 Giorgia Palladino1,2
Giorgia Palladino1,2 Elena Biagi1,2
Elena Biagi1,2 Simone Rampelli1
Simone Rampelli1 Margherita Musella1,2
Margherita Musella1,2 Federica D’Amico3,4
Federica D’Amico3,4 Silvia Turroni1
Silvia Turroni1 Patrizia Brigidi4
Patrizia Brigidi4 Gian Marco Luna5
Gian Marco Luna5 Marco Candela1,2*
Marco Candela1,2*- 1Unit of Microbiome Science and Biotechnology, Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy
- 2Fano Marine Center, The Inter-Institute Center for Research on Marine Biodiversity, Resources and Biotechnologies, Fano, Italy
- 3Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy
- 4Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy
- 5Institute for Marine Biological Resources and Biotechnology, National Research Council (IRBIM-CNR), Ancona, Italy
Increasing evidence indicates that host-associated microbial communities play a key role in the biology of marine eukaryotic organisms. Amongst them, Corallimorpharia are extensively found on reefs, carpeting vast reef areas, where they can exert important roles as habitat forming holobionts, being at the base of complex trophic webs. Here we explore the bacterial community structure, and its changes across different seasons, associated with the jewel anemone Corynactis viridis, an anthozoan Cnidaria that is widely distributed in the northeastern Atlantic Ocean and the Mediterranean Sea. Samples were collected in the North Adriatic Sea in three seasons and the community composition was studied using 16S rDNA sequencing. We show that C. viridis-associated microbial communities are unique and significantly different from those in the surrounding seawater. Interestingly, we observe remarkable changes in the C. viridis microbiome according to seasonality. In particular, the C. viridis microbiome is capable of rearranging its overall ecological structure with the winter-summer transition, moving from an oligotrophic anaerobic community to a heterotrophic ecosystem, with the propensity to ferment proteins and complex polysaccharides. Our findings demonstrate that C. viridis has a unique associated microbiota and suggest that this is capable of adapting to seasonal changes in the host physiology, by establishing a microbiome-host interaction process whose relevance to C. viridis has yet to be determined.
Introduction
The Cnidaria are a large, diverse, and ecologically important group of organisms that are widely distributed in marine environments, where they represent important habitat-forming organisms (Marzinelli et al., 2018). The study of Cnidaria have gained increasing importance in recent years due to the growing awareness of their vulnerability to anthropogenic stressors, with obvious cascade impacts at the ecosystem level (Gerhardt, 2002; Rocha et al., 2014). As all marine animals, Cnidarians live as “holobionts,” as a result of a close interaction with a complex microbial community (O’Brien et al., 2019), defined as microbiome (Berg et al., 2020). The “holobionts” microbiomes are inherent to the host physiology, being capable of providing the host with ecological functions crucial for its health (Pita et al., 2018). Particularly, several studies aimed at characterizing Cnidaria-associated microbes are now available, providing important information on their role in nutrition, defense, immunity and development (Littman et al., 2009; Liu et al., 2018; Pollock et al., 2018; Ziegler et al., 2019). This awareness also highlighted the possible importance of the Cnidaria microbiomes as a determinant of their adaptive or maladaptive response to stressors, including growing climate-related and anthropogenic-induced stressors already impacting the ocean environment (Apprill, 2017). Indeed, it has been proposed that variations in Cnidaria microbiome can be employed as early bio-indicator of both environmental and anthropogenic stressors (Stabili et al., 2018), making the study of Cnidarians microbiomes, and its variations, a crucial point at the conjunction between animal and environmental health. However, the vast majority of the studies on Cnidaria microbiomes are focused on corals, with a lack of information on all the other organisms belonging to this phylum. To the best of our knowledge, only a limited number of pioneering studies on non-coral Cnidaria have been performed (Di Camillo et al., 2012; Har et al., 2015; Murray et al., 2016; Brown et al., 2017) and no studies have been conducted to describe the microbes associated with Corallimorpharia, non-calcifying, close relatives of scleractinian corals (Lin et al., 2016). Although scleractinian corals are the most important reef builders, Corallimorpharia can also be extensively found on reefs either as solitary individuals or as colonies that may carpet vast reef areas, where they can exert important roles as habitat forming halobionts, being at the base of complex trophic chains (Kuguru et al., 2004). It is thus important to fill this knowledge gap, providing basic knowledge on the structure of the Corallimorpharia microbiome and on its possible role in host physiology and health. In this scenario, we aim to investigate the community structure, and its seasonal variations, of the microbes associated with the jewel anemone Corynactis viridis, an anthozoan Cnidaria belonging to the order Corallimorpharia. C. viridis is a brightly colored marine invertebrate similar in body form to a sea anemone. Its typical habitat encompasses the northeastern Atlantic Ocean, including Scotland, Ireland, and the southern and western coasts of Great Britain, the southwestern coasts of continental Europe and the Mediterranean Sea. It is found on rocks, caves or beneath overhangs sheltered from the light forming dense aggregations, and its depth range goes from the lower shore down to about 80 m (Hill and White, 2008). Specifically, in our work, we characterize the microbial communities associated with C. viridis from 30 individuals collected in three different seasons (winter 2018, spring and summer 2019) by next-generation sequencing (NGS) of the V3-V4 region of the 16S rRNA gene. According to our data C. viridis possesses a characteristic associated microbiome whose inherent plasticity is possibly functional for the adaptation to seasonal changes.
Materials and Methods
Sample Collection and DNA Extraction
Corynactis viridis individuals were collected at the “Il Paguro” site by the diving association “Dive Planet” (Rimini, Italy). “Il Paguro” is the wreck of a methane extraction platform located around 12 nautical miles offshore Ravenna, Italy. The wreck extends from a depth of 10 m in its most superficial part to 35 m, and it hosts a great variety of marine wildlife. In order to regulate the scuba activity on the site and preserve its coral reef-like ecosystem, an Italian association responsible for the site (“Associazione Paguro”) has been established and, starting from July the 21st, 1995, the area was declared “Area of biological protection” by the Italian Ministry of Agricultural Resources. In February 2010, “Il Paguro” was declared “Site of community interest” by the Emilia-Romagna region (Rinaldi and Rambelli, 2004). “Associazione Paguro” kindly provided permission to sample C. viridis within the “Il Paguro” site for scientific purposes only and Italian Cost Guard was made aware of the sampling activity. We used satellite maps from NASA (available at https://giovanni.gsfc.nasa.gov/giovanni/) to provide some data on the main environmental variables of the sampling site, including temperature and chlorophyll a to evidence eutrophication occurrence (Ignatiades, 2005). Ten anemone individuals were collected at three different time points in December 2018 (winter sampling), May 2019 (spring sampling) and July 2019 (summer sampling), for a total of 30 individuals (Supplementary Figure 1). For each site, 2 L of seawater were also sampled at the same depth as the anemones, by using previously sterilized polypropylene bottles. Anemones were collected using knives previously sterilized with alcohol, by placing 1–3 rocks hosting different individuals in sterile disposable plastic containers to avoid contamination. The picking of anemones from the respective rocks was performed once returned to the laboratory (within a few hours of sampling at sea), under a vertical laminar airflow cabinet using sterile tweezers and scalpels. Each individual was placed in a single 1.5-mL microtube and stored at −80°C until further processing. Seawater samples were filtered using vacuum filtration with MF-Millipore (Darmstadt, Germany) Membrane filters with 0.45-μm pore size, 47-mm diameter and mixed cellulose esters membrane. Each 2 L sample of seawater was filtered on a single filter, which was rolled on itself using sterilized forceps and placed in a 2-mL microtube, then stored at −80°C until further processing.
Total microbial DNA extraction was performed for each individual anemone (weighing approximately 0.15–0.20 g) using the DNeasy PowerBiofilm Kit (Qiagen, Hilden, Germany) with a modified protocol. In particular, the homogenization step was performed using a FastPrep instrument (MP Biomedicals, Irvine, CA, United States) at 5.5 movements per sec for 1 min and, before the elution step, an incubation of 5 min at 4°C was performed. Seawater microbial DNA extraction was carried out using the DNeasy PowerWater Kit (Qiagen) following the manufacturer’s instructions. DNA samples were quantified using NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, United States) and stored at -20°C until further processing.
16S rRNA Gene Amplification and Sequencing
Amplification of the V3-V4 hypervariable region of the 16S rRNA gene was performed in 50-μL volumes containing 25 ng of microbial DNA, 2X KAPA HiFi HotStart ReadyMix (Roche, Basel, Switzerland), and 200 nmol/L of 341F and 785R primers carrying Illumina overhang adapter sequences. The thermal cycle consisted of 3 min at 95°C, 30 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, and a final 5-min step at 72°C (Turroni et al., 2016). PCR reactions were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, United States). Nextera technology was used to prepare indexed libraries by limited-cycle PCR reaction. After a further clean-up step as described above, libraries were normalized to 4 nM and pooled. The sample pool was denatured with 0.2 N NaOH and diluted to 6 pM with a 20% PhiX control. Sequencing was performed on Illumina MiSeq platform using a 2 × 250 bp paired end protocol, according to the manufacturer’s instructions (Illumina, San Diego, CA, United States).
Bioinformatics and Statistics
Raw sequences were processed using a pipeline combining PANDAseq (Masella et al., 2012) and QIIME 2 (Bolyen et al., 2019). High-quality reads (min/max length = 350/550 bp) were binned into amplicon sequence variants (ASVs) using DADA2 (Callahan et al., 2016), as previously described (D’Amico et al., 2019). Taxonomy was assigned using the VSEARCH algorithm (Rognes et al., 2016) and the Silva database (December 2017 release) (Quast et al., 2013). The sequences assigned to the eukaryotic host (chloroplasts and mitochondria) as well as the unassigned ones were discarded. The ASV table was rarefied up to 1733 reads per sample, corresponding to the lowest read number observed among the samples. Alpha diversity was evaluated using three different metrics: Faith’s Phylogenetic Diversity (PD_whole_tree) (Faith, 1992), Chao1 index for microbial richness and number of observed ASVs. The unweighted UniFrac distance was used to build Principal Coordinates Analysis (PCoA) plots.
Statistical analysis was performed using R software version 3.6.11. PCoA, bacterial co-abundance groups (CAGs), box-and-whisker plots and Venn diagrams were generated using “Stats”2, “Made4” (Culhane et al., 2005), “vegan”3, “boxplot” (Murrell, 2018) and “VennDiagram”4 packages. Unweighted UniFrac distance was used for PCoA, and the significance of separation was tested by permutation test with pseudo F-ratio (function “Adonis” in “vegan”). Bacterial groups with the largest contribution to the ordination space were found by using the function envfit of the R package vegan on the family relative abundances. Significant data separation was assessed by Kruskal–Wallis test or Wilcoxon rank-sum test, based on the data. When necessary, p-values were corrected for multiple testing with Benjamini-Hochberg method, with a false discovery rate (FDR) ≤ 0.05 considered as statistically significant.
For graphical representation, anemone microbial phyla whose relative abundance was less than 0.5% in at least 10% of the samples were filtered out under the name “other phyla.” The same was done for microbial families and genera with a relative abundance lower than 2% in at least 10% of the samples (“other families” and “other genera,” respectively). For the generation of CAGs, only bacterial genera present in at least 10% of the samples with relative abundance ≥ 0.5% were considered. Co-abundance was evaluated by the Kendall correlation test and displayed with hierarchical Ward-linkage clustering based on the Spearman correlation coefficient (Claesson et al., 2012). Wiggum plot networks were created using Cytoscape software5 (Smoot et al., 2011), where the circle size represents the bacterial abundance and the connections between nodes represent significant Kendall correlations between genera (FDR ≤ 0.05). For Venn diagrams, the ASVs present in at least one sample of each season group were taken into account. We processed all the 4,623 ASVs obtained from DADA2 binning of high-quality reads through the R package VennDiagram, in order to produce a visual output of the ASVs sharing between seasons. The FASTA files of the ASV sequences shared between all seasons or couples of seasons were blasted against the 16S rRNA database through BLASTN algorithm (Altschul et al., 1990).
Results
C. viridis and Seawater Microbiota
A total of 30 individuals of C. viridis were sampled in three different seasons in December 2018 (winter), May 2019 (spring) and July 2019 (summer) at the “Il Paguro” site in the North Adriatic Sea. The monthly satellite maps of temperature and surface chlorophyll a from NASA for each sampling period are provided in Supplementary Figure 2. Briefly, seawater temperature during the three sampling times variated between 10–12°C in winter, 15–17°C in spring, and 26–28°C in summer. Chlorophyll a concentration was always > 2 mg/m3 (ranging between 2.2 and 3.6), classifying the sampling region as eutrophic (Ignatiades, 2005; Ferreira et al., 2011). For each time point, 10 individuals and 2 L of seawater were collected at a depth of 20 m. From each sample, the microbiome compositional structure was obtained by NGS sequencing of the V3–V4 hypervariable region of the 16S rRNA gene, resulting in 252,160 high-quality reads and an average of 7,641.212 ± 5,177.348 (mean ± SD) reads per sample. High-quality reads were binned into 4,623 ASVs.
At first, we compared the alpha diversity between C. viridis and seawater communities. Although not significant, our data show a trend toward an overall higher microbial richness in seawater than in anemone, as measured by Chao1 metrics and number of observed ASV (Wilcoxon rank-sum test controlled for multiple testing using FDR, p-value > 0.05) (Supplementary Figure 3).
We next explored the community composition in C. viridis and seawater at different phylogenetic levels (Figure 1). At the phylum level, the C. viridis microbiota is characterized by five dominant phyla, Proteobacteria (mean relative abundance ± SD, 35.3 ± 14.4%), Firmicutes (15.3 ± 20.8%), Tenericutes (15.3 ± 21.3%), Planctomycetes (10.3 ± 8.4%) and Bacteroidetes (10.0 ± 9.1%). Less abundant phyla are represented by Actinobacteria (4.0 ± 4.3%), Cyanobacteria (2.7 ± 4.4%), Verrucomicrobia (1.9 ± 1.6%) and Chloroflexi (1.7 ± 2.2%). The most represented families are Spiroplasmataceae (15.2 ± 21.3%), Planctomycetaceae (9.8 ± 8.1%) and Rhodobacteraceae (7.1 ± 5.5%). Subdominant families are Flavobacteriaceae (4.5 ± 3.0%), Clostridiales family XII (2.9 ± 5.0%), Vibrionaceae (2.9 ± 3.8%) and Clostridiaceae (2.5 ± 4.0%). At the genus level, the dominant taxon is Spiroplasma (15.2 ± 21.3%), while less represented genera are Fusibacter (2.9 ± 5.0%), Blastopirellula (2.5 ± 2.6%) and Pir4 lineage (2.5 ± 3.5%). On the other hand, seawater shows a distinct microbial structure already at the phylum level, being dominated by Proteobacteria (57.1 ± 0.75), Bacteroidetes (19.6 ± 2.3%), Cyanobacteria (7.6 ± 2.7%) and Verrucomicrobia (6.2 ± 2.8%), with Planctomycetes (2.6 ± 2.2%), Actinobacteria (2.5 ± 2.5%) and Firmicutes (1.6 ± 2.8%) as subdominant taxa. The most represented families are Flavobacteriaceae (10.9 ± 3.3%), family I of Cyanobacteria subsection I (7.6 ± 2.8%) and Rhodobacteraceae (6.9 ± 2.9%), while Halieaceae (3.5 ± 3.8%), Verrucomicrobiaceae (3.0 ± 2.0%) and Rhodospirillaceae (3.0 ± 1.2%) are subdominant. Synechococcus (5.3 ± 5.3%) is the most abundant classified genus, while Tenacibaculum (2.5 ± 2.2%) the subdominant one.
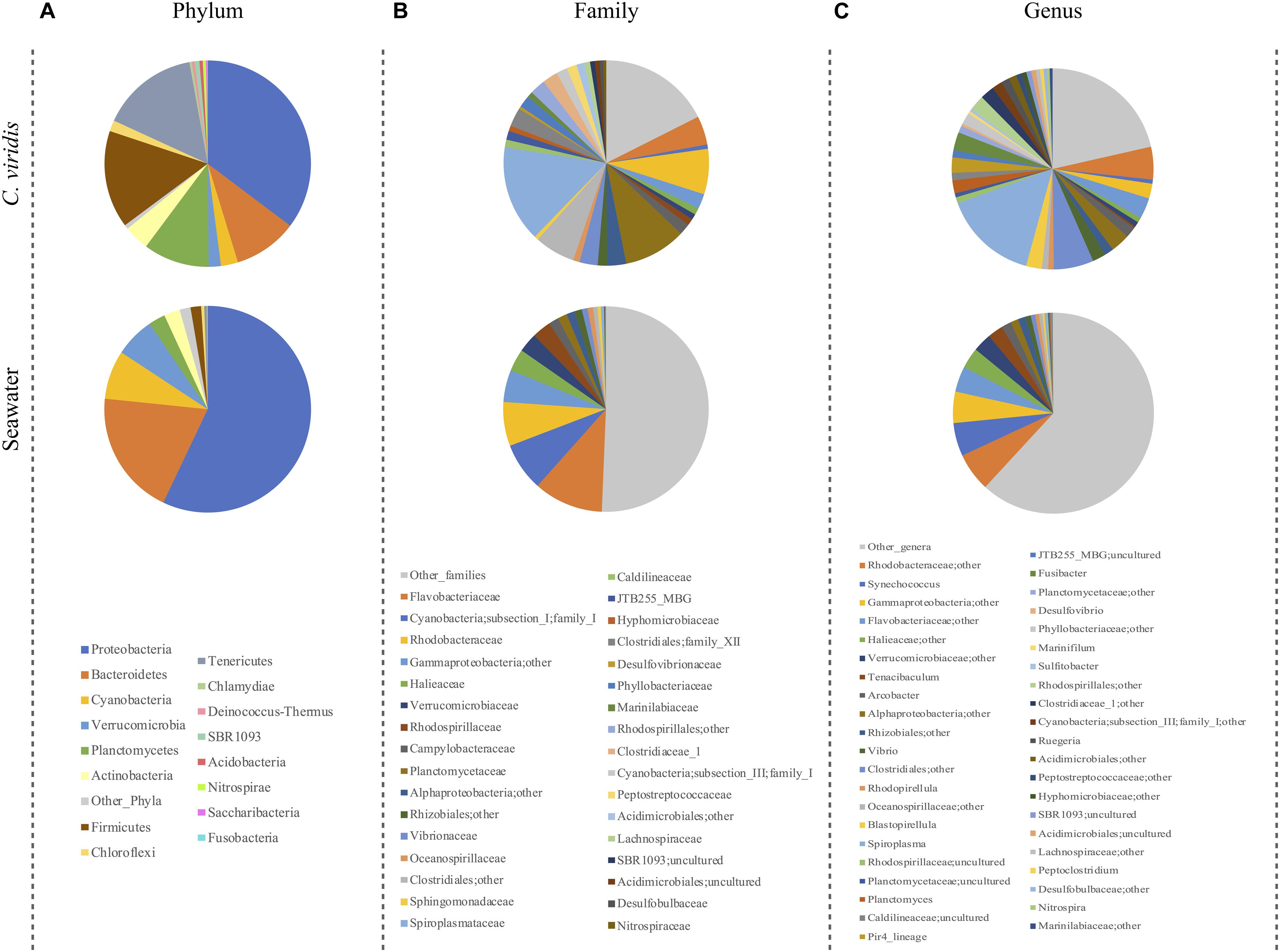
Figure 1. Microbiota compositionof C. viridis and seawater. Pie charts summarizing the phylum (A), family (B) and genus-level (C) microbiota composition of C. viridis and seawater. Only phyla with relative abundance >0.5% in at least 10% of samples, and families and genera with relative abundance >2% in at least 10% of samples are represented.
In order to highlight the overall compositional differences between C. viridis and seawater microbial communities, a PCoA of the unweighted UniFrac distances was carried out. As expected, the C. viridis microbiome significantly segregates from that of seawater and such a segregation was found to be robust to seasonality (permutation test with pseudo-F ratio, p-value ≤ 0.01) (Supplementary Figure 4). To identify the bacterial families most contributing to the separation, their relative abundance was superimposed on the PCoA plots. According to our findings, the families Planctomycetaceae, Spiroplasmataceae, and Clostridiales family XII are the most characteristic of the C. viridis microbiome, but with a seasonality-specific pattern. Conversely, Flavobacteriaceae and family I of subsection I in the phylum Cyanobacteria are the most distinguishing of the seawater samples.
Seasonal Trends in the C. viridis Microbiome
We tested whether the C. viridis-associated microbial communities varied by season. To do this, we first assessed the existence of significant differences among seasons for alpha diversity. All metrics consistently indicated greater microbial diversity in winter and summer, compared to spring (Kruskal–Wallis test controlled for multiple testing using FDR, p-value ≤ 0.05) (Figure 2A). We next explored seasonal patterns in the community structure by analyzing beta diversity. Interestingly, the unweighted UniFrac-based PCoA revealed a sharp segregation of the microbiome structures through seasonality (permutation test with pseudo F-ratio, p-value ≤ 0.001) (Figure 2B). Conversely, seawater showed only a limited variation across the different seasons. When comparing the relative abundance of C. viridis microbial families among seasons (Figure 3), we found that during the winter, Caldilineaceae, Nitrospiraceae, and Planctomycetaceae were significantly more abundant, whereas the Vibrionaceae family was largely underrepresented. On the other hand, the spring samples were characterized by a higher proportion of the families Rhodobacteraceae, Rhodospirillaceae, Campylobacteraceae, Oceanospirillaceae, and Spiroplasmataceae. Finally, the relative abundance of Marinilabiaceae, family I of Cyanobacteria subsection III, Clostridiaceae 1, family XII of Clostridiales, Lachnospiraceae, Peptostreptococcaceae, Desulfobulbaceae, and Desulfovibrionaceae was significantly higher during the summer.
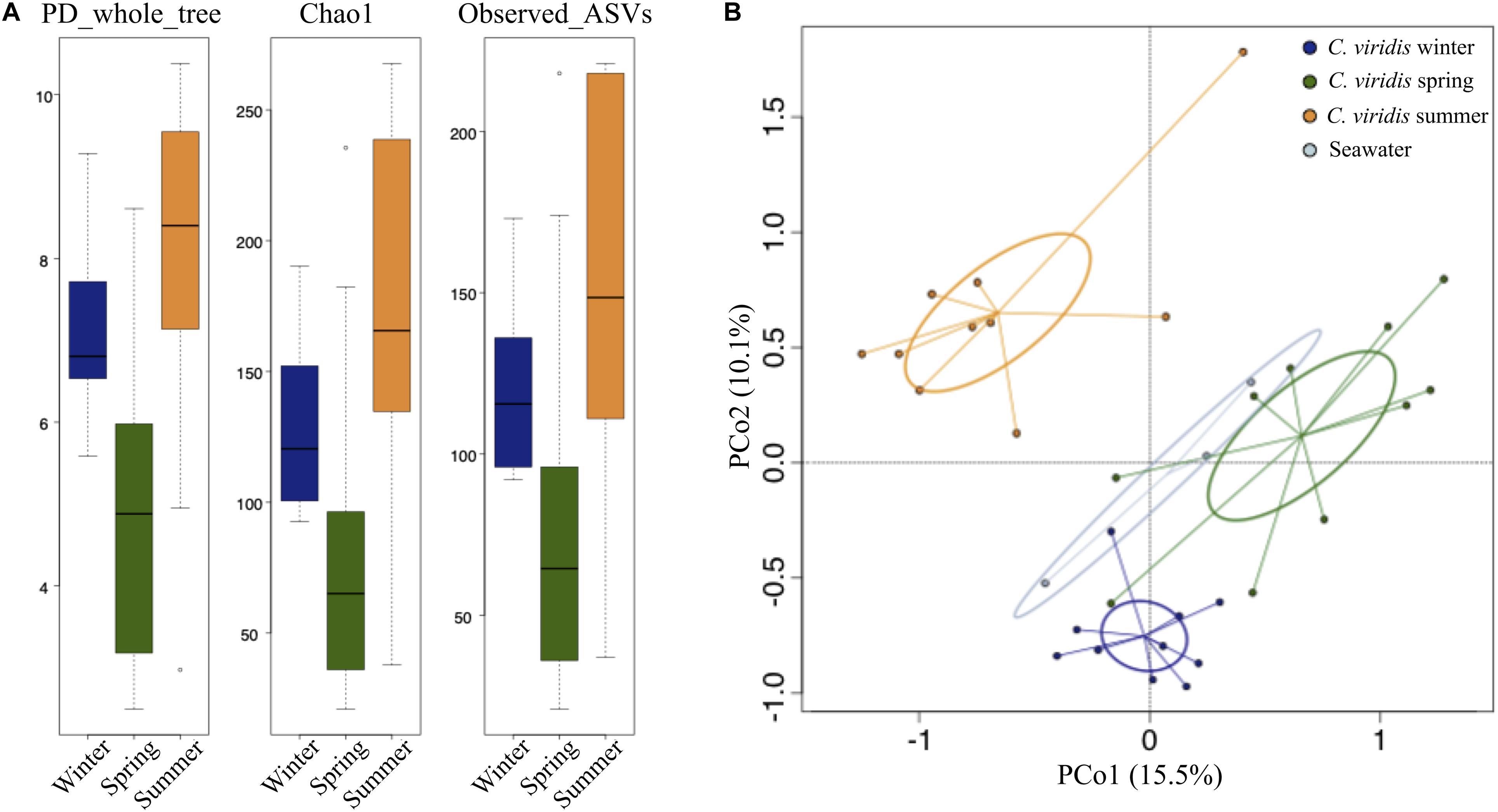
Figure 2. Alpha and beta diversity of the C. viridis microbiota. (A) Box-and-whisker distribution of the Faith’s Phylogenetic Diversity (PD_whole_tree), Chao1 index for microbial richness and number of observed ASVs, calculated for winter (dark blue), spring (dark green) and summer (orange) anemone samples. According to all metrics, greater microbial diversity was observed in winter and summer than in spring (Kruskal–Wallis test controlled for multiple testing using FDR, p-value ≤ 0.05). (B) Principal Coordinates Analysis (PCoA) based on unweighted UniFrac distances showing the variation of the anemone microbiota across seasons (same color code as in A) and seawater samples (light blue) (permutation test with pseudo F-ratio, p-value ≤ 0.001). The first and second principal components (PCo1 and PCo2) are plotted and the percentage of variance in the dataset explained by each axis is reported. Ellipses include 95% confidence area based on the standard error of the weighted average of sample coordinates.
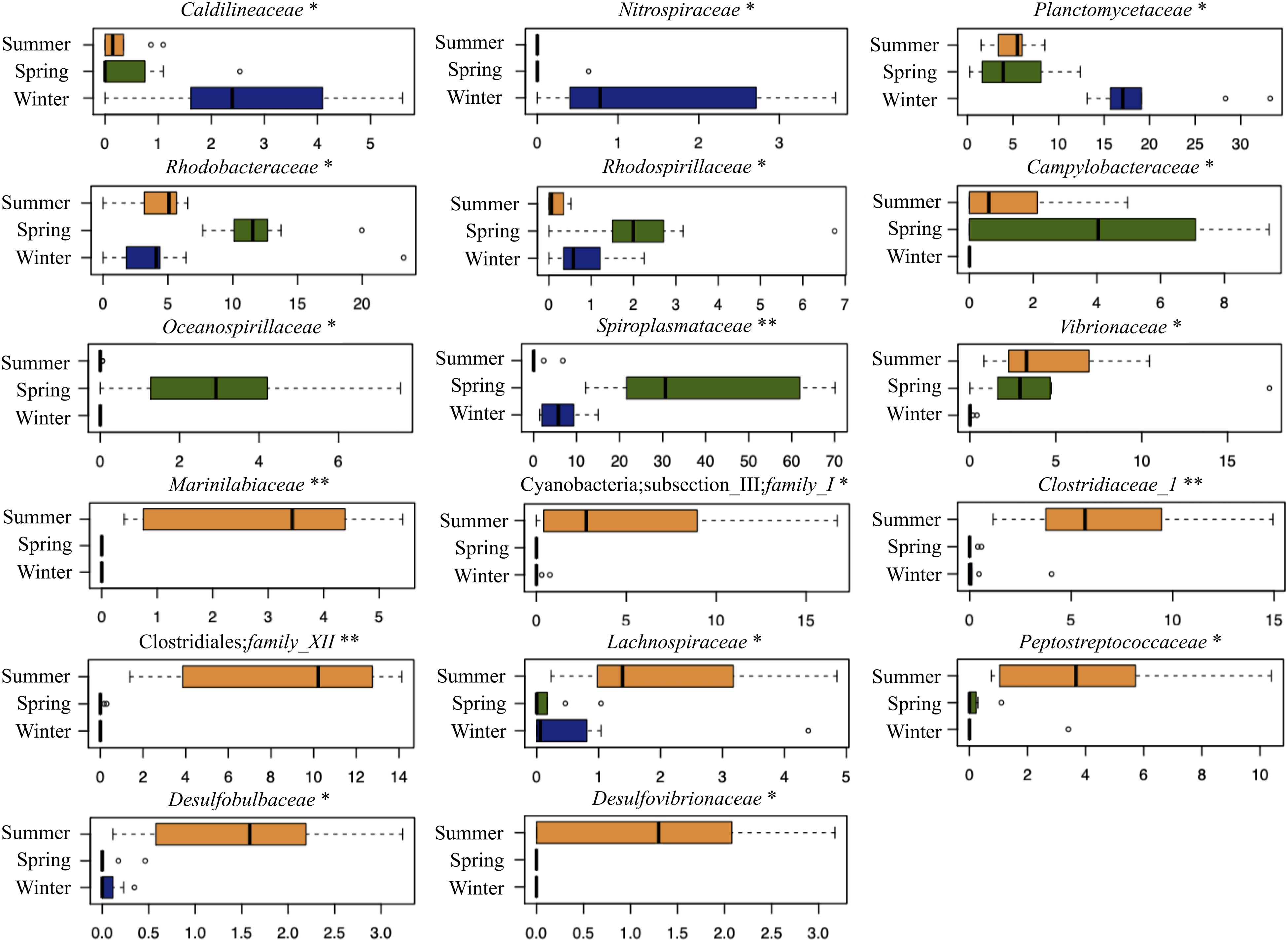
Figure 3. C. viridis-associated bacterial families differently represented across seasons. Box-and-whisker plots showing the relative abundance distribution of bacterial families in the different seasons (winter, dark blue; spring, dark green; and summer, orange). The central box represents the distance between the 25th and 75th percentiles. The median is marked with a black line. Whiskers identify the 10th and 90th percentiles. ∗p-value ≤ 0.05; ∗∗p-value ≤ 0.01; Kruskal–Wallis test controlled for multiple testing using FDR.
To gain further insights into the C. viridis-associated microbiome, we explored its topological variation by clustering the bacterial genera into CAGs. Three CAGs were identified, and the prevalence and connections between the represented genera were obtained by a Wiggum plot network analysis (Supplementary Figure 5). Interestingly, we observed a peculiar declination of the C. viridis CAGs based on seasonality. CAGs were named according to the genus showing the higher overabundance in a seasonal-dependent pattern: “Mycobacterium CAG,” “Colwellia CAG,” and “Desulfovibrio CAG” (Figure 4). Winter communities were characterized by the “Mycobacterium CAG”, which also included Nitrospira, Planctomyces, Blastopirellula, Pir4 lineage, OM60 (NOR5) clade and Lysinibacillus. Conversely, the “Colwellia CAG,” including Pseudoalteromonas, Aquibacter, Tenacibaculum, Spiroplasma, Vibrio, Sulfitobacter, Pseudofulvibacter, and Arcobacter, dominated the spring microbiomes. Finally, the summer communities were characterized by the “Desulfovibrio CAG,” including Peptoclostridium, Marinifilum, Cetobacterium, Maribacter, Rhodopirellula, Fusibacter, Streptomyces, and Truepera.
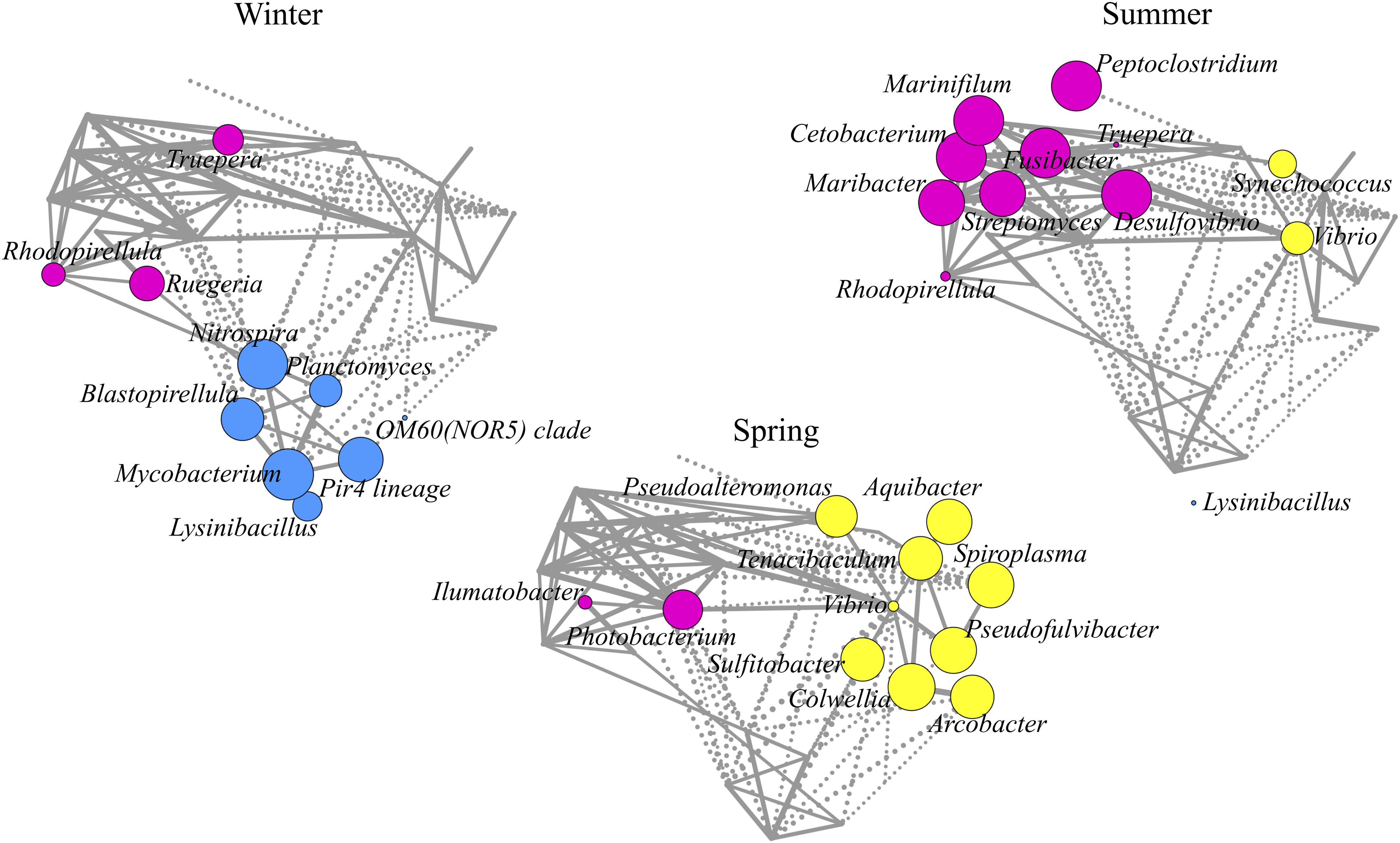
Figure 4. Declination of C. viridis co-abundance groups according to seasonality. Co-abundance groups (CAGs) are named according to the dominant genus in each group: Mycobacterium, Colwellia, and Desulfovibrio. Each node represents a bacterial genus and its size is proportional to its overabundance on the average value within the population. The connections between nodes represent positive (solid lines) and negative (dashed lines) Kendall correlations between genera (FDR ≤ 0.05).
C. viridis Core Microbiome
Finally, we investigated whether C. viridis possesses a core microbiome, which is stably associated with the host and thus does not change with the seasons. As shown in Figure 5, only four ASVs were shared among all three seasons, whereas nine ASVs were shared between winter and spring samples, 18 ASVs between winter and summer samples, and seven ASVs between spring and summer samples, for a total of 38 shared ASVs in the three sampling seasons. According to a BLAST analysis, the four ASVs shared among the three seasons were all assigned to the bacterial family Spiroplasmataceae (Table 1). More detailed information on BLAST alignment for each ASV is provided in Supplementary Table 1.
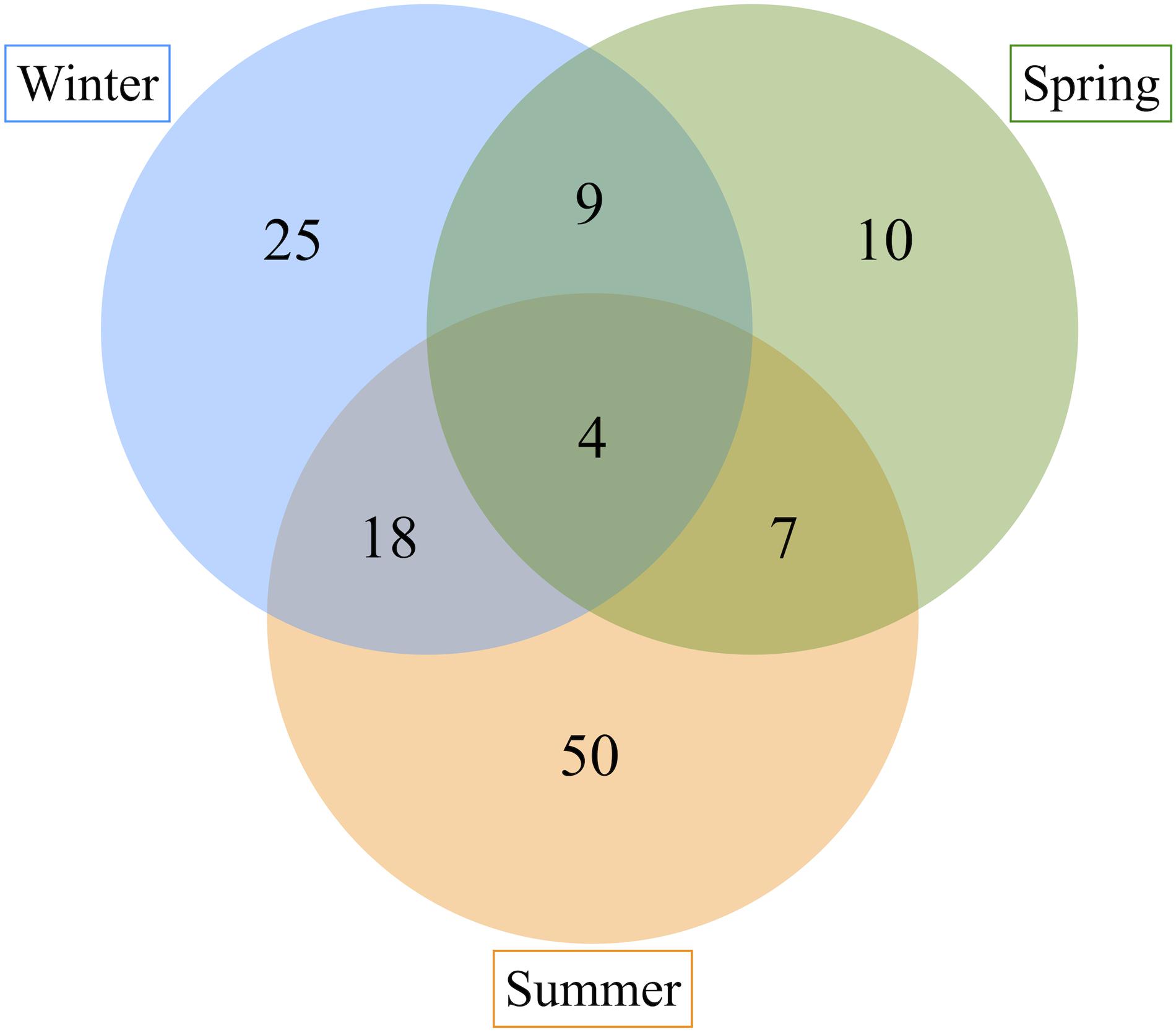
Figure 5. ASVs sharing among C. viridis bacterial communities across seasons. Venn diagram showing the number of ASVs shared between the C. viridis bacterial communities in different seasons. Four ASVs are shared among all the three seasons, nine ASVs between winter and spring samples, 18 between winter and summer samples and seven between spring and summer samples.
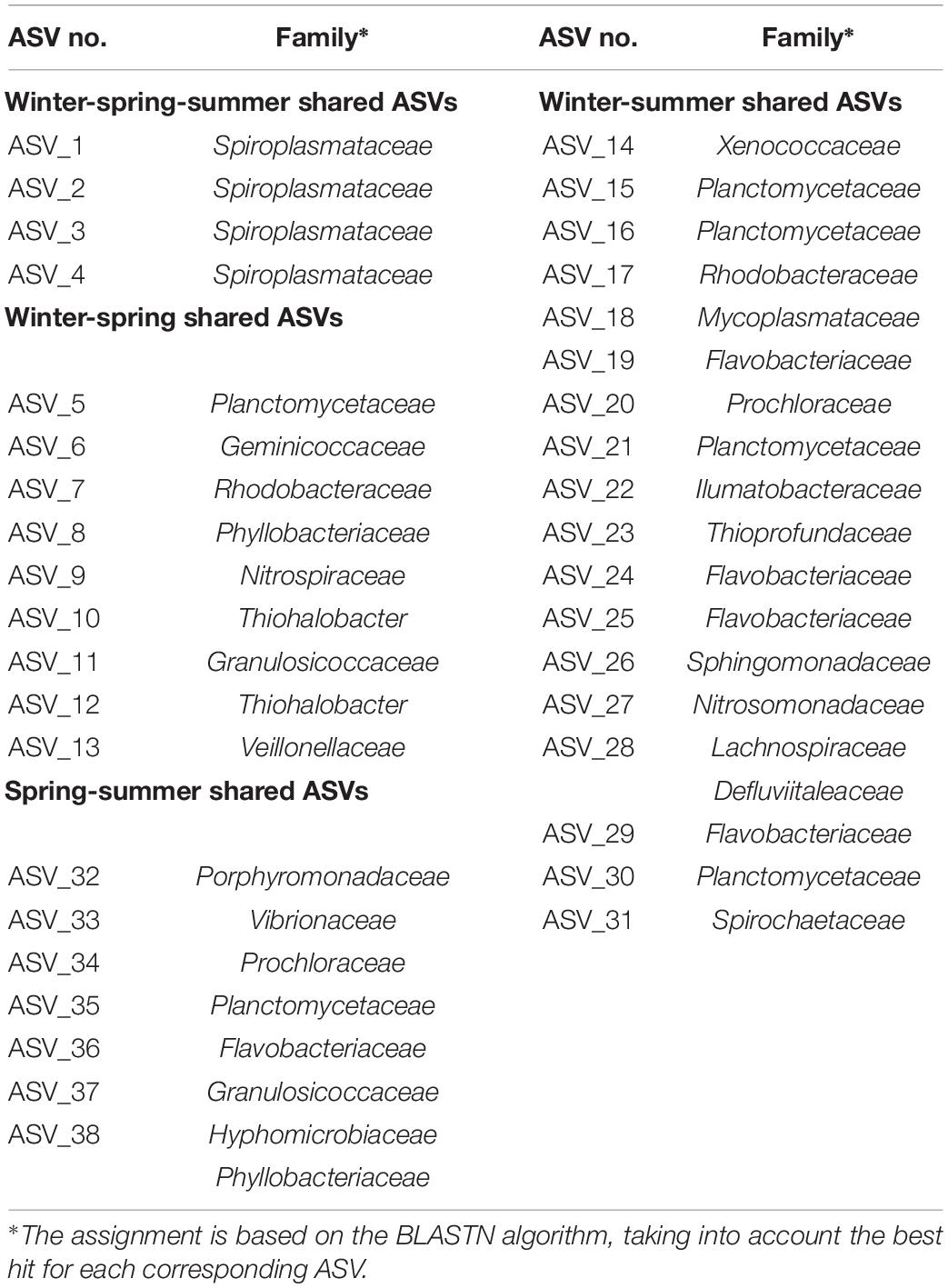
Table 1. ASVs sharing among C. viridis-associated microbial communities in winter, spring, and summer.
Discussion
Given the fundamental role that microorganisms play in the functioning of eukaryotic hosts, investigating the diversity, community composition and seasonal changes of the host-associated microbial communities is crucial to understand the physiological significance of the association, also in light of anthropic threats. We show here that the microbial communities associated with C. viridis are unique and significantly different from those found in the surrounding seawater. In particular, C. viridis microbiome includes microorganisms belonging to nine main phyla, of which Proteobacteria, Firmicutes, Tenericutes, Planctomycetes, and Bacteroidetes are the dominant (relative abundance – r.a. – between 10 and 35%) and Actinobacteria, Cyanobacteria, Verrucomicrobia, and Chloroflexi are the subdominant (r.a. from 1.7 to 4%). Statistical analysis indicated that Spiroplasmataceae, Planctomycetaceae, and Clostridiales family XII are the families that most differentiate the C. viridis and seawater communities, allowing us to hypothesize, at least for these taxa, a non-neutral selection process from the surrounding environment. Supporting our findings, the communities associated with another sea anemone (i.e., Nematostella vectensis) show an overall phylum-level composition that well resembles what we observed for C. viridis, including Proteobacteria, Bacteroidetes, Firmicutes, Tenericutes, and Planctomycetes among the dominant phyla (Har et al., 2015). However, at lower taxonomic levels (e.g., family and genus levels), the microbial composition of different anthozoan cnidarians drastically changes (Murray et al., 2016; Brown et al., 2017), demonstrating a robust host-specific profile as already observed for other marine holobionts (Pita et al., 2018; Wilkins et al., 2019). Particularly, regarding Cnidaria, a robust specie-specific microbiome profile was demonstrated in three highly abundant and widespread Indo-Pacific species (Acropora aculeus, Mycedium elephantotus, and Pachyseris speciosa), with very few bacterial phylotypes being shared between the three coral species (Hernandez-Agreda et al., 2018).
While the bacterioplankton communities in seawater only show slight variations across different seasons, the C. viridis-associated microbiota considerably changes, as previously shown for the coral-associated microbiome (Chen et al., 2011; Sharp et al., 2017; Cai et al., 2018). According to our data, C. viridis is capable of rearranging its associated microbial community along the transition from winter to spring and summer. In particular, the winter communities appear enriched in oligotrophic anaerobic microorganisms, such as Planctomycetaceae, Caldilineaceae and Nitrospiraceae, which are commonly found in other marine holobionts (Lee et al., 2018). Bacteria belonging to these taxa have been implicated in the cycling of important elements such as nitrogen, sulfur and iron, and provide key ecological functions to the marine ecosystem (Clum et al., 2009; Zhang et al., 2017, 2018). Though being characterized by a different pattern in the dominant bacterial families, the spring communities show a structure similar to that observed in winter, being dominated by taxa such as Rhodospirillaceae, Rhodobacteraceae, and Oceanospirillaceae, microorganisms that are known to be anaerobic oligotrophic components of the microbiome of marine holobionts and play important roles in carbon and sulfur cycling (Pujalte et al., 2014; Cortés-Lara et al., 2015; Tinta et al., 2019). During the summer, C. viridis communities drastically increase their biodiversity and change their composition. Indeed, with the spring-summer transition, C. viridis-associated communities show dominance of heterotrophic anaerobic microorganisms, which generally populate the holobiont digestive tract. In particular, the C. viridis microbiome in summer is characterized by higher relative abundance of Lachnospiraceae, Clostridiaceae and Desulfovibrionaceae, well-known primary and secondary fermenters that populate the gastrointestinal tract of a wide range of terrestrial and marine holobionts, including mammalians (Rausch et al., 2019). During the summer, we also observed an increase in Vibrionaceae members that have been frequently detected as symbionts of marine holobionts and recently hypothesized to be involved in regulating host developmental processes (Tinta et al., 2019). Finally, although the Spiroplasmataceae family increases its relative abundance during spring, this taxon is the one that is most consistently present in all seasons. Being endosymbiotic commensals of several marine holobionts, such as the jellyfishes Aurelia aurita, Cotylorhiza tuberculata, and Pelagia noctiluca (Cortés-Lara et al., 2015; Weiland-Bräuer et al., 2015), members of this family might thus represent a stable component of the C. viridis communities and play an important physiological role for the host health and survival. Further experiments should demonstrate the role of these organisms in the holobiont functioning.
Conclusion
In conclusion, here we demonstrate that C. viridis possesses a characteristic host-associated microbiome. These anemone-associated microbial communities show significant variations with the seasons, moving from an anaerobic community cycling essential nutrients in winter to a community dominated by anaerobic heterotrophs, more specialized in fermenting proteins and complex polysaccharides, in summer. We hypothesize that seasonal change in community composition may reflect the corresponding seasonal changes in the host physiology. Indeed, in the phylum Cnidaria, the release of particulate and dissolved organic matter (including carbon and nitrogen) in the form of mucus is significantly higher in summer (Kurihara et al., 2018). During the summer, Cnidaria typically produce and release more mucus, which can be used as a food source by holobiont microorganisms, thus supporting the transition to a more heterotrophic fermentative microbial community (Wright et al., 2019). Supporting our hypothesis, analogous shifts in the associated microbial community have been shown for Astrangia Poculata in the winter to summer transition, when the emergence from the host quiescence makes available to the associated microbiome host substrates supporting the heterotrophic growth (Sharp et al., 2017). A similar adaptive microbiome response to seasonal changes has been observed also for Isopora palifera (Chen et al., 2011). According to the authors, during the winter - when available nitrogen is limited - the corals enrich nitrogen fixing microorganisms in the associated communities. These microbes then decrease in the summer, when exogenous nitrogen sources become available. Although our findings led us to hypothesize an important role of the C. viridis microbiome for host biology, and vice versa, more evidences in this direction to prove it conclusively are still needed. For instance, the functional relevance of the observed seasonal changes for host physiology and health must be dissected. The study of the C. viridis microbes relationship will certainly benefit from further extension to other geographical sites and from matching the patterns of phylogenetic changes with the corresponding functional variations in the holobiont.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA (accession: PRJNA674749).
Author Contributions
GP and MC conceived and designed the study and wrote the manuscript. GP performed the lab work, including DNA extraction and 16S rRNA gene amplification, and library preparation for sequencing, with the help of MM and FD’A. GP and SR analyzed the data. EB, GML, ST, and PB reviewed and edited the manuscript. All authors read and approved the final version.
Funding
This work was supported by the “Controlling Microbiomes Circulations for Better Food Systems” (CIRCLES) project, funded by the European Union’s Horizon 2020 Research and Innovation Program under grant agreement no. 818290. This publication reflects only the author’s view and the Agency is not responsible for any use that may be made of the information it contains.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study represents partial fulfillment of the requirements for the Ph.D. thesis of GP at the Ph.D. course of Innovative Technologies and Sustainable Use of Mediterranean Sea Fishery and Biological Resources (FishMed – University of Bologna, Italy). We would like to thank Associazione Paguro (Ravenna, Italy) and Dive Planet (Rimini, Italy) for their primary contribution to the sample collection and for the pictures of “Il Paguro.” Finally, we would also like to acknowledge Dr. Pierluigi Penna for his help with the measurement of environmental parameters.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.627585/full#supplementary-material
Footnotes
- ^ https://www.r-project.org/
- ^ https://cran.r-project.org/web/packages/STAT/index.html
- ^ https://cran.r-project.org/web/packages/vegan/index.html
- ^ https://cran.r-project.org/web/packages/VennDiagram/index.html
- ^ http://www.cytoscape.org/
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Apprill, A. (2017). Marine animal microbiomes: toward understanding host–microbiome interactions in a changing ocean. Front. Mar. Sci. 4:222. doi: 10.3389/fmars.2017.00222
Berg, G., Rybakova, D., Fischer, D., Cernava, T., Vergès, M. C., and Charles, T. (2020). Correction to: microbiome definition re-visited: old concepts and new challenges. Microbiome 8:119. doi: 10.1186/s40168-020-00905-x
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brown, T., Otero, C., Grajales, A., Rodriguez, E., and Rodriguez-Lanetty, M. (2017). Worldwide exploration of the microbiome harbored by the cnidarian model, Exaiptasia pallida (Agassiz in Verrill, 1864) indicates a lack of bacterial association specificity at a lower taxonomic rank. PeerJ 5:e3235. doi: 10.7717/peerj.3235
Cai, L., Zhou, G., Tong, H., Tian, R. M., Zhang, W., Ding, W., et al. (2018). Season structures prokaryotic partners but not algal symbionts in subtropical hard corals. Appl. Microbiol. Biotechnol. 102, 4963–4973. doi: 10.1007/s00253-018-8909-5
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, C. P., Tseng, C. H., Chen, C. A., and Tang, S. L. (2011). The dynamics of microbial partnerships in the coral Isopora palifera. ISME J. 5, 728–740. doi: 10.1038/ismej.2010.151
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O’Connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. doi: 10.1038/nature11319
Clum, A., Nolan, M., Lang, E., Glavina Del Rio, T., Tice, H., Copeland, A., et al. (2009). Complete genome sequence of Acidimicrobium ferrooxidans type strain (ICP). Stand. Genomic Sci. 1, 38–45. doi: 10.4056/sigs.1463
Cortés-Lara, S., Urdiain, M., Mora-Ruiz, M., Prieto, L., and Rosselló-Móra, R. (2015). Prokaryotic microbiota in the digestive cavity of the jellyfish Cotylorhiza tuberculata. Syst. Appl. Microbiol. 38, 494–500. doi: 10.1016/j.syapm.2015.07.001
Culhane, A. C., Thioulouse, J., Perrière, G., and Higgins, D. G. (2005). MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics 21, 2789–2790. doi: 10.1093/bioinformatics/bti394
D’Amico, F., Biagi, E., Rampelli, S., Fiori, J., Zama, D., Soverini, M., et al. (2019). Enteral nutrition in pediatric patients undergoing hematopoietic SCT promotes the recovery of gut microbiome homeostasis. Nutrients 11:2958. doi: 10.3390/nu11122958
Di Camillo, C. G., Luna, G. M., Bo, M., Giordano, G., Corinaldesi, C., and Bavestrello, G. (2012). Biodiversity of prokaryotic communities associated with the ectoderm of Ectopleura crocea (Cnidaria, Hydrozoa). PLoS One 7:e39926. doi: 10.1371/journal.pone.0039926
Faith, D. P. (1992). Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. doi: 10.1016/0006-3207(92)91201-3
Ferreira, J. G., Andersen, J. H., Borja, A., Bricker, S. B., Camp, J., Da Silva, M. C., et al. (2011). Overview of eutrophication indicators to assess environmental status within the European marine strategy framework directive. Estuar. Coast. Shelf Sci. 93, 117–131. doi: 10.1016/j.ecss.2011.03.014
Gerhardt, A. (2002). Bioindicator species and their use in biomonitoring. Environ. Monit. 1, 77–123.
Har, J. Y., Helbig, T., Lim, J. H., Fernando, S. C., Penn, K., Reitzel, A. M., et al. (2015). Microbial diversity and activity in the Nematostella vectensis holobiont: insights from 16S rRNA gene sequencing, isolate genomes, and a pilot-scale survey of gene expression. Front. Microbiol. 6:818. doi: 10.3389/fmicb.2015.00818
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., Herrera, C., and Ainsworth, T. D. (2018). Rethinking the coral microbiome: simplicity exists within a diverse microbial biosphere. mBio 9:e00812-18. doi: 10.1128/mBio.00812-18
Hill, J., and White, N. (2008). Biology and Sensitivity Key Information Sub-programme. The Marine Life Information Network (MarLIN). Available at: https://www.marlin.ac.uk (accessed November 9, 2020).
Ignatiades, L. (2005). Scaling the trophic status of the Aegean Sea, eastern Mediterranean. J. Sea Res. 54, 51–57. doi: 10.1016/j.seares.2005.02.010
Kuguru, B. L., Mgaya, Y. D., Öhman, M. C., and Wagner, G. M. (2004). The reef environment and competitive success in the Corallimorpharia. Mar. Biol. 145, 875–884. doi: 10.1007/s00227-004-1376-9
Kurihara, H., Ikeda, N., and Umezawa, Y. (2018). Diurnal and seasonal variation of particle and dissolved organic matter release by the coral Acropora tenuis. PeerJ 6:e5728. doi: 10.7717/peerj.5728
Lee, M. D., Kling, J. D., Araya, R., and Ceh, J. (2018). Jellyfish life stages shape associated microbial communities, while a core microbiome is maintained across all. Front. Microbiol. 9:1534. doi: 10.3389/fmicb.2018.01534
Lin, M. F., Chou, W. H., Kitahara, M. V., Chen, C. L., Miller, D. J., and Forêt, S. (2016). Corallimorpharians are not “naked corals”: insights into relationships between Scleractinia and Corallimorpharia from phylogenomic analyses. PeerJ 4:e2463. doi: 10.7717/peerj.2463
Littman, R. A., Willis, B. L., Pfeffer, C., and Bourne, D. G. (2009). Diversities of coral-associated bacteria differ with location, but not species, for three acroporid corals on the great barrier reef. FEMS Microbiol. Ecol. 68, 152–163. doi: 10.1111/j.1574-6941.2009.00666.x
Liu, Y. C., Huang, R. M., Bao, J., Wu, K. Y., Wu, H. Y., Gao, X. Y., et al. (2018). The unexpected diversity of microbial communities associated with black corals revealed by high-throughput Illumina sequencing. FEMS Microbiol. Lett. 365:10.1093/femsle/fny167. doi: 10.1093/femsle/fny167
Marzinelli, E. M., Qiu, Z., Dafforn, K. A., Johnston, E. L., Steinberg, P. D., and Mayer-Pinto, M. (2018). Coastal urbanisation affects microbial communities on a dominant marine holobiont. NPJ Biofilms Microbiomes 4:1. doi: 10.1038/s41522-017-0044-z
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G., and Neufeld, J. D. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinform. 13:31. doi: 10.1186/1471-2105-13-31
Murray, A. E., Rack, F. R., Zook, R., Williams, M. J., Higham, M. L., Broe, M., et al. (2016). Microbiome composition and diversity of the ice-dwelling sea anemone. Edwardsiella andrillae. Integr. Comp. Biol. 56, 542–555. doi: 10.1093/icb/icw095
O’Brien, P. A., Webster, N. S., Miller, D. J., and Bourne, D. G. (2019). Host-microbe coevolution: applying evidence from model systems to complex marine invertebrate holobionts. mBio 10, e02241–18.
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6:46. doi: 10.1186/s40168-018-0428-1
Pollock, F. J., McMinds, R., Smith, S., Bourne, D. G., Willis, B. L., Medina, M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9:4921. doi: 10.1038/s41467-018-07275-x
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R., and Macián, M. C. (2014). “The Family Rhodobacteraceae,” in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, ed. E. Rosenberg (Berlin: Springer Berlin Heidelberg), 439–512. doi: 10.1007/978-3-642-30197-1_377
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rausch, P., Rühlemann, M., Hermes, B. M., Doms, S., Dagan, T., Dierking, K., et al. (2019). Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 7:133. doi: 10.1186/s40168-019-0743-1
Rocha, J., Coelho, F. J., Peixe, L., Gomes, N. C., and Calado, R. (2014). Optimization of preservation and processing of sea anemones for microbial community analysis using molecular tools. Sci. Rep. 4:6986. doi: 10.1038/srep06986
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Sharp, K. H., Pratte, Z. A., Kerwin, A. H., Rotjan, R. D., and Stewart, F. J. (2017). Season, but not symbiont state, drives microbiome structure in the temperate coral Astrangia poculata. Microbiome 5:120. doi: 10.1186/s40168-017-0329-8
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., and Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. doi: 10.1093/bioinformatics/btq675
Stabili, L., Parisi, M. G., Parrinello, D., and Cammarata, M. (2018). Cnidarian interaction with microbial communities: from aid to animal’s health to rejection responses. Mar. Drugs 16:296. doi: 10.3390/md16090296
Tinta, T., Kogovšek, T., Klun, K., Malej, A., Herndl, G. J., and Turk, V. (2019). Jellyfish-associated microbiome in the marine environment: exploring its biotechnological potential. Mar. Drugs 17:94. doi: 10.3390/md17020094
Turroni, S., Fiori, J., Rampelli, S., Schnorr, S. L., Consolandi, C., Barone, M., et al. (2016). Fecal metabolome of the Hadza hunter-gatherers: a host-microbiome integrative view. Sci. Rep. 6:32826. doi: 10.1038/srep32826
Weiland-Bräuer, N., Neulinger, S. C., Pinnow, N., Künzel, S., Baines, J. F., and Schmitz, R. A. (2015). Composition of bacterial communities associated with aurelia aurita changes with compartment, life stage, and population. Appl. Environ. Microbiol. 81, 6038–6052. doi: 10.1128/AEM.01601-15
Wilkins, L., Leray, M., O’Dea, A., Yuen, B., Peixoto, R. S., Pereira, T. J., et al. (2019). Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 17:e3000533. doi: 10.1371/journal.pbio.3000533
Wright, R. M., Strader, M. E., Genuise, H. M., and Matz, M. (2019). Effects of thermal stress on amount, composition, and antibacterial properties of coral mucus. PeerJ 7:6849. doi: 10.7717/peerj.6849
Zhang, B., Xu, X., and Zhu, L. (2017). Structure and function of the microbial consortia of activated sludge in typical municipal wastewater treatment plants in winter. Sci. Rep. 7:17930. doi: 10.1038/s41598-017-17743-x
Zhang, B., Yu, Q., Yan, G., Zhu, H., Xu, X. Y., and Zhu, L. (2018). Seasonal bacterial community succession in four typical wastewater treatment plants: correlations between core microbes and process performance. Sci. Rep. 8:4566. doi: 10.1038/s41598-018-22683-1
Keywords: Cnidaria, Corallimorpharia, marine holobionts, microbiome, seasonality, 16S rRNA sequencing
Citation: Palladino G, Biagi E, Rampelli S, Musella M, D’Amico F, Turroni S, Brigidi P, Luna GM and Candela M (2021) Seasonal Changes in Microbial Communities Associated With the Jewel Anemone Corynactis viridis. Front. Mar. Sci. 8:627585. doi: 10.3389/fmars.2021.627585
Received: 09 November 2020; Accepted: 13 January 2021;
Published: 02 February 2021.
Edited by:
Russell T. Hill, University of Maryland, Baltimore County, United StatesReviewed by:
Angela Cuttitta, National Research Council (CNR), ItalyLuigi Vezzulli, University of Genoa, Italy
Copyright © 2021 Palladino, Biagi, Rampelli, Musella, D’Amico, Turroni, Brigidi, Luna and Candela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Candela, bWFyY28uY2FuZGVsYUB1bmliby5pdA==