and Admixture (bottom).
Discussion
This is the first population genetic structure study conducted in bull sharks using SNP markers. The major finding of this study is that the insular bull shark population from Fiji is genetically distinct from its continental counterparts. Therefore, the data presented here supports the alternative hypothesis that biogeographic barriers to dispersal result in a heterogeneous distribution of the neutral genetic diversity between island bull sharks compared to populations sampled from continental shelves across the Indo-Pacific region.
Genetic Diversity
The sampling regime and SNP marker set used herein possessed the ability to resolve relatively low levels of differentiation enough to define bull shark population structure (Table 3). Based on SNP data, levels of observed heterozygosity (Ho = 0.128–0.214) in bull sharks are comparable to other tropically distributed sharks, such as the gray reef shark (Carcharhinus amblyrhynchos) (0.139–0.312) (Momigliano et al., 2017) and the Galapagos shark (C. galapagensis) (0.188–0.193) (Pazmiño et al., 2017).
Population Genetic Structure
All five analyses examining population structuring and connectivity (FST, AMOVA, DAPC, Admixture and PopAssign) failed to detect any barriers to gene flow among the six continental a priori populations (from the Pacific and Indian Oceans) in this study. Initially this might be surprising, given the large distances between the sampled populations. However, a recent study by Pirog et al. (2019) incorporating 25 microsatellites and using DAPC and fastStructure analyses also detected large ocean wide clusters including all western Atlantic populations in one and western Indian Ocean and Pacific sites in the other. Additionally, to date, no ocean basin subdivision has been detected for bull sharks (Karl et al., 2011; Tillett et al., 2012; Testerman, 2014). This is not surprising since tracking studies indicate bull sharks are capable of long-distance movements of up to 1,506 km along coastlines (Carlson et al., 2010) and can traverse open-ocean expanses of 2,000 km (Lea et al., 2015). However, genetic connectivity does not necessarily equate to demographic connectivity, as the exchange of only a few individual migrants per generation is enough to homogenize the gene pool even of distantly located populations (Waples, 1998). Low levels of migration among locations in turn are likely to be insufficient to compensate localized declines in abundance, but rare excursions can have important consequences for population viability by prevention of inbreeding.
However, the same five analyses all detected differentiation between all continental populations and Fiji. Significant pairwise FST values ranged from 0.034 to 0.044 between all locations (including New Caledonia) and Fiji. While values for New Caledonia were significant, they were smaller ranging between 0.016 and 0.024 and no significant differentiation was detected between Indonesia (Banda Aceh) and New Caledonia (FST = 0.001); locations which are separated by thousands of kilometers of ocean. These smaller values and conflicting patterns may be a result of the small sample size (N = 8) of the New Caledonia population and may not reflect the true genetic structure. The DAPC results clearly differentiated Fiji from the rest of the populations, with New Caledonia clustering well within the continental cluster. Further support comes from Admixture and fastStructure, which both identified an optimal k of two with all continental locations (including New Caledonia) in one cluster and Fiji samples in the other. Furthermore, given a k of two, PopAssign was able to assign accurately the same individuals to each of the two populations that Admixture allocated. Thus, the corroboration of these independent analyses clearly indicates genetic heterogeneity and divergence between the insular population of Fiji and the other continental demes.
Bull sharks have a continuous distribution across South Africa, Indonesia and Australia (Ebert et al., 2013). The presence of continuous continental shelf waters off those landmasses and the absence of physical barriers along the shoreline habitats of Indo-Australia may facilitate dispersal in this coastal shark species. This is also suggested by the differences between South Africa and Western Australia but not with eastern Australia (the FST between SAF and WAS is the same as between SAF and EAS). Thus, this lack of migration to Fiji from the surrounding continental populations suggests that bull shark migration across wide expenses of deep oceanic waters is an extremely rare event. The lack of differentiation between SAF samples and the rest of the continental sites despite the deep waters of the Indian Ocean is likely due to the continuous coastal habitats extending from Africa to southern Asia to the Sunda Shelf in Indonesia, allowing for the avoidance of deep water trenches and a near continuous migration route.
Management
The genetic differentiation of the Fiji bull sharks is a robust finding and makes this population of special interest due to its genetic and geographic isolation. Indonesia and Western Australia appear to be sites of transition for continental associated bull sharks. Dispersal limitations due to vast ocean expanses has implications for the degree of genetic (and demographic) connectivity among populations, and therefore the spatial scale of their management (Waples and Gaggiotti, 2006). For example, genetically isolated populations such as presumably the Fiji bull sharks could benefit from local protection of large, fecund individuals (Gwinn et al., 2015). Also, uncovering potential mating and nursery areas of high conservation value (Heupel et al., 2007) and fine-scale genetic connectivity is key to further population genetic studies (Mourier and Planes, 2013). Consequently, a comparison between information generated by nuclear DNA (i.e., from both parents, relevant for kinship studies) with results derived from mtDNA (maternal, relevant for studies on philopatry) would help to define populations relevant for management. Additionally, the previously reported female philopatry implies that subpopulations within oceanic basins are likely to be independent demographically over ecological timescales, and that further work to identify discrete matrilineal populations along continuous coastlines throughout the species range is imperative to formulating effective management policies.
Data Availability Statement
The data presented in this manuscript are permanently archived at the Dryad Digital Repository: doi: 10.5061/dryad.0vt4b8gwf.
Ethics Statement
The animal study was reviewed and approved by University Research Ethics Committee of the University of the South Pacific. All shipping procedures of shark tissue samples were conducted under the relevant import and export permits issued by the Australian Government, Department of Agriculture and Water Resources to Diversity Arrays Technology Pty Ltd, Canberra, Australia and the Ministry of Fisheries and Forests in Fiji. Bull shark tissues from Papua New Guinea came from the CSIRO/ACIAR/PNG-NFA FIS/2012/102 ‘Sustainable management of the shark resources of Papua New Guinea: socioeconomic and biological characteristics of the fishery’. Handling of live shark specimens were approved under the Research Permit issued by the Fiji Ministry of Education, Heritage and Arts to Beqa Adventure Divers and performed in accordance with relevant guidelines and regulations.
Author Contributions
KG, JB, MS, and CR designed the study. CR wrote the project proposal. CR and KG obtained funding. JB, MS, SA, and EC provided samples. KG, AM, BS, and SA ran the analysis. KG created the figures and wrote the first draft of the manuscript. All authors contributed to the write up of the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The project was supported by the Ausbildungs-Stiftung fuer den Kanton Schwyz und die Bezirke See und Gaster (Kanton St. Gallen, Switzerland), the Shark Foundation Switzerland and Pacific Scholarship for Excellence in Research & Innovation of the University of the South Pacific. Grant SRT/F1006-R1001-71502-663 was given to CR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Great thanks are owed to all assisting research colleagues, fishermen and the staff from Beqa Adventure Divers in Fiji. We authors thank Dr. Will White (CSIRO) for the bull shark tissue samples from Papua New Guinea via collaboration with a PNG-NFA ACIAR study, and the observers from commercial fishing vessels and artisanal surveys who collected the samples. We thank Crispen Wilson and Les Noble for sharing samples collected in South Africa and Indonesia. KG thanks the Ausbildungs-Stiftung fuer den Kanton Schwyz und die Bezirke See und Gaster (Kanton St. Gallen), the Shark Foundation Switzerland and Pacific Scholarship for Excellence in Research & Innovation of The University of the South Pacific. We thank Mike Neumann and Ian Campbell for continued advice during the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.586015/full#supplementary-material
References
Ahonen, H., Harcourt, R., and Stow, A. (2009). Nuclear and mitochondrial DNA reveals isolation of imperilled grey nurse shark populations (Carcharias taurus). Mol. Ecol. 18, 4409–4421. doi: 10.1111/j.1365-294x.2009.04377.x
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Allendorf, F. W., England, P. R., Luikart, G., Ritchie, P. A., and Ryman, N. (2008). Genetic effects of harvest on wild animal populations. Trends Ecol. Evol. 23, 327–337. doi: 10.1016/j.tree.2008.02.008
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N., and Bonhomme, F. (1996). GENETIX 4.05, Logiciel Sous Windows TM Pour la Génétique des Populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000. (Montpellier: Université de Montpellier).
Benavides, M. T., Feldheim, K. A., Duffy, C. A., Wintner, S., Braccini, J. M., Boomer, J., et al. (2011). Phylogeography of the copper shark (Carcharhinus brachyurus) in the southern hemisphere: implications for the conservation of a coastal apex predator. Mar. Freshw. Res. 62, 861–869. doi: 10.1071/mf10236
Benestan, L., Gosselin, T., Perrier, C., Sainte-Marie, B., Rochette, R., and Bernatchez, L. (2015). RAD genotyping reveals fine-scale genetic structuring and provides powerful population assignment in a widely distributed marine species, the American lobster (Homarus americanus). Mol. Ecol. 24, 3299–3315. doi: 10.1111/mec.13245
Bernard, A. M., Feldheim, K. A., Heithaus, M. R., Wintner, S. P., Wetherbee, B. M., and Shivji, M. S. (2016). Global population genetic dynamics of a highly migratory, apex predator shark. Mol. Ecol. 25, 5312–5329. doi: 10.1111/mec.13845
Brunnschweiler, J., Queiroz, N., and Sims, D. (2010). Oceans apart? Short-term movements and behaviour of adult bull sharks Carcharhinus leucas in Atlantic and Pacific Oceans determined from pop-off satellite archival tagging. J. Fish Biol. 77, 1343–1358. doi: 10.1111/j.1095-8649.2010.02757.x
Brunnschweiler, J. M., and Baensch, H. (2011). Seasonal and long-term changes in relative abundance of bull sharks from a tourist shark feeding site in Fiji. PLoS One 6:e16597. doi: 10.1371/journal.pone.0016597
Carlson, J. K., Ribera, M. M., Conrath, C. L., Heupel, M. R., and Burgess, G. H. (2010). Habitat use and movement patterns of bull sharks Carcharhinus leucas determined using pop-up satellite archival tags. J. Fish Biol. 77, 661–675. doi: 10.1111/j.1095-8649.2010.02707.x
Carson, A. R., Smith, E. N., Matsui, H., Brækkan, S. K., Jepsen, K., Hansen, J.-B., et al. (2014). Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinformatics 15:125. doi: 10.1186/1471-2105-15-125
Castro, A., Stewart, B., Wilson, S., Hueter, R., Meekan, M., Motta, P., et al. (2007). Population genetic structure of Earth’s largest fish, the whale shark (Rhincodon typus). Mol. Ecol. 16, 5183–5192. doi: 10.1111/j.1365-294x.2007.03597.x
Castro, J. I. (1983). The Sharks of North American Waters. College Station, TX: Texas A. & M. University Press.
Chang, C. C., Chow, C. C., Tellier, L., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7.
Chapman, D. D., Feldheim, K. A., Papastamatiou, Y. P., and Hueter, R. E. (2015). There and back again: a review of residency and return migrations in sharks, with implications for population structure and management. Annu. Rev. Mar. Sci. 7, 547–570. doi: 10.1146/annurev-marine-010814-015730
Chen, K.-Y., Marschall, E. A., Sovic, M. G., Fries, A. C., Gibbs, H. L., and Ludsin, S. A. (2018). assignPOP: an R package for population assignment using genetic, non-genetic, or integrated data in a machine-learning framework. Methods Ecol. Evol. 9, 439–446. doi: 10.1111/2041-210x.12897
Compagno, L. J. (2001). Sharks of the World: An Annotated and Illustrated Catalogue of Shark Species Known To Date, Vol. 2. Rome: FAO.
Cowen, R. K., Lwiza, K. M., Sponaugle, S., Paris, C. B., and Olson, D. B. (2000). Connectivity of marine populations: Open or closed? Science 287, 857–859. doi: 10.1126/science.287.5454.857
Cowen, R. K., and Sponaugle, S. (2009). Larval dispersal and marine population connectivity. Annu. Rev. Mar. Sci. 1, 443–466. doi: 10.1146/annurev.marine.010908.163757
Cowman, P. F., and Bellwood, D. R. (2013). Vicariance across major marine biogeographic barriers: temporal concordance and the relative intensity of hard versus soft barriers. Proc. R. Soc. B Biol. Sci. 280:20131541. doi: 10.1098/rspb.2013.1541
Crandall, E., Riginos, C., Bird, C., Liggins, L., Treml, E., Beger, M., et al. (2019). The molecular biogeography of the Indo-Pacific: testing hypotheses with multispecies genetic patterns. Glob. Ecol. Biogeogr. 28, 943–960. doi: 10.1111/geb.12905
Daly, R., Smale, M. J., Cowley, P. D., and Froneman, P. W. (2014). Residency patterns and migration dynamics of adult bull sharks (Carcharhinus leucas) on the east coast of southern Africa. PLoS One 9:e109357. doi: 10.1371/journal.pone.0109357
Daly-Engel, T. S., Grubbs, R. D., Feldheim, K. A., Bowen, B. W., and Toonen, R. J. (2010). Is multiple mating beneficial or unavoidable? Low multiple paternity and genetic diversity in the shortspine spurdog Squalus mitsukurii. Mar. Ecol. Prog. Ser. 403, 255–267. doi: 10.3354/meps08417
Dulvy, N. K., Fowler, S. L., Musick, J. A., Cavanagh, R. D., Kyne, P. M., Harrison, L. R., et al. (2014). Extinction risk and conservation of the world’s sharks and rays. eLife 3:e00590.
Duncan, K. M., and Holland, K. N. (2006). Habitat use, growth rates and dispersal patterns of juvenile scalloped hammerhead sharks Sphyrna lewini in a nursery habitat. Mar. Ecol. Prog. Ser. 312, 211–221. doi: 10.3354/meps312211
Duncan, K. M., Martin, A. P., Bowen, B. W., and De Couet, H. G. (2006). Global phylogeography of the scalloped hammerhead shark (Sphyrna lewini). Mol. Ecol. 15, 2239–2251. doi: 10.1111/j.1365-294X.2006.02933.x
Ebert, D. A., Fowler, S. L., and Compagno, L. J. (2013). Sharks of the World: A Fully Illustrated Guide. Plymouth: Wild Nature Press.
Espinoza, M., Heupel, M. R., Tobin, A. J., and Simpfendorfer, C. A. (2016). Evidence of partial migration in a large coastal predator: Opportunistic foraging and reproduction as key drivers? PLoS One 11:e0147608. doi: 10.1371/journal.pone.0147608
Excoffier, L., and Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Feldheim, K. A., Gruber, S. H., Dibattista, J. D., Babcock, E. A., Kessel, S. T., Hendry, A. P., et al. (2014). Two decades of genetic profiling yields first evidence of natal philopatry and long-term fidelity to parturition sites in sharks. Mol. Ecol. 23, 110–117. doi: 10.1111/mec.12583
Foll, M., and Gaggiotti, O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Gagnaire, P. A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., et al. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evol. Appl. 8, 769–786. doi: 10.1111/eva.12288
Galarza, J. A., Carreras-Carbonell, J., Macpherson, E., Pascual, M., Severine, R., Turner, G. F., et al. (2009). The influence of oceanographic fronts and early-life-history traits on connectivity among littoral fish species. Proc. Natl. Acad. Sci. U.S.A. 106, 1473–1478. doi: 10.1073/pnas.0806804106
Gondro, C., Lee, S. H., Lee, H. K., and Porto-Neto, L. R. (2013). “Quality control for genome-wide association studies,” in Genome-Wide Association Studies and Genomic Prediction, eds C. Gondro, J. van der Werf, and B. Hayes (London: Springer).
Grant, V. (1985). The Evolutionary Process: A Critical Review Of Evolutionary Theory. New York, NY: Columbia University Press.
Gwinn, D. C., Allen, M. S., Johnston, F. D., Brown, P., Todd, C. R., and Arlinghaus, R. (2015). Rethinking length-based fisheries regulations: the value of protecting old and large fish with harvest slots. Fish Fish. 16, 259–281. doi: 10.1111/faf.12053
Hare, M. P., Guenther, C., and Fagan, W. F. (2005). Nonrandom larval dispersal can steepen marine clines. Evolution 59, 2509–2517. doi: 10.1554/05-150.1
Hauser, L., and Carvalho, G. R. (2008). Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish. 9, 333–362. doi: 10.1111/j.1467-2979.2008.00299.x
Heupel, M. R., Carlson, J. K., and Simpfendorfer, C. A. (2007). Shark nursery areas: concepts, definition, characterization and assumptions. Mar. Ecol. Prog. Ser. 337, 287–297. doi: 10.3354/meps337287
Heupel, M. R., Simpfendorfer, C. A., Espinoza, M., Smoothey, A. F., Tobin, A., and Peddemors, V. (2015). Conservation challenges of sharks with continental scale migrations. Front. Mar. Sci. 2:12. doi: 10.3389/fmars.2015.00012
Hoelzel, A. R., Shivji, M. S., Magnussen, J., and Francis, M. P. (2006). Low worldwide genetic diversity in the basking shark (Cetorhinus maximus). Biol. Lett. 2, 639–642. doi: 10.1098/rsbl.2006.0513
Jombart, T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart, T., and Ahmed, I. (2011). adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi: 10.1093/bioinformatics/btr521
Karl, S., Castro, A., Lopez, J., Charvet, P., and Burgess, G. (2011). Phylogeography and conservation of the bull shark (Carcharhinus leucas) inferred from mitochondrial and microsatellite DNA. Conserv. Genet. 12, 371–382. doi: 10.1007/s10592-010-0145-1
Keeney, D., Heupel, M., Hueter, R., and Heist, E. (2005). Microsatellite and mitochondrial DNA analyses of the genetic structure of blacktip shark (Carcharhinus limbatus) nurseries in the northwestern Atlantic, Gulf of Mexico, and Caribbean Sea. Mol. Ecol. 14, 1911–1923. doi: 10.1111/j.1365-294x.2005.02549.x
Keeney, D. B., and Heist, E. J. (2006). Worldwide phylogeography of the blacktip shark (Carcharhinus limbatus) inferred from mitochondrial DNA reveals isolation of western Atlantic populations coupled with recent Pacific dispersal. Mol. Ecol. 15, 3669–3679. doi: 10.1111/j.1365-294X.2006.03036.x
Kelly, R. P., and Eernisse, D. J. (2007). Southern hospitality: a latitudinal gradient in gene flow in the marine environment. Evolution 61, 700–707. doi: 10.1111/j.1558-5646.2007.00055.x
Lea, J., Humphries, N., Clarke, C., and Sims, D. (2015). To Madagascar and back: long-distance, return migration across open ocean by a pregnant female bull shark Carcharhinus leucas. J. Fish Biol. 87, 1313–1321. doi: 10.1111/jfb.12805
Leis, J. M. (2002). Pacific coral-reef fishes: the implications of behaviour and ecology of larvae for biodiversity and conservation, and a reassessment of the open population paradigm. Environ. Biol. Fish. 65, 199–208.
Lotterhos, K. E., and Whitlock, M. C. (2014). Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 23, 2178–2192. doi: 10.1111/mec.12725
Marie, A. D., Herbinger, C., Fullsack, P. M., and Rico, C. (2019). First reconstruction of kinship in a scalloped hammerhead shark aggregation reveals the mating patterns and breeding sex ratio. Front. Mar. Sci. 6:676. doi: 10.3389/fmars.2019.00676
Martin, A. P., Naylor, G. J., and Palumbi, S. R. (1992). Rates of mitochondrial DNA evolution in sharks are slow compared with mammals. Nature 357, 153–155. doi: 10.1038/357153a0
Mengoni, A., and Bazzicalupo, M. (2002). The statistical treatment of data and the analysis of molecular variance (AMOVA) in molecular microbial ecology. Ann. Microbiol. 52, 95–102.
Momigliano, P., Harcourt, R., Robbins, W., Jaiteh, V., Mahardika, G., Sembiring, A., et al. (2017). Genetic structure and signatures of selection in grey reef sharks (Carcharhinus amblyrhynchos). Heredity 119, 142–153. doi: 10.1038/hdy.2017.21
Morin, P. A., Luikart, G., and Wayne, R. K. (2004). SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 19, 208–216. doi: 10.1016/j.tree.2004.01.009
Mourier, J., and Planes, S. (2013). Direct genetic evidence for reproductive philopatry and associated fine-scale migrations in female blacktip reef sharks (Carcharhinus melanopterus) in French Polynesia. Mol. Ecol. 22, 201–214. doi: 10.1111/mec.12103
Müller, J., and Henle, F. G. J. (1839). Systematische Beschreibung der Plagiostomen. Berlin. 29–102.
Nance, H. A., Klimley, P., Galvan-Magana, F., Martinez-Ortiz, J., and Marko, P. B. (2011). Demographic processes underlying subtle patterns of population structure in the scalloped hammerhead shark, Sphyrna lewini. PLoS One 6:e0021459. doi: 10.1371/journal.pone.0021459
Ovenden, J. R., Kashiwagi, T., Broderick, D., Giles, J., and Salini, J. (2009). The extent of population genetic subdivision differs among four co-distributed shark species in the Indo-Australian archipelago. BMC Evol. Biol. 9:40. doi: 10.1186/1471-2148-9-40
Palsbøll, P. J., Berube, M., and Allendorf, F. W. (2007). Identification of management units using population genetic data. Trends Ecol. Evol. 22, 11–16. doi: 10.1016/j.tree.2006.09.003
Palumbi, S. R. (1994). Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 25, 547–572. doi: 10.1146/annurev.es.25.110194.002555
Pazmiño, D. A., Maes, G. E., Simpfendorfer, C. A., Salinas-de-León, P., and van Herwerden, L. (2017). Genome-wide SNPs reveal low effective population size within confined management units of the highly vagile Galapagos shark (Carcharhinus galapagensis). Conserv. Genet. 18, 1151–1163. doi: 10.1007/s10592-017-0967-1
Pirog, A., Ravigné, V., Fontaine, M. C., Rieux, A., Gilabert, A., Cliff, G., et al. (2019). Population structure, connectivity, and demographic history of an apex marine predator, the bull shark Carcharhinus leucas. Ecol. Evol. 9, 12980–13000. doi: 10.1002/ece3.5597
Portnoy, D. S., McDowell, J. R., Heist, E. J., Musick, J. A., and Graves, J. E. (2010). World phylogeography and male-mediated gene flow in the sandbar shark, Carcharhinus plumbeus. Mol. Ecol. 19, 1994–2010. doi: 10.1111/j.1365-294x.2010.04626.x
Raj, W. R., Stephens, M., and Pritchard, J. K. (2014). fastStructure: variational inference of population structure in large SNP data sets. Genetics 197, 573–589. doi: 10.1534/genetics.114.164350
Rice, W. R. (1989). Analyzing tables of statistical tests. Evolution 43, 223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x
Roberts, C. M. (1997). Connectivity and management of Caribbean coral reefs. Science 278, 1454–1457. doi: 10.1126/science.278.5342.1454
Rousset, F. (2008). genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106. doi: 10.1111/j.1471-8286.2007.01931.x
Schneider, S., Roessli, D., and Excoffier, L. (2000). Arlequin Ver. 2.0: A Software for Population Genetics Data Analysis. Geneva: University of Geneva.
Schultz, J., Feldheim, K., Gruber, S., Ashley, M., McGovern, T., and Bowen, B. (2008). Global phylogeography and seascape genetics of the lemon sharks (genus Negaprion). Mol. Ecol. 17, 5336–5348. doi: 10.1111/j.1365-294x.2008.04000.x
Selkoe, K. A., Aloia, C. C., Crandall, E. D., Iacchei, M., Liggins, L., Puritz, J. B., et al. (2016). A decade of seascape genetics: contributions to basic and applied marine connectivity. Mar. Ecol. Prog. Ser. 554, 1–19. doi: 10.3354/meps11792
Sequeira, A., Mellin, C., Meekan, M., Sims, D., and Bradshaw, C. (2013). Inferred global connectivity of whale shark Rhincodon typus populations. J. Fish Biol. 82, 367–389.
Simpfendorfer, C., and Milward, N. (1993). Utilization of a tropical bay as a nursery area by sharks of the families Carcharhinidae and Sphyrnidae. Environ. Biol. Fish. 37, 337–345. doi: 10.1007/bf00005200
Sodré, D., Rodrigues-Filho, L. F., Souza, R. F., Rêgo, P. S., Schneider, H., Sampaio, I., et al. (2012). Inclusion of South American samples reveals new population structuring of the blacktip shark (Carcharhinus limbatus) in the western Atlantic. Genet. Mol. Biol. 35, 752–760. doi: 10.1590/s1415-47572012005000062
Speed, C. W., Field, I. C., Meekan, M. G., and Bradshaw, C. J. A. (2010). Complexities of coastal shark movements and their implications for management. Mar. Ecol. Prog. Ser. 408, 275–293. doi: 10.3354/meps08581
Testerman, C. B. (2014). Molecular Ecology of Globally Distributed Sharks. Ph.D. dissertation, Nova Southeastern Universit, Fort Lauderdale, FL.
Tillett, B. J., Meekan, M. G., Field, I. C., Thorburn, D. C., and Ovenden, J. R. (2012). Evidence for reproductive philopatry in the bull shark Carcharhinus leucas. J. Fish Biol. 80, 2140–2158. doi: 10.1111/j.1095-8649.2012.03228.x
van der Hilst, R. (1995). Complex morphology of subducted lithosphere in the mantle beneath the Tonga trench. Nature 374, 154–157. doi: 10.1038/374154a0
Venables, W. N., Smith, D. M., and the R Development Core Team (2003). An Introduction to R, Version, 1(0). London: Network Theory Limited.
Vignaud, T., Clua, E., Mourier, J., Maynard, J., and Planes, S. (2013). Microsatellite analyses of blacktip reef sharks (Carcharhinus melanopterus) in a fragmented environment show structured clusters. PLoS One 8:e61067. doi: 10.1371/journal.pone.0061067
Wang, J. (2011). COANCESTRY: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol. Ecol. Resour. 11, 141–145. doi: 10.1111/j.1755-0998.2010.02885.x
Waples, R. S. (1998). Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J. Hered. 89, 438–450. doi: 10.1093/jhered/89.5.438
Keywords: island populations, connectivity, coastal sharks, shark reef marine reserve, population genomics, dispersal barriers
Citation: Glaus KBJ, Appleyard SA, Stockwell B, Brunnschweiler JM, Shivji M, Clua E, Marie AD and Rico C (2020) Insights Into Insular Isolation of the Bull Shark, Carcharhinus leucas (Müller and Henle, 1839), in Fijian Waters. Front. Mar. Sci. 7:586015. doi: 10.3389/fmars.2020.586015
Received: 22 July 2020; Accepted: 19 November 2020;
Published: 14 December 2020.
Edited by:
Lorenzo Zane, University of Padua, ItalyReviewed by:
Ka Yan Ma, Sun Yat-sen University, ChinaSimo Njabulo Maduna, Norwegian Institute of Bioeconomy Research (NIBIO), Norway
Copyright © 2020 Glaus, Appleyard, Stockwell, Brunnschweiler, Shivji, Clua, Marie and Rico. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kerstin B. J. Glaus, a2Vyc3Rpbl9nbGF1c0BvdXRsb29rLmNvbQ==
†These authors have contributed equally to this work