 Arthur Escalas*
Arthur Escalas* Jean-Christophe Auguet
Jean-Christophe Auguet Amandine Avouac
Amandine Avouac Raphaël SeguinAntoine GradelLucie Borrossi
Raphaël SeguinAntoine GradelLucie Borrossi Sébastien Villéger
Sébastien Villéger
- MARBEC, Université de Montpellier, CNRS, IRD, IFREMER, Montpellier, France
Animals have been developing key associations with micro-organisms through evolutionary processes and ecological diversification. Hence, in some host clades, phylogenetic distance between hosts is correlated to dissimilarity in microbiomes, a pattern called phylosymbiosis. Teleost fishes, despite being the most diverse and ancient group of vertebrates, have received little attention from the microbiome perspective and our understanding of its determinants is currently limited. In this study, we assessed the gut microbiome of 12 co-occurring species of teleost representing a large breadth of ecological diversity and originating from a single family (i.e., the Sparidae). We tested how host evolutionary history, diet composition and morphological traits are related to fish gut microbiome. Despite fish species having different microbiomes, there is no phylosymbiosis signal in this fish family, but gut length and diet had a strong influence on the microbiome. We revealed that the only species with a specialized herbivorous diet, Sarpa salpa had a 3.3 times longer gut than carnivorous species and such a long gut favor the presence of anaerobic bacteria typical of herbivorous gut microbiomes. Hence, dietary uniqueness is paired with both unique gut anatomy and unique microbiome.
Introduction
All animals evolved and diversified within a microbial world, and consequently developed a myriad of associations with micro-organisms (McFall-Ngai et al., 2013). The abundant and diverse microbial communities present within and on the surface of animal bodies, i.e., the microbiomes, play a central role in the fitness of their host, providing benefits that include successful development of the immune system and protection against pathogens, but also enhanced food processing and nutrient absorption (McKenney et al., 2018a; Moran et al., 2019). These long histories of animal-microbes interactions resulted for many clades in phylosymbiosis, i.e., the eco-evolutionary pattern where phylogenetically related hosts tend to have more similar microbiomes than distantly related ones (Brooks et al., 2016; Lim and Bordenstein, 2019). Further, phylosymbiosis signal in the gut microbiome was shown to be stronger when hosts share a recent common ancestor (i.e., intra clade) than when shared ancestry is more ancient (i.e., inter clades) (Groussin et al., 2017). Phylosymbiosis has been evidenced in many animals, notably those for which vertical transmission of the microbiome has been reported, such as in the gut of insects (Brucker and Bordenstein, 2012; Haddow et al., 2013; Sanders et al., 2014; Brooks et al., 2016), and mammals (Phillips et al., 2012; Sanders et al., 2014), or on the skin of mammals (Ross et al., 2018) and elasmobranchs (Doane et al., 2020). However, evidences are far less pronounced in animals with horizontal microbiome transmission, with the report of weak to no support for skin phylosymbiosis in amphibians (Bletz et al., 2017) and marine fishes (Chiarello et al., 2018, 2019; Doane et al., 2020).
Marine teleost fishes gather >10,000 species and >100 families, and a large range of ecological strategies, with for instance diet ranging from herbivory to top-predators even within some families (e.g., Labridae, Sparidae). However, to date only a few studies have assessed the microbiome of several fish species from a single ecosystem using sequencing-based approaches (Givens et al., 2015; Miyake et al., 2015; Sullam et al., 2015; Sylvain et al., 2019; Ruiz-Rodríguez et al., 2020). Consequently, the presence or absence of gut phylosymbiosis in the gut microbiome of marine fishes has never been tested. However, a case of co-phylogeny, i.e., congruent phylogeny between hosts and individual members of their microbiome, has been reported between Acanthuridae (i.e., surgeonfishes) and the bacteria Epulopiscium spp., a genus from the class Clostridia in the Firmicutes phylum (Miyake et al., 2015, 2016). This specific host-microbes association between a bacterial clade and a fish family could still be coupled with low association between the whole microbiome and host phylogeny, especially when considering fishes from different families.
Beside hosts identity and long term association with micro-organisms that result in phylosymbiosis, the composition of the gut microbiome in marine teleosts is influenced by many factors (Sullam et al., 2012; Nikouli et al., 2020). Environment plays a role by influencing bacterioplankton communities, notably for marine fishes who continuously drink seawater to maintain osmolarity (Baldisserotto et al., 2019). This flow of environmental micro-organisms contributes to the early stages of gut microbial colonization and will influence microbiome composition at later stages (Egerton et al., 2018; Nikouli et al., 2019). Hosts diet also shapes the gut microbiome of fishes as food composition influences the type of resources available to gut micro-organisms and places the hosts in contact with the microbiomes of their prey (Sullam et al., 2012). Additionally, fishes have specific gut morphological traits that depend on their diet with, for instance, the presence of a gizzard in some species that consume sediment or a differentiated hindgut in herbivores which result in anaerobic conditions that favor certain micro-organisms (Mountfort et al., 2002; Clements et al., 2009; Egerton et al., 2018). Despite these recent advances, the overall role of diet and anatomy in shaping the gut microbiome still remains to be unfolded, especially within families where ecological diversification occurred.
In this study, we assessed the gut microbiome of 12 species from a single family (Sparidae) representing a large breadth of diet and sampled from a single location (i.e., the North-Western Mediterranean Sea). We then assessed how host evolutionary history, diet and gut anatomy are related to gut microbiome.
Materials and Methods
Samples Collection
Fishes were collected in the shallow habitats (2–10 m) of the North-Western coast of Gulf of Lions (Hérault, France, Mediterranean Sea) between June 2018 and February 2019. Fishes were caught at night by artisanal fishermen at the end of the night using gillnets, stored on ice on the boat and were processed within 3 h after being caught. Fish were dissected using tools cleaned with 70° ethanol, the gut was stretched and a picture was taken for morphometric measurements. The last third of the gut, before the cloaca, was squeezed out on a piece of parafilm paper using a sterile pipette. If this gut content was made of digested food (i.e., not only mucus) it was then homogenized and collected in a 3 mL cryotube before storage at −80°C until DNA extraction. Gut microbiome from a total of 48 adult individuals from 12 Sparidae species were sampled (2 to 8 individuals per species, Figure 1 and Supplementary Table 1).
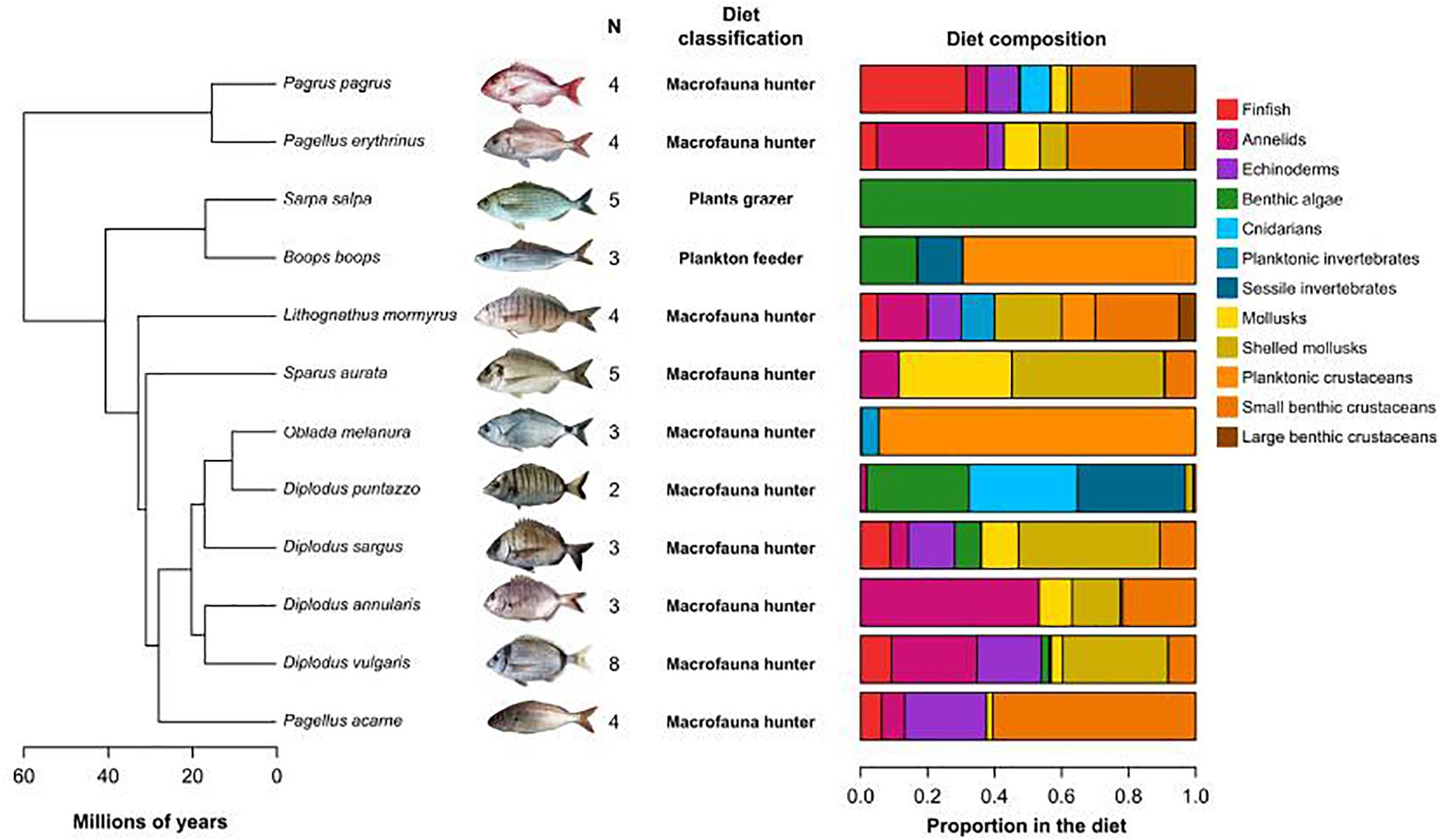
Figure 1. Phylogenetic relatedness and diet of the 12 studied species. Phylogeny of the Sparidae used in this study (from Santini et al., 2014). N is the number of individuals analyzed per species. Diet classification and proportions of food items in the diet were collected in FishBase.
Phylogenetic, Dietary and Morphological Dissimilarity Between Hosts
Phylogenetic distances between fish species were estimated as the cophenetic distance extracted from the only multi-locus time-calibrated phylogeny of Sparidae (Santini et al., 2014; Figure 1).
Diet of all species was recovered from food items listed in FishBase (Froese and Pauly, 2003), selecting only reports from the Mediterranean Sea and only included adults records. FishBase uses different levels of food items classification and we used the level that allows discriminating between groups of plants and invertebrates. This included the following food items: benthic algae, finfish, planktonic crustaceans, planktonic invertebrates, small benthic crustaceans, large benthic crustaceans, sessile invertebrates, echinoderms, annelids, cnidarians, mollusks, and shelled mollusks (Figure 1). The records were averaged for each species and resulted in a matrix containing the proportions of each item in the diet of each species. Species were classified into three groups according to their main type of prey (plants, plankton, and macrofauna). The prey proportion matrix was used to compute diet dissimilarity between species using the Bray-Curtis index.
Gut morphology was characterized using gut length (mm) and gut length relative to fish standard length (from snout to basis of the caudal fin). Fishes morphological features were measured at the individual level and species-level traits were estimated as the average across individuals from that species (Figure 1). Inter-species morphological dissimilarity was computed using Euclidean distance. Phylogenetic conservatism of gut traits was tested using Abouheif test (Abouheif, 1999), which is designed to detect phylogenetic autocorrelation in a quantitative trait, as implemented in the adephylo package (Jombart et al., 2010).
DNA Extraction
DNA extractions were performed in the molecular biology platforms of the MARBEC laboratory (UMR 9190)1 and at the GenSeq platform2, using the Qiagen MagAttract PowerSoil DNA KF Kit, selected for its compliance with the Earth Microbiome Project (Marotz et al., 2017). Extractions were performed in 96 wells plates in which three wells were left empty to serve as negative controls and three wells were loaded using standard mock communities (ZymoBIOMICS Microbial Community DNA Standards II, Zymo Research). These standards were used to evaluate the quality of our sample processing pipeline and to identify potential contaminants from the reagents. Extraction wells were loaded with one spatula of homogenized gut content (∼0.25 g). DNA extraction protocol followed the manufacturer instructions. It included a chemical lysis consisting of 60 μL of manufacturer provided SL solution heated at 60°C and added before a bead beating step, which corresponded to 10 min at 20 Hz using a Tissue Lyzer, then the plate was flipped and shake again for 10 min at 20 Hz. DNA recovery was based on magnetic beads and automated with a Kingfisher Flex robot. DNA was eluted in 200 μL of elution buffer before quantification of DNA quantity and quality using a NanoDrop 8000 spectrometer. Extracted DNA was stored at 4°C until PCR amplification, which was done the next day.
PCR Amplification
PCR amplification was done using primers selected for their compliance with the Earth Microbiome Project (Parada et al., 2016): 515F-Y (5′-GTGYCAGCMGCCGCGGTAA) and 926R (5′-CCGYCAATTYMTTTRAGTTT). The targeted sequence was 411 bp and corresponded to the V3–V4 regions of the prokaryotic 16S rRNA gene. PCR amplification was carried out in 96 well plates in triplicate for each DNA extract and was done in a 25 μL reaction volume. The PCR mix consisted of 9.75 μL of water, 0.75 μL of DMSO, 0.5 μL of each primer (concentration), 12.5 μL of Phusion ready-to-use Taq mix (Phusion High-Fidelity PCR Master Mix with GC Buffer), and 1 μL of DNA. The PCR cycle consisted of 35 cycles of 10 s denaturation at 98°C, 1 min annealing at 58°C, and 1 min 30 s of extension at 72°C. Final extension was held for 10 min at 72°C before keeping the reaction at 4°C. The success of PCR amplification was checked on 2% agarose gel in TAE buffer and using a 100 bp DNA ladder. The wells left empty during DNA extraction served as negative controls for contamination of the PCR reactions. PCR triplicates were pooled and stored at −20°C before sequencing. Amplicons library was constructed by the Genotoul platform3 and sequencing was carried out using an Illumina MiSeq (2 × 250 bp) sequencer.
Amplicon Sequencing and Sequences Processing
The demultiplexed reads were processed using the R software environment and the package dada2 (Callahan et al., 2016). Briefly, the quality of the reads for each sample was inspected using graphic representations of their quality scores and reads were filtered based on their length and quality. Amplicon sequence variants (ASVs) were inferred using the dada algorithm (Divisive Amplicon Denoising Algorithm) after pooling dereplicated reads from all samples. Then, forward and reverse reads were merged and chimeric sequences were removed. After these steps, a third of the original sequences were kept for further analyses, on average. The taxonomic classification of ASVs was performed with the naive Bayesian RDP classifier implemented in dada2 and using the SILVA reference database nr_V132 (10.5281/zenodo.1172783). As many ASVs were not affiliated at the genus level, ASVs sequences were blasted against the NCBI 16S rRNA bacterial and archaeal database, and the best hit was used to assign ASVs at the genus level.
Several data cleaning steps were performed to remove poorly characterized and rare ASVs. First, the mock communities and blank samples were used to identify contamination from the reagents (e.g., extraction kit, polymerase), and these ASVs were removed from the data set (e.g., Ralstonia, Rhizobium). Second, ASVs not assigned to the bacterial domain, unclassified at the Class level, assigned to chloroplasts or mitochondria were filtered out. Third, ASVs present in less than two samples were removed.
Amplicon sequence variants sequences were aligned using mafft implemented in Qiime2 before being inserted in the Greengene reference phylogenetic tree (Janssen et al., 2018). The tree was then ultrametricized using pathd84.
Identification of the Core Microbiome
The core microbiome of Sparidae fishes, i.e., the most frequent and/or abundant bacterial members of their microbiome, was estimated for the 12 Sparidae species altogether using the core identification algorithm from Magurran and Henderson (2003). This method accounts for both ASVs occurrence and abundance across communities and is based on the comparison of the observed abundance-occurrence distribution of ASVs with a random distribution under a stochastic Poisson model. We selected two individuals from each species in order to give equal weight to each species and the procedure was repeated 1000 times using different combinations of individuals for the species represented by more than two individuals. Then, we defined the size of the core as the median number of core ASVs (n = 842 ASVs) observed across all 1000 combinations and thus the core microbiome of Sparidae was defined as the 842 ASVs that were considered as core the highest number of times across the 1000 replicates. The abundance-occurrence distribution of ASVs across samples and ASVs identified as core are presented in Supplementary Figure 1.
Relationships Between Microbiome Dissimilarity and Host Ecological, Morphological and Phylogenetic Dissimilarities
In order to test for phylosymbiosis signal and identify the determinants of the core microbiome in Sparidae fishes, we computed microbiome dissimilarity using complementary diversity indices at several ranks of the bacterial taxonomy. Inter-species dissimilarities were estimated after taking the average taxa abundance across all the individuals from each species.
We used six dissimilarity indices from the Hill numbers framework (Chao et al., 2014), as these combine taxonomic and phylogenetic dissimilarities within a common mathematical framework. In addition, Hill numbers allow the estimation of composition and structure dissimilarity by giving more or less weight to taxa abundances, depending on the value of a parameter, the order q. These six indices were computed for six ranks across the bacterial taxonomy (Phylum, Class, Order, Family, Genus, and ASVs). For that, all the ASVs were aggregated at a given taxonomic rank and we computed dissimilarity on aggregated data. In a second approach, we tested whether intra-taxa dissimilarity was related with host dissimilarity. Here, the dataset was split into individual bacterial taxa for the highest taxonomic ranks (Phylum, Class, and Order) and dissimilarity was computed using only the ASVs from the focus taxa. For this analysis we used only the taxa that were represented by at least 10 ASVs (N = 31 taxa corresponding to 7 phyla, 10 classes, and 14 orders).
We used Mantel tests implemented in the R package ecodist (Goslee and Urban, 2007) to test the relationships between microbiome and host (phylogenetic, dietary, and morphological) dissimilarity matrices.
Identification of Gut Microbiome Determinants in Sparidae Fishes
Differences between species and diet types in terms of gut microbiome structure were tested using multivariate Welch MANOVA (Wd test, Hamidi et al., 2019) implemented in the R package MicEco (Russel, 2020). This approach has the advantage, over classically used PERMANOVA, of being robust to heteroscedasticity in the data, i.e., differences in multivariate groups dispersion. Association of host species and diet types with particular bacterial taxa was assessed using the LEfSe approach (Segata et al., 2011).
We used constrained analysis on principal coordinates (CAP analysis, Anderson and Willis, 2003) to determine which of the diet, gut anatomy and host phylogeny explained the better the differences in microbiome structure. Then, we identified which microbial taxa correlated with particular diet items or traits. Again, this was done for all the combinations of dissimilarity indices and bacterial taxonomic ranks. CAP models were defined to explain microbiome dissimilarity by (i) the proportions of the 12 dietary items, (ii) the values of the two morphological traits, and (iii) the phylogenetic relatedness of the hosts. This later was included as phylogenetic eigenvectors resulting from the decomposition of the cophenetic distance matrix between hosts performed using the R package PVR (Diniz-Filho et al., 1998; Santos, 2018). The individual effect of each explaining variable was tested using ANOVA and the anova.cca function in R.
Results
Phylogenetic Relationships and Morphological Similarity of Mediterranean Sparidae
The 12 studied Sparidae species shared a common ancestor around 60 million years ago and separated in two main branches which do not contain recent (<12 Mya) speciation events (Figure 1). Two genera contained more than one species, Diplodus (n = 4) and Pagellus (n = 2), but while the former is monophyletic, the latter is spread in the two main branches of the family tree. From a dietary aspect, ten species are considered as macrofauna hunters according to FishBase (including all Diplodus and Pagellus species), but this rough classification blurs a high dissimilarity in terms of diet composition, with mostly zooplankton for Oblada melanura to omnivory on various proportion of crustacean, annelids, and/or echinoderms among other species (Figure 1). The two remaining species have a unique diet: Boops boops is a selective plankton feeder and Sarpa salpa is a strict herbivore feeding on benthic macrophytes. Set aside S. salpa, only B. boops and Diplodus puntazzo consume more than 15% of plant material. B. boops is the closest species to S. salpa (divergence ∼18.4 Mya) and those two species also have the highest values for all gut traits (Supplementary Table 2). Gut length and relative gut length showed a significant phylogenetic signal (Abouheif test, p-value < 0.05, Supplementary Table 3 and Supplementary Figure 2) with longer guts in the Boops-Sarpa clade.
Diversity of the Core Microbiome of Mediterranean Sparidae
The 842 ASVs that composed the core gut microbiome of Sparidae fishes from the Gulf of Lion represented 20.3% of the total number of ASVs and 51.1% of the total number of sequences in the whole dataset (3.2 + −0.9% of ASVs and 52.6 + −14.4% of sequences per species, on average). The core microbiome gathered 14 bacterial phyla, 21 classes, 48 orders, 66 families, and 98 genera. The most dominant phyla were Proteobacteria, Bacteroidetes, and Firmicutes with 322, 205, and 184 ASVs, respectively. Four phyla had more than 10 ASVs: Planctomycetes (40), Verrucomicrobia (35), Tenericutes (17), and Actinobacteria (13). The eight remaining phyla were represented by 1 to 8 ASVs.
Three dominant phyla represented 84.3% of the sequences, with 39.7% for Proteobacteria, 25.2% for Firmicutes, and 19.4% for Bacteroidetes (Supplementary Figure 3). All the other phyla represented less than 5%. At the class level, Gammaproteobacteria was the most abundant (25.5%), followed by Bacteroidia (19.4%), Clostridia (18.3%), Deltaproteobacteria (8.7%), Alphaproteobacteria (5.5%), Verrucomicrobiae (4.1%), Bacilli (3.8%), Mollicutes (3.2%), Erysipelotrichia (3.3%), and Oxyphotobacteria (2.6%). Dominant bacterial family and genus markedly varied among species with only no genus and four families (Rikenellaceae, Lachnospiraceae, Desulfovibrionaceae, and Vibrionaceae) that reached 10% of relative abundance in more than two species (Table 1). Hence, gut microbiome structure differed significantly between host species, with 26 out of 36 tests (6 bacterial taxonomic ranks times 36 dissimilarity indices) being significant (Wd test, p-value < 0.05, Supplementary Figure 4). The F-values associated were low, 2.5 ± 1.9 on average, and the p-values tended to decrease at lower bacterial taxonomic ranks. Gut microbiome structure also differed significantly between the three diet types with 32 out of 36 tests being significant (Wd test, p-value < 0.05, 29 tests had a p-value < 0.01) and the F-values of those tests were 13 ± 12.1.
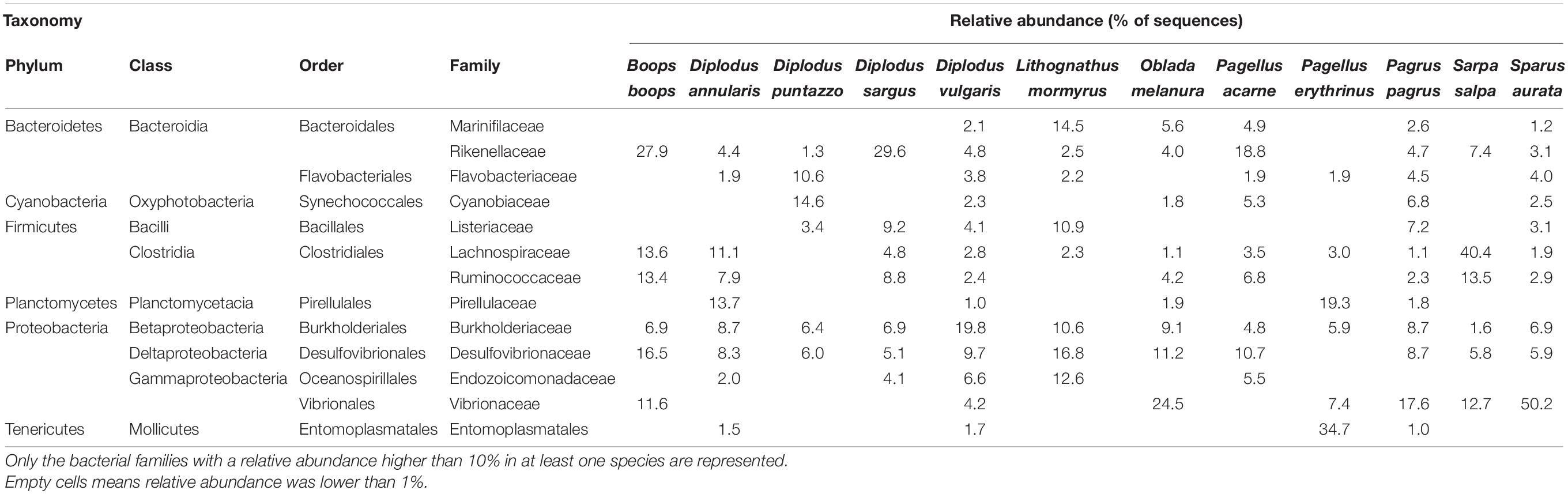
Table 1. Relative abundance of the main bacterial families in the gut microbiome of Mediterranean Sparidae.
Microbial Taxa Associated With Host Species and Diet Types
Most of the 189 key ASVs identified by the LEfSe analysis (Table 2) were associated with S. salpa (n = 26), its closest relative B. boops (n = 17) and Pagellus erythrinus (n = 11), or with selective plankton feeding (n = 71) and aquatic plants grazing (n = 96). The other species (maximum 3 ASVs) and macrofauna hunting (4 ASVs) were associated with a much lower number of ASVs. S. salpa and its diet were mostly associated with members of the Clostridiales (Firmicutes) from the families Lachnospiraceae (notably the genus Tyzerella) and Ruminococcaceae (Angelakisella, Ruminiclostridium), the Bacteroidetes family Rikenellaceae (genera Alistipes and Rikenella) and several Desulfovibrionaceae ASVs. B. boops and its diet were associated with the Firmicutes family Ruminococcaceae (genera UCG-008) and the Lachnospiraceae Tyzerella, the Rikenellaceae (genera Alistipes and Rikenella) and the genus Desulfovibrio. P. erythrinus was mostly associated with Planctomycetes from the Pirellulaceae family (Blastopirellula and Rhodopirellula) but also Alphaproteobacteria (Ahrensia, Roseobacter).

Table 2. Bacterial taxa associated with Sparidae species and diet types.
Relationships Between Host and Gut Microbiome Dissimilarity
We did not observe any phylosymbiosis signal as phylogenetic distance between fish species was not significantly correlated with their microbiome dissimilarity for any of the 36 combinations of dissimilarity indices and bacterial taxonomy ranks tested (Mantel tests, p-values > 0.05, Figure 2). Diet dissimilarity was related to microbiome dissimilarity in only one out of 36 tests, i.e., using taxonomic microbiome dissimilarity computed on presence-absence data at the bacterial order level (q = 0). Morphological dissimilarity based on gut traits was significantly related to microbiome dissimilarity in five instances (out of 72 Mantel tests, p-values < 0.05), all with abundance-weighted phylogenetic dissimilarity (q = 2) computed at higher ranks of bacterial taxonomy (Phylum = 1, Class = 2, Order = 2).
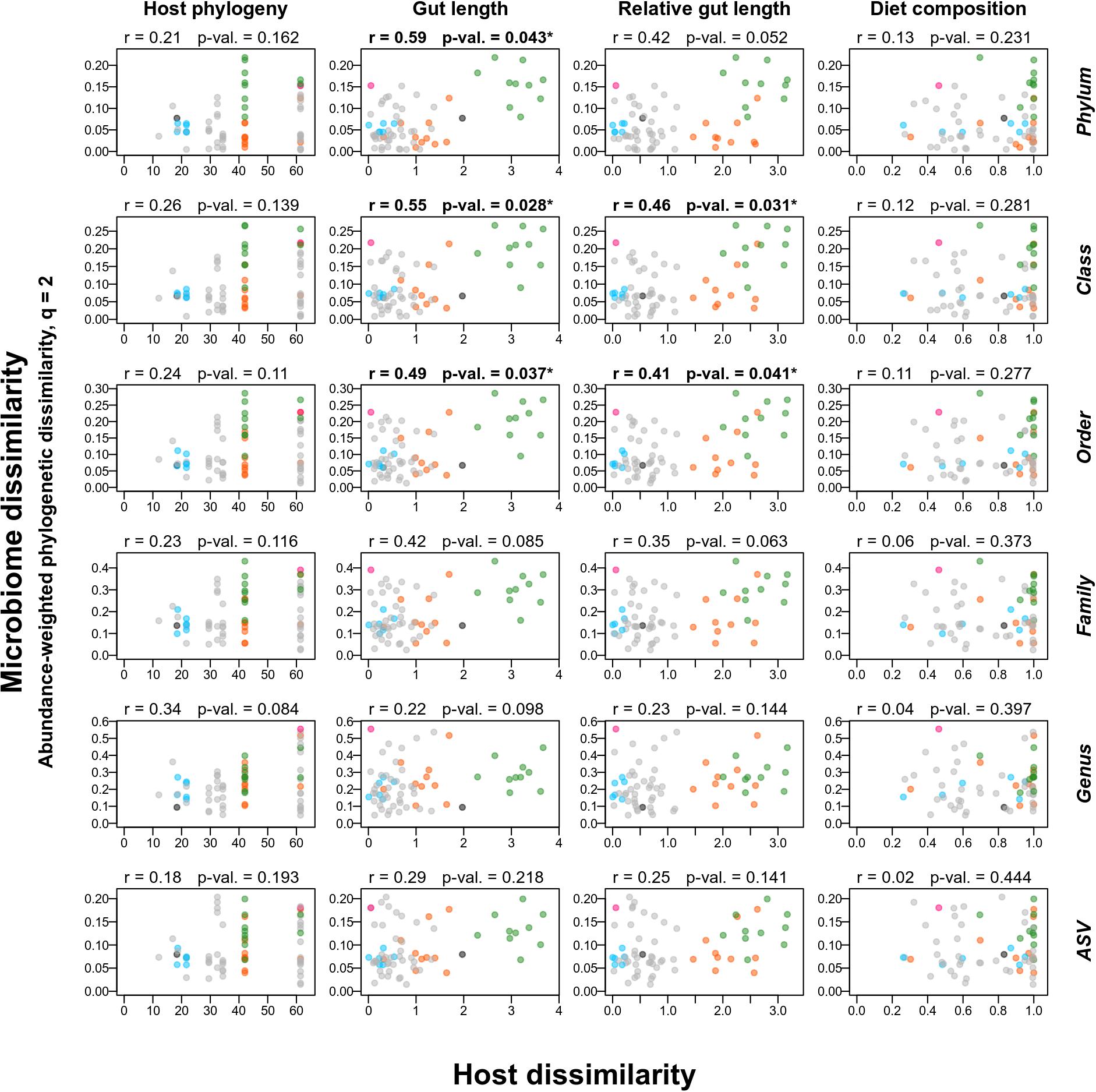
Figure 2. Gut traits explain differences in the phylogenetic structure of the microbiome better than host phylogeny or diet. Relationships between dissimilarity of the gut microbiome (Y axes) and phylogenetic, morphological, and dietary dissimilarity (columns) between host fishes (X axis) using Mantel tests (for each subplot: r = correlation between X and Y variables and p-value = p-value of the Mantel test, bolded r and p-values correspond to significant relationships). Gut microbiome dissimilarity, here estimated using abundance-weighted Hill numbers phylogenetic index with q = 2, was computed at six different levels of taxonomic resolution (row names on the right of the figure). Each point represents a pair of fish species. Pairs within the Diplodus genus are depicted in blue, pair within the Pagellus genus is depicted in pink, pair between Sarpa salpa and Boops boops is depicted in black and other pairs that included S. salpa or B. boops are depicted in green and orange, respectively.
We found 11 significant relationships between intra-taxa abundance-weighted (q = 2) phylogenetic dissimilarity (Supplementary Figure 5) and host dissimilarity (phylogenetic, diet and morphological). Half of these relationships were negative (6/13) and, overall, the strength of the relationships was weak. Only one significant relationship involved host phylogenetic distance, which was found to be negatively related with dissimilarity in the order Desulfovibrionales. The strongest association was found between phylogenetic dissimilarity within the Bacillales order and Bacilli class (Firmicutes phylum) and dissimilarity in gut traits.
Association Between Sparidae Microbiome, Gut Morphological Traits, Diet Items, and Host Phylogeny
More than half (n = 19, 53%) of the CAP models testing the relationship between gut traits, diet composition, host phylogeny, and microbiome structure were significant (i.e., p-value < 0.05 in 19 out of 36 models, Supplementary Table 4). The first two axes of the significant CAP models explained between 41 and 91% of the variation in gut microbiome (Supplementary Figure 6). Among these 19 significant CAP models, the most influential variable was by far gut length, as it exhibited the highest number of significant relationships with microbiome components (n = 19) and the highest F-values (Supplementary Table 4). The proportion of Echinoderms and Mollusks in the diet was the second and third most influential traits, followed by relative gut length. Phylogenetic eigenvectors did not explain significant proportion of the variance in any of the CAP models.
Hereafter, we focus on the results obtained using abundance-weighted taxonomic dissimilarity (q = 1) as this method yielded a significant CAP model five out of six taxonomic ranks (i.e., not at the Class) and explained a high proportion of variance in microbiome composition, on average (88 ± 8%). The first CAP axis explained 26.2% of the variation and mostly separated the species with a long gut that do not consume Mollusks, i.e., S. salpa, from the other species (Figure 3A). The second CAP axis explained 18.6% of the variation and separated two species, P. erythrinus and B. boops, from the others.
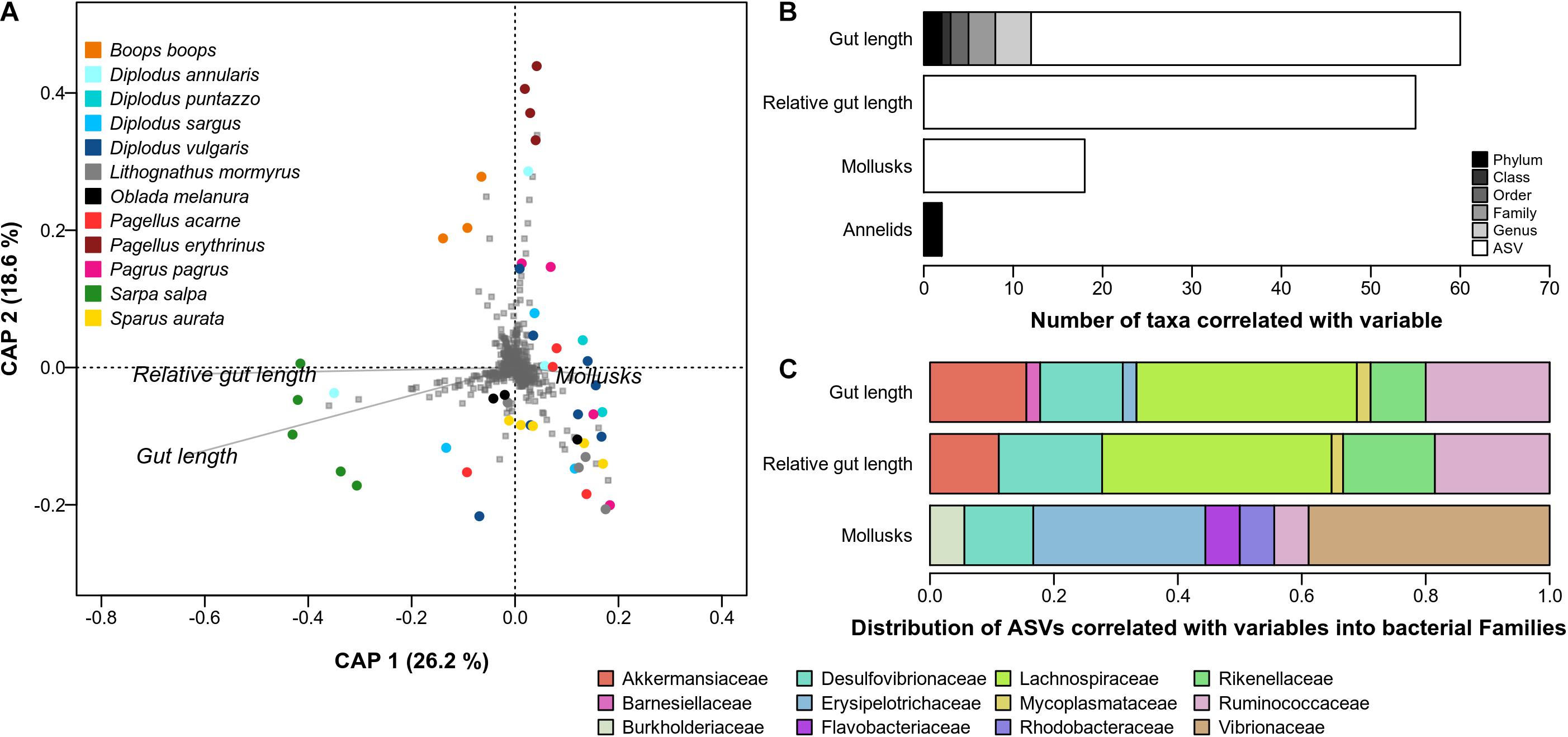
Figure 3. Association between microbiome, gut morphological traits and diet items in Sparidae. (A) Constrained analysis on principal coordinates (CAP) analysis. Fish individuals are represented by colored dots, gut traits and diet items are represented by italicized labels and arrows, bacterial ASVs are represented by gray squares. Dissimilarity between microbiomes was estimated using abundance-weighted taxonomic dissimilarity (Hill diversity with q = 1). Only the variables identified as explaining a significant proportion of total microbiome variation are depicted (ANOVA.CCA, p-value < 0.05). (B) Number of taxa whose abundance are significantly correlated with variables that explained a significant proportion of microbiome variation. ASVs correspond to panel (A) while other ranks correspond to plots in Supplementary Figure 4. (C) The ASVs associated with explaining variables (white bars in panel B) were taxonomically assigned at the Family level and their proportion were estimated.
We found 131 significant correlations between gut traits or the proportions of diet items and the abundance of bacterial taxa at different ranks. Only four were negative, which corresponded to the negative association between gut length and Proteobacteria, Betaproteobacteriales and Burkholderiaceae, and between Annelids and Firmicutes. Gut length was the variable that correlated with the highest number of bacterial taxa (Figure 3B), i.e., 60 including 48 ASVs, and was the only variable associated with taxa across all the taxonomic ranks (i.e., phylum to ASVs). The strongest correlations included those with the Clostridia class and the Clostridiales order (r = 0.83, p-value < 0.001), the Lachnospiraceae family (r = 0.81, p-value < 0.001) and one of its ASV (r = 0.87, p-value < 0.001). The other traits/items were mostly associated with ASVs, 55 for relative gut length and 18 for Mollusks.
Gut length was mostly associated with Firmicutes from the Clostridia class (Figure 3C), notably the Lachnospiraceae Tyzerella (8 ASVs), and several Ruminococcaceae (9 ASVs). The genera Akkermansia (7 ASVs, Verrucomicrobia) and Alistipes (4 ASVs, Bacteroidetes) were also well represented. Desulfovibrionaceae (6 ASVs, Deltaproteobacteria) and Rikenellaceae (4 ASVs) were also identified. Relative gut length was associated with ASVs from similar families and genus: Tyzerella (12 ASVs), Alistipes (7 ASVs), Akkermansia (6 ASVs). Mollusks were associated with two main phyla. Firmicutes, mostly from the Erysipelotrichaceae (5 ASVs), and Gammaproteobacteria (8 ASVs).
Discussion
Composition of the Core Microbiome of Sparidae From the Mediterranean Sea
The core microbiome of the 12 Sparidae species studied here was typical of what is reported for marine fishes (Sullam et al., 2012; Egerton et al., 2018), i.e., a dominance of Proteobacteria, Bacteroidetes, and Firmicutes, with these three phyla representing more than 84% of the sequences. Our results also confirm that the genus Vibrio, albeit dominant on the mucus, skin and gills of teleost (Chiarello et al., 2018; Ruiz-Rodríguez et al., 2020), represents only a small proportion of their gut microbiome. Regarding Sparidae’ microbiome, the literature is highly skewed toward one species, Sparus aurata, which is used as a model in aquaculture (Dimitroglou et al., 2010; Estruch et al., 2015; Nikouli et al., 2019; Rosado et al., 2019), while microbiomes of most other Sparidae have not been described in the wild, with the exception of a recent study (Ruiz-Rodríguez et al., 2020). To our knowledge, our study provides the first report of the gut microbiome composition for five species: B. boops, D. puntazzo, Diplodus sargus, Lithognathus mormyrus, and Pagellus acarne. The structure of the gut microbiome significantly differed between the 12 studied species, with no genus and only four families (Rikenellaceae, Lachnospiraceae, Desulfovibrionaceae, and Vibrionaceae) representing more than 10% of the sequences in more than two host species. Species-specific gut microbiomes have been reported for other marine fishes both within and between families (Miyake et al., 2015; Ruiz-Rodríguez et al., 2020), but some studies also reported the absence of inter-specific difference because of high inter-individual variability in the gut microbiome (Nikouli et al., 2020).
It is worth mentioning that the method used here to identify the members of the core microbiome was based on the abundance-occurrence distribution of ASVs (Fillol et al., 2016; Jeanbille et al., 2016) and not on arbitrarily defined occurrence or abundance thresholds. This could explain the high number of ASVs identified as core (n = 842) compared with other studies (e.g., n = 15 in Sullam et al., 2015; 0 < n < 110 in Chiarello et al., 2018; n = 7 in Nikouli et al., 2020). Indeed, in our method, even ASVs observed in a low number of species (low occurrence) but that were consistently abundant within individuals from these species were identified as core.
Weak Support for Phylosymbiosis in the Gut Microbiome of Sparidae Fishes
Overall, we found little support for the existence of gut phylosymbiosis in Sparidae fishes, i.e., we did not observe significant relationships between host phylogenetic relatedness and microbiome dissimilarity (using both Mantel tests and CAP analysis). Such result is in agreement with the observation of weak phylosymbiosis signal in skin microbiome of teleost fishes (Chiarello et al., 2018; Doane et al., 2020; Sylvain et al., 2020). In our case, the absence of phylosymbiosis, despite inter-species differences, is driven by the fact that the 10 carnivorous species are spread on the two main branches of the Sparidae phylogeny while they have similar gut traits and microbiome. For instance, P. erythrinus and Pagrus pagrus are phylogenetically distant but have a microbiome similar to those of most of the other Mediterranean Sparidae (i.e., the Diplodus cluster, the other Pagellus species, S. aurata and L. mormyrus). This microbiome convergence in carnivorous Sparidae resulted in species pairs with small microbiome dissimilarity despite high phylogenetic distance (i.e., points in the bottom right of the plots in Figure 2) that blurred the phylosymbiosis signal. On the contrary, the outlier regarding diet, gut traits and microbiome (S. sarpa) is not a phylogenetic outlier, which resulted in species pairs with small phylogenetic distance and strong microbiome dissimilarity (i.e., green points in Figure 2).
Determinants of the Gut Microbiome in Mediterranean Sparidae
Gut morphology and diet were the strongest determinants of the gut microbiome in Mediterranean Sparidae. These relationships were mostly driven by the fact that S. salpa, and to a lesser extent B. boops, were simultaneously microbiome and gut outliers. The importance of gut features in shaping the microbiome has been proposed to explain the observation of similar microbiome composition in wild and domesticated populations of zebrafish that experienced very different dietary and environmental conditions (Roeselers et al., 2011). Here, gut length and relative gut length were found to be conserved in host phylogeny, with one clade composed of S. salpa an B. boops having significantly longer guts than other Sparidae. While the relative gut length of these two species are 2.5 and 2.1 times higher than those of carnivorous species, respectively, their absolute gut length are 3.3 times longer for S. salpa (591 mm against an average of 177 ± 56 mm for the ten carnivores) and only 1.8 times longer for B. boops (329 mm) which is a much smaller species. These two species also appeared as outliers regarding their trophic ecology, with uncommon diet classification and a low diversity diet compared with the other studied species, along with the consumption of algae in S. salpa.
One limitation of our study lies in the use of stomach content data collected in other studies from the same region and recovered from FishBase. Individual analysis of stomach content was not feasible as the studied species are mostly diurnal and the fishes were caught during the night so their stomach was empty. Another approach to get the identity of ingested preys at the individual level corresponds to the metabarcoding of the gut content and feces (Soininen et al., 2009; Kartzinel et al., 2015), but this approach does not provide quantitative estimates of the ingested preys, contrarily with gut content analyses (Parravicini et al., 2020).
Sarpa salpa and B. boops, with their unique diet specialization (plant grazer and plankton feeder, respectively) and their extremely long guts, host bacterial taxa that were scarce in the other ten species (classified as macrofauna hunters). These taxa belong to three families of the Clostridiales order in particular (Lachnospiraceae and Ruminococcaceae), which are obligate anaerobes well-known for their associations with several herbivorous hosts (Llewellyn et al., 2014; Moran et al., 2019) including fishes (Clements et al., 2009; Sullam et al., 2012; Jones et al., 2018). In addition, S. salpa and B. boops traits and microbiome were associated with two genera known for their anaerobic lifestyle, Akkermansia and Alistipes, and more generally with their respective phyla, Verrucomicrobia and Bacteroidetes, also known to be abundant in the gut of herbivorous fishes (Sullam et al., 2012; Miyake et al., 2015; Ruiz-Rodríguez et al., 2020).
Herbivory in general is associated with modifications of the gut to accommodate the microbes involved in the digestion of plant-based material (Moran et al., 2019), either with differentiated parts (e.g., rumen, hindgut) or more simply through an elongation of the digestive tract that favor more anaerobic conditions within the distal part of the gut. Longer guts also increase food transit time, a necessary step to allow the fermentation process to take place and extract nutrients from plant material (Clements and Choat, 1995). In these regards, S. salpa is truly unique among the studied Sparidae as its gut is twice longer than the one of its closest, and yet distant, relative B. boops (divergence ∼18.4 Mya), allowing a long transit time (∼18 h, Goldenberg and Erzini, 2014). Furthermore, contrarily with B. boops that only consumes a limited proportion of plants/algae, S. salpa is specialized on these resources and consequently provides the raw material required for fermentation of plant polysaccharides to SCFAs (butyrate, acetate) by gut anaerobes (Boutard et al., 2014). As a result, Clostridiales represented 56% of the microbiome in S. salpa and only 30% in B. boops, while Lachnospiraceae represented 40 and 14% of the total, respectively. Lachnospiraceae is the most abundant taxa in the cow rumen where it represents more than a third of the sequences (Seshadri et al., 2018) and participates to plant fermentation as they are particularly efficient in cellulose degradation (Schwarz, 2001; Biddle et al., 2013).
The multifaceted ecological uniqueness exhibited by S. salpa has been reported in other clades such as the giant and red pandas, that are bamboo-eating specialists within a group of carnivores. Similarly, recent studies revealed that their typical straight, short and non-differentiated carnivore guts limit their ability to host the typical diversified microbiome of herbivores (Guo et al., 2020), contrarily with the bamboo lemur (Hapalemur griseus), a bamboo specialist hosting a much more diversified microbiome (McKenney et al., 2018b) thanks to its omnivorous digestive tract and a very long transit time (>30 h).
Conclusion
In this study, we highlight the presence of a dietary, anatomical and microbiological outlier within a carnivorous family of teleost fishes, the Sparidae. We revealed that the diet originality of the strict herbivore S. salpa is supported by a longer gut which increases the food transit time as well as anaerobic conditions favoring bacteria clades able to degrade plant materials. A key challenge is now to assess whether S. salpa’ microbiome is comparable with the one of typically herbivorous fish clades such as Kyphosidae, Acanthuridae or Siganidae, both in terms of composition and efficiency to help the host assimilate nutrients. This comparison is particularly urgent to address because two Lessepsian rabbitfishes are spreading in the Eastern part of the Mediterranean basin and led in many places to overgrazing of macrophytes (Vergés et al., 2014).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Data Dryad, with the following doi: 10.5061/dryad.kprr4xh35.
Ethics Statement
Ethical review and approval was not required for the animal study because the fish individuals used for this study were already dead when collected from local fishermen.
Author Contributions
AE, J-CA, AA, RS, and SV collected the samples. RS, AG, and LB provided fish traits data. AA and AE performed the molecular lab work. AE analyzed the data, performed the statistical analyses, and wrote the manuscript with the help of J-CA and SV. SV, AE, and J-CA conceived the study. All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This project was funded by the French ANR (grant ANR-17-CE32-0003) through the EXOFISHMED project (https://exofishmed.cnrs.fr).
Acknowledgments
We are grateful to Denis Pons and Jean-Pierre Gritti for their help with fish sampling, Marie Gislard from the GenoToul sequencing platform and Corinne Bouvier, Elsa Gadoin, and Marie-Charlotte Cheutin for their assistance in samples preparation. We would like to thank the two reviewers and the editor who helped to improve this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.622883/full#supplementary-material
Footnotes
References
Abouheif, E. (1999). A method for testing the assumption of phylogenetic independence in comparative data. Evol. Ecol. Res. 1, 895–909. doi: 10.1007/s11295-009-0238-5
Anderson, M. J., and Willis, T. J. (2003). Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84, 511–525. doi: 10.1890/0012-9658(2003)084[0511:caopca]2.0.co;2
Baldisserotto, B., Mancera, J. M., and Kapoor, B. G. (2019). Fish Osmoregulation. Boca Raton, FL: CRC Press. doi: 10.1201/b10994
Biddle, A., Stewart, L., Blanchard, J., and Leschine, S. (2013). Untangling the genetic basis of fibrolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 5, 627–640. doi: 10.3390/d5030627
Bletz, M. C., Archer, H., Harris, R. N., McKenzie, V. J., Rabemananjara, F. C. E., Rakotoarison, A., et al. (2017). Host ecology rather than host phylogeny drives amphibian skin microbial community structure in the biodiversity hotspot of Madagascar. Front. Microbiol. 8:1530. doi: 10.3389/fmicb.2017.01530
Boutard, M., Cerisy, T., Nogue, P. Y., Alberti, A., Weissenbach, J., Salanoubat, M., et al. (2014). Functional diversity of carbohydrate-active enzymes enabling a bacterium to ferment plant biomass. PLoS Genet. 10:e1004773. doi: 10.1371/journal.pgen.1004773
Brooks, A. W., Kohl, K. D., Brucker, R. M., van Opstal, E. J., and Bordenstein, S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14:e2000225. doi: 10.1371/journal.pbio.2000225
Brucker, R. M., and Bordenstein, S. R. (2012). The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution (N.Y.) 66, 349–362. doi: 10.1111/j.1558-5646.2011.01454.x
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chao, A., Chiu, C.-H., and Jost, L. (2014). Unifying species diversity, phylogenetic diversity, functional diversity, and related similarity and differentiation measures through hill numbers. Annu. Rev. Ecol. Evol. Syst. 45, 297–324. doi: 10.1146/annurev-ecolsys-120213-091540
Chiarello, M., Auguet, J. C., Bettarel, Y., Bouvier, C., Claverie, T., Graham, N. A. J., et al. (2018). Skin microbiome of coral reef fish is highly variable and driven by host phylogeny and diet. Microbiome 6:147. doi: 10.1186/s40168-018-0530-4
Chiarello, M., Paz-Vinas, I., Veyssière, C., Santoul, F., Loot, G., Ferriol, J., et al. (2019). Environmental conditions and neutral processes shape the skin microbiome of European catfish (Silurus glanis) populations of Southwestern France. Environ. Microbiol. Rep. 11, 605–614. doi: 10.1111/1758-2229.12774
Clements, K. D., and Choat, J. H. (1995). Fermentation in tropical marine herbivorous fishes. Physiol. Zool. 68, 355–378. doi: 10.1086/physzool.68.3.30163774
Clements, K. D., Raubenheimer, D., and Choat, J. H. (2009). Nutritional ecology of marine herbivorous fishes: ten years on. Funct. Ecol. 23, 79–92. doi: 10.1111/j.1365-2435.2008.01524.x
Dimitroglou, A., Merrifield, D. L., Spring, P., Sweetman, J., Moate, R., and Davies, S. J. (2010). Effects of mannan oligosaccharide (MOS) supplementation on growth performance, feed utilisation, intestinal histology and gut microbiota of gilthead sea bream (Sparus aurata). Aquaculture 300, 182–188. doi: 10.1016/j.aquaculture.2010.01.015
Diniz-Filho, J. A. F., De SanT’Ana, C. E. R., and Bini, L. M. (1998). An eigenvector method for estimating phylogenetic inertia. Evolution (N.Y.) 52, 1247–1262. doi: 10.1111/j.1558-5646.1998.tb02006.x
Doane, M. P., Morris, M. M., Papudeshi, B., Allen, L., Pande, D., Haggerty, J. M., et al. (2020). The skin microbiome of elasmobranchs follows phylosymbiosis, but in teleost fishes, the microbiomes converge. Microbiome 8:93. doi: 10.1186/s40168-020-00840-x
Egerton, S., Culloty, S., Whooley, J., Stanton, C., and Ross, R. P. (2018). The gut microbiota of marine fish. Front. Microbiol. 9:873. doi: 10.3389/fmicb.2018.00873
Estruch, G., Collado, M. C., Peñaranda, D. S., Tomás Vidal, A., Jover Cerdá, M., Pérez Martínez, G., et al. (2015). Impact of fishmeal replacement in diets for gilthead sea bream (Sparus aurata) on the gastrointestinal microbiota determined by pyrosequencing the 16S rRNA gene. PLoS One 10:e0136389. doi: 10.1371/journal.pone.0136389
Fillol, M., Auguet, J. C., Casamayor, E. O., and Borrego, C. M. (2016). Insights in the ecology and evolutionary history of the miscellaneous crenarchaeotic group lineage. ISME J. 10, 665–677. doi: 10.1038/ismej.2015.143
Froese, R., and Pauly, D. (2003). Fish Base 2000: Concepts, Design and Data Sources. Los Banos: ICLARM, 344.
Givens, C. E., Ransom, B., Bano, N., and Hollibaugh, J. T. (2015). Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 518, 209–223. doi: 10.3354/meps11034
Goldenberg, S. U., and Erzini, K. (2014). Seagrass feeding choices and digestive strategies of the herbivorous fish Sarpa salpa. J. Fish Biol. 84, 1474–1489. doi: 10.1111/jfb.12371
Goslee, S. C., and Urban, D. L. (2007). The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 22, 1–19. doi: 10.18637/jss.v022.i07
Groussin, M., Mazel, F., Sanders, J. G., Smillie, C. S., Lavergne, S., Thuiller, W., et al. (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 8:14319. doi: 10.1038/ncomms14319
Guo, W., Chen, Y., Wang, C., Ning, R., Zeng, B., Tang, J., et al. (2020). The carnivorous digestive system and bamboo diet of giant pandas may shape their low gut bacterial diversity. Conserv. Physiol. 8:coz104. doi: 10.1093/conphys/coz104
Haddow, A. D., Guzman, H., Popov, V. L., Wood, T. G., Widen, S. G., Haddow, A. D., et al. (2013). First isolation of Aedes flavivirus in the Western Hemisphere and evidence of vertical transmission in the mosquito Aedes (Stegomyia) albopictus (Diptera: Culicidae). Virology 440, 134–139. doi: 10.1016/j.virol.2012.12.008
Hamidi, B., Wallace, K., Vasu, C., and Alekseyenko, A. V. (2019). W d∗ -test: robust distance-based multivariate analysis of variance. Microbiome 7:51. doi: 10.1186/s40168-019-0659-9
Janssen, S., McDonald, D., Gonzalez, A., Navas-Molina, J. A., Jiang, L., Xu, Z. Z., et al. (2018). Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3, e00021–18. doi: 10.1128/msystems.00021-18
Jeanbille, M., Gury, J., Duran, R., Tronczynski, J., Agogué, H., Ben Saïd, O., et al. (2016). Response of core microbial consortia to chronic hydrocarbon contaminations in coastal sediment habitats. Front. Microbiol. 7:1637. doi: 10.3389/fmicb.2016.01637
Jombart, T., Balloux, F., and Dray, S. (2010). adephylo: new tools for investigating the phylogenetic signal in biological traits. Bioinformatics 26, 1907–1909. doi: 10.1093/bioinformatics/btq292
Jones, J., DiBattista, J. D., Stat, M., Bunce, M., Boyce, M. C., Fairclough, D. V., et al. (2018). The microbiome of the gastrointestinal tract of a range-shifting marine herbivorous fish. Front. Microbiol. 9:2000. doi: 10.3389/fmicb.2018.02000
Kartzinel, T. R., Chen, P. A., Coverdale, T. C., Erickson, D. L., Kress, W. J., Kuzmina, M. L., et al. (2015). DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. Proc. Natl. Acad. Sci. U.S.A. 112, 8019–8024. doi: 10.1073/pnas.1503283112
Lim, S. J., and Bordenstein, S. R. (2019). An introduction to phylosymbiosis. PeerJ 7:e27879v2. doi: 10.7287/peerj.preprints.27879v2
Llewellyn, M. S., Boutin, S., Hoseinifar, S. H., and Derome, N. (2014). Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 5:207. doi: 10.3389/fmicb.2014.00207
Magurran, A. E., and Henderson, P. A. (2003). Explaining the excess of rare species in natural species abundance distributions. Nature 422, 714–716. doi: 10.1038/nature01547
Marotz, C., Amir, A., Humphrey, G., Gaffney, J., Gogul, G., and Knight, R. (2017). DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques 62, 290–293. doi: 10.2144/000114559
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C. G., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
McKenney, E. A., Koelle, K., Dunn, R. R., Yoder, A. D., and Yoder, K. K. R. R. D. A. D. (2018a). The ecosystem services of animal microbiomes. Mol. Ecol. 27, 2164–2172. doi: 10.1111/mec.14532
McKenney, E. A., Maslanka, M., Rodrigo, A., and Yoder, A. D. (2018b). Bamboo specialists from two mammalian orders (Primates, Carnivora) share a high number of low-abundance gut microbes. Microb. Ecol. 76, 272–284. doi: 10.1007/s00248-017-1114-8
Miyake, S., Ngugi, D. K., and Stingl, U. (2015). Diet strongly influences the gut microbiota of surgeonfishes. Mol. Ecol. 24, 656–672. doi: 10.1111/mec.13050
Miyake, S., Ngugi, D. K., and Stingl, U. (2016). Phylogenetic diversity, distribution, and cophylogeny of giant bacteria (Epulopiscium) with their surgeonfish hosts in the Red Sea. Front. Microbiol. 7:285. doi: 10.3389/fmicb.2016.00285
Moran, N. A., Ochman, H., and Hammer, T. J. (2019). Evolutionary and ecological consequences of gut microbial communities. Annu. Rev. Ecol. Evol. Syst. 50, 451–475. doi: 10.1146/annurev-ecolsys-110617-062453
Mountfort, D. O., Campbell, J., and Clements, K. D. (2002). Hindgut fermentation in three species of marine herbivorous fish. Appl. Environ. Microbiol. 68, 1374–1380. doi: 10.1128/AEM.68.3.1374-1380.2002
Nikouli, E., Meziti, A., Antonopoulou, E., Mente, E., and Kormas, K. A. (2019). Host-associated bacterial succession during the early embryonic stages and first feeding in farmed gilthead sea bream (Sparus aurata). Genes (Basel) 10:483. doi: 10.3390/genes10070483
Nikouli, E., Meziti, A., Smeti, E., Antonopoulou, E., Mente, E., and Kormas, K. A. (2020). Gut microbiota of five sympatrically farmed marine fish species in the Aegean Sea. Microb. Ecol. 81, 460–470. doi: 10.1007/s00248-020-01580-z
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Parravicini, V., Casey, J., Schiettekatte, N. M., Brandl, S., Pozas-Schacre, C., Carlot, J., et al. (2020). Global gut content data synthesis and phylogeny delineate reef fish trophic guilds. bioRxiv [Preprint] doi: 10.1101/2020.03.04.977116
Phillips, C. D., Phelan, G., Dowd, S. E., McDonough, M. M., Ferguson, A. W., Delton Hanson, J., et al. (2012). Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627. doi: 10.1111/j.1365-294X.2012.05568.x
Roeselers, G., Mittge, E. K., Stephens, W. Z., Parichy, D. M., Cavanaugh, C. M., Guillemin, K., et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J. 5, 1595–1608. doi: 10.1038/ismej.2011.38
Rosado, D., Pérez-Losada, M., Severino, R., Cable, J., and Xavier, R. (2019). Characterization of the skin and gill microbiomes of the farmed seabass (Dicentrarchus labrax) and seabream (Sparus aurata). Aquaculture 500, 57–64. doi: 10.1016/j.aquaculture.2018.09.063
Ross, A. A., Müller, K. M., Scott Weese, J., and Neufeld, J. D. (2018). Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc. Natl. Acad. Sci. U.S.A. 115, E5786–E5795. doi: 10.1073/pnas.1801302115
Ruiz-Rodríguez, M., Scheifler, M., Sanchez-Brosseau, S., Magnanou, E., West, N., Suzuki, M., et al. (2020). Host species and body site explain the variation in the microbiota associated to wild sympatric mediterranean teleost fishes. Microb. Ecol. 80, 212–222. doi: 10.1007/s00248-020-01484-y
Sanders, J. G., Powell, S., Kronauer, D. J. C., Vasconcelos, H. L., Frederickson, M. E., and Pierce, N. E. (2014). Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol. Ecol. 23, 1268–1283. doi: 10.1111/mec.12611
Santini, F., Carnevale, G., and Sorenson, L. (2014). First multi-locus timetree of seabreams and porgies (Percomorpha: Sparidae). Ital. J. Zool. 81, 55–71. doi: 10.1080/11250003.2013.878960
Santos, T. (2018). PVR: Phylogenetic Eigenvectors Regression and Phylogentic Signal-Representation Curve. Available online at: https://cran.r-project.org/package=PVR (accessed May 30, 2018).
Schwarz, W. H. (2001). The cellulosome and cellulose degradation by anaerobic bacteria. Appl. Microbiol. Biotechnol. 56, 634–649. doi: 10.1007/s002530100710
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Seshadri, R., Leahy, S. C., Attwood, G. T., Teh, K. H., Lambie, S. C., Cookson, A. L., et al. (2018). Cultivation and sequencing of rumen microbiome members from the Hungate1000 collection. Nat. Biotechnol. 36, 359–367. doi: 10.1038/nbt.4110
Soininen, E. M., Valentini, A., Coissac, E., Miquel, C., Gielly, L., Brochmann, C., et al. (2009). Analysing diet of small herbivores: the efficiency of DNA barcoding coupled with high-throughput pyrosequencing for deciphering the composition of complex plant mixtures. Front. Zool. 6:16. doi: 10.1186/1742-9994-6-16
Sullam, K. E., Essinger, S. D., Lozupone, C. A., O’Connor, M. P., Rosen, G. L., Knight, R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sullam, K. E., Rubin, B. E., Dalton, C. M., Kilham, S. S., Flecker, A. S., and Russell, J. A. (2015). Divergence across diet, time and populations rules out parallel evolution in the gut microbiomes of Trinidadian guppies. ISME J. 9, 1508–1522. doi: 10.1038/ismej.2014.231
Sylvain, F., Holland, A., Audet-Gilbert, É, Luis Val, A., and Derome, N. (2019). Amazon fish bacterial communities show structural convergence along widespread hydrochemical gradients. Mol. Ecol. 28, 3612–3626. doi: 10.1111/mec.15184
Sylvain, F. -É, Holland, A., Bouslama, S., Audet-Gilbert, É, Lavoie, C., Luis Val, A., et al. (2020). Fish skin and gut microbiomes show contrasting signatures of host species and habitat. Appl. Environ. Microbiol. 86, e00789–20. doi: 10.1128/aem.00789-20
Keywords: fish gut microbiome, Sparidae, phylosymbiosis, morphological traits, diet, herbivory, ecological outlier
Citation: Escalas A, Auguet J-C, Avouac A, Seguin R, Gradel A, Borrossi L and Villéger S (2021) Ecological Specialization Within a Carnivorous Fish Family Is Supported by a Herbivorous Microbiome Shaped by a Combination of Gut Traits and Specific Diet. Front. Mar. Sci. 8:622883. doi: 10.3389/fmars.2021.622883
Received: 11 November 2020; Accepted: 25 January 2021;
Published: 15 February 2021.
Edited by:
Alfonso Benítez-Páez, Principe Felipe Research Center (CIPF), SpainReviewed by:
Konstantinos Ar. Kormas, University of Thessaly, GreeceFotini Kokou, Wageningen University and Research, Netherlands
Copyright © 2021 Escalas, Auguet, Avouac, Seguin, Gradel, Borrossi and Villéger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arthur Escalas, YXJ0aHVyLmVzY2FsYXNAZ21haWwuY29t