 Anne Tourneroche1
Anne Tourneroche1 Raphaël Lami2*
Raphaël Lami2* Gaëtan Burgaud3
Gaëtan Burgaud3 Isabelle Domart-Coulon1
Isabelle Domart-Coulon1 Wei Li4
Wei Li4 Claire Gachon1,5
Claire Gachon1,5 Marc Gèze1,6
Marc Gèze1,6 Dominique Boeuf2
Dominique Boeuf2 Soizic Prado1
Soizic Prado1- 1Unité Molécules de Communication et Adaptation des Micro-organismes (UMR 7245), Muséum National d'Histoire Naturelle, Centre National de la Recherche Scientifique (CNRS), Paris, France
- 2Sorbonne Université, CNRS, Laboratoire de Biodiversité et Biotechnologies Microbiennes (LBBM), Observatoire Océanologique de Banyuls sur Mer, Avenue Pierre Fabre, Banyuls-sur-Mer, France
- 3Univ Brest, Laboratoire Universitaire de Biodiversité et Ecologie Microbienne, Plouzané, France
- 4College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 5Culture Collection of Algae and Protozoa, Scottish Association for Marine Science, Scottish Marine Institute, Oban, United Kingdom
- 6Centre de Microscopie et d'Imagerie numérique (CeMIm) Muséum National d'Histoire Naturelle, Paris, France
The sugar kelp Saccharina latissima dominates many temperate coastal ecosystems, plays key ecological roles and presents important economic potential. However, its microbiota remains poorly investigated, although it could play an important role in algal fitness. In this study, we combined high throughput Illumina-based DNA sequencing and Fluorescence In Situ Hybridization to perform a culture-independent investigation of the S. latissima bacterial and fungal microbiota. Up to 600 bacterial and 100 fungal Amplicon Sequence Variants were identified per algal individual, revealing diverse bacterial and fungal communities associated to S. latissima. Overall, bacterial communities were dominated by Proteobacteria, Actinobacteria, and Bacteroidetes, in particular Hyphomonadaceae and Cyclobacteriaceae. Fungal communities were dominated by Ascomycota and Basidiomycota, in particular Mycosphaerellaceae, Psathyrellaceae, and Bulleribasidiaceae. Our results also revealed a variable distribution of S. latissima microbiota, as two adjacent tissue samples typically contained distinct fungal and bacterial assemblages, and CARD-FISH analysis detected microbial endosymbionts (with a few epibionts). Complementary analyses showed that despite achieving a good sequencing coverage for each tissue sample, the unexpected diversity and variability of ASVs made the definition of a core fungal and bacterial microbiota difficult, and highlights novel avenues to overcome the limitations of current surface-sterilization and metabarcoding protocols.
Introduction
The sugar kelp Saccharina latissima (formerly Laminaria saccharina) is distributed in cold to temperate waters of the Northern hemisphere. This large brown alga colonizes sheltered to medium wave-exposed coastal areas, where it can form kelp forests (Moy and Christie, 2012). Along with other Laminariales, this species plays key ecological roles in coastal environments, especially by providing shelter and nutrients for a wide diversity of marine organisms (Bartsch et al., 2008). Large scale declines of the population of S. latissima have been recently observed, e.g., along the Norwegian coast, raising concerns about the fate of such coastal ecosystems and their associated biodiversity in the current context of global change (Moy and Christie, 2012). S. latissima is also one of the fastest-growing European kelp species, with a high carbohydrate content and significant economic potential for moving toward a seaweed-based economy (Skjermo et al., 2014). Traditionally, this species was used as biosourced fertilizer and animal feed in agriculture, and other applications are currently evaluated, such as potential source of biofuel (Adams et al., 2009; Vivekanand et al., 2012) and of highly antioxidative compounds for cosmetics (Mesnildrey et al., 2012; Skjermo et al., 2014; Hermund et al., 2018). S. latissima may also be an efficient bioremediation agent limiting eutrophication from fish-farming (Handå et al., 2013).
As other multicellular eukaryotes, marine macroalgae grow and develop in close interactions with microbial communities. Some of these microorganisms, the so-called “epiphytes” colonize the algal surfaces and various studies also unveiled the presence of bacterial and fungal “endophytes” (Hollants et al., 2013; Flewelling et al., 2015). These closely associated microbial communities form the algal microbiota, and their association with the host forms a meta-organism also named “holobiont” (Egan et al., 2013). A growing number of studies have emphasized the key role of microbial symbionts on the development and defense of their macroalgal host (Egan et al., 2013; Tapia et al., 2016; Saha and Weinberger, 2019). For instance, bacterial morphogens are required to support the normal development of Ulva intestinalis and Ulva mutabilis (Ghaderiardakani et al., 2017). Also, dysbiosis, an emerging concept in the field of marine (micro)biology, highlights the link between shifting microbiota composition and the host health status (Egan and Gardiner, 2016). As a result, the characterization of the microbial communities associated with healthy algae appears as an essential step toward a better understanding of the importance of such interactions and thus a better control of the algal life cycle in the context of seaweed farming.
Several studies have suggested the existence of host-specific bacterial communities in some algal species (Goecke et al., 2010; Barott et al., 2011; Hollants et al., 2011; Lachnit et al., 2011). In S. latissima, our recent studies of the cultivable microbial communities highlighted the capacity of fungal strains to produce compounds active against algal pathogens (Vallet et al., 2018), and the ability of bacterial strains to communicate by quorum sensing signaling (Tourneroche et al., 2019). Similarly, some authors reported the expression of specific metabolites (Flewelling et al., 2015) or enzymes (Martin et al., 2015) by culturable seaweed-associated bacteria. Nevertheless, a comprehensive work on Ectocarpus highlighted a notable discrepancy between the composition of the bacterial microbiota retrieved by metabarcoding and the taxonomic identification of isolated strains, suggesting that many abundant bacteria are uncultivable and conversely, that some cultivable bacteria may have a very low abundance in algal tissues (KleinJan et al., 2017).
In S. latissima, an early study described a large bacterial diversity using DNA fingerprinting methods (Denaturing Gradient Gel Electrophoresis, DGGE) coupled with 16S rRNA gene based clone libraries (Staufenberger et al., 2008). This study detected 45 different phylotypes, affiliated to Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria. Consistently with most of the literature on other algae, this study highlighted that the alga-associated bacterial communities were distinct from those in seawater (Bengtsson et al., 2010; Michelou et al., 2013). Regarding fungi, however, from a combination of various datasets, Panzer et al. (2015) pointed out no significant difference between the composition of fungal communities associated with marine plant/algae and the ones found in seawater.
Interestingly, it has also been highlighted that the bacterial communities associated with the youngest parts of the algae were the most specific ones (Staufenberger et al., 2008). Using the DGGE approach, Lachnit et al. showed that the epibacterial communities of S. latissima varied between different individuals, but this variability was lower than the differences between S. latissima and other algal species, suggesting some specificity in the bacterial associations (Lachnit et al., 2011).
Endosymbiotic associations between bacteria and marine macroalgae have been reported for over 40 years. Intracellular endophytic bacteria have been microscopically observed in the vacuolar and cytoplasmic regions of various green algae and characterized molecularly within the green algae Bryopsis and Caulerpa (Hollants et al., 2011; Aires et al., 2015). Intracellular Rickettsiales have also been described in Mesostigma viride and eustigmatophytes (Yang et al., 2016; Yurchenko et al., 2018). These studies used Fluorescence In Situ Hybridization (FISH) to visualize endophytic bacteria in alga structures with a culture -and PCR- independent approach. In Bryopsis as well as eustigmatophytes, previous studies suggest a close metabolic cooperation between the host and its intracellular bacteria (Yurchenko et al., 2018; Zan et al., 2019).
In this study, we aimed at better characterizing the S. latissima microbiota through a DNA-based metabarcoding approach combined with CARD-FISH. We used amplicon sequencing of 16S rRNA genes and ITS (Internal Transcribed Spacer) genomic fragments to unveil the diversity and spatial structure of bacterial and fungal communities associated with S. latissima, while the presence of bacterial symbionts was visualized by CARD-FISH experiments.
Materials and Methods
Sampling and DNA Extraction
Algal sampling were performed in July 2017 along the west coast of Scotland between the mainland and Seil island (56° 30′ N; −5° 58′ W) at low tide. Five visually healthy S. latissima growing a few meters away from each other were collected, along with five samples of 200 mL of surrounding sea water. For each algal individual, a large piece of the youngest part of the blade was cut with a sterile scalpel (11.5 cm2) and rinsed three times with sterile seawater. Samples were kept on ice during transportation (<3 h). Back to the lab, under sterile conditions, two contiguous pieces of tissue (1 × 2.5 × 0.4 cm; i.e., 1 cm3) were cut from each algal blade and stored at −80°C for later DNA extraction. Also we decided to test the effect of a commonly used surface sterilization method on microbial communities. The remaining blade fragments were subjected to a surface sterilization protocol (successive baths of 70% EtOH for 30 s, 0.1% NaClO for 30 s and three times sterile seawater for 30 s (Vallet et al., 2018). Then, two contiguous pieces of tissue (1 × 2.5 × 0.4 cm each; i.e., 1 cm3) were cut from each surface-sterilized sample and stored at −80°C for later DNA extraction, whereas one other piece (5 mm wide) was cut in order to obtain both transverse and longitudinal tissue sections that were immediately placed in aldehyde fixative for later CARD-FISH analyses (fixation protocol described below). The five collected seawater samples were pooled (1 L total) to average the community composition of the seawater and filtered through a 0.22 μm filter which was stored at −80°C for later DNA extraction. Total DNA extraction of algal tissues and filter was performed using a CTAB protocol modified from Doyle and Doyle (Doyle and Doyle, 1987).
PCR Amplification, Illumina Based-Amplification of 16S rRNA Genes, and ITS2 Intergenic Sequences
The primer pair 799F (5′-AACMGGATTAGATACCCKG-3′) and 1,193R (5′-ACGTCATCCCCACCTTCC-3′) (Vieira et al., 2016) was selected for this experimentation, as it avoids amplification of bacteria-like 16S rRNA genes chloroplastic sequences (Beckers et al., 2016). PCR amplifications were carried out in 20 μL reaction mixtures containing 2 μL of DNA template (with a concentration of 5 ng/μL), 0.2 mM dNTPmix, 0.5 μM of each primer, 4 μL of Phusion HF Buffer, and 0.2 μL of high-fidelity polymerase Phusion (Thermo Fisher Scientific, MA, USA). The PCR cycling conditions were 98°C for 2 min followed by 30 cycles of 98°C for 20 s; 53°C for the first set of primers, 54°C for the second set of primers and for 30 s; 72°C for 30 sec, and a final extension step of 72°C for 10 min. The quality of PCR amplifications was verified by gel electrophoresis (2% agarose). PCR products were then sent to Mr DNA for MiSeq analysis (http://www.mrdnalab.com, Shallowater, TX, USA) where they were sequenced twice along independent runs (technical replicates).
As preliminary experiments revealed that a classic ITS2-based PCR approach did not yield significant amplifications, a nested PCR approach was set up to amplify the fungal microbiota. The first PCR was performed using the primer pair ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (Rämä et al., 2016) and for the second PCR the primer pair 5.8S-FUN (5′-AACTTTYRRCAAYGGATCWCT-3′) and ITS4-FUN (5′-AGCCTCCCGCTTATTGATATGCTTAART-3′) (Taylor et al., 2016) was used. First PCR amplifications were carried out in 10 μL reaction mixtures containing 2 μL of DNA template (at 5 ng/μL), 0.2 mM dNTP mix, 0.5 μM of each primer, 2 μL of Phusion HF Buffer and 0.1 μL of high-fidelity polymerase Phusion (Thermo Fisher Scientific, MA, USA). The first PCR cycling conditions were 98°C for 30 s followed by 35 cycles of 98°C for 10 s, 57°C for 30 s and 72°C for 30 s, and a final extension step of 72°C for 5 min. The second PCR amplifications were carried out in 20 μL reaction mixtures containing 2 μL of first PCR product diluted 1:100, 0.2 mM dNTPmix, 0.5 μM of each primer, 4 μL of Phusion HF Buffer, and 0.2 μL of high-fidelity polymerase Phusion (Thermo Fisher Scientific, Waltham, MA, USA). The second PCR cycling conditions were 98°C for 30 s followed by 30 cycles of 98°C for 10 s, 58°C for 30 s, and 72°C for 30 s, and a final extension step of 72°C for 5 min. The quality of the PCR amplifications was verified by gel electrophoresis. PCR products were then sent to Mr DNA for MiSeq analysis (http://www.mrdnalab.com, Shallowater, TX, USA) and sequenced twice (technical replicates, Supplementary Data Sheet 1).
All the following steps were conducted using routine Mr DNA based MiSeq-based pipelines. Briefly, PCR products were barcoded, pooled together in equal proportions and purified. Paired-end sequencing (2 × 250 pb) of bacterial 16S rRNA genes fragments and fungal ITS genomic fragments were performed on the Illumina MiSeq sequencing platform (Illumina, San Diego, USA). The raw paired-end reads from 16S rRNA and ITS2 genes fragments were separately assembled and reoriented using the MrDNA pipeline, and then, further steps were conducted at the lab using our bioinformatic pipelines. Reads abundance for 16S rRNA genes was in the 9,035–105,596 range, while reads abundance for ITS genomic fragments was in the 16,669–70,629 range.
Sequence Dataset Analysis
Briefly, for 16SrRNA based sequences, adapter sequences were trimmed in the raw FastQ files with Cutadapt v1.2.1 (Martin, 2011). The DADA2 (Callahan et al., 2016) protocol (https://benjjneb.github.io/dada2/tutorial.html) was then followed up to the production of a non-chimeric ASVs table (<4% of chimeras per sample were detected). Taxonomic assignment (min boot 80) was based on the GreenGenes reference database (v.13.8) (DeSantis et al., 2006). This taxonomic assignation also permitted to remove from the dataset all sequences that were affiliated with chloroplastic 16S rRNA. For ITS sequences, the same pipeline was used, excepted that taxonomic assignments were conducted using the latest available version of the UNITE database (v. 8.0) (Abarenkov et al., 2010). The computation of distance matrices based on ASV abundances was performed using the Gower distance metric (Kuczynski et al., 2010). Both Alpha- et Beta-diversity analysis were conducted using the R software package phyloseq, as well as the preparation of all graphical representations (McMurdie and Holmes, 2013). Sequences were submitted in the NCBI Sequences Read Archive (PRJNA602593).
Catalyzed Reporter Deposition Fluorescence In Situ Hybridization (CARD-FISH)
The pieces of algal thalli (3 mm × 3 mm each) were fixed in 4% paraformaldehyde in 50 mM HEPES (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid) buffer, pH 7.4 for 12 h at 4°C. After dehydration through a graded (30–70%) ethanol series, the algal tissue was infiltrated by London Resin White embedding medium (London Resin, UK). Samples were oriented in molds for cuts perpendicular to the algal surface, and allowed to polymerize at 45°C for 4 days. Semi-thin sections (1 μm) were then cut using a diamond knive on an Ultracut microtome (Reichert-Jung, Germany) and collected on Superfrost Plus™ slides (Thermo Fisher Scientific, MA, USA) at the MNHN Electron Microscopy platform. Alternately, completely ethanol-dehydrated tissue samples were embedded in Paraplast paraffin, cut to 5 μm sections with a Leica microtome equipped with a disposable razor blade, collected on Superfrost Plus™ slides, and used after deparaffinization with xylene.
After rehydration in phosphate buffer (PBS pH 7.4), the sections (either 1 μm resin-embedded or 5 μm deparaffinized) were first incubated in a solution of hydrogen peroxide (0.09%) for 10 min to inactivate endogenous peroxidases then rinsed three times in Tris-HCl 20 mM (pH 8). To allow DNA oligonucleotidic probes to access the bacterial 16S rRNA, the sections were then successively incubated in HCl (10 min, 10 mM), in lysozyme (15 min, 37°C, 10 mg/mL) and in proteinase K (15 min, 37°C, 1 μg/ml), including three rinses in Tris-HCl 20 mM (pH 8) after each step. Hybridization was performed with an equimolar mixture of the eubacterial probes EUB338 I (5′-GCT GCC TCC CGT AGG AGT-3′) (Amann et al., 1990), EUB338 II (5′-GCA GCC ACC CGT AGG TGT-3′) (Daims et al., 1999), and EUB338 III (5′-GCT GCC ACC CGT AGG TGT-3′) (Daims et al., 1999) with 1 μL probe mix (50 ng/μL) in 149 μL hybridization buffer (0.9 M NaCl, 20 mM TrisHCl pH 8.0, 0.01% SDS, 1% blocking reagent, 10% dextran sulfate, 35% (v/v) formamide). Negative controls were performed using a non-EUB probe (5′-CTC CTA CGG GAG GCA GC-3′) (Amann et al., 1990). All probes were purchased conjugated with horseradish peroxidase from http://www.biomers.net/ (Germany). Sections were incubated at 42°C for 3 h, then washed in pre-warmed washing buffer (5 mM EDTA, 42 mM NaCl, 20 mM TrisHCl pH 8.0, and 0.05% SDS) at 43°C for 15 min then rinsed three times in PBS 10 mM (pH 7.4). Sections were incubated in the dark for 15 min at 37°C in amplification buffer (1 mL) containing 2 μL of Alexa488-labeled tyramides (Invitrogen) and 10 μL 3% hydrogen peroxide. After incubation, the sections were rinsed three times with PBS for 2 min and counterstained for 10 min with DAPI, 10 μg/mL. Then the sections were rinsed twicewith PBS for 2 min, once with H2O for 2 min, air-dried in the dark then mounted in Fluoroshield (Sigma-Aldrich, St. Louis, MO, USA). The semi-thin resin sections were observed with a Nikon eclipse TE300 (Nikon, Tokyo, Japan) wide-field epifluorescence microscope fitted with a mercury lamp and dichroic filter sets to collect DAPI and Alexa488 signals, at the CeMIM photonic microscopy platform of the French Museum National d'Histoire Naturelle. The 5 μm deparaffinized sections were observed with a Zeiss AxioObserver LSM880 Airyscan confocal microscope with a Plan Apochromatic x40 NA 1.3 oil DIC objective, with images acquired simultaneously for Dapi (Diode laser 0, 2% Ex 405 nm, Em 405–495 nm) and Alexa488 (Argon laser 0,5% Ex 488 nm, Em 505–550 nm) fluorochromes. Exposure and gain settings per each channel were kept constant for probe EUBmix and control non-EUB treatments, that were placed on serial sections on the same slide. Images acquired in Zstack series (0.2 μm step) were denoised with Airyscan SR processing algorithm and Maximum intensity projections were obtained to visualize the bacterial morphotypes across the section 5 μm depth. Five independent Card-FISH experiments were performed on 1 or 5 μm sections from adjacent tissue blocks sampled from three different algae (Alga 2, 3, and 5). Serial (5 μm) sections from Alga 2, cut at 2 tissue depths separated by ~200 μm, were tested independently in separate CardFISH experiments, to increase the explored tissue volume.
Results
Effect of a Common Sterilization Protocol on Microbial Diversity
We compared the bacterial and fungal diversity before and after surface sterilization (Figures 1A,B), based on a commonly used methodology to assess cultivable endophytic microbial communities [successive baths of 70% EtOH for 30 s, 0.1% NaClO for 30 s, and three times sterile seawater for 30 s (Vallet et al., 2018)].
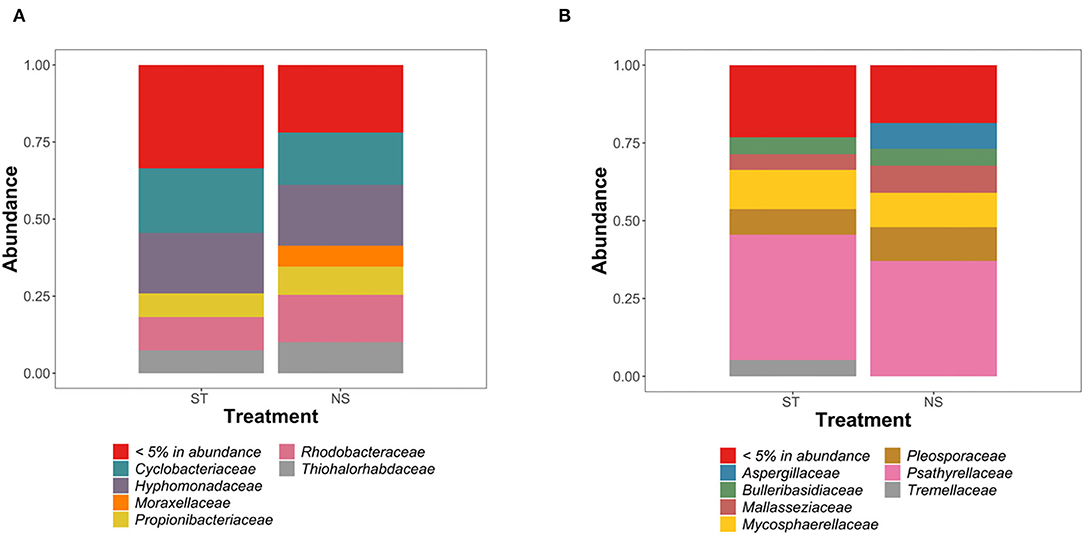
Figure 1. Analysis of the composition of bacterial (A) and fungal (B) communities in “sterilized” (ST) and “non-sterilized” (NS) algal samples.
When considering the 16SrRNA dataset, sterilized samples presented a Chao-1 index of 137–377, compared to 129–205.4 for non-sterilized samples. The Simpson index was 0.71–0.982 for sterilized samples similar to 0.73–0.982 in non-sterilized samples. The Shannon index was 2.08–4.63 in sterilized samples, similar to 2.24–4.57 in non-sterilized samples. Overall, Shannon, Simpson and Chao-1 indexes were not significantly different between surface sterilized and non-sterilized samples (p-value > 0.05; ANOVA, Tukey post-hoc test).
When considering the ITS genomic fragment datasets, sterilized samples presented a Chao-1 index which was between 9 and 28 for sterilized samples, compared to 8–32 for non-sterilized samples. The Shannon index was between 0.75 and 2.09 for sterilized samples, and 0.15–2.10 in non-sterilized samples. The Simpson index was 0.02–0.81 in sterilized samples, and 0.47–0.64 in non-sterilized samples. Overall, Shannon, Simpson, and Chao-1 indexes were not significantly different between surface sterilized and non-sterilized samples (p-value > 0.05; ANOVA, Tukey post-hoc test).
Community analysis revealed similar fungal and bacterial colonization of sterilized and non-sterilized samples, with two exceptions (Figures 1A,B). In bacterial communities, Moraxellaceae appeared sensitive to the sterilization treatment, as no related 16S rRNA sequences were detected in “sterile” samples. A similar pattern was observed in fungal communities for Aspergillaceae (Figures 1A,B). Despite these small differences, statistical analysis confirmed the absence of significant differences in the diversity of bacterial and fungal communities between the surface-sterilized and the non-sterilized samples (p-value > 0.05; ANOVA, Tukey post-hoc test).
Patterns of Bacterial and Fungal Diversity in S. latissima Algae
Alpha-diversity analysis revealed the presence of diverse bacterial communities associated with algal tissues (Figure 2A). We observed between 130 and 270 bacterial ASVs in each piece of tissue (all five algal individuals considered). Chao-1 and rarefaction curves analysis confirmed a wide diversity of bacteria associated to S. latissima (Supplementary Data Sheet 2). Similar observations were made with the Shannon (between 2.1 and 4.6 for algal samples) and Simpson indexes (between 0.708 and 0.982 for algal samples) (Figure 3A). Statistical analysis confirmed that seawater samples (300–428 ASVs, Shannon index of 5.1–5.4, and Simpson index of 0.987–0.988) were significantly more diverse than each piece of algal tissue sampled (p-value < 0.05; ANOVA, Tukey post-hoc test).
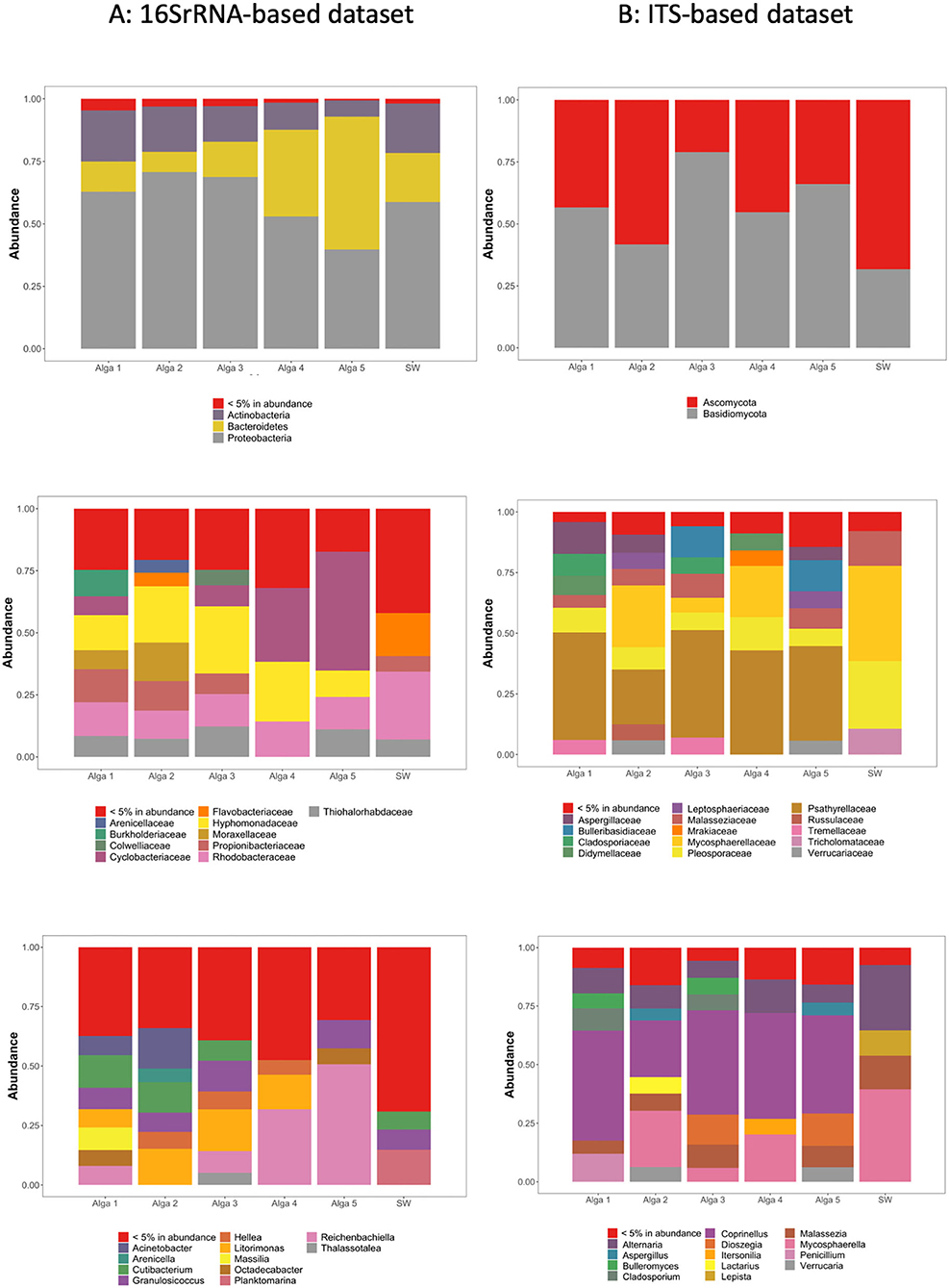
Figure 2. Relative abundance of the most abundant ASVs in our dataset, clustered according to their taxonomic assignation within each S. latissima individual and seawater sample (SW) based on 16S rRNA genes relative abundance (A) and ITS genomic fragments relative abundance (B). Top panel: Assignation at the phyla level. Middle panel: Assignation at the family level. Bottom panel: Assignation at the genus level.
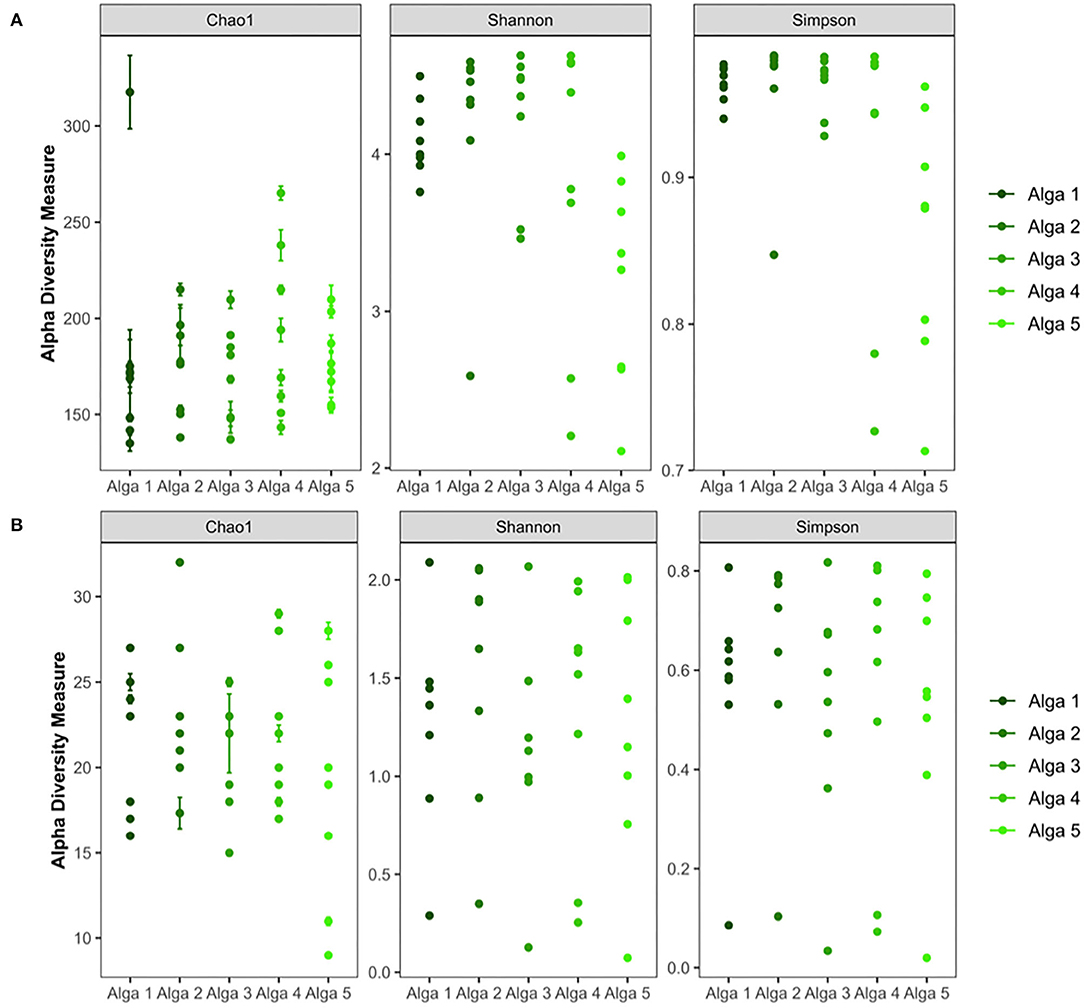
Figure 3. Alpha-diversity analysis of bacterial (A) and fungal (B) communities of the Saccharina latissima microbiota.
Alpha-diversity analysis also highlighted the presence of diverse fungal communities associated with algal tissues (5–32 ASV) (Figure 2B). Chao-1 and rarefaction curves analysis confirmed a wide diversity of fungi associated to S. latissima (Supplementary Data Sheet 2). The Shannon index was in between 0.2–2.25 and Simpson index in between 0.02–0.83 (Figure 3B). Statistical analysis confirmed that seawater sample (18–31 ASVs, Shannon index of 2.1–2.2, and Simpson index of 0.79–0.82) was equally diverse than most of pieces of algal tissue sampled (p-value > 0.05; ANOVA, Tukey post-hoc test).
Composition of Bacterial Communities in Each Alga and in the Nearby Seawater
In the light of the above results, community composition was assessed after aggregation and rarefaction of the samples originating from the same alga (Figure 2A). Regarding environmental seawater, when considering the most abundant ASVs (>5% abundance), the seawater bacterial community appeared dominated by Proteobacteria (58.4% of total sequences), Bacteroidetes (20.1%), and Actinobacteria (19.6%). At the family level, the seawater samples were dominated by Rhodobacteraceae (27.1%), Flavobacteriaceae (17.1%), and Thiohalorhabdaceae (7.0%) (Figure 2A).
The bacterial communities associated with algal tissues appeared quantitatively very variable among individual algae. Actinobacteria represented between 6.4 and 20.3% of total sequences, Bacteroidetes between 8.1 and 53.1%, and Proteobacteria between 39.7 and 70.5% (Figure 2A). The ASVs representing <5% of total sequences abundance contained bacterial taxa affiliated to Armatimonadetes, Chlamydiae, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Dependentiae, FBP, Fibrobacteres, Firmicutes, Fusobacteria, Gemmatimonadetes, Marinimicrobia (SAR406_clade), Nitrospirae, Patescibacteria, Planctomycetes, Spirochaetes, Verrucomicrobia, and WPS-2 phyla.
The most abundant ASVs were assigned to only a few types of bacterial families. Indeed, ASVs related to Hyphomonadaceae were abundant in all sampled individuals, ranging from 10.6 to 24.8% of sequences depending on the alga. Cyclobacteriaceae represented between 0 and 8.7%, except in Alga 4 and 5 where this family reached 29.7 and 48.0% of sequences, respectively. Also, a few other bacterial families were frequently found in S. latissma: Moraxellaceae (up to 15.4%), Propionibacteraceae (up to 13.0%), Rhodobacteraceae (11.3–14.3%), Thiohalorhabdaceae (up to 12.1%), Burkholderiaceae (up to 10.7%), Colwelliaceae (up to 6.1%), and Flavobacteriaceae (up to 5.4%) (Figure 2A).
Composition of Fungal Communities in Each Alga
When considering the most abundant ASVs in the seawater (>5% in total abundance), the fungal community appeared dominated by Ascomycota (54.6%) and Basidiomycota (45.3%) (Figure 2B). At the family level, seawater samples were dominated by diverse groups of fungi, including Mycosphaerellaceae (39.2%), Pleosporaceae (26.2%), Malasseziaceae (15.2%), Tricholomataceae (10.9%), and Psathyrellaceae (4.3%). Ceratobasidiaceae, Bulleribasidiaceae, Leptosphaeriaceae, Verrucariaceae, Tremellaceae, and Didymellaceae were also detected, but at much lower abundance (below 1.4%) (Figure 2B).
The fungal communities associated with algal tissues appeared different although as diverse as in seawater. Algal communities were dominated by Psathyrellaceae (39–44.9% depending on the sampled algae). Also, we found in lower abundances Pleosporaceae (6.7–13.7% depending on the sampled algae), Cladosporiaceae (0.07–8.9%), Mycosphaerellaceae (2.6–25.9%), Malasseziaceae (3.7–10.8%), Aspergillaceae (0.3–12.6%), Bulleribasidiaceae (0.2–12.7%), Didymellaceae (0.2–6.3%), Leptosphaeriaceae (0.2–7.35%), Mrakiaceae (0.12–6.75%), Russulaceae (0.05–6.4%), Tremellaceae (0.1–6.9%), Tricholomataceae (0.15–10.9%) and Verrucariaceae (0.2–6.2%) (Figure 2B).
A Global Beta-Diversity Analysis and an Attempt to Define a Core Microbiota
We then examined the degree of similarity between each algal sample (in terms of total ASVs composition) using a NMDS representation (Figure 4). Interestingly, when considering either the 16SrRNA based dataset or the ITS genomic fragments-based dataset, the samples originating from the same alga did not cluster together, and are frequently more closely related to samples from a different alga, revealing a high patchiness of bacterial and fungal communities (Figure 4).
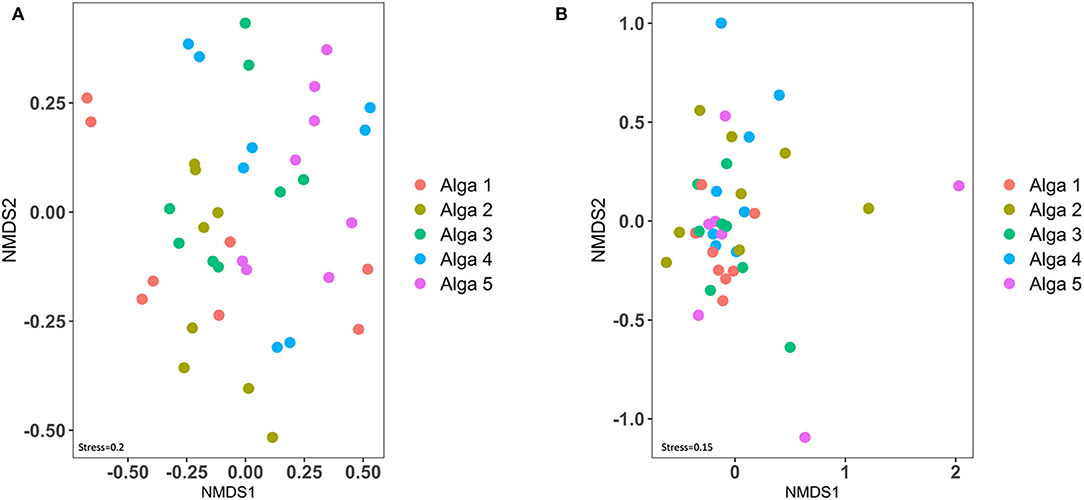
Figure 4. Beta-diversity analysis of bacterial (A) and fungal (B) communities of the Saccharina latissima microbiota.
To further investigate a potential core microbiota, we hypothesized that a small fragment of algal tissue might only collect a limited representation of the microbiota diversity. To test this hypothesis, we virtually pooled and rarefied, one by one and for each alga, the four tissue samples (4 × 1 cm3 = 4 cm3 in total) (Supplementary Tables 1, 2). Following this methodology, we found that the overall bacterial and fungal diversity observed in each alga was much higher compared to the one found in seawater, and inter-individual differences were also observable. Between 1,024 and 1,034 bacterial ASVs were observed in each alga, compared with the 428 ASVs detected in the seawater (1 L); and between 67 and 93 fungal ASVs were observed in each alga, against 31 in the seawater. Interestingly, the number of observed ASVs as well as Chao-1 values continuously increased while virtually adding new pieces of tissues in our analysis, showing that we did not collect the whole bacterial and fungal diversity of the S. latissima microbiota. However, these results demonstrate that the whole bacterial and fungal community in the total sampled 4 cm3 volume of algal tissue appeared much more diverse compared to the one of the 1 L sampled seawater. This result reveals that surfaces commonly sampled on kelps [i.e., up to 1 cm2 (Staufenberger et al., 2008; Weigel and Pfister, 2019)] may reveal some interesting biological patterns, but are insufficient to retrieve the complete composition of the Saccharina latissima microbiota.
Therefore, there are great challenges to correctly define a core bacterial and fungal community for S. latissima macroalgae. Indeed, using a threshold level at 0.001 in terms of relative sequence abundance and a prevalence of 80%, the bacterial core microbiota of each alga (i.e., using the ASV lists generated for all tissue samples originating from the same alga) appeared composed of Cutibacterium sp., Robignitomaculum sp., Hellea sp., Reichenbachiella sp., Litorimonas sp., and the fungal core microbiota contained Coprinellus sp. and Dioszegia sp. However, each of these core taxa represents <1% of all sequences in each tissue sample taken individually.
Visualization of Bacteria Within the Tissues of S. latissima
CARD-FISH with a universal bacterial probe mix (EUB338 I + EUB338 II + EUB338 III), highlighted the presence of endophytic bacteria within S. latissima tissues (Figure 5). Bacterial morphotypes positive to the bacterial probe mix were observed consistently but at low abundance in all 5 μm (paraffin) or 1 μm (LR White) thick tissue sections from Alga 2 (sampled at two depths separated by ~200 μm) and from Alga 3 (Figure 5 in paraffin sections). Fewer signals were observed in tissue sections of Alga 5 (in LR White 1 μm sections, data not shown). Note that the EUB probe-mix also bound to plastid 16S RNA inside chloroplasts, which were preferentially distributed in the algal cortex, and had however distinct size and shapes compared to the bacterial cocci or rod-shaped morphotypes. Controls with non-EUB non-sense probe were negative (Figure 5D). Positively-labeled bacteria were detected preferentially in the cortical tissue of the algae, with less abundant signal in the medulla. The bacteria were located between algal cells or in the cytoplasm away from the chloroplasts, with a spatially heterogeneous distribution, either as isolated morphotypes or as small aggregates of 2–6 bacterial cells (circled in white in Figures 5C,E). Some positively-labeled epiphytic bacteria were also observed at the surface of the epidermis (Figure 5B) however at much lower abundance than the endophytic bacteria.
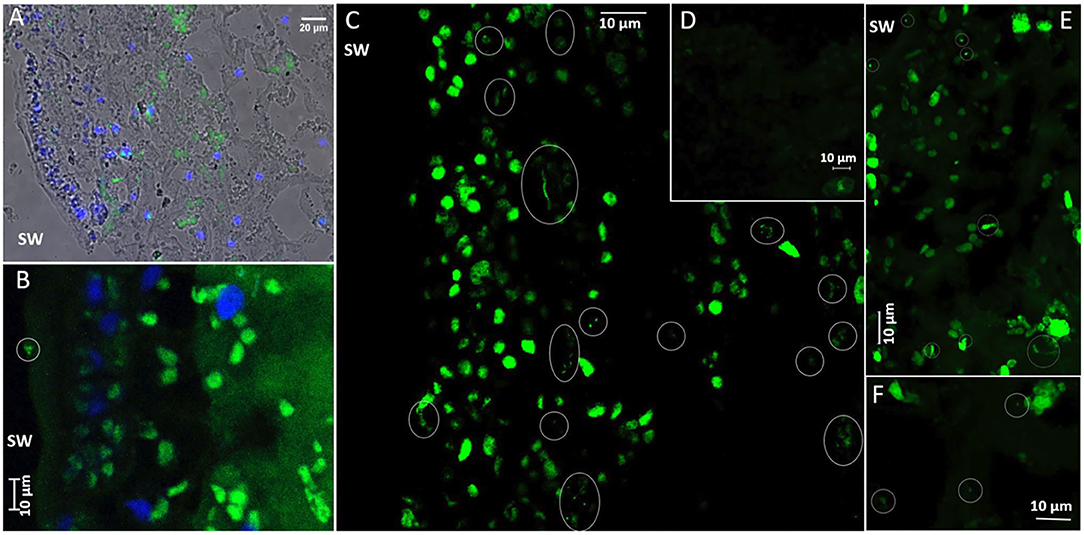
Figure 5. CARD-FISH visualization of bacteria in Saccharina latissima tissue. Ellipses mark the position of bacteria. (A) Overview of bacteria in S. latissima alga 3S1 (histological section 5 μm), with overlayed Transmission, Dapi-stained nuclei (blue) and Alexa488 (green) EUBmix probe positive bacteria in wide-field epifluorescence microscopy (Nikon Eclipse TE300, x40 oil objective). (B) A few bacteria at the surface of S. latissima alga 2S2 (histological section slide L2), imaged with Zeiss LSM880 in conventional mode (Maximum Intensity Projection of 1.5 μm Zstack series with 0.5 μm step increments). Chloroplasts in the cortex (with distinctive larger shape) are also labeled with the EUBmix probe. (C) Diverse bacterial morphotypes (green) in S. latissima alga 2S2 (histological section slide L5), visualized in superresolution mode in confocal microscopy (Zeiss LSM880 Airyscan processing, maximum Intensity Projection of 3.2 μm Zstack series with 0.2 μm step increments, x40 oil NA1.3 DIC objective). Clusters of long thin morphotypes are preferentially distributed in the cortex, along with a few isolated or clustered cocci and short and thick bacilli. (D) Negative control with Alexa488-HRP non-sense probe non-EUB in S. latissima alga 2S2 (histological section slide L2) visualized in superresolution mode (Zeiss LSM880 Airyscan processing, Maximum Intensity Projection of 4.4 μm Zstack series with 0.2 μm step increments). (E) Irregular distribution of bacterial morphotypes from the surface (upper left) to the cortex (lower right) of S. latissima alga 2S2 (histological section slide L2) (Zeiss LSM880 Airyscan processing, Maximum Intensity Projection of 5.6 μm Zstack series with 0.2 μm step increments). (F) Very few bacterial morphotypes deeper in the medulla of S. latissima alga 2S2 (histological section slide L2) (Zeiss LSM880 Airyscan processing, Maximum Intensity Projection of 1.4 μm Zstack series with 0.2 μm step increments).
Discussion
It is now well-established that the seaweed microbiota is of major importance for their host, impacting their development, nutrition and capacity to fight pathogens (Salaün et al., 2012; Singh and Reddy, 2014), with active exploration of its composition in diverse macroalgal models [i.e., among others: (KleinJan et al., 2017; Serebryakova et al., 2018)]. Here, to the best of our knowledge, we report the first culture-independent snapshot of Saccharina latissima fungal and bacterial communities, combining data from high-throughput amplicon sequencing of bacterial and fungal ribosomal markers and CARD-FISH visualization of bacterial morphotypes.
CARD-FISH analyses revealed an unexpectedly spatially highly heterogeneous distribution of bacterial endosymbionts within the algal tissue. Bacteria were mainly observed within the inner tissues (cortex and medulla), although some rare FISH signals were also observed at the surface of the epidermis, suggesting a relatively good efficiency of our surface-sterilization protocol. However, as shown by the similar bacterial diversity retrieved from surface-sterilized or intact algal thalli, this classic sterilization protocol, based on rinses in ethanol 70% then sodium hypochloride at the concentration of 0.1%, seems unable to efficiently degrade DNA (Kemp and Smith, 2005; Vallet et al., 2018). Thus, our results highly suggest avoiding the use of this widespread approach to sterilize the surface of macroalgae, when the goal is to specifically target the endophytic communities using DNA sequencing.
To further explore the composition of the whole S. latissima microbiota (both its endophytic and epiphytic compartments), we performed a metabarcoding approach targeting both fungal and bacterial communities. In order to avoid excessive amplification of chloroplastic 16S rRNA gene reads we used the primer pair 799F/1193R (Beckers et al., 2016). Primer pair selection to target the broadest possible bacterial diversity remains an open discussion (Thijs et al., 2017). However, a detailed comparison between all available primer sets remains difficult as most studies do not use an ASVs-based diversity analysis but instead rely on OTU-based approaches, known to generate spurious taxonomic units (Callahan et al., 2016). Here, our results point out that the selected primer pair 799F/1193R is suitable to examine the bacterial microbiota associated to (S. latissima) algal tissues, as we detected less that 5% of 16S rDNA chloroplastic sequences using this methodology.
Importantly, our results unveiled that both the bacterial and fungal communities were spatially organized as highly diverse patches within the sampled algal tissues. In fact, our data revealed that whilst good sequencing coverage allowed to detect the diversity present in our samples, we did not retrieve all the bacterial and fungal community present within each alga. Therefore, the high variability observed between tissue sample replicates, and between individuals, may obscure meaningful trends, and the incomplete sampling hinders the definition of a core microbiota (Hernandez-Agreda et al., 2017) for this large-sized macroalga. Moving forward, it will be important to incorporate this within-tissue spatial variability into experimental designs, especially as existing studies have typically focused on the variability between organs, seasons, and populations (Staufenberger et al., 2008; Bengtsson et al., 2012). Taking into account this dimension—typically using a hierarchical sampling design—will allow to define at which scale the microbiota might be constant and potentially resilient to change (see examples in Rahel, 1990; Kendall and Widdicombe, 1999). It will also allow to tackle more complex questions, such as the possibility that macroalgal microbiota may be structured in dynamic patches, as opposed to an equilibrium (Wu and Loucks, 1995).
Despite these caveats, our results do highlight the very high diversity of fungal and bacterial communities able to colonize S. latissima. Few studies previously attempted to address this question. In his culture-based inventory of S. latissima, Staufenberger et al. (2008) for example depicted the presence of bacteria on one individual alga that were not found on other algae. Considering only one individual, large variations of bacterial diversity were also recorded between different tissues of Sargassum muticum (Serebryakova et al., 2018) Some authors suggested that such pattern is typical of holobiont's communities structuration (Burke et al., 2011a,b). Our observation highlights the necessity to ascertain more precisely the spatial and temporal scales at which core microbiota should be best defined.
We recorded important differences between the seaweed-associated microbiota and the surrounding seawater microbiota. This observation could be indicative of a selection process whereby the seaweed would have the capacity to specifically recruit, at least part of, its microbiota. Such result appears consistent with the recent “microbial gardening” hypothesis, suggesting the selection of beneficial epibacterial strains by the seaweed holobiont, which might be also involved in the molecular dialog between both the alga and its bacterial microbiota (Kessler et al., 2018). Seaweeds use chemical compounds to communicate with microbes and manipulate bacterial community composition and abundance (Nylund et al., 2011; da Gama et al., 2014; Saha and Weinberger, 2017), also to deter pathogens (Saha and Weinberger, 2019). Similarly, the richness of bacterial assemblages associated to Cystoseira was measured significantly higher compared to the surrounding seawater (Mancuso et al., 2016). The stochastic recruitment of functionally redundant species, as well as the influence of abiotic and biotic factors (especially the existence of interactions within the algal host and interactions within the microbiota) have been proposed to explain those patterns (Burke et al., 2011a,b; Egan et al., 2013; Kessler et al., 2018). These observations with ours suggest that the degree of colonization is, at least partially, linked with the type of metabolites released by the algae, which may favor the accumulation or the elimination or reduction of specific bacterial species (Salaün et al., 2012; Paix et al., 2019, 2020).
The bacterial communities of S. latissima were dominated by the Proteobacteria, Bacteroidetes and Actinobacteria phyla. Although their proportions varied according to the samples, this observation is consistent with previous published reports focusing either on epibiotic or endophytic communities (Wang et al., 2007; Hollants et al., 2013; Martin et al., 2015; KleinJan et al., 2017). The most important difference was observed for Planktomycetes which were not abundant in our samples but found in large proportion in many other studies using culture dependent or culture-independent studies on other types of algae (Serebryakova et al., 2018). Planctomycetes are indeed frequently associated to macroalgae, and thus were found growing on Laminaria hyperborea, Macrocystis pyrifera, Ulva australis, and many others (Longford et al., 2007; Bengtsson and Øvreås, 2010; Bengtsson et al., 2012; Michelou et al., 2013). Nevertheless, this bacterial group is probably subjected to important seasonal variations, as mostly found in summer in Sargassum muticum (Serebryakova et al., 2018). Such seasonal variability could explain the low abundance of this phyla in our samples.
At the family level, Rhodobacteraceae and the closely related phylogenetic group of Hyphomonadaceae were well-represented in S. latissima microbiota in coherence with previous culture-based inventories of the S. latissima microbiota (Staufenberger et al., 2008) and other algae (Lupette et al., 2016; Serebryakova et al., 2018; Parrot et al., 2019; Paix et al., 2020). Rhodobacteraceae are frequently detected on diverse types of marine surfaces. These pioneer biofilm-forming colonizers were sometimes depicted as pathogens, but other members of this family are known to be involved in key physiological activities, like nitrogen fixation (Dang et al., 2008). In addition, we found some Flavobacteriaceae in the endophytic community of two of the sampled algae. This presence was not surprising, as previously detected within the endophytic community of Bryopsis (Hollants et al., 2011) and also because Flavobacteria are known to be abundant marine algal-associated sugar consumers (Kirchman, 2002). Nevertheless, the presence of Rhodobacteraceae and Flavobacteriaceae in the S. latissima holobiont appears to be of the rare common patterns with other algae's microbiota. Indeed Pseudoalteromonadaceae, Pseudomonadaceae, Alteromonadaceae, Sphingomonadaceae, Vibrionaceae, Granulosicoccaceae, Saprospiraceae, Sphingobacteriaceae are frequently found in the epibiotic community of Gracilaria sp. (Rhodophyta), Laminaria sp.(Heterokontophyta), and Ulva sp. (Chlorophyta)(Florez et al., 2017) but are among the rare members of the community in the present S. latissima. We could hypothesize based on these observations that the S. latissima microbiota described here presents specific features compared to other types of (brown) algae.
Most of the studies reviewed in Hollants et al. (2013) -and on which our comparisons are based- relied on culture-dependent studies, and thus probably not representative of the “real” bacterial diversity. Indeed, it is known that a large fraction of marine communities are not currently cultivable (Amann et al., 1995). Few inventories of algal microbiota based on high-throughput sequencing are available in the literature and thus, our study paves the way for the future research focusing on the core microbiota of brown algae and reinforce the need to pursue these investigations based on DNA-based sequencing of algal holobionts. However, considering the importance and the diversity of S. latissima holobiont, this study paradoxically also reinforces the need to pursue continuous and important effort to isolate new bacterial models using innovative cultured based approaches, pertinent to address the role of the holobiont in algal physiology, like those conducted on the brown algae Ectocarpus using extinction-dilution approaches (KleinJan et al., 2017).
Regarding fungal communities, a dominance of both Ascomycota (54.6%) and Basidiomycota (45.3%) was reported. Previous studies relying on high-throughput sequencing approaches to describe the fungal communities associated with macroalgae also reported a predominance of Ascomycota, followed by Basidiomycota (Agusman and Danqing, 2017; Wainwright et al., 2017). Moreover, data from culture-dependent approaches of fungal communities associated with macroalgae also reported a majority of Ascomycota (reviewed in Flewelling et al., 2015). Interestingly, as in our dataset, the genus Cladosporium has been frequently reported from diverse macroalgae, using culture-dependent and -independent approaches (Flewelling et al., 2015; Wainwright et al., 2017) which seems to confirm their ubiquitous nature. Also, using DNA Stable Isotope Probing, Cladosporium was determined as part of the mycoplankton with capabilities to assimilate algal-derived particulate organic matter, highlighting their non-trivial roles in the marine environment (Cunliffe et al., 2017).
All these results highlighted that the variation of bacterial richness associated with S. latissima tissues is also due to rare ASVs, which appears consistent with previous studies on brown algae (Bengtsson et al., 2012; Stratil et al., 2013). Especially, Stratil et al. (2013) suggested that the proportions of a few OTUs (including three from the “rare biosphere”) may vary according to the environmental conditions, and that those OTUs may also highly contribute to dynamic interactions between the host and its microbiota and thus might govern and regulate key physiological features of both the microbiota and its host. Collectively, all this information reinforces the need to collect more data about the potential physiological and functional roles of the “rare biosphere” (e.g., chemical defenses).
Although additional studies with more replicates are needed to confirm our results, they question the concept of core microbiota, or at least the methodology to be used to define it in macroalgae. Indeed, studies using DGGE and cultivable approaches suggested that a core community which is specifically adapted to an algal host–associated lifestyle could be defined, at least at higher taxonomic level (Staufenberger et al., 2008; Tujula et al., 2010; Hollants et al., 2013). As in the present study, the work of Burke et al. led a contradictory hypothesis by showing that there was no consistent core community of bacterial species colonizing the surface of U. australis and suggested that many different bacterial and fungal species are able to colonize algal tissues (Burke et al., 2011b). Overall those results confirmed the need to combine functional approaches (Burke et al., 2011a; Florez et al., 2017) and underscore the issue of the total extent of tissue to sample to draw conclusions on such large macroalgae.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA602593.
Author Contributions
AT, RL, and SP designed the experiment. AT, ID-C, MG, and SP collected samples and performed experiments. RL, GB, WL, and DB analyzed sequences datasets. All authors significantly contributed to data interpretation and manuscript writing.
Funding
This work has received funding by UKRI GlobalSeaweed-STAR (BB/P027806/1), the European Union's Horizon 2020 Framework Programme under grant agreement No 727892, and under the Marie Skłodowska-Curie grant agreement No 642575. This work was also funded by ASSEMBLE (227799), ASSEMBLE+ (730984), and EMBRC (262280) grants.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MS declared a past co-authorship with several of the authors CG and SP.
Acknowledgments
We acknowledged the Scottish Association for Marine Science (SAMS) and the Biological Station of Roscoff for hosting the field missions. We were grateful to the Bio2Mar platform for providing technical help and support. Fluorescence imaging facilities were provided by the CeMIm platform at the MNHN. We thanked both reviewers for constructive comments that help to significantly improve the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.587566/full#supplementary-material
Supplementary Data Sheet 1. The 16S rRNA bacteria community analysis based on the sequencing primer pair 515F/926R in all samples sequenced in this study and raw data comparison between the two sequencing runs for both 16S rRNA and ITS genomic fragments datasets.
Supplementary Data Sheet 2. Rarefaction curves build from of 16S rRNA and ITS genomic fragments-based datasets.
Supplementary Table 1. Number of observed ASV and Chao-1 values when increasing the number of collected algal pieces in our 16S rRNA genes-based analysis.
Supplementary Table 2. Number of observed ASV and Chao-1 values when increasing the number of collected algal pieces in our ITS genes fragments-based analysis.
Abbreviations
ASV, Amplicon Sequence Variant; CARD-FISH, CAtalyzed Reporter Deposition Fluorescence In Situ Hybridization; DGGE, Denaturing Gradient Gel Electrophoresis; ITS, Internal Transcribed Spacer; OTU, Operational Taxonomic Unit.
References
Abarenkov, K., Henrik Nilsson, R., Larsson, K., Alexander, I. J., Eberhardt, U., Erland, S., et al. (2010). The UNITE database for molecular identification of fungi-recent updates and future perspectives. New Phytol. 186, 281–285. doi: 10.1111/j.1469-8137.2009.03160.x
Adams, J. M., Gallagher, J. A., and Donnison, I. S. (2009). Fermentation study on Saccharina latissima for bioethanol production considering variable pre-treatments. J. Appl. Phycol. 21, 569–574. doi: 10.1007/s10811-008-9384-7
Agusman, A., and Danqing, F. (2017). Fungal community structure of macroalga Ulva intestinalis revealed by Miseq sequencing. Squalen Bull. Mar. Fish. Postharvest Biotechnol. 12, 99–106. doi: 10.15578/squalen.v12i3.315
Aires, T., Moalic, Y., Serrao, E. A., and Arnaud-Haond, S. (2015). Hologenome theory supported by cooccurrence networks of species-specific bacterial communities in siphonous algae (Caulerpa). FEMS Microbiol. Ecol. 91:fiv067. doi: 10.1093/femsec/fiv067
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A. (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Env. Microbiol. 56, 1919–1925. doi: 10.1128/AEM.56.6.1919-1925.1990
Amann, R. I., Ludwig, W., and Schleifer, K.-H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1128/MMBR.59.1.143-169.1995
Barott, K. L., Rodriguez-Brito, B., Janouškovec, J., Marhaver, K. L., Smith, J. E., Keeling, P., et al. (2011). Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral Montastraea annularis: microbial diversity on benthic algae and corals. Environ. Microbiol. 13, 1192–1204. doi: 10.1111/j.1462-2920.2010.02419.x
Bartsch, I., Wiencke, C., Bischof, K., Buchholz, C. M., Buck, B. H., Eggert, A., et al. (2008). The genus Laminaria sensu lato: recent insights and developments. Eur. J. Phycol. 43, 1–86. doi: 10.1080/09670260701711376
Beckers, B., Op De Beeck, M., Thijs, S., Truyens, S., Weyens, N., Boerjan, W., et al. (2016). Performance of 16s rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 7:650. doi: 10.3389/fmicb.2016.00650
Bengtsson, M., Sjøtun, K., and Øvreås, L. (2010). Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat. Microb. Ecol. 60, 71–83. doi: 10.3354/ame01409
Bengtsson, M. M., and Øvreås, L. (2010). Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol. 10:261. doi: 10.1186/1471-2180-10-261
Bengtsson, M. M., Sjøtun, K., Lanzén, A., and Øvreås, L. (2012). Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J. 6:2188. doi: 10.1038/ismej.2012.67
Burke, C., Steinberg, P., Rusch, D., Kjelleberg, S., and Thomas, T. (2011a). Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. U.S.A. 108, 14288–14293. doi: 10.1073/pnas.1101591108
Burke, C., Thomas, T., Lewis, M., Steinberg, P., and Kjelleberg, S. (2011b). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5:590. doi: 10.1038/ismej.2010.164
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13:581. doi: 10.1038/nmeth.3869
Cunliffe, M., Hollingsworth, A., Bain, C., Sharma, V., and Taylor, J. D. (2017). Algal polysaccharide utilisation by saprotrophic planktonic marine fungi. Fungal Ecol. 30, 135–138. doi: 10.1016/j.funeco.2017.08.009
da Gama, B. A., Plouguerné, E., and Pereira, R. C. (2014). The antifouling defence mechanisms of marine macroalgae. Adv. Bot. Res. 71, 413–440. doi: 10.1016/B978-0-12-408062-1.00014-7
Daims, H., Brühl, A., Amann, R., Schleifer, K. H., and Wagner, M. (1999). The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444. doi: 10.1016/S0723-2020(99)80053-8
Dang, H., Li, T., Chen, M., and Huang, G. (2008). Cross-ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Appl. Environ. Microbiol. 74, 52–60. doi: 10.1128/AEM.01400-07
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Doyle, J. J., and Doyle, J. L. (1987). A Rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Egan, S., and Gardiner, M. (2016). Microbial dysbiosis: rethinking disease in marine ecosystems. Front. Microbiol. 7:991. doi: 10.3389/fmicb.2016.00991
Egan, S., Harder, T., Burke, C., Steinberg, P., Kjelleberg, S., and Thomas, T. (2013). The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Flewelling, A. J., Currie, J., Gray, C. A., and Johnson, J. A. (2015). Endophytes from marine macroalgae: promising sources of novel natural products. Curr. Sci. 109, 88–111.
Florez, J. Z., Camus, C., Hengst, M. B., and Buschmann, A. H. (2017). A functional perspective analysis of macroalgae and epiphytic bacterial community interaction. Front. Microbiol. 8:2561. doi: 10.3389/fmicb.2017.02561
Ghaderiardakani, F., Coates, J. C., and Wichard, T. (2017). Bacteria-induced morphogenesis of Ulva intestinalis and Ulva mutabilis (Chlorophyta): a contribution to the lottery theory. FEMS Microbiol. Ecol. 93:fix094. doi: 10.1093/femsec/fix094
Goecke, F., Labes, A., Wiese, J., and Imhoff, J. (2010). Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 409, 267–299. doi: 10.3354/meps08607
Handå, A., Forbord, S., Wang, X., Broch, O. J., Dahle, S. W., Størseth, T. R., et al. (2013). Seasonal- and depth-dependent growth of cultivated kelp (Saccharina latissima) in close proximity to salmon (Salmo salar) aquaculture in Norway. Aquaculture 414, 191–201. doi: 10.1016/j.aquaculture.2013.08.006
Hermund, D. B., Sivasubramaniam, N., Neerup, R., Thomsen, B. R., and Jacobsen, C. (2018). “Antioxidant characterization and cosmetic application of extracts from brown alga Saccharina latissima,” in Seaweed Health Conf. - GMIT Galway Irel. 24 June 2018 - 27 June 2018 (Galway).
Hernandez-Agreda, A., Gates, R. D., and Ainsworth, T. D. (2017). Defining the core microbiome in corals' microbial soup. Trends Microbiol. 25, 125–140. doi: 10.1016/j.tim.2016.11.003
Hollants, J., Leliaert, F., De Clerck, O., and Willems, A. (2013). What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol. Ecol. 83, 1–16. doi: 10.1111/j.1574-6941.2012.01446.x
Hollants, J., Leroux, O., Leliaert, F., Decleyre, H., De Clerck, O., and Willems, A. (2011). Who is in there? Exploration of endophytic bacteria within the siphonous green seaweed bryopsis (Bryopsidales, Chlorophyta). PLoS ONE 6:e26458. doi: 10.1371/journal.pone.0026458
Kemp, B. M., and Smith, D. G. (2005). Use of bleach to eliminate contaminating DNA from the surface of bones and teeth. Forensic Sci. Int. 154, 53–61. doi: 10.1016/j.forsciint.2004.11.017
Kendall, M., and Widdicombe, S. (1999). Small scale patterns in the structure of macrofaunal assemblages of shallow soft sediments. J. Exp. Mar. Biol. Ecol. 237, 127–140. doi: 10.1016/S0022-0981(98)00224-X
Kessler, R. W., Weiss, A., Kuegler, S., Hermes, C., and Wichard, T. (2018). Macroalgal–bacterial interactions: role of dimethylsulfoniopropionate in microbial gardening by Ulva (Chlorophyta). Mol. Ecol. 27, 1808–1819. doi: 10.1111/mec.14472
Kirchman, D. L. (2002). The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100. doi: 10.1016/S0168-6496(01)00206-9
KleinJan, H., Jeanthon, C., Boyen, C., and Dittami, S. M. (2017). Exploring the cultivable Ectocarpus microbiome. Front. Microbiol. 8:2456. doi: 10.3389/fmicb.2017.02456
Kuczynski, J., Liu, Z., Lozupone, C., McDonald, D., Fierer, N., and Knight, R. (2010). Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods 7, 813–819. doi: 10.1038/nmeth.1499
Lachnit, T., Meske, D., Wahl, M., Harder, T., and Schmitz, R. (2011). Epibacterial community patterns on marine macroalgae are host-specific but temporally variable:epiphytic communities on macroalgae. Environ. Microbiol. 13, 655–665. doi: 10.1111/j.1462-2920.2010.02371.x
Longford, S., Tujula, N., Crocetti, G., Holmes, A., Holmström, C., Kjelleberg, S., et al. (2007). Comparisons of diversity of bacterial communities associated with three sessile marine eukaryotes. Aquat. Microb. Ecol. 48, 217–229. doi: 10.3354/ame048217
Lupette, J., Lami, R., Krasovec, M., Grimsley, N., Moreau, H., Piganeau, G., et al. (2016). Marinobacter dominates the bacterial community of the Ostreococcus tauri phycosphere in culture. Front. Microbiol. 7:1414. doi: 10.3389/fmicb.2016.01414
Mancuso, F. P., D'Hondt, S., Willems, A., Airoldi, L., and De Clerck, O. (2016). Diversity and temporal dynamics of the epiphytic bacterial communities associated with the canopy-forming seaweed cystoseira compressa (Esper) Gerloff and Nizamuddin. Front. Microbiol. 7:476. doi: 10.3389/fmicb.2016.00476
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martin, M., Barbeyron, T., Martin, R., Portetelle, D., Michel, G., and Vandenbol, M. (2015). The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-polysaccharide-degrading bacteria. Front. Microbiol. 6:1487. doi: 10.3389/fmicb.2015.01487
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Mesnildrey, L., Jacob, C., Frangoudes, K., Reunavot, M., and Lesueur, M. (2012). Seaweed Industry in France. Report Interreg program NETALGAE. Agrocampus Ouest, France.
Michelou, V. K., Caporaso, J. G., Knight, R., and Palumbi, S. R. (2013). The ecology of microbial communities associated with Macrocystis pyrifera. PLoS ONE 8:e67480. doi: 10.1371/annotation/48e29578-a073-42e7-bca4-2f96a5998374
Moy, F. E., and Christie, H. (2012). Large-scale shift from sugar kelp (Saccharina latissima) to ephemeral algae along the south and west coast of Norway. Mar. Biol. Res. 8, 309–321. doi: 10.1080/17451000.2011.637561
Nylund, G. M., Weinberger, F., Rempt, M., and Pohnert, G. (2011). Metabolomic assessment of induced and activated chemical defence in the invasive red alga Gracilaria vermiculophylla. PLoS ONE 6:e29359. doi: 10.1371/journal.pone.0029359
Paix, B., Carriot, N., Barry-Martinet, R., Greff, S., Misson, B., Briand, J., et al. (2020). A multi-omics analysis suggests links between the differentiated surface metabolome and epiphytic microbiota along the thallus of a Mediterranean seaweed holobiont. Front. Microbiol. 11:494. doi: 10.3389/fmicb.2020.00494
Paix, B., Othmani, A., Debroas, D., Culioli, G., and Briand, J. (2019). Temporal covariation of epibacterial community and surface metabolome in the Mediterranean seaweed holobiont Taonia atomaria. Environ. Microbiol. 21, 3346–3363. doi: 10.1111/1462-2920.14617
Panzer, K., Yilmaz, P., Weiß, M., Reich, L., Richter, M., Wiese, J., et al. (2015). Identification of habitat-specific biomes of aquatic fungal communities using a comprehensive nearly full-length 18S rRNA dataset enriched with contextual data. PLoS ONE 10:e0134377. doi: 10.1371/journal.pone.0134377
Parrot, D., Blümel, M., Utermann, C., Chianese, G., Krause, S., Kovalev, A., et al. (2019). Mapping the surface microbiome and metabolome of brown seaweed Fucus vesiculosus by amplicon sequencing, integrated metabolomics and imaging techniques. Sci. Rep. 9, 1–17. doi: 10.1038/s41598-018-37914-8
Rahel, F. J. (1990). The hierarchical nature of community persistence: a problem of scale. Am. Nat. 136, 328–344. doi: 10.1086/285101
Rämä, T., Davey, M. L., Nordén, J., Halvorsen, R., Blaalid, R., Mathiassen, G. H., et al. (2016). Fungi sailing the Arctic ocean: speciose communities in North Atlantic driftwood as revealed by high-throughput amplicon sequencing. Microb. Ecol. 72, 295–304. doi: 10.1007/s00248-016-0778-9
Saha, M., and Weinberger, F. (2017). Boom and bust path of Gracilaria: Loss of bacterial “friends” can limit the invasion success of a seaweed host? Phycologia 56:165.
Saha, M., and Weinberger, F. (2019). Microbial “gardening” by a seaweed holobiont: surface metabolites attract protective and deter pathogenic epibacterial settlement. J. Ecol. 107, 2255–2265. doi: 10.1111/1365-2745.13193
Salaün, S., La Barre, S., Santos-Goncalvez, M. D., Potin, P., Haras, D., and Bazire, A. (2012). Influence of exudates of the kelp Laminaria Digitata on biofilm formation of associated and exogenous bacterial epiphytes. Microb. Ecol. 64, 359–369. doi: 10.1007/s00248-012-0048-4
Serebryakova, A., Aires, T., Viard, F., Serrão, E. A., and Engelen, A. H. (2018). Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PLoS ONE 13:e0206734. doi: 10.1371/journal.pone.0206734
Singh, R. P., and Reddy, C. R. K. (2014). Seaweed-microbial interactions: key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 88, 213–230. doi: 10.1111/1574-6941.12297
Skjermo, J., Aasen, I. M., Arff, J., Broch, O. J., Carvajal, A. K., Christie, H. C., et al. (2014). A New Norwegian Bioeconomy Based on Cultivation and Processing of Seaweeds: Opportunities and RandD Needs. Trondheim: SINTEF Fisheries and Aquaculture.
Staufenberger, T., Thiel, V., Wiese, J., and Imhoff, J. F. (2008). Phylogenetic analysis of bacteria associated with Laminaria saccharina: bacteria associated with Laminaria saccharina. FEMS Microbiol. Ecol. 64, 65–77. doi: 10.1111/j.1574-6941.2008.00445.x
Stratil, S. B., Neulinger, S. C., Knecht, H., Friedrichs, A. K., and Wahl, M. (2013). Temperature-driven shifts in the epibiotic bacterial community composition of the brown macroalga Fucus vesiculosus. Microbiologyopen 2, 338–349. doi: 10.1002/mbo3.79
Tapia, J. E., González, B., Goulitquer, S., Potin, P., and Correa, J. A. (2016). Microbiota influences morphology and reproduction of the brown alga Ectocarpus sp. Front. Microbiol. 7:382. doi: 10.3389/fmicb.2016.00197
Taylor, D. L., Walters, W. A., Lennon, N. J., Bochicchio, J., Krohn, A., Caporaso, J. G., et al. (2016). Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl. Environ. Microbiol. 82, 7217–7226. doi: 10.1128/AEM.02576-16
Thijs, S., Op De Beeck, M., Beckers, B., Truyens, S., Stevens, V., Van Hamme, J. D., et al. (2017). Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Front. Microbiol. 8:494. doi: 10.3389/fmicb.2017.00494
Tourneroche, A., Lami, R., Hubas, C., Blanchet, E., Vallet, M., Escoubeyrou, K., et al. (2019). Bacterial–fungal interactions in the kelp endomicrobiota drive autoinducer-2 quorum sensing. Front. Microbiol. 10:1693. doi: 10.3389/fmicb.2019.01693
Tujula, N. A., Crocetti, G. R., Burke, C., Thomas, T., Holmström, C., and Kjelleberg, S. (2010). Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J. 4:301. doi: 10.1038/ismej.2009.107
Vallet, M., Strittmatter, M., Murúa, P., Lacoste, S., Dupont, J., Hubas, C., et al. (2018). Chemically-mediated interactions between macroalgae, their fungal endophytes, and protistan pathogens. Front. Microbiol. 9:3161. doi: 10.3389/fmicb.2018.03161
Vieira, C., Engelen, A. H., Guentas, L., Aires, T., Houlbreque, F., Gaubert, J., et al. (2016). Species specificity of bacteria associated to the brown seaweeds Lobophora (Dictyotales, Phaeophyceae) and their potential for induction of rapid coral bleaching in Acropora muricata. Front. Microbiol. 7:316. doi: 10.3389/fmicb.2016.00316
Vivekanand, V., Eijsink, V. G. H., and Horn, S. J. (2012). Biogas production from the brown seaweed Saccharina latissima: thermal pretreatment and codigestion with wheat straw. J. Appl. Phycol. 24, 1295–1301. doi: 10.1007/s10811-011-9779-8
Wainwright, B. J., Zahn, G. L., Spalding, H. L., Sherwood, A. R., Smith, C. M., and Amend, A. S. (2017). Fungi associated with mesophotic macroalgae from the ‘Au‘au Channel, west Maui are differentiated by host and overlap terrestrial communities. PeerJ. 5:e3532. doi: 10.7717/peerj.3532
Wang, G., Shuai, L., Li, Y., Lin, W., Zhao, X., and Duan, D. (2007). Phylogenetic analysis of epiphytic marine bacteria on Hole-Rotten diseased sporophytes of Laminaria japonica. J. Appl. Phycol. 20, 403–409. doi: 10.1007/s10811-007-9274-4
Weigel, B. L., and Pfister, C. A. (2019). Successional dynamics and seascape-level patterns of microbial communities on the canopy-forming kelps Nereocystis luetkeana and macrocystis pyrifera. Front. Microbiol. 10:346. doi: 10.3389/fmicb.2019.00346
Wu, J., and Loucks, O. L. (1995). From balance of nature to hierarchical patch dynamics: a paradigm shift in ecology. Q. Rev. Biol. 70, 439–466. doi: 10.1086/419172
Yang, C. Y., Song, G. J., Zhu, Q., Liu, S. J., and Xia, C. H. (2016). The influence of bacterial quorum-sensing inhibitors against the formation of the diatom-biofilm. Chem. Ecol. 32, 169–181. doi: 10.1080/02757540.2015.1120722
Yurchenko, T., Ševčíková, T., Pribyl, P., El Karkouri, K., Klimeš, V., Amaral, R., et al. (2018). A gene transfer event suggests a long-term partnership between eustigmatophyte algae and a novel lineage of endosymbiotic bacteria. ISME J. 12, 2163–2175. doi: 10.1038/s41396-018-0177-y
Keywords: algae, fungi, bacteria, holobiont, endophyte, biofilm, microbiota, Saccharina latissima
Citation: Tourneroche A, Lami R, Burgaud G, Domart-Coulon I, Li W, Gachon C, Gèze M, Boeuf D and Prado S (2020) The Bacterial and Fungal Microbiota of Saccharina latissima (Laminariales, Phaeophyceae). Front. Mar. Sci. 7:587566. doi: 10.3389/fmars.2020.587566
Received: 26 July 2020; Accepted: 17 November 2020;
Published: 23 December 2020.
Edited by:
David William Waite, Ministry for Primary Industries, New ZealandReviewed by:
Tomislav Cernava, Graz University of Technology, AustriaMahasweta Saha, Plymouth Marine Laboratory, United Kingdom
Copyright © 2020 Tourneroche, Lami, Burgaud, Domart-Coulon, Li, Gachon, Gèze, Boeuf and Prado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raphaël Lami, cmFwaGFlbC5sYW1pQG9icy1iYW55dWxzLmZy